

Abstract
The contribution of CD4+ T cells to protective or pathogenic immune responses to SARS-CoV-2 infection remains unknown. Here, we present single-cell transcriptomic analysis of >100,000 viral antigen-reactive CD4+ T cells from 40 COVID-19 patients. In hospitalized patients compared to non-hospitalized patients, we found increased proportions of cytotoxic follicular helper cells and cytotoxic T helper (TH) cells (CD4-CTLs) responding to SARS-CoV-2 and reduced proportion of SARS-CoV-2-reactive regulatory T cells (TREG). Importantly, in hospitalized COVID-19 patients, a strong cytotoxic TFH response was observed early in the illness, which correlated negatively with antibody levels to SARS-CoV-2 spike protein. Polyfunctional TH1 and TH17 cell subsets were underrepresented in the repertoire of SARS-CoV-2-reactive CD4+ T cells compared to influenza-reactive CD4+ T cells. Together, our analyses provide insights into the gene expression patterns of SARS-CoV-2-reactive CD4+ T cells in distinct disease severities.
Keywords: human, SARS-CoV-2, COVID-19, CD4+ T cells, single-cell RNA sequencing, scRNA-seq, antigen-reactive T cell enrichment, ARTE, TFH, TREG, cytotoxic, CD4-CTLs
Graphical Abstract
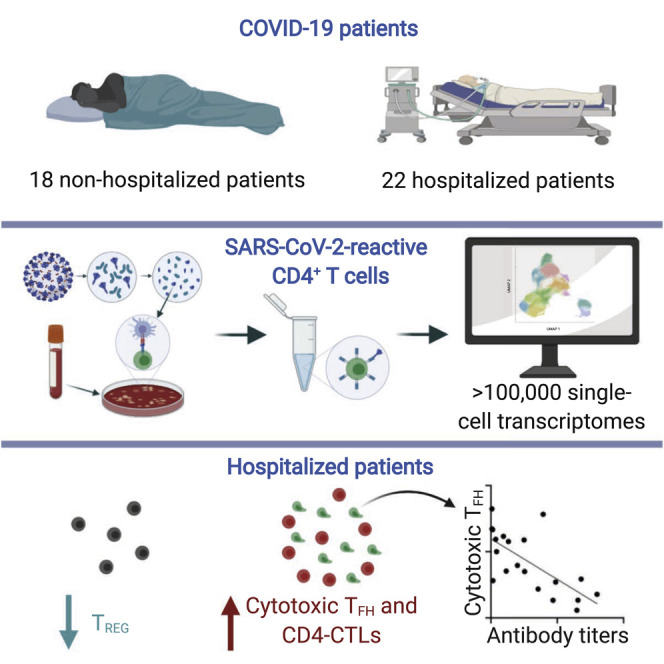
Analyses of CD4+ T cells from 40 COVID-19 patients show that hospitalization is associated with increased cytotoxic follicular helper cells and cytotoxic T helper cells and a reduction in regulatory T cells.
Introduction
Coronavirus disease 2019 (COVID-19) is causing substantial mortality, morbidity, and societal disruption (Tay et al., 2020; Vabret et al., 2020), and effective vaccines and therapeutics may take several months or years to become available. A substantial number of patients become life-threateningly ill, and the mechanisms responsible for causing severe acute respiratory distress syndrome (SARS) in COVID-19 are not well understood. Therefore, there is an urgent need to understand the key players driving protective and pathogenic immune responses in COVID-19 (Vabret et al., 2020). This knowledge may help devise better therapeutics and vaccines for tackling the current pandemic. CD4+ T cells are key orchestrators of anti-viral immune responses, either by enhancing the effector functions of other immune cell types like cytotoxic CD8+ T cells, NK cells, and B cells or through direct killing of infected cells (Sallusto, 2016). Recent studies in patients with COVID-19 have verified the presence of CD4+ T cells that are reactive to SARS-CoV-2 (Braun et al., 2020; Thieme et al., 2020; Grifoni et al., 2020). However, the nature and types of CD4+ T cell subsets that respond to SARS-CoV-2 and the roles these subsets play in driving protective or pathogenic immune responses remain elusive. Here, we have analyzed single-cell transcriptomes of virus-reactive CD4+ T cells to determine associations with severity of COVID-19 illness and to compare the molecular properties of SARS-CoV-2-reactive CD4+ T cells to other common respiratory virus-reactive CD4+ T cells from healthy control subjects.
