

Abstract
Structure-based virtual screening (SBVS) relies on classical scoring functions that often fail to reliably discriminate binders from non-binders. In this work, we present a high-throughput protein-ligand complex MD simulations that uses the output from AutoDock Vina to improve docking results in distinguishing active from decoy ligands in DUD-E (directory of useful decoy, enhanced) dataset. MD trajectories are processed by evaluating ligand binding stability using RMSD (root-mean-square deviations). We select 56 protein targets (of 7 different protein classes) and 560 ligands (280 actives, 280 decoys) and show 22% improvement in ROC AUC (area under the curve, receiver operating characteristics curve), from an initial value of 0.68 (AutoDock Vina) to a final value of 0.83. MD simulation demonstrates a robust performance across all 7 different protein classes. In addition, some predicted ligand binding modes are moderately refined during MD simulations. These results systematically validate the reliability of physics-based approach to evaluate protein-ligand binding interactions.
Graphical Abstract
Introduction
Structure-based virtual screening (SBVS) is an important early-phase step in drug discovery workflow that is widely used in the development of new therapeutic agents.1, 2 Molecular docking is a method of choice for SBVS to predict binding mode and binding energy of a small molecule compound to the binding site of a target protein. The increasing availability of high-resolution three-dimensional structures of holo target proteins and rapid advances in computational techniques have contributed to many successful results of SBVS in the past two decades. Most recently, Lyu and colleagues ran ultra-large library docking and discovered new chemotypes for AmpC β-lactamase and D4 dopamine receptor.3 A few of the previous success cases include STX-0119 for lymphoma, novel histamine H4 receptor ligands, and novel Pim-1 kinase inhibitors.4–6 In practice, SBVS is attractive because it is capable of quickly screening large compound libraries. In addition, SBVS has been shown to reasonably reproduce experimental binding modes using standard docking programs: for example, AutoDock Vina with 78% success rate in self-docking (i.e., docking of a native ligand back to its receptor protein crystal structure).7 Despite this, a major limitation to docking methodology is still the scoring function that aims to approximate binding affinity and discriminate binders from non-binders.8
Over the past few years, numerous approaches have been developed to improve the scoring function in SBVS. The most notable ones are machine-learning-based methods that show significant improvements in screening for real binders from a given library of compounds. Recently, water and ligand stability were incorporated into machine-learning scoring functions that successfully boosted AutoDock Vina docking results.9 Enhanced performance in scoring function using machine-learning was persuasively reported by Wojcikowski and colleagues using DUD-E (database of useful decoys – enhanced) dataset.10, 11 More recently, Yasuo and Sekijima added interaction-energy-based learning to further improve scoring functions tested on the same DUD-E benchmark set.12 Another notable case was shown by Pereira and colleagues using deep neural network that achieved significant improvements on docking results.13 While various types of machine-learning methods have shown success cases in improving the scoring function of SBVS, this approach is still relatively new and actively growing.14 And a few problems persist in these machine-learning-based methods, including judicious choice to obtain a balance between model overfitting to a training set and keeping it generalizable.15
Physics-based molecular dynamics (MD) simulations is a powerful methodology in biophysical research that provides invaluable structural and dynamics information of protein-ligand interactions at the atomic level. D.E. Shaw research lab showed a long and unbiased MD simulation that captured the process of a small molecule recognizing and binding to a protein target.16 The binding site was correctly identified by the ligand and the binding mode was essentially identical to the experimentally determined crystal structure. The MD approach effectively includes entropy effects and the role of water molecules during the binding process, unlike machine-learning-based methods. MD has also been sparingly used before and is continually being used today to evaluate and improve SBVS docking results.17 We had previously shown that short equilibration of MD simulations could better filtered near-native pose prediction as compared to a docking scoring function for MDM2 and MDMX inhibitors.18 Analyzing the ligand stability, we showed that correctly predicted binding modes from docking results tend to be more stable than incorrectly bound ligands during MD simulations. Similarly, Cavalli and colleagues showed that short MD simulations were able to discriminate stable near-native poses from decoy poses of propidium on HuAChE (human acetylcholinetransferase).19 MD was also used by Park and colleagues to demonstrate the differences in binding site flexibility between CDK4 and CDK2 that explained selective binding of CDK4 inhibitors.20 Ogrizek and colleagues used MD to obtain various snapshots for docking that resulted in better discrimination between active from inactive compounds.21 Recently, Radom and colleagues showed that for protein-protein docking complexes, MD simulations successfully discriminated correct models from decoy models.22 They revealed that correct models were kinetically stable, whereas decoys were unstable and detached from each other during MD simulations. Nevertheless, relative to docking techniques, MD simulations are still more computationally intensive, challenging, and expensive. These limitations often discourage drug discovery researchers to actively incorporate MD simulations into their workflows. However, recent advances in computational power, decreasing cost, and user-friendly tools have alleviated most of the limitations associated with MD simulations.23 A few examples include the use of GPUs (graphics processing units) to accelerate MD simulations and web-based GUI (graphical user interface) tools to facilitate system set up.24–26
In this study, we present a high-throughput protein-ligand complex MD simulations method that integrates the functionality of CHARMM-GUI (http://www.charm-gui.org) web server to improve docking results in discriminating small molecule binders from non-binders.27, 28 Using AutoDock Vina, we obtain docked protein-ligand complex structures and process them using a python script that uses CHARMM-GUI to automate the production of MD simulation system and input files for each structure in a high-throughput manner.7 Post-processing of MD data is simply based on ligand-RMSD (calculating ligand-RMSD relative to the initial docking structure by aligning the protein structure but not the ligand) to assess ligand stability and distinguish small molecule binders from non-binders. We evaluate our method on 56 target proteins of 7 different protein classes selected from DUD-E dataset based on their crystal structure resolution.11 For each target, we randomly select 10 compounds, 5 of which are active and the other 5 are decoys. Our results show consistently better AUC ROC (area under the curve, receiver operating characteristics curve) compared to docking results. Overall, we achieve AUC of 0.83 compared to 0.68 (AutoDock Vina). In addition, we show that MD simulation can relax and slightly improve the overall binding mode predicted from AutoDock Vina, in some cases. These results indicate a promising high-throughput MD simulation protocol that can be integrated with docking from SBVS to improve its scoring function and binding mode results.
Methods
We conducted all-atom MD simulations in explicit solvents on 56 protein targets from DUD-E dataset.11 DUD-E dataset consists of 102 diverse protein targets and most of them have pharmacological precedence. This dataset has been frequently used as a benchmark to test proposed docking/scoring methodologies.10, 12, 13 In this study, we only included protein targets that have crystal structure resolutions of 2 Å or better and excluded transmembrane protein targets. It reduced our targets to 56 proteins, which still maintain the diversity of protein classes from the original dataset, including 7 protein categories with 14 kinases, 10 proteases, 6 nuclear receptors, 2 ion channels (non-transmembrane domains), 2 cytochrome P450, 18 other enzymes, and 4 miscellaneous proteins. For each protein target, we randomly selected 5 active compounds and 5 decoy compounds to compare the ability of docking program and MD simulation in discriminating active from decoy compounds. The native ligand for each target protein was also used as a control for this experiment. The complete list of protein targets and selected ligands are listed in Table S1. In addition, we calculated Tanimoto coefficients of the active compounds (Table S2) and the decoy compounds (Table S3). Tanimoto coefficients were calculated using F2 fingerprint option on Open Babel 2.4.1.29 Tanimoto score distributions in Figure S1 shows that the majority of compounds used in this study are not similar to one another.
The workflow of our method is presented in Figure S2 and further described in the following paragraphs. We chose AutoDock Vina as our docking program, because it has been shown to be one of the best performing docking programs that is freely available to researchers in academia.7, 30 AutoDock Vina provides a scoring function that relies on knowledge-based potentials and available empirical information to rank ligands docked to protein structures. Protein receptors and ligands were prepared using default criteria from AutoDock Vina.7 Receptors were treated as rigid, while the ligands were flexible. The search space for docking of ligands was determined based on the coordinate of the native ligand from the crystal structure. A cubic box search space with 22.5 Å edge in each direction was added for ligand docking (i.e., flexible ligand and rigid receptor). For the purpose of running a smooth high-throughput workflow, we only selected the top scoring docked output for each protein-ligand complex.
