

Abstract
Gut microbial β-glucuronidases have the ability to deconjugate glucuronides of some drugs, thus have been considered as an important drug target to alleviate the drug metabolites-induced gastrointestinal toxicity. In this study, thiazolidin-2-cyanamide derivatives containing 5-phenyl-2-furan moiety (1–13) were evaluated for inhibitory activity against Escherichia coli β-glucuronidase (EcGUS). All of them showed more potent inhibition than a commonly used positive control, d-saccharic acid 1,4-lactone, with the IC50 values ranging from 1.2 µM to 23.1 µM. Inhibition kinetics studies indicated that compound 1–3 were competitive type inhibitors for EcGUS. Molecular docking studies were performed and predicted the potential molecular determinants for their potent inhibitory effects towards EcGUS. Structure–inhibitory activity relationship study revealed that chloro substitution on the phenyl moiety was essential for EcGUS inhibition, which would help researchers to design and develop more effective thiazolidin-2-cyanamide type inhibitors against EcGUS.
Keywords: Structure–inhibitory activity relationships, thiazolidin-2-cyanamide derivatives, Escherichia coliβ-glucuronidase, β-glucuronidase inhibitors, CPT-11-induced toxicity
1. Introduction
Hepatic glucuronidation is an important process of drug detoxication and a major pathway of phase II xenobiotic biotransformation in the human body1–3. However, many drugs can be excreted into the intestinal lumen after they are glucuronidated and usually can be reactivated by gut microbial β-glucuronidases. Unfortunately, sometimes the reactivation can cause severe gastrointestinal damage, for example, hydrolysis of metabolites from irinotecan (CPT-11) or carboxylic acid-containing non-steroidal anti-inflammatory drugs (NSAIDs) lead to severe late diarrhoea in patients receiving therapy4–7.
Interestingly, recent studies demonstrated that gut microbial β-glucuronidase inhibitors, such as Escherichia coli β-glucuronidase (EcGUS) inhibitors, could significantly attenuate gastrointestinal toxicity caused by CPT-11 and NSAIDs8,9. With the deepening of research on gut microbial β-glucuronidase inhibitors, more and more natural and synthetic EcGUS inhibitors have been reported10. Previous studies reported many synthetic EcGUS inhibitors, for example, an antidepressant, amoxapine, was identified as a potent EcGUS inhibitor11, Salar et al.12 evaluated the inhibitory effects of twelve thiadiazole derivatives towards EcGUS with IC50 values ranging from 3.10 µM to 35.40 µM, Taha et al.13 reported that oxadiazole coupled-thiadiazole derivatives as potent EcGUS inhibitors and the most active inhibitor with an IC50 value of 0.96 µM. Interestingly, all these types of compounds contain an extremely similar moiety, a phenyl group substituted with a heterocycle. Molecular docking studies further demonstrated that both the phenyl and heterocyclic groups interacted with the corresponding pocket residues via π–π stacking, and the heterocyclic nitrogen, sulphur and/or oxygen increased hydrogen bonding capability of these compounds for pocket binding14–16. The structure of 5-phenyl-2-furan is very similar to the above mentioned structural units. Additionally, our previous studies have reported that the derivatives of 5-phenyl-2-furan showed broad-spectrum bioactivities, such as antibacterial, antitumor, and anti-inflammatory activities17–20. In 2018, we synthesised a series of thiazolidin-2-cyanamide derivatives, which also contained 5-phenyl-2-furan moiety and could reduce the disease symptoms of Xanthomonas oryzae pv. oryzae on the rice cultivar IR2421.
Therefore, in this study, 13 thiazolidin-2-cyanamide derivatives containing 5-phenyl-2-furan moiety were selected and subjected to evaluate their inhibitory effects on EcGUS. p-Nitrophenyl-β-d-glucuronide acid (PNPG) was used as the probe substrate for EcGUS. Then the inhibitory behaviours of these compounds against EcGUS were characterised and their structure–inhibitory activity relationships were explored. Finally, molecular docking studies were performed to predict the molecular determinants of thiazolidin-2-cyanamide derivatives against EcGUS.