Results
CD4+ T Cell Responses in COVID-19 Illness
To capture CD4+ T cells responding to SARS-CoV-2 in patients with COVID-19 illness, we employed the antigen-reactive T cell enrichment (ARTE) assay (Bacher et al., 2013, 2016, 2019; Schmiedel et al., 2018) that relies on in vitro stimulation of peripheral blood mononuclear cells (PBMCs) for 6 h with overlapping peptide pools targeting the immunogenic domains of the spike and membrane proteins of SARS-CoV-2 (see STAR Methods; Thieme et al., 2020). Following in vitro stimulation, SARS-CoV-2-reactive CD4+ memory T cells were isolated based on the expression of cell surface markers (CD154 and CD69) that reflect recent engagement of the T cell receptor (TCR) by cognate major histocompatibility complex (MHC)-peptide complexes (Figure S1 A). In the context of acute COVID-19 illness, CD4+ T cells expressing activation markers have been reported in the blood (Braun et al., 2020; Thevarajan et al., 2020); such CD4+ T cells, presumably activated in vivo by endogenous SARS-CoV-2 viral antigens, were also captured during the ARTE assay, thereby enabling us to study a comprehensive array of CD4+ T cell subsets responding to SARS-CoV-2. We sorted > 300,000 SARS-CoV-2-reactive CD4+ T cells from > 1.3 billion PBMCs isolated from a total of 40 patients with COVID-19 illness (22 hospitalized patients with severe illness, 9 of whom required intensive care unit [ICU] treatment, and 18 non-hospitalized subjects with relatively milder disease; Figures 1A and 1B and Tables S1A and S1B). In addition to expressing CD154 and CD69, sorted SARS-CoV-2-reactive CD4+ T cells co-expressed other activation-related cell surface markers like CD38, CD137 (4-1BB), CD279 (PD-1), and HLA-DR (Figures 1C and S1B and Table S1C).
Figure S1.

CD4+ T Cell Responses in COVID-19 Illness, Related to Figure 1
(A) Gating strategy to sort: lymphocytes size-scatter gate, single cells (Height versus Area forward scatter (FSC)), live, CD3+ CD4+ memory (CD45RA+ CCR7+ naive cells excluded) activated CD154+ CD69+ cells. Surface expression of activation markers was analyzed on memory CD4+ T cells.
(B) Representative FACS plots (left) showing surface expression of PD-1 and CD38 in memory CD4+ T cells ex vivo and in CD154+ CD69+ memory CD4+ T cells following 6 h of stimulation, post-enrichment (CD154-based). (Middle) Plots depicting percentage of CD154+ CD69+ memory CD4+ T cells expressing PD-1 or CD38 following stimulation and post-enrichment (CD154-based) in 17 hospitalized and 18 non-hospitalized COVID-19 patients. (Right) Plot showing the total number of sorted CD154+ CD69+ memory CD4+ T cells per million PBMCs; data are mean ± SEM.
(C) Representative FACS plots showing surface staining of CD154 and CD69 in memory CD4+ T cells stimulated for 6 h with individual virus megapools, pre-enrichment (top) and post-enrichment (CD154-based) (bottom) in healthy non-exposed subjects. (Right) Percentage of memory CD4+ T cells co-expressing CD154 and CD69 following stimulation with individual virus megapools (pre-enrichment); data are mean ± SEM.
(D) Representative FACS plots (left) showing surface staining of CD154 in memory CD4+ T cells stimulated with Influenza megapool, pre-enrichment in healthy subjects pre and/or post-vaccination. (Right) Percentage of memory CD4+ T cells expressing CD154 following stimulation with Influenza megapool (pre-enrichment); data are mean ± SEM.