Protein-ligand complex structures were processed using a python script (Script S1) to automate browser actions for the production of MD system and input files using CHARMM-GUI.27, 28 This script was inspired by a script that utilizes the Solution Builder module for building systems that contain various combinatorial ligand structures from the CHARMM-GUI Ligand Reader and Modeler module.31 Topology and parameter files for the ligands were generated using the latest CGenFF via CHARMM-GUI.32, 33 Protein-ligand structures were solvated in a cubic box using TIP3P water models that extended 10 Å from the protein.34 The systems were neutralized using K+ and Cl− ions. Periodic boundary conditions were applied to the systems in the NPT (constant particle number, pressure, and temperature) ensemble using Langevin thermostat.35 The particle-mesh Ewald method was used for electrostatic interactions.36 Force-based switching function was used to truncate nonbonded interactions over 10 and 12 Å.37 Hydrogen atoms were constrained using the SHAKE algorithm.38 We selected to use CHARMM36m force field for all of the simulations.39 Equilibration and production runs were carried out using OpenMM program.40 Our systems were minimized for 5,000 steps using the steepest descent method followed by 1-ns equilibration with NVT (constant particle number, volume, and temperature) setting. During equilibration, the ligands were positionally restrained while the proteins were allowed to relax. Restraints were removed for subsequent production runs that were carried out at 303.5 K and 1 atm with an integration time step of 2 fs. For each protein-ligand complex, we ran 3 × 100-ns production runs, each started with the same initial structure but using different initial velocity random seeds. To evaluate binding stability of a ligand to its receptor protein, for each snapshot, we first aligned the protein structure only and calculated the ligand RMSD to capture RMSD from ligand’s translation and rotation during the simulation. The proteins were superimposed based on their entire heavy atom coordinates using CHARMM. For each protein-ligand complex we averaged the ligand RMSD throughout the simulation time (100 ns) and obtained a single ligand RMSD value.
We used PBEQ Solver (Poisson-Boltzmann equation solver) in CHARMM-GUI to calculate and visualize electrostatic potential surface for proteins.27, 41, 42 Subsequently, we overlaid the native ligand binding mode from each crystal structure as well as decoy ligands to visually assess their binding modes and interactions.
In order to evaluate the conformational changes underwent by the active ligands during MD simulation, we performed binding pose comparisons with available crystal structures. Since we had randomly chosen ligands for this study without considering the availability of their experimental structures, we had to conduct another set of simulations that include protein-ligand complexes with crystal structures. We chose one protein from each of the 7 different protein classes and randomly selected 5 active ligands for each target structure. However, for Cytochrome P450, we could only find crystal structures with 3 different ligands. The complete list of the 7 protein targets and their ligand crystal structures are listed in Table S4. We obtained additional 33 ligands and docked these ligands using AutoDock Vina to the 7 selected protein targets. In Vina, ligands were flexible and receptors were rigid. The search space for ligand docking was based on the crystal structure coordinate of the ligand in the target protein. The search space was determined with 22.5 Å edge cubic box from the center of mass of the crystal ligand. After docking, we calculated ligand RMSD relative to available crystal structures (Table S4), by aligning protein structures only and calculating their ligand RMSD (crystal structure against predicted docking pose). After MD simulations, we obtained the last MD trajectory snapshot (a single protein-ligand complex structure) and calculated the ligand RMSD relative to the crystal structure (crystal structure against MD result), by superimposing the protein structures only and calculating their ligand RMSD.
Our simulations were conducted using 1 RTX 2080TI GPU (graphics processing unit) and 1 CPU (core processing unit). The simulation time for the smallest system with 107 residues (23,310 atoms total) was 11.9 hours for 100 ns. The biggest system with 540 residues (57,164 atoms) required 26.6 hours for 100 ns (Table S5). MD simulation’s speed is orders of magnitude higher than computational docking, where AutoDock Vina docked a molecule to a target within 2 minutes using 4 CPUs.
Results and Discussion
Short Molecular Dynamics Simulations effectively distinguish decoys from active binders
Docking results from randomly selected 280 active compounds and 280 decoy compounds on 56 target protein (10 compounds per target) show a ROC AUC of 0.683 in its ability to distinguish active from decoy compounds using the AutoDock Vina scoring function (Figure 1). The histogram distribution of active and decoy compounds across the docking scores shows that both have a roughly unimodal distribution. There is a lot of overlap between active and decoy docking scores, where both have similar peaks at −8.5 and −8 kcal/mol, respectively (Figure 1A). As a result, it is difficult to discriminate active from decoy compounds just based on their docking scores. ROC plot suggests an optimal cutoff (to minimize false positive while maximizing true positive rate) at a docking score of −8 kcal/mol. At this cutoff, the true positive rate (of correctly identifying active compounds with < −8 kcal/mol) is 61%, while false positive rate (of incorrectly identifying decoy compounds as active with < −8 kcal/mol) is 36% (Figure 1C).
Figure 1.
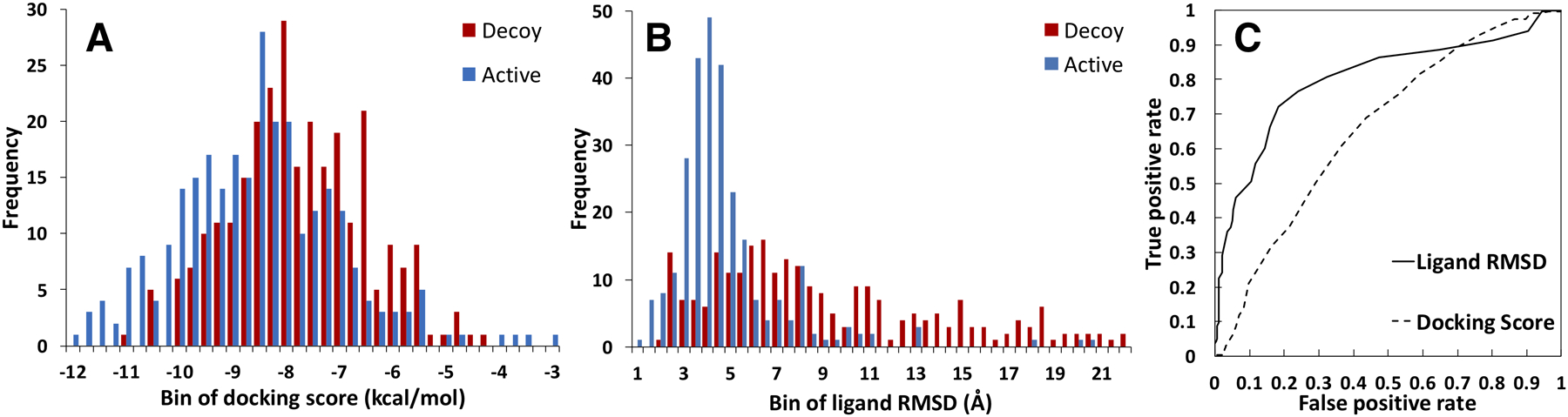
Histogram distributions of (A) docking scores and (B) ligand RMSD for 560 protein-ligand complex structures. Proteins bound to decoy compounds are shown in red and active compounds are shown in blue. (C) ROC plots comparing docking scores with ligand RMSDs from MD simulations. The AUC values are 0.832 (ligand RMSD) and 0.683 (docking score).