2. Materials and methods
2.1. Materials
Thirteen thiazolidin-2-cyanamide derivatives containing 5-phenyl-2-furan moiety were provided by Prof. Zi-Ning Cui from South China Agricultural University (Guangzhou, China). p-Nitrophenyl-β-d-glucuronide acid (PNPG), d-saccharic acid-1,4-lactone (DSL), and dimethyl sulfoxide (DMSO) were supplied by Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s phosphate-buffered saline (PBS) was provided by Life Technologies (Carlsbad, CA, USA). Imidazole, kanamycin, and isopropyl β-d-1-thiogalactopyranoside (IPTG) were supplied by Biosharp (Hefei, China). Recombinant E. coli BL21 (DE3) harbouring pET28a-EcGUS was generously provided by Prof. Ru Yan from the University of Macau (Macau, China). Deionised water was purified by a Milli-Q purification system (Millipore, Bedford, MA, USA). Purities were all >98%.
2.2. General synthetic procedure
The synthetic route of title compounds was shown in Figure 1. The key intermediate I was synthesised from substituted aniline by Meerwein arylation reaction according to the reported procedure22,23. A mixture of 5-substituted phenyl-2-furancarboxylic acid I and thionyl chloride was refluxed in anhydrous toluene at 80 °C for 3 h to afford the 5-phenyl-2-furancarbonyl chloride, which was added into 2-cyanoiminoradical-1, 3-thiazolidine in refluxing anhydrous acetonitrile in presence of an equivalent amount of potassium carbonate at 75 °C for 3–6 h to afford the title compounds in moderate or good yields (for the details, see Supplementary Materials). The structures were also further confirmed by X-ray single-crystal analysis and a perspective view of the compound 6 (CCDC No.: 1565820) was shown in Figure 2.
Figure 1.
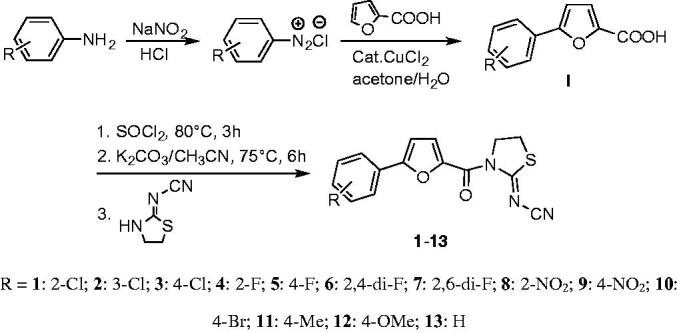
General synthetic procedure for title compounds 1–13. R = 1: 2-Cl; 2: 3-Cl; 3: 4-Cl; 4: 2-F; 5: 4-F; 6: 2,4-di-F; 7: 2,6-di-F; 8: 2-NO2; 9: 4-NO2; 10: 4-Br; 11: 4-Me; 12: 4-OMe; 13: H.
Figure 2.

Single crystal structure of compound 6.
2.3. Enzyme preparation
EcGUS was prepared according to our previous report24. The recombinant strain were incubated in 200 mL of LB broth (Tryptone, 10 g/L; yeast extract, 5 g/L; NaCl, 10 g/L; pH 7.0) containing 1% kanamycin at 220 rpm and 37 °C until OD600 reached 0.5–0.6. Afterward, IPTG (final concentration, 0.5 mM) was added and the culture was incubated at 220 rpm and 16 °C for 16 h to induce the expression of β-glucuronidase. The cells were collected by centrifugation and suspended in PBS buffer (pH 7.4), and then applied to extract the enzyme by sonication. Finally, pure EcGUS was obtained from the cell-free extracts through a Ni-NTA column (EMD Millipore Corp., MA, USA). Protein concentration was determined using the BCA Protein Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s instruction, and its purity was determined by SDS-PAGE.