(E) Representative FACS plots showing surface staining of CD154 in memory CD4+ T cells stimulated with Influenza megapool, post-enrichment (CD154-based), in healthy subjects pre and/or post-vaccination
Figure 1.

CD4+ T Cell Responses in COVID-19 Illness
(A) Study overview.
(B) Representative FACS plots showing surface staining of CD154 (CD40L) and CD69 in memory CD4+ T cells stimulated for 6 h with SARS-CoV-2 peptide pools, post-enrichment (CD154-based), in 22 hospitalized and 18 non-hospitalized COVID-19 patients (left), and summary of numbers of cells sorted (right); data are mean ± SEM.
(C) Representative FACS plots (left) showing surface expression of CD137 (4-1BB) and HLA-DR in memory CD4+ T cells ex vivo (without in vitro stimulation) and in CD154+ CD69+ memory CD4+ T cells following stimulation, post-enrichment (CD154-based). (Right) Percentage of CD154+ CD69+ memory CD4+ T cells expressing CD137 (4-1BB) or HLA-DR in 17 hospitalized and 18 non-hospitalized COVID-19 patients; data are mean ± SEM.
Recent evidence from studies in non-exposed individuals (blood sample obtained pre-COVID-19 pandemic) indicates pre-existing SARS-CoV-2-reactive CD4+ T cells, possibly indicative of human coronavirus (HCoV) cross-reactivity. Such cells are observed in up to 50% of the subjects studied (Braun et al., 2020; Grifoni et al., 2020; Le Bert et al., 2020). To capture such SARS-CoV-2-reactive CD4+ T cells, likely to be coronavirus (CoV)-reactive, we screened healthy non-exposed subjects and isolated CD4+ T cells responding to SARS-CoV-2 peptide pools from 4 subjects with highest responder frequency (Figures 1A and S1C and Table S1D). Next, for defining the CD4+ T cell subsets and their properties that distinguish SARS-CoV-2-reactive cells from other common respiratory virus-reactive CD4+ T cells, we isolated CD4+ T cells responding to peptide pools specific to influenza hemagglutinin protein (FLU-reactive cells, see STAR Methods) from 8 additional healthy subjects who provided blood samples before and/or after influenza vaccination (Figures 1A, S1D, and S1E and Tables S1D and S1E). CD4+ T cells responding to peptide pools specific to other common respiratory viruses like human parainfluenza (HPIV) and human metapneumovirus (HMPV) were also isolated from healthy subjects (Figure S1C and Tables S1D and S1F). In total, we interrogated the transcriptome and TCR sequence of >100,000 viral-reactive CD4+ T cells from 53 subjects (Figures 1A, S2 A, and S2B and Tables S2A–S2E).
Figure S2.

SARS-CoV-2-Reactive CD4+ T Cells Are Enriched for TFH Cells and CD4-CTLs, Related to Figure 2
(A) Number of genes recovered for each 10x library sequenced.
(B) Proportion of cells in each cluster for the 6 batches of donors.
(C) Donut charts show proportion of individual virus-reactive CD4+ T cells per cluster for different viruses. Notable clusters are highlighted.
(D) Violin plots showing enrichment patterns of TH17, IFN response, TFH, and CD4-CTLs gene signatures for each cluster. Color indicates mean signature score of cells within a cluster.
(E) Violin plots showing normalized expression level (log2(CPM+1)) of select TH1, TH17, IFN response, TFH and CD4-CTL marker transcripts in designated clusters compared to an aggregation of remaining cells (Rest). Color indicates the percentage of cells expressing indicated transcript.
(F) Scatterplot displaying co-expression level (log2(CPM+1)) of IL2 and TNF transcripts in IFNG-expressing, virus-reactive memory CD4+ T cells in cluster 1. Numbers indicate percentage of cells in each quadrant.
(G) Gene set enrichment analysis (GSEA) for TH17, IFN response, cell cycling, TFH and CD4-CTL signature genes in a given cluster compared to the rest of the cells; ∗p < 0.05; ∗∗∗p < 0.01; ∗∗∗p < 0.001.