The best complex model (with the highest docking score from Vina) for each protein-ligand pair is used as a starting structure for 100-ns MD simulation. In order to set up this process in a high-throughput manner, only the best docking output is used. MD data are processed by calculating the ligand RMSD (from the starting structure) to evaluate binding stability. The ROC AUC result from ligand RMSD is 0.832 (Figure 1). The histogram shows that active compounds have a unimodal distribution centering at 4 Å, while decoy compounds have a skewed-right distribution, showing a lot of ligands leaving the binding site during MD simulations (Figure 1B). It is clear that the majority of decoy ligands can be distinguished from active ligands based on the distribution of their ligand RMSDs. The optimal ROC plot cutoff is at a RMSD of 5.5 Å, where the true positive rate is 79% with a false positive rate of 24%. AUC improvement from AutoDock Vina scoring function (AUC of 0.683) to MD simulation ligand RMSD (AUC of 0.832) is 22%. Using a deep convolution neural network, Pereira and colleagues performed their method on 8 randomly chosen protein targets from DUD-E dataset. Their overall result showed the same AUC from AutoDock Vina as ours (0.68), and their method only improved that to 0.77, which amounted to 13% improvement.13
The early stage of our workflow, which is directly using the output binding mode from docking, is similar to any other machine-learning-based methods to improve protein-ligand model complex structures.10, 12, 13, 22 The limitation of this approach is that incorrect binding modes from docking for active compounds have the potential to deteriorate the ability of MD simulation to correctly identify active compounds. Our results show that a few active ligands are detached from the binding site during the simulation, suggesting that they might have been docked incorrectly at the binding site (Figure 1B). This problem has also been mentioned in machine-learning-based methods. For example, Pereira and colleagues reported that some poor quality docking output binding modes negatively affected the performance of their method to improve docking results.13
Ligand RMSD scoring from MD shows robust performance across different protein classes
In total, we have 56 target proteins from 7 protein classes that include 14 kinases, 10 proteases, 6 nuclear receptors, 2 ion channels, 2 cytochrome P450, 18 other enzymes, and 4 miscellaneous proteins. Table 1 summarizes the AUC for each protein class and Figure 2 shows their ROC plots. Through MD simulations and ligand RMSD evaluation, we observe similar distribution of ROC AUC values (around an average value of 0.832) across different protein classes. All but one of the protein classes have AUC values of 0.8 and above. The exception is the nuclear receptor protein class that has an average AUC of 0.783. One possible explanation is that nuclear receptor proteins have binding pockets that are closed and buried, making them difficult to be evaluated using ligand RMSD through MD simulations. Deeply buried (active or decoy) ligands in closed binding pockets tend to stay in the binding sites throughout the simulations, making it more difficult to distinguish between active and decoy ligands. Nevertheless, ligand RMSD improves the overall AUC of nuclear receptor proteins from 0.622 (docking scoring) to 0.783 (Table 1, Figure 2).
Table 1.
Summary of AUC ROC from docking results using AutoDock Vina and ligand RMSD using MD for DUD-E dataset†
| total ligands | AUC | ||||||
|---|---|---|---|---|---|---|---|
| protein class | total proteins | actives | decoys | docking | 100-ns MD | 50-ns MD | 10-ns MD |
| total | 56 | 280 | 280 | 0.683 | 0.832 | 0.840 | 0.807 |
| kinase | 14 | 70 | 70 | 0.812 | 0.806 | 0.836 | 0.800 |
| protease | 10 | 50 | 50 | 0.626 | 0.824 | 0.822 | 0.805 |
| other enzymes | 18 | 90 | 90 | 0.665 | 0.834 | 0.826 | 0.801 |
| nuclear receptor | 6 | 30 | 30 | 0.622 | 0.783 | 0.778 | 0.804 |
| ion channel | 2 | 10 | 10 | 0.950 | 0.970 | 0.980 | 0.910 |
| cytochrome P450 | 2 | 10 | 10 | 0.550 | 0.890 | 0.900 | 0.835 |
| miscellaneous | 4 | 20 | 20 | 0.482 | 0.910 | 0.963 | 0.803 |
Improved cases of AUC values using MD simulations are highlighted in bold
Figure 2.
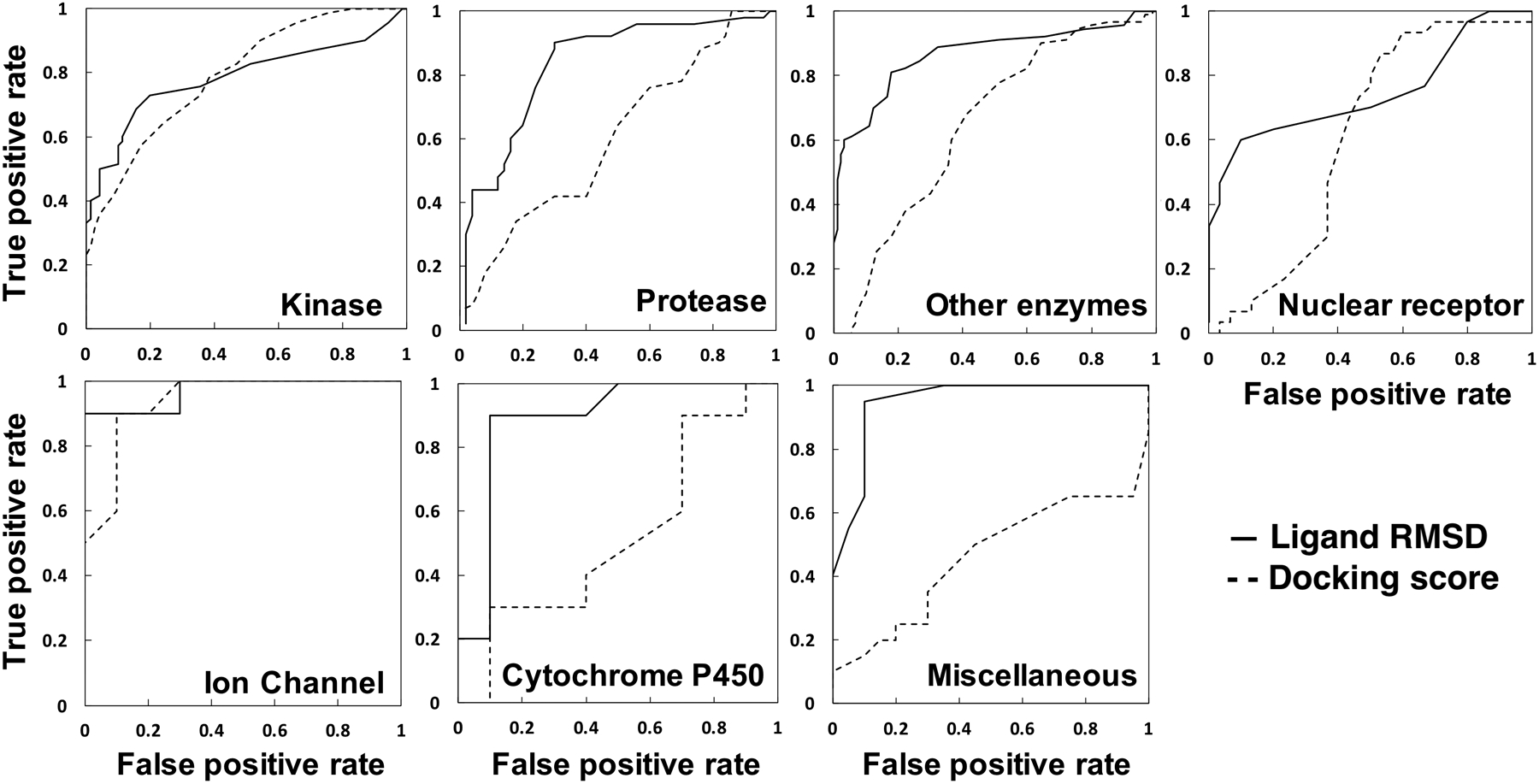
ROC plots comparing docking scores with ligand RMSDs from MD simulations for 14 protein kinases, 10 proteases, 18 other enzymes, 6 nuclear receptors, 2 ion channels, 2 Cytochrome P450 proteins, and 4 miscellaneous proteins.