2.4. Enzyme inhibition assays
Thirteen thiazolidin-2-cyanamide derivatives were subjected to screening for potent EcGUS inhibitor. The inhibitory effects of these compounds were determined by measuring the PNP formation generated from PNPG by EcGUS. Briefly, the assays were conducted in 96-well flat-bottomed tissue culture plates (Nunc, Denmark) with a total volume of 100 µL which consisted of 10 µL EcGUS (final concentration, 2 µg/mL), 70 µL PBS buffer (pH 7.4), 10 µL test compound or DSL (final concentration, 10 µM) and 10 µL PNPG (final concentration, 250 µM). DSL was used as a positive control, and 1%DMSO was used as blank control. All reactions were performed in triplicate and the produced PNP was monitored by measuring absorbance at 405 nm in a plate reader (SpectraMax Plus384, Molecular Devices, CA, USA) after incubating at 37 °C for 30 min. The PNP concentration was determined from a standard curve (0, 10, 20, 40, 60, 80, 100, 120 µM PNP) in PBS buffer (pH 7.4). Relative activity was defined as a percent of PNP formation in the presence of test compound within 30 min compared with the blank control group25.
2.5. Inhibition kinetic analysis
IC50 values of the 13 thiazolidin-2-cyanamide derivatives against EcGUS were investigated using similar assays as described in Section 2.4. The reaction conditions are as follows: 10 µL pure enzyme (final concentration, 2 µg/mL), 70 µL PBS buffer (pH 7.4), 10 µL test compound or DSL (final concentration, 0.001–5000 µM) and 10 µL PNPG (final concentration, 250 µM) incubating at 37 °C for 30 min. The IC50 values were calculated by plotting the concentration–response curve using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA).
To further characterise the inhibition behaviour of discovered inhibitors, multiple concentrations of compounds (0.5–12 µM) or DSL (50–100 µM) and PNPG (0.2, 0.3, 0.5, and 1.0 mM) were applied to determine the corresponding reaction rates. The inhibition type was evaluated through determining the intersection point in Lineweaver–Burk plots, and the Ki values were calculated by nonlinear regression of different inhibition types using the GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA) 25–27.
2.6. Molecular docking studies
Molecular docking studies were conducted to explore the molecular determinants of discovered inhibitors against EcGUS using MOE (Version 2014. 09, Chemical Computing Group Inc., Montreal, Canada). Docking studies were performed according to our previous report25. Briefly, the X-ray crystal structure of EcGUS (PDB ID: 3K4D) was retrieved from the Protein Data Bank and prepared using MOE. Each inhibitor was docked into the active site of EcGUS using the Triangular Matching docking method and 30 conformations of each ligand–protein complex were generated according to the docking score. Docking poses were visually inspected to analyse the interactions of inhibitors with binding pocket residues.
2.7. Statistical analysis
All experiments were carried out in triplicate and repeated at least two times. All data were expressed as mean ± standard deviation (SD). The IC50 values were defined as the concentration of inhibitor that is required for 50% inhibition and evaluated by nonlinear regression using GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA). The inhibition type was determined according to the intersection location of Lineweaver–Burk plot: competitive inhibition, the intersection at the y-axis; uncompetitive inhibition, parallel lines; non-competitive inhibition, the intersection at the x-axis; mixed inhibition, the intersection in first or second quadrant28.
3. Results
3.1. Screening of potent EcGUS inhibitor
The inhibitory effects of 13 thiazolidin-2-cyanamide derivatives were determined and presented in Table 1. Among 13 compounds (compound 1–13) tested, all of them showed more potent inhibitory effects against EcGUS than DSL at 10 µM, a known positive control. Compound 2 showed the highest inhibition towards EcGUS with the inhibition rate of 95.2 ± 0.3%, followed by compounds 3 and 1, whose inhibition rates were 80.2 ± 1.8% and 77.2 ± 0.6%, respectively (Table 1). The inhibition rates of compounds 4–13 ranged from 31.4% to 68.7%, all larger than that of DSL (11.6 ± 0.6%; Table 1).
Table 1.