SARS-CoV-2-Reactive CD4+ T Cells Are Enriched for TFH Cells and CD4-CTLs
Analysis of the single-cell transcriptomes of all viral-reactive CD4+ T cells from all subjects revealed 13 CD4+ T cell subsets that clustered distinctly, reflecting their unique transcriptional profiles (Figures 2 A–2D and Table S2F). Strikingly, a number of clusters were dominated by cells reactive to particular viruses (Figures 2B and S2C). For example, the vast majority of cells in clusters 1 and 10 were FLU-reactive (>65%), whereas cells in clusters 0, 5, 6, 7, and 12 mainly consisted of SARS-CoV-2-reactive CD4+ T cells (>70%) from COVID-19 patients (Figures 2B and S2C). Conversely, cells in clusters 2, 3, 4, 8, and 9 were not preferentially enriched for reactivity to any given virus (Figures 2B and S2C). These findings suggest that distinct viral infections generate CD4+ T cell subsets with distinct transcriptional programs, although the timing of survey (acute illness versus past infection) will also contribute to their cellular states. Our data highlight substantial heterogeneity in the nature of CD4+ T cells generated in response to different viral infections on the one hand and shared features on the other.
Figure 2.

SARS-CoV-2-Reactive CD4+ T Cells Are Enriched for TFH Cells and CD4-CTLs
(A) Single-cell transcriptomes of sorted CD154+ CD69+ memory CD4+ T cells following 6 h stimulation with virus-specific peptide megapools are displayed by uniform manifold approximation and projection (UMAP). Seurat-based clustering of 102,230 cells colored based on cluster type.
(B) UMAPs showing memory CD4+ T cells for individual virus-specific megapool stimulation conditions (left), and normalized proportions of each virus-reactive cells per cluster is shown (right).
(C) Heatmap showing expression of the most significantly enriched transcripts in each cluster (see Table S2F). Seurat marker gene analysis (comparison of cluster of interest versus all other cells). The top 200 transcripts are shown based on adjusted P value < 0.05, log2 fold change > 0.25 and > 10% difference in the percentage of cells expressing selected transcript between two groups of cells compared.
(D) Plot shows average expression (color scale) and percent of expressing cells (size scale) for selected marker gene transcripts in each cluster.
(E) Violin plots showing normalized expression level (log2(CPM+1)) of TFH (top), TH1 (middle), and TH17 (bottom) marker transcripts in designated clusters compared to an aggregation of remaining cells (Rest). Color indicates percentage of cells expressing indicated transcript.
(F) UMAP showing TFH, CD4-CTL, TH17, and interferon (IFN) response signature scores for each cell.
The clusters enriched for FLU-reactive CD4+ T cells (clusters 1 and 10) displayed features suggestive of polyfunctional T helper (TH)1 cells which have been associated with protective anti-viral immune responses (Seder et al., 2008). Such features include the expression of transcripts encoding for the cytokines linked to polyfunctionality such as IFN-γ, IL-2, and TNFα , and several other cytokines and chemokines like IL-3, CSF2, IL-23A, and CCL20 (Figures 2D, 2E, S2E, and S2F). SARS-CoV-2-reactive CD4+ T cells were underrepresented in these clusters (cluster 1 and 10, < 2%) when compared to FLU-reactive cells (> 70%) or HMPV- and HPIV-reactive cells (~5%–20%) (Figure S2C). Furthermore, SARS-CoV-2-reactive CD4+ T cells in cluster 1 expressed significantly lower levels of IFNG and IL2 transcripts when compared to FLU-reactive cells (Table S2G). Together, these data suggested a failure to generate robust polyfunctional TH1 cells in SARS-CoV-2 infection. A similar pattern was also observed in SARS-CoV-2-reactive CD4+ T cells from healthy non-exposed subjects (Figures 2B and S2C) but not for HPIV- or HMPV-reactive CD4+ T cells, suggesting the defect in generating polyfunctional TH1 cells may be a common feature for coronaviruses, although further studies specifically analyzing HCoV-reactive CD4+ T cells in healthy individuals will be required to verify this.