The AutoDock Vina scoring function combines knowledge-based potentials with available empirical data, so it relies on a machine-learning predictive nature to discriminate active from decoy compounds.1, 7 An apparent disadvantage to this approach is that the scoring function can be biased toward certain protein classes with the most available empirical data and/or most popular drug target classes. Our results show that docking outputs from AutoDock Vina tend to favor kinase (AUC of 0.812) and ion channels (AUC of 0.950) in terms of discriminating active from decoy compounds using its scoring function. Meanwhile, miscellaneous proteins (including fatty acid binding protein, heat shock protein, leukocyte adhesion glycoprotein, and inhibitor of apoptosis) have an average AUC of 0.482. This AUC value means that docking scoring function cannot discriminate active from decoy compounds for this set of proteins. Other enzymes, proteases, nuclear receptors, and cytochrome P450 have an average AUC of 0.665, 0.626, 0.622, and 0.550, respectively (Table 1, Figure 2). This problem of scoring functions being biased toward specific protein families have been acknowledged and discussed in a few previous reports.10, 43, 44 Schneider discussed that specific scoring function calibrated for a data set is usually preferred, because of their varying predictive accuracy across different protein families.43 Recently, using AutoDock, Ramirez and Caballero showed that its scoring function favored B-Raf kinase over MAO-B and thrombin.44 Taken together, having MD simulations as a step after docking to re-assess docked complexes using physics-based theoretical considerations appears to be a more objective and attractive approach. MD simulations incorporate the effects of solvation, flexibility of both protein target and ligand, and better evaluation of non-bonded interactions, which ultimately account for induced fit.
Shorter MD simulation runs can still consistently improve docking results
We observe that most ligands with incorrect binding modes and decoy compounds show signs of binding instability when evaluating their ligand RMSD over simulation time (Figure 3). In general, ligands that have to undergo major binding adjustments of their initial docked conformation end up leaving the binding site. During MD simulation, docked ligands change their binding interactions with the proteins to adjust to physics-based potential energy functions from MD force field. Unfavorable electrostatics and van Der Waals interactions result in unbinding of ligands from the proteins. We calculate the AUC ROC by only taking shorter MD trajectory and compare with the AUC from the full 100 ns. Interestingly, shorter MD simulations, 10 ns (AUC of 0.807) and 50 ns (AUC of 0.840), show similar ability to distinguish active from decoy compounds based on ligand RMSD (Table 1). It has been observed before that short simulations can more effectively evaluate the stability of bound ligand than the scoring functions from docking.17–20 We previously showed that several hundreds of picoseconds simulations could evaluate ligand binding stability in MDM2 better than AutoDock Vina scoring function.18 It is encouraging to notice that shorter simulations can effectively improve docking results, because it can save resources, time, and be efficiently incorporated into a high-throughput workflow.
Figure 3.
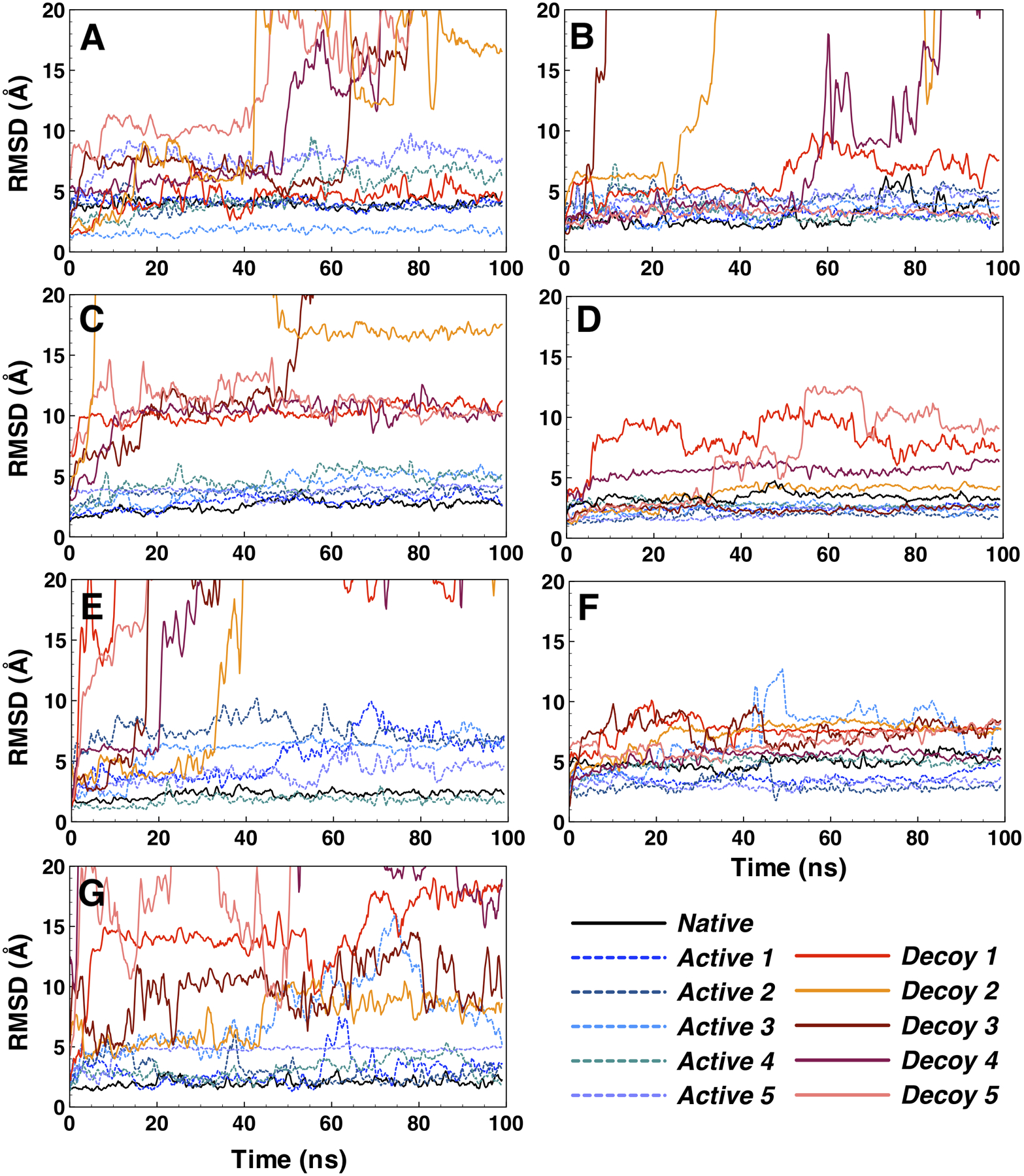
Stability of bound ligands evaluated using ligand RMSD for representative proteins from 7 different protein classes. (A) kinase: PDB 2of2, (B) protease: PDB 1w7x, (C) other enzyme: PDB 2b8t, (D) nuclear receptor: PDB 1mv9, (E) ion channel: PDB 1vso, (F) cytochrome P450: PDB 3nxu, and (G) miscellaneous: PDB 3hl5.
We use DUD-E dataset, which provides decoy compounds that have similar physical properties to known active compounds, but are different in their 2D-topology.7 One of the advantages of using MD simulation is that the protein-ligand interactions are depicted as a function of simulation time. In Figure 3, ligand RMSD of representatives from each of the 7 protein classes show clear differences in ligand binding stabilities between active and decoys compounds. During the simulation, we observe that false/weak interactions between decoy ligands and protein targets tend to be short-lived, and some ligands detach from their binding sites. In contrast, stronger interactions between active ligands (and native ligands self-docked back to its binding site) and their protein receptors stay stable throughout the simulation. For example, in the case of tyrosine protein kinase (PDB 2of2), a representative of the kinase class, ligand RMSDs for 4 active compounds are stable, while all but one decoy compounds are detached from the protein during the simulation (Figure 3A). Binding instabilities are more pronounced as the simulation proceeds to 100-ns, where decoy ligands only start to drift away from the binding site at around 40 ns. Native ligand, furanopyridine 8 binds to tyrosine protein kinase (PDB 2of2) with its pyridine and piperidine rings filling the negative potential binding pocket (Figure 4).45 Ligand decoy 5 that detaches from the binding site at 40 ns shows unstable binding with carbonyl group that fills the negative potential binding pocket (Figure 3A, S3).