Chemical structures of 13 thiazolidin-2-cyanamide derivatives and the inhibitory activity against EcGUS-mediated PNPG hydrolysis.
| No | R | Molecular weight (Da) | Inhibition rate at 10 μM | IC50 (μM) | Ki (μM) | Inbibition type |
|---|---|---|---|---|---|---|
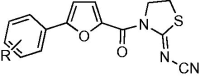 | ||||||
| 1 | 2-Cl | 331.77 | 77.2 ± 0.6% | 5.7 ± 0.6 | 3.5 ± 0.3 | Competitive |
| 2 | 3-Cl | 331.77 | 95.2 ± 0.3% | 1.2 ± 0.2 | 0.7 ± 0.1 | Competitive |
| 3 | 4-Cl | 331.77 | 80.2 ± 1.8% | 4.7 ± 0.6 | 2.0 ± 0.4 | Competitive |
| 4 | 2-F | 315.32 | 61.6 ± 0.6% | 11.9 ± 1.5 | ND | ND |
| 5 | 4-F | 315.32 | 66.5 ± 1.2% | 7.3 ± 0.7 | ND | ND |
| 6 | 2,4-di-F | 333.31 | 43.9 ± 0.7% | 23.1 ± 1.5 | ND | ND |
| 7 | 2,6-di-F | 333.31 | 64.1 ± 1.2% | 9.3 ± 1.1 | ND | ND |
| 8 | 2-NO2 | 342.33 | 31.4 ± 1.8% | 6.1 ± 0.2 | ND | ND |
| 9 | 4-NO2 | 342.33 | 40.3 ± 3.2% | 22.2 ± 2.5 | ND | ND |
| 10 | 4-Br | 376.23 | 68.7 ± 1.8% | 10.8 ± 0.2 | ND | ND |
| 11 | 4-Me | 311.36 | 33.5 ± 2.5% | 22.3 ± 2.6 | ND | ND |
| 12 | 4-OMe | 327.36 | 49.6 ± 0.6% | 18.5 ± 2.1 | ND | ND |
| 13 | H | 297.33 | 61.8 ± 1.3% | 10.7 ± 0.5 | ND | ND |
| d-Saccharic acid 1,4-lactone | 210.14 | 11.6 ± 0.6% | 67.3 ± 0.6 | 30.9 ± 2.2 | ND | |
ND: not detect.
3.2. IC50 values of thiazolidin-2-cyanamide derivatives against EcGUS
The effects of the concentrations of the inhibitors on PNPG-hydrolyzing activity of EcGUS were presented in the dose-dependent inhibition curves. The curves confirmed all 13 thiazolidin-2-cyanamide derivatives displayed the more potent inhibitory effects on EcGUS with IC50 values ranging from 1.2 µM to 23.1 µM than DSL (IC50 = 67.3 ± 0.6 µM; Figure 3). Among them, compounds 2 and 6 displayed the most and worst potent inhibition against EcGUS with IC50 values evaluated as 1.2 ± 0.2 µM and 23.1 ± 1.5 µM, respectively. The IC50 values of compounds 1, 3, 5, 7, and 8 were less than 10 µM, suggesting that they are potent EcGUS inhibitors.
Figure 3.

The dose-dependent inhibition curves of inhibitors on PNPG-hydrolyzing activity of EcGUS. (A) Compounds 1–8; (B) compounds 9–13, and DSL. All data were expressed as mean ± standard deviation of triplicate reactions.
3.3. Structure–inhibitory activity relationships of thiazolidin-2-cyanamide derivatives against EcGUS
As shown in Table 1, all thiazolidin-2-cyanamide derivatives, compounds 1–13, have different substituted phenyl moiety and demonstrated varied inhibitory potency against EcGUS. For example, compounds 2 with a meta-chloro-substituted phenyl moiety (IC50 = 1.2 ± 0.2 µM) showed more potent inhibition against EcGUS than compound 1 and 3 (ortho-chloro-substitution or para-chloro-substitution on the phenyl moiety, IC50 = 5.7 ± 0.6 µM and 4.7 ± 0.6 µM), and compound 13 (no substitution on the phenyl moiety, IC50 = 10.7 ± 0.5 µM), suggesting that the chloro substitution on the phenyl moiety was essential for the potent inhibition of EcGUS and meta-chloro-substitution was more beneficial than ortho-chloro-substitution or para-chloro-substitution for EcGUS inhibition.