Figure 4.
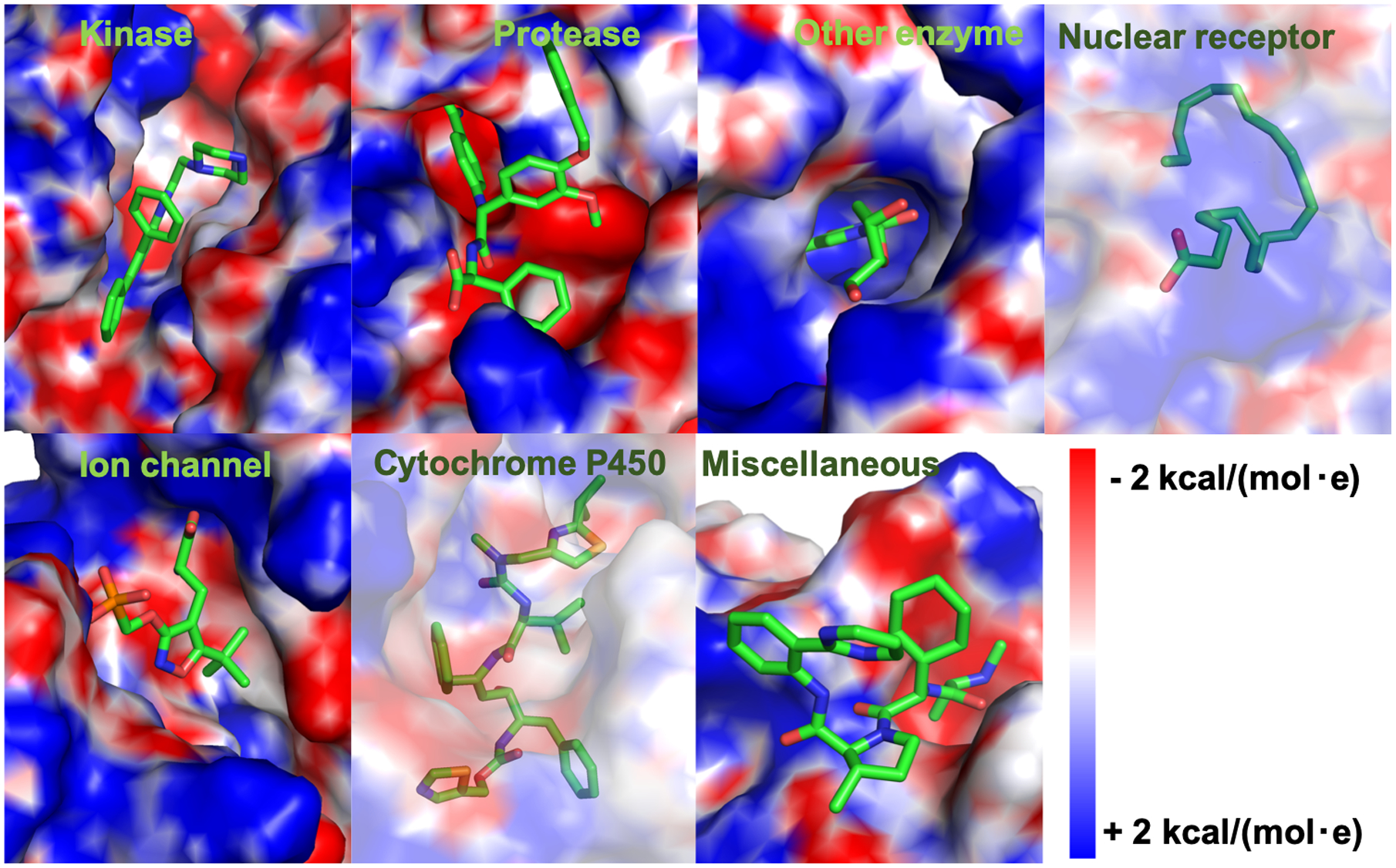
Surface representations showing electrostatic potentials of holo structures with their native ligand binding modes for representative proteins from 7 different classes. Kinase: tyrosine protein kinase, PDB 2of2. Protease: blood coagulation factor VIIa, PDB 1w7x. Other enzyme: thymidine kinase, PDB 2b8t. Nuclear receptor: retinoid x receptor, PDB 1mv9. Ion channel: glutamate receptor, PDB 1vso. Cytochrome P450: human CP450 3A4, PDB 3nxu. Miscellaneous: inhibitor of apoptosis, PDB 3hl5. A transparent surface is used to show ligands that are buried in nuclear receptor and cytochrome P450.
Protease representative (PDB 1w7x) shows good results of discriminating active from decoy compounds based on their ligand RMSD. All 5 active ligands and the native ligand show stable binding throughout the simulation. However, only one of the decoy compounds is stable, 3 of them detach from the binding pocket and one shows high fluctuations toward the end of the simulation (Figure 3B). Protease binding site contains an evolutionary conserved S1 pocket that accommodates big ring structures from small molecules.46 The native ligand contains a benzamidine that fits in S1 pocket and forms a salt bridge with Asp189 at the base of the binding pocket (Figure 4).47 This binding mode stabilizes the native ligand binding throughout the simulation time. In contrast, ligand decoy 2 does not contain a moiety that binds at S1 pocket (Figure S3), resulting in weaker binding affinity and unbinding in MD simulation.
Other enzymes class is represented by thymidine kinase (PDB 2b8t) that shows clear distinctions between active and decoy compounds binding stability. All 5 of active compounds are stable, showing similar fluctuations to the native ligand. However, all 5 of decoy compounds get displaced from the binding pocket within the first 10 ns of the simulation time (Figure 3C). Thymidine kinase has a specific lasso-like loop in its binding site that stabilizes the binding of thymidine moiety (Figure 4).48 This binding specificity does not seem to tolerate decoy compounds with different topology to bind at the binding pocket (Figure S3).
Ion channel protein class is represented by the ionotropic glutamate receptor (PDB 1vso) that demonstrates clear differences in binding stability of active and decoy compounds. All decoy compounds are detached from the binding site by 40 ns. Two of them leave in the first 5 ns (Figure 3E). In contrast, all active compounds stay in the binding site. Although, most of the active compounds appear to undergo binding adjustments at the binding pocket with fluctuations in their ligand RMSDs. We observe that the binding pocket has an opened architecture that allows for more movements of bound ligand. The binding site of the ligand-binding-domain from glutamate receptor adopts a clamshell-like conformation with S1 and S2 amino acid sequences that loosely sandwich a bound ligand (Figure 4).49, 50 Ligand decoy 5 contains a big three-ring structure that ends up detaching from the binding site (Figure S3).
The next two classes of proteins, nuclear receptors and cytochrome P450, are represented by RXR retinoid receptor (PDB 1mv9) and CYP3A4 (PDB 3nxu), respectively. The binding pockets in both structures are closed and buried, which substantially limit the ability of bound ligands to escape (Figure 4). The ligand binding site of a nuclear receptor is enclosed in a deep pocket that is formed by stable structures of the surrounding helices.51 In addition, ligand binding has been shown to be largely stabilized by strong hydrophobic interactions and extensive hydrogen-bonding networks that closely contour the shape of specific native ligands. The binding pocket of cytochrome P450 is closed up, but of a significantly bigger volume as compared to nuclear receptor.52 In general, bound ligands in these binding pockets show smaller and more stable ligand RMSD relative to other protein classes (Figure 3D, 3F). At the same time, they show that active ligands have better ligand stability relative to decoy ligands, making it possible to discriminate the two. Ligand decoy 1 for nuclear receptor and decoy 3 for cytochrome P450 appear to bind loosely at the binding site and show more fluctuations during MD simulation (Figure S3).
The remaining proteins are classified into the miscellaneous class, which is represented by an inhibitor of apoptosis, IAP (PDB 3hl5). It is a relatively small protein (136 residues) with a flat and shallow binding site.53 Its native ligand CS3 shows a hydrogen bond that forms between the terminal amine group of the ligand and Asp320 on the surface binding grove of IAP (Figure 4). At the same binding groove, ligand decoy 1 positions a hydroxyl terminal that results in unstable binding (Figure S3). The AutoDock Vina scoring function cannot discriminate active from decoy compounds for this protein (and others in this class), but MD simulation can effectively discriminate binders from non-binders for this protein (Table 1, Figure 3G). All decoy ligands are shown to escape the binding site, and all but one of the active ligands show stable binding throughout the simulation.
Short MD simulations show limited improvement of ligand binding modes for active compounds
Self-docking is the practice of computational docking of native ligand back to its receptor protein crystal structure. And, cross-docking is the practice of docking a ligand to a receptor protein that originally contains a different ligand. It is commonly accepted that most docking programs can produce good self-docking results across different dataset. A successful docking result generally pass the criterion of having ligand RMSD < 2 Å.54 AutoDock Vina was shown to have 78% success rate in self-docking for 190 protein-ligand complex structures in the PDBbind dataset.7 Recently, we tested AutoDock Vina self-docking on 40 structures from Astex list and showed 82.5% success rate.55 Here, 79% success rate is obtained from self-docking of 56 DUD-E proteins. However, success rate for cross-docking has been low due to the specific variations in the local shape of the binding site crystal structure that fits its native ligand.44
Here, we explore the ability of MD simulations to refine the predicted ligand binding modes from cross-docking results. Unfortunately, most of the active ligands from DUD-E dataset do not have crystal structures that we can use as reference. To solve this, we collected active ligands with available crystal structures. Using the same workflow, we selected 7 proteins, each one from the 7 different protein classes (Table S4). For each target, we selected 5 ligands, except for CP450 that only has 3 ligands with crystal structures. For cross-docking, we extracted ligands from these crystal structures and performed docking of these ligands to 7 target proteins from DUD-E. The resulting ligand binding poses are assessed by calculating their ligand RMSDs from their crystal structures. Our results show that short MD simulations can moderately improve the predicted ligand binding modes from docking (Figure 5). The average improvement is 0.85 Å (from initial value of 5.21 to a final value of 4.36 Å). Predicted ligand binding modes from docking that have < 2 Å ligand RMSD relative to their crystal structures do not show significant changes in binding modes during MD simulations. Their average change is an increase of 0.08 Å from docking to MD simulation, suggesting that ligands with good initial binding modes tend to stay stable throughout MD simulation runs. Ligands with initial predicted binding modes between 2–5 Å relative to their crystal structures show moderate improvements of 0.69 Å on average. The rest of the ligands with > 6 Å RMSD away from their crystal structures do not show any significant improvements that can bring them below 5 Å RMSD, suggesting that the changes in their ligand RMSDs during MD simulations are random and not significant.
Figure 5.
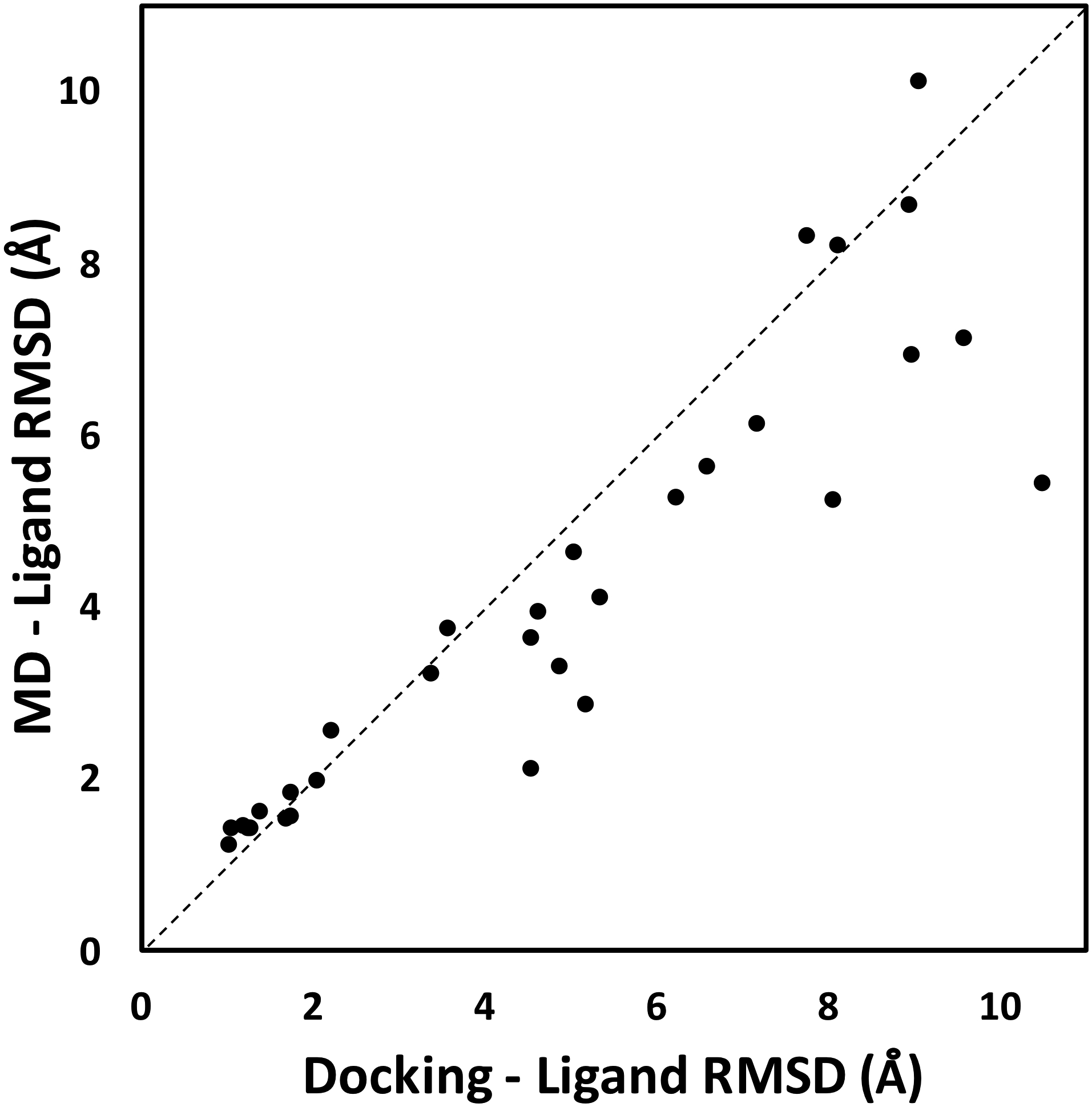
Ligand RMSD values comparing initial docking results with final conformations obtained from MD simulations against their crystal structures for 33 protein-ligand complexes. In many cases, MD simulations improve the predicted ligand binding modes from docking results. The average improvement is 0.85 Å.