It also can be seen from Table 1 that compound 3 with a para-chloro-substituted phenyl moiety (IC50 = 4.7 ± 0.6 µM) ranked the most potent inhibitor among compounds with a para-substituted phenyl moiety, including compound 5 (IC50 = 7.3 ± 0.7 µM), 9 (IC50 = 22.2 ± 2.5 µM), 10 (IC50 = 10.8 ± 0.2 µM), 11 (IC50 = 22.3 ± 2.6 µM), 12 (IC50 = 18.5 ± 2.1 µM), and 13 (IC50 = 10.7 ± 0.5 µM), indicating that effect of substitution group on EcGUS inhibition are as follows: Cl > F > Br ≈ H > OMe > NO2 ≈ Me. In other words, chloro and fluorine substitutions at the para position of the phenyl moiety are beneficial for EcGUS inhibition, while methoxy, methyl and nitro group substitutions are unbeneficial.
Similarly, after comparing the difference in structure and inhibitory activity of compounds 1, 4, 8, and 13, it could be easily found that chloro substitution at the ortho position of the phenyl moiety is beneficial for EcGUS inhibition, while fluorine substitution is unfavourable. Interestingly, the IC50 values of compound 5 (para-fluorine substitution on the phenyl moiety), 7 (2,6-difluorine substitution on the phenyl moiety), 4 (ortho-fluorine substitution on the phenyl moiety), and 6 (2,4-difluorine substitution on the phenyl moiety) were determined as 7.3 ± 0.7 µM, 9.3 ± 1.1 µM, 11.9 ± 1.5 µM and 23.1 ± 1.1 µM, respectively, demonstrating that para-fluorine substitution is more beneficial than ortho-fluorine substitution on the phenyl moiety for EcGUS inhibition, and fluorine substitution at the 2-position of the phenyl moiety of compound 5 is unfavourable for EcGUS inhibition, while fluorine substitution at the 6-position of the phenyl moiety of compound 4 is beneficial for EcGUS inhibition.
3.4. Inhibition behaviours of thiazolidin-2-cyanamide derivatives against EcGUS
The inhibition behaviours of three potent EcGUS inhibitors, compounds 1, 2, 3, and DSL were further investigated. As shown in Figure 4, Lineweaver–Burk plots indicated that compounds 1, 2, 3 were competitive inhibitors for EcGUS as judged from the intersection point at the y positive axis, implying that the binding areas of these compounds on EcGUS may be overlapped with the binding area of the substrate, PNPG, while DSL was mixed inhibitors for EcGUS as judged from the intersection point at the second quadrant. Besides, the Ki values of compounds 1, 2, 3, and DSL also were determined as 3.5 µM, 0.7 µM, 2.0 µM, and 30.9 µM, respectively (Table 1). In summary, the results demonstrated that compounds 1, 2 and 3 can be as potent competitive inhibitors against EcGUS with Ki values less than 3.5 µM.
Figure 4.

The Lineweaver–Burk plots of (A) compound 1, (B) compound 2, and (C) compound 3 against EcGUS. All data were expressed as mean ± standard deviation of triplicate reactions.
3.5. Molecular docking simulations
To predict the molecular determinants of thiazolidin-2-cyanamide derivatives 1–3 in inhibiting EcGUS, molecular docking studies were performed using MOE. As shown in Figure 5, PNPG, compound 1, 2, and 3 could be well docked into the active site of EcGUS. PNPG formed hydrogen bond interactions with His330, Asn412, Tyr472, Glu505 and Arg562 (Figure 5(A)), while hydrogen bonds for compound 1 were found with residues His330, Tyr472, Asn566 and Lys568, and the binding interactions were formed with the phenolic or thiazolidine groups (Figure 5(B)). Ligand interactions between compounds 2 and 3 with EcGUS were nearly the same to each other, and also very similar to that of compound 1, except that one more hydrogen bond interaction was observed in compounds 2 and 3. In detail, the binding area of compound 2 was highly overlapped with that of PNPG (Figure 5(D)), and it developed five hydrogen bonds with catalytic residues His330, Tyr472, Arg562, Asn566 and Lys568 (Figure 5(C,F)), which may be associated with the more potent inhibitory potential than compund 1.
Figure 5.