Overall, most of the changes in ligand RMSDs are small. Each protein-ligand complex was simulated for 100 ns, which is not enough to refine significantly incorrect binding poses (e.g., ligand RMSD > 5 Å). This is because ligand binding modes in MD simulations can get trapped in a local minimum (in energy landscape), which requires a significant amount of time to escape. In particular for cross-docking binding pose, two conformations need to change to allow for the correction of binding pose, including the protein binding site and the ligand. Binding site structures of the same protein that binds different ligands can be significantly different due to induced fit.56 In a rugged energy landscape with many available local minima, it can be quite difficult for a short MD simulation to correctly identify the global minimum. We expect that significantly longer simulations may result in better improvements, as suggested by previous MD runs by DE Shaw group that successfully docked ligand dasatinib on its target Src kinase.16
Ligand binding modes from docking show a significant majority at 64% with incorrect binding poses (> 2 Å). At the same time, ROC AUC values are reasonably good from both Vina (0.68) and MD (0.83) methods to discriminate active from inactive compounds, suggesting that reproducing crystal-like binding poses is not necessary. In our simulations, we observe that most incorrect binding poses of active compounds contain some favorable interactions between the ligand and the binding site residues that are maintained during the simulations. Conversely, most decoy compounds lack favorable interactions and unbind during the simulations (Figure S3). In general, cross-docking results have been shown to be relatively poor compared to self-docking.44, 56 Ramirez and Caballero recently showed that cross-docking of 30 ligands to 3 different targets (B-Raf, thrombin, and monoamine oxidase B) resulted in bad ligand poses with > 3 Å RMSD relative to their crystal structures.44 Our results suggest that severely incorrect predicted binding modes cannot be sufficiently refined during 100 ns MD simulation. However, some moderately incorrect binding modes can be refined with short MD simulation. There are a few approaches that can be taken to avoid poor cross-docking results. One approach is to filter ligands based on their similarity to the bound ligand in the target structure (similarity selection) suggested by Sutherland and colleagues.56 They found that cross-docking accuracy increased when ligands that are similar to the native ligand are used for docking. Another approach is to include side chain residue flexibility in docking experiments to allow for flexible receptor docking. Adding receptor flexibility has been shown to increase docking success rate using AutoDockFR.57 A different approach that considers receptor flexibility is ensemble docking, where an ensemble of representative structures is used to account for differences in binding site structures.58 Ensemble docking has also been shown to improve cross-docking outputs.59
Conclusions
It is widely accepted that standard scoring functions from molecular docking programs are still having difficulties in discriminating binders from non-binders. Here, we propose a high-throughput protein-ligand complex MD simulations method to improve docking results and more accurately distinguish binders from non-binders. MD simulation effectively incorporates the flexibility of both receptor and ligand, more accurately describes their interactions, and adds solvation effects, which more realistically depicts protein-ligand binding process. Therefore, MD simulation is an attractive method that has been shown to improve docking results for a few protein targets.17–20, 22, 60 To the best of our knowledge, this is the first study using MD simulations that systematically use a large set of 56 diverse target proteins and 560 ligands from DUD-E dataset. Our high-throughput method shows consistent improvements in discriminating active from decoy compounds compared to docking scores. Using 100-ns MD simulation as a step after AutoDock Vina docking produces an average AUC of 0.83 from its initial value of 0.68 using docking score. MD results show robust performance across different protein classes without favoring a particular protein class as observed in docking results. Shorter MD simulations of 50 ns and 10 ns show similar improvements compared to running 100 ns simulations. Furthermore, we show that some of the ligand binding modes of active compounds can be moderately refined during MD simulations.
We expect that our high-throughput protocol will make it easier to prepare simulation systems and inputs for virtual screening of protein-ligand complexes using CHARMM-GUI.27, 28 Our results can serve as a benchmark that showcase the ability of physics-based MD simulations in helping to discriminate binders from non-binders across a large number of 56 target proteins in DUD-E. At the same time, a few limitations persist, including the accuracy of ligand force field parameters and computational cost in running virtual screening using MD simulations for thousands of ligands. As described in the introduction, many machine-learning-based methods have been applied to docking results to boost their scoring functions.10, 12–14 Going forward, it appears that we can take advantage of our benchmark MD dataset for machine-learning training that can be applied to the rest of the ligand dataset in DUD-E. For example, we can include receptor flexibility information from MD to discriminate binders from non-binders. Various types of regularly occurring stable interactions throughout MD simulations can also be useful information for machine-learning. We expect that this next step will effectively alleviate limitations that we have with our current protocol.
Supplementary Material
Acknowledgement
This work has been supported by NIH GM126140 and XSEDE MCB070009. We thank to Hui Sun Lee for a helpful discussion.
Footnotes
Supporting Information
The Supporting Information is available free of charge.
Methods; Table S1, List of protein targets and ligands; Table S2, Tanimoto scores between the active compounds; Table S3, Tanimoto scores between the decoy compounds; Table S4, List of ligands with crystal structures selected for cross-docking; Table S5, MD simulation (GPU hours) for the smallest and biggest systems in this study; Figures S1, Histogram distributions of Tanimoto scores from active compounds and decoy compounds; Figure S2, schematic workflow; Figure S3, Surface representations with bound decoy ligands for representative proteins from 7 different classes; Script S1, python script for high-throughput protein-ligand complex MD simulations using CHARMM-GUI webserver.
The authors declare no competing financial interests.
References
- 1.Lionta E; Spyrou G; Vassilatis DK; Cournia Z, Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem 2014, 14, 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liao C; Sitzmann M; Pugliese A; Nicklaus MC, Software and resources for computational medicinal chemistry. Future Med Chem 2011, 3, 1057–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; Tolmachev AA; Shoichet BK; Roth BL; Irwin JJ, Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsuno K; Masuda Y; Uehara Y; Sato H; Muroya A; Takahashi O; Yokotagawa T; Furuya T; Okawara T; Otsuka M; Ogo N; Ashizawa T; Oshita C; Tai S; Ishii H; Akiyama Y; Asai A, Identification of a New Series of STAT3 Inhibitors by Virtual Screening. ACS Med Chem Lett 2010, 1, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiss R; Kiss B; Konczol A; Szalai F; Jelinek I; Laszlo V; Noszal B; Falus A; Keseru GM, Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J Med Chem 2008, 51, 3145–3153. [DOI] [PubMed] [Google Scholar]
- 6.Ren JX; Li LL; Zheng RL; Xie HZ; Cao ZX; Feng S; Pan YL; Chen X; Wei YQ; Yang SY, Discovery of novel Pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on SVM model, pharmacophore, and molecular docking. J Chem Inf Model 2011, 51, 1364–1375. [DOI] [PubMed] [Google Scholar]
- 7.Trott O; Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jorgensen WL, The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [DOI] [PubMed] [Google Scholar]
- 9.Lu J; Hou X; Wang C; Zhang Y, Incorporating Explicit Water Molecules and Ligand Conformation Stability in Machine-Learning Scoring Functions. J Chem Inf Model 2019, 59, 4540–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wojcikowski M; Ballester PJ; Siedlecki P, Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep 2017, 7, 46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mysinger MM; Carchia M; Irwin JJ; Shoichet BK, Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yasuo N; Sekijima M, Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J Chem Inf Model 2019, 59, 1050–1061. [DOI] [PubMed] [Google Scholar]
- 13.Pereira JC; Caffarena ER; Dos Santos CN, Boosting Docking-Based Virtual Screening with Deep Learning. J Chem Inf Model 2016, 56, 2495–2506. [DOI] [PubMed] [Google Scholar]
- 14.Carpenter KA; Cohen DS; Jarrell JT; Huang X, Deep learning and virtual drug screening. Future Med Chem 2018. [DOI] [PMC free article] [PubMed]
- 15.Hawkins DM, The problem of overfitting. J Chem Inf Comput Sci 2004, 44, 1–12. [DOI] [PubMed] [Google Scholar]
- 16.Shan Y; Kim ET; Eastwood MP; Dror RO; Seeliger MA; Shaw DE, How does a drug molecule find its target binding site? J Am Chem Soc 2011, 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alonso H; Bliznyuk AA; Gready JE, Combining docking and molecular dynamic simulations in drug design. Med Res Rev 2006, 26, 531–568. [DOI] [PubMed] [Google Scholar]
- 18.Lee HS; Jo S; Lim HS; Im W, Application of binding free energy calculations to prediction of binding modes and affinities of MDM2 and MDMX inhibitors. J Chem Inf Model 2012, 52, 1821–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavalli A; Bottegoni G; Raco C; De Vivo M; Recanatini M, A computational study of the binding of propidium to the peripheral anionic site of human acetylcholinesterase. J Med Chem 2004, 47, 3991–3999. [DOI] [PubMed] [Google Scholar]
- 20.Park H; Yeom MS; Lee S, Loop flexibility and solvent dynamics as determinants for the selective inhibition of cyclin-dependent kinase 4: comparative molecular dynamics simulation studies of CDK2 and CDK4. Chembiochem 2004, 5, 1662–1672. [DOI] [PubMed] [Google Scholar]
- 21.Ogrizek M; Turk S; Lesnik S; Sosic I; Hodoscek M; Mirkovic B; Kos J; Janezic D; Gobec S; Konc J, Molecular dynamics to enhance structure-based virtual screening on cathepsin B. J Comput Aid Mol Des 2015, 29, 707–712. [DOI] [PubMed] [Google Scholar]
- 22.Radom F; Pluckthun A; Paci E, Assessment of ab initio models of protein complexes by molecular dynamics. PLoS Comput Biol 2018, 14, e1006182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganesan A; Coote ML; Barakat K, Molecular dynamics-driven drug discovery: leaping forward with confidence. Drug Discov Today 2017, 22, 249–269. [DOI] [PubMed] [Google Scholar]
- 24.Stone JE; Hardy DJ; Ufimtsev IS; Schulten K, GPU-accelerated molecular modeling coming of age. J Mol Graph Model 2010, 29, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nobile MS; Cazzaniga P; Tangherloni A; Besozzi D, Graphics processing units in bioinformatics, computational biology and systems biology. Brief Bioinform 2017, 18, 870–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo S; Cheng X; Lee J; Kim S; Park SJ; Patel DS; Beaven AH; Lee KI; Rui H; Park S; Lee HS; Roux B; MacKerell AD Jr.; Klauda JB; Qi Y; Im W, CHARMM-GUI 10 years for biomolecular modeling and simulation. J Comput Chem 2017, 38, 1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 28.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; Jo S; Pande VS; Case DA; Brooks CL 3rd; MacKerell AD Jr.; Klauda JB; Im W, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput 2016, 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR, Open Babel: An open chemical toolbox. J Cheminform 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z; Sun H; Yao X; Li D; Xu L; Li Y; Tian S; Hou T, Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys 2016, 18, 12964–12975. [DOI] [PubMed] [Google Scholar]
- 31.Kim S; Lee J; Jo S; Brooks CL 3rd; Lee HS; Im W, CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J Comput Chem 2017, 38, 1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr., CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu W; He X; Vanommeslaeghe K; MacKerell AD Jr., Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J Comput Chem 2012, 33, 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983, 79, 926–935. [Google Scholar]
- 35.Allen MP, Tildesley DJ, Computer Simulations of liquids. Clarendon Press: Oxford: 1987. [Google Scholar]
- 36.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG, A Smooth Particle Mesh Ewald Method. J Chem Phys 1995, 103, 8577–8593. [Google Scholar]
- 37.Steinbach PJ; Brooks BR, New Spherical-Cutoff Methods for Long-Range Forces in Macromolecular Simulation. Journal of Computational Chemistry 1994, 15, 667–683. [Google Scholar]
- 38.Barth E; Kuczera K; Leimkuhler B; Skeel RD, Algorithms for Constrained Molecular-Dynamics. Journal of Computational Chemistry 1995, 16, 1192–1209. [Google Scholar]
- 39.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr., CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jo S; Vargyas M; Vasko-Szedlar J; Roux B; Im W, PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res 2008, 36, W270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Im W; Beglov D; Roux B, Continuum Solvation Model: computation of electrostatic forces from numerical solutions to the Poisson-Boltzmann equation. Comput Phys Commun 1998, 111, 59–75. [Google Scholar]
- 43.Schneider G, Virtual screening: an endless staircase? Nat Rev Drug Discov 2010, 9, 273–276. [DOI] [PubMed] [Google Scholar]
- 44.Ramirez D; Caballero J, Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin MW; Newcomb J; Nunes JJ; Bemis JE; McGowan DC; White RD; Buchanan JL; DiMauro EF; Boucher C; Faust T; Hsieh F; Huang X; Lee JH; Schneider S; Turci SM; Zhu X, Discovery of novel 2,3-diarylfuro[2,3-b]pyridin-4-amines as potent and selective inhibitors of Lck: synthesis, SAR, and pharmacokinetic properties. Bioorg Med Chem Lett 2007, 17, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 46.Perona JJ; Craik CS, Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J Biol Chem 1997, 272, 29987–29990. [DOI] [PubMed] [Google Scholar]
- 47.Zbinden KG; Obst-Sander U; Hilpert K; Kuhne H; Banner DW; Bohm HJ; Stahl M; Ackermann J; Alig L; Weber L; Wessel HP; Riederer MA; Tschopp TB; Lave T, Selective and orally bioavailable phenylglycine tissue factor/factor VIIa inhibitors. Bioorg Med Chem Lett 2005, 15, 5344–5352. [DOI] [PubMed] [Google Scholar]
- 48.Kosinska U; Carnrot C; Eriksson S; Wang L; Eklund H, Structure of the substrate complex of thymidine kinase from Ureaplasma urealyticum and investigations of possible drug targets for the enzyme. FEBS J 2005, 272, 6365–6372. [DOI] [PubMed] [Google Scholar]
- 49.Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R, Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hald H; Naur P; Pickering DS; Sprogoe D; Madsen U; Timmermann DB; Ahring PK; Liljefors T; Schousboe A; Egebjerg J; Gajhede M; Kastrup JS, Partial agonism and antagonism of the ionotropic glutamate receptor iGLuR5: structures of the ligand-binding core in complex with domoic acid and 2-amino-3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid. J Biol Chem 2007, 282, 25726–25736. [DOI] [PubMed] [Google Scholar]
- 51.Bain DL; Heneghan AF; Connaghan-Jones KD; Miura MT, Nuclear receptor structure: implications for function. Annu Rev Physiol 2007, 69, 201–220. [DOI] [PubMed] [Google Scholar]
- 52.Lewis DF, Structural characteristics of human P450s involved in drug metabolism: QSARs and lipophilicity profiles. Toxicology 2000, 144, 197–203. [DOI] [PubMed] [Google Scholar]
- 53.Ndubaku C; Varfolomeev E; Wang L; Zobel K; Lau K; Elliott LO; Maurer B; Fedorova AV; Dynek JN; Koehler M; Hymowitz SG; Tsui V; Deshayes K; Fairbrother WJ; Flygare JA; Vucic D, Antagonism of c-IAP and XIAP proteins is required for efficient induction of cell death by small-molecule IAP antagonists. ACS Chem Biol 2009, 4, 557–566. [DOI] [PubMed] [Google Scholar]
- 54.Bursulaya BD; Totrov M; Abagyan R; Brooks CL 3rd, Comparative study of several algorithms for flexible ligand docking. J Comput Aided Mol Des 2003, 17, 755–763. [DOI] [PubMed] [Google Scholar]
- 55.Guterres H; Lee HS; Im W, Ligand-Binding-Site Structure Refinement Using Molecular Dynamics with Restraints Derived from Predicted Binding Site Templates. J Chem Theory Comput 2019, 15, 6524–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutherland JJ; Nandigam RK; Erickson JA; Vieth M, Lessons in molecular recognition. 2. Assessing and improving cross-docking accuracy. J Chem Inf Model 2007, 47, 2293–2302. [DOI] [PubMed] [Google Scholar]
- 57.Ravindranath PA; Forli S; Goodsell DS; Olson AJ; Sanner MF, AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput Biol 2015, 11, e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong CF, Flexible receptor docking for drug discovery. Expert Opin Drug Discov 2015, 10, 1189–1200. [DOI] [PubMed] [Google Scholar]
- 59.Osguthorpe DJ; Sherman W; Hagler AT, Generation of receptor structural ensembles for virtual screening using binding site shape analysis and clustering. Chem Biol Drug Des 2012, 80, 182–193. [DOI] [PubMed] [Google Scholar]
- 60.Sakano T; Mahamood MI; Yamashita T; Fujitani H, Molecular dynamics analysis to evaluate docking pose prediction. Biophys Physicobiol 2016, 13, 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.