Ligand interactions of (A) PNPG, (B) compound 1, (C) compound 2 and (D) compound 3 with EcGUS. (E) Stereo diagram of PNPG combined with compound 2 in the active site of EcGUS. (F) Binding mode of compound 2 in the active site of EcGUS.
4. Discussion
Gut bacterial β-glucuronidases have been proved to play important roles in CPT-11-induced intestinal toxicity, and EcGUS inhibitors have shown great potential in alleviating the toxicity. Previously identified EcGUS inhibitors usually contain heterocyclic moiety and compounds with a furan ring frequently show various bioactivities. Therefore, we evaluated the inhibitory activities of our previous synthesised thiazolidin-2-cyanamide derivatives containing 5-phenyl-2-furan moiety towards EcGUS and revealed that 13 synthetic thiazolidin-2-cyanamide derivatives were potent EcGUS inhibitors with IC50 values ranging from 1.2 µM to 23.1 µM, which were much more potent than a positive control, d-saccharic acid 1,4-lactone. Structure–activity relationship analysis suggested that chloro and fluorine substitutions at the para position of the phenyl moiety in thiazolidin-2-cyanamide derivatives are beneficial for EcGUS inhibition, while methoxy, methyl and nitro group substitutions are unbeneficial. Inhibition behaviour analysis indicated that compounds 1, 2, and 3 were identified as competitive EcGUS inhibitors with Ki values less than 3.5 µM and the potential molecular determinants were predicted by molecule docking studies.
Recently, 49 thiadiazole derivatives were synthesised and 38 of them were evaluated as potent bovine liver β-glucuronidase inhibitors with IC50 values ranging from 2.16 ± 0.01 µM to 58.06 ± 1.60 µM by Khan’s research group. Structure–activity relationship analysis also indicated that chloro substitution at the phenyl moiety in thiadiazole derivatives is beneficial for β-glucuronidase inhibition29–31. Since EcGUS shared high sequence similarity with bovine liver β-glucuronidase32 and our synthetic thiazolidin-2-cyanamides derivatives also exhibited high similarity in structure with thiadiazole derivatives, we predicted that thiazolidin-2-cyanamides derivatives may also show potent inhibition towards bovine liver β-glucuronidase and thiadiazole derivatives may be potential EcGUS inhibitors, which warrant further studies.
In the present study, we focus on investigating the effects of different substitution groups on the phenyl moiety of the thiadiazole-2-cyanamide derivatives on EcGUS inhibition and revealed that different substitution groups greatly affect the inhibitory potency towards EcGUS, and meta-chloro-substitution was most beneficial for EcGUS inhibition among all substitution groups tested. The effects of substitution on furan or thiazolidin moiety worth further studies.
When Wallace et al.32 first confirmed that selectively inhibition of bacterial β-glucuronidases could alleviate CPT-11-induced toxicity in 2010, they also identified four uncompetitive EcGUS inhibitors. Scutellarein and caffeic acid ethyl ester were found to function as potent competitive inhibitors for EcGUS28,33. Our previous study found that sanggenon C and kuwanon G exhibited mixed-type inhibition against EcGUS, while amoxapine demonstrated uncompetitive inhibition against EcGUS25. In this study, we characterised the inhibition behaviours of compounds 1, 2, and 3, and figured out that they were competitive EcGUS inhibitors. These findings demonstrated the complex interactions between EcGUS and the inhibitors, whose influence on the in vivo efficacy needs detailed analysis.
In conclusion, the present study identified 13 synthetic thiazolidin-2-cyanamide derivatives as potent EcGUS inhibitors and found that chloro and fluorine substitutions at the C4-position of the phenyl moiety of the furan are beneficial for EcGUS inhibition. Our findings are beneficial for developing more potent EcGUS inhibitors from thiazolidin-2-cyanamide derivatives and the in vivo efficacy of discovered EcGUS inhibitors also warrant further study.
Supplementary Material
Acknowledgements
The authors thank Prof. Yan Ru (University of Macau, Macau, China) for providing us the recombinant Escherichia coli BL21(DE3) for Escherichia coli β-glucuronidase preparation.
Funding Statement
This work was financially supported by the National Key Research and Development Program [2017YFE0103100], International Science and technology cooperation program in Guangdong [2020A0505100048] and the National 111 Project [No. D17012].
Ethical approval
This research did not include any human subjects and animal experiments.
Disclosure statement
The authors declare that there are no conflicts of interest.
References
- 1.Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J 2003;3:136–58. [DOI] [PubMed] [Google Scholar]
- 2.de Graaf M, Boven E, Scheeren HW, et al. Beta-glucuronidase-mediated drug release. Curr Pharm Des 2002;8:1391–403. [DOI] [PubMed] [Google Scholar]
- 3.Garcia C, Barriga A, Diaz JC, et al. Route of metabolization and detoxication of paralytic shellfish toxins in humans. Toxicon 2010;55:135–44. [DOI] [PubMed] [Google Scholar]
- 4.Cheng KW, Tseng CH, Tzeng CC, et al. Pharmacological inhibition of bacterial β-glucuronidase prevents irinotecan-induced diarrhea without impairing its antitumor efficacy in vivo. Pharmacol Res 2019;139:41–9. [DOI] [PubMed] [Google Scholar]
- 5.Alexander JL, Wilson ID, Teare J, et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol 2017;14:356–65. [DOI] [PubMed] [Google Scholar]
- 6.LoGuidice A, Wallace BD, Bendel L, et al. Pharmacologic targeting of bacterial β-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J Pharmacol Exp Ther 2012;341:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tobin P, Clarke S, Seale JP, et al. The in vitro metabolism of irinotecan (CPT-11) by carboxylesterase and β-glucuronidase in human colorectal tumours. Br J Clin Pharmacol 2006;62:122–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace BD, Roberts AB, Pollet RM, et al. Structure and inhibition of microbiome β-Glucuronidases essential to the alleviation of cancer drug toxicity. Chem Biol 2015;22:1238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saitta KS, Zhang C, Lee K, et al. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 2014;44:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paul A, Nosipho C, Nagaraju K, et al. Therapeutic signifificance of β-glucuronidase activity and its inhibitors: a review. Eur J Med Chem 2020;187:111921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong R, Liu T, Zhu X, et al. Old drug new use-amoxapine and its metabolites as potent bacterial β-glucuronidase inhibitors for alleviating cancer drug toxicity. Clin Cancer Res 2014;20:3521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salar U, Khan KM, Syed S, et al. Synthesis, in vitro β-glucuronidase inhibitory activity and in silico studies of novel (E)-4-Aryl-2-(2-(pyren-1-ylmethylene)hydrazinyl)thiazoles. Bioorg Chem 2017;70:199–209. [DOI] [PubMed] [Google Scholar]
- 13.Taha M, Imran S, Alomari M, et al. Synthesis of oxadiazole-coupled-thiadiazole derivatives as a potent β-glucuronidase inhibitors and their molecular docking study. Bioorg Med Chem 2019;27:3145–55. [DOI] [PubMed] [Google Scholar]
- 14.Taha M, Ismail NH, Imran S, et al. Synthesis of novel benzohydrazone-oxadiazole hybrids as β-glucuronidase inhibitors and molecular modeling studies. Bioorg Med Chem Lett 2015;23:7394–404. [DOI] [PubMed] [Google Scholar]
- 15.Taha M, Baharudin MS, Ismail NH, et al. Synthesis and in silico studies of novel sulfonamides having oxadiazole ring: as β-glucuronidase inhibitors. Bioorg Chem 2017;71:86–96. [DOI] [PubMed] [Google Scholar]
- 16.Zawawi NK, Taha M, Ahmat N, et al. Novel 2,5-disubtituted-1,3,4-oxadiazoles with benzimidazole backbone: a new class of β-glucuronidase inhibitors and in silico studies. Bioorg Med Chem 2015;23:3119–25. [DOI] [PubMed] [Google Scholar]
- 17.Rani M, Yusuf M, Khan SA, et al. Synthesis, studies and in-vitro antibacterial activity of N-substituted 5-(furan-2-yl)-phenyl pyrazolines. Arab J Chem 2015;8:174–80. [Google Scholar]
- 18.Huo JQ, Ma LY, Zhang Z, et al. Synthesis and biological activity of novel N-(3-furan-2-yl-1-phenyl-1H-pyrazol-5-yl) amides derivatives. Chin Chem Lett 2016;27:1547–50. [Google Scholar]
- 19.Tao H, Tian H, Jiang S, et al. Synthesis and biological evaluation of 1,3,4-thiadiazole derivatives as type III secretion system inhibitors against Xanthomonas oryzae. Pestic Biochem Physiol 2019;160:87–94. [DOI] [PubMed] [Google Scholar]
- 20.Cui ZN, Li XH, Nishida Y.. Synthesis and bioactivity of novel carvacrol and thymol derivatives containing 5-phenyl-2-furan. Lett Drug Des Discov 2014;11:877–85. [Google Scholar]
- 21.Xiang XW, Tao H, Jiang S, et al. Synthesis and bioactivity of thiazolidin-2-cyanamide derivatives against type III secretion system of Xanthomonas oryzae on rice. Pestic Biochem Physiol 2018;149:89–97. [DOI] [PubMed] [Google Scholar]
- 22.Cui ZN, Li Y, Ling Y, et al. New class of potent antitumor acylhydrazone derivatives containing furan. Eur J Med Chem 2010;45:5576–84. [DOI] [PubMed] [Google Scholar]
- 23.Jiang S, He M, Xiang XW, et al. Novel S-thiazol-2-yl-furan-2-carbothioate derivatives as potential T3SS inhibitors against Xanthomonas oryzae on rice. J Agric Food Chem 2019;67:11867–76. [DOI] [PubMed] [Google Scholar]
- 24.Wei B, Wang PP, Yan ZX, et al. Characteristics and molecular determinants of a highly selective and efficient glycyrrhizin-hydrolyzing β-glucuronidase from Staphylococcus pasteuri 3I10. Appl Microbiol Biotechnol 2018;102:9193–205. [DOI] [PubMed] [Google Scholar]
- 25.Wei B, Yang W, Yan ZX, et al. Prenylflavonoids sanggenon C and kuwanon G from mulberry (Morus alba L.) as potent broad-spectrum bacterial β-glucuronidase inhibitors: biological evaluation and molecular docking studies. J Funct Foods 2018;48:210–9. [Google Scholar]
- 26.Xin H, Qi XY, Wu JJ, et al. Assessment of the inhibition potential of Licochalcone A against human UDP-glucuronosyltransferases. Food Chem Toxicol 2016;90:112–22. [DOI] [PubMed] [Google Scholar]
- 27.Lv X, Wang XX, Hou J, et al. Comparison of the inhibitory effects of tolcapone and entacapone against human UDP-glucuronosyltransferases. Toxicol Appl Pharmacol 2016;301:42–9. [DOI] [PubMed] [Google Scholar]
- 28.Weng ZM, Wang P, Ge GB, et al. Structure-activity relationships of flavonoids as natural inhibitors against E. coli β-glucuronidase. Food Chem Toxicol 2017;109:975–83. [DOI] [PubMed] [Google Scholar]
- 29.Salar U, Taha M, Ismail NH, et al. Thiadiazole derivatives as new class of β-glucuronidase inhibitors. Bioorg Med Chem 2016;24:1909–18. [DOI] [PubMed] [Google Scholar]
- 30.Taha M, Almandil NB, Rashid U, et al. 2,5-Disubstituted thiadiazoles as potent β-glucuronidase inhibitors; synthesis, in vitro and in silico studies. Bioorg Chem 2019;91:103126. [DOI] [PubMed] [Google Scholar]
- 31.Khan KM, Karim A, Saied S, et al. Evaluation of the thiazole Schiff bases as β-glucuronidase inhibitors and their in silico studies. Mol Divers 2014;18:295–306. [DOI] [PubMed] [Google Scholar]
- 32.Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 2010;330:831–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li XN, Hua LX, Zhou TS, et al. Cinnamic acid derivatives: inhibitory activity against Escherichia coli β-glucuronidase and structure-activity relationships. J Enzym Inhib Med Chem 2020;35:1372–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.