Other clusters that were relatively underrepresented for SARS-CoV-2-reactive CD4+ T cells included clusters 2 and 8, which were both enriched for TH17 signature genes, with cluster 2 highly enriched for cells expressing IL17A and IL17F transcripts, thus representing bona fide TH17 cells (Figures 2B–2F and S2C–S2E and Table S2F). TH17 cells have been associated with protective immune responses in certain models of viral infections (Acharya et al., 2016; Wang et al., 2011); however, in other contexts they have been shown to promote viral disease pathogenesis (Acharya et al., 2016; Ma et al., 2019). Therefore, the functional relevance of an impaired TH17 response in COVID-19 is not clear and requires further investigation.
Clusters that were evenly distributed across all viral-specific CD4+ T cells include clusters 3 and 4. Cluster 3 displayed a transcriptional profile consistent with enrichment of interferon (IFN)-response genes (IFIT3, IFI44L, ISG15, MX2, OAS1), and cluster 4 was enriched for CCR7, IL7R, and TCF7 transcripts, likely representing central memory CD4+ T cell subset (Figures 2B–2F and S2C–S2E and Table S2F). Cluster 12, which expressed high levels of transcripts linked to cell cycle genes MKI67 and CDK1, also contained a large proportion of SARS-CoV-2-reactive CD4+ T cells (Figures 2B–2D), indicative of actively proliferating cells responsive to SARS-CoV-2 antigens. Cluster 6, also dominated by SARS-CoV-2-reactive CD4+ T cells, was characterized by high levels of PRF1, GZMB, GZMH, GNLY, and NKG7 transcripts, which encode for molecules linked to cytotoxicity (Patil et al., 2018) (Figures 2B–2F and S2C–S2E and Table S2F). Gene set enrichment analysis (GSEA) showed significant positive enrichment of signature genes for cytotoxicity in clusters 6 and 9 (Figure S2G and Table S2H), confirming these clusters represent cytotoxic CD4+ T cells (CD4-CTLs).
Clusters 0, 5, and 7, which were colocalized in the uniform manifold approximation and projection (UMAP) plot, were dominated by SARS-CoV-2-reactive CD4+ T cells (Figures 2A and 2B). Cells in these clusters were uniformly enriched for transcripts encoding for cytokines, surface markers, and transcriptional coactivators associated with T follicular helper (TFH) cell function (CXCL13, IL21, CD200, BTLA, and POU2AF1) (Locci et al., 2013) (Figures 2B–2F and S2C–S2E and Table S2F). Independent GSEA showed significant positive enrichment of TFH signature genes in these clusters, confirming that cells in these clusters represent circulating TFH cells (Figure S2G and Table S2H). Bona fide TFH cells reside in the germinal center; however, TFH cells have been described in the blood where increased numbers have been reported during viral infections and following vaccinations (Bentebibel et al., 2013; Koutsakos et al., 2018; Smits et al., 2020). Thus, the increase in circulating SARS-CoV-2-reactive TFH subsets observed in patients with COVID-19 is consistent with published reports in acute infections. Overall, our single-cell transcriptomic analysis revealed substantial differences in the nature of CD4+ T cell responses to viral infections and highlight subsets that are specifically enriched or depleted in COVID-19 illness.
SARS-CoV-2-Reactive CD4+ T Cell Subsets Associated with Disease Severity
We next assessed if the proportions of SARS-CoV-2-reactive CD4+ T cells in any cluster were greater or lower in hospitalized COVID-19 patients when compared to non-hospitalized patients. Unsupervised clustering of patients, based on the proportions of SARS-CoV-2-reactive CD4+ T cells in different clusters, showed that patients with an increased proportion of TFH cells in cluster 0 clustered distinctly from those with increased proportions of TFH cells in cluster 5 or CD4-CTL cells (cluster 6) (Figure 3 A). The total frequency of SARS-CoV-2-reactive CD4+ T cells with a TFH profile (cluster 0, 5, and 7) was not significantly different between hospitalized and non-hospitalized COVID-19 patients (Figure 3B). However, the relative proportion of TFH cells in cluster 5 was significantly greater in hospitalized patients (severe disease) compared to non-hospitalized patients (mild disease), and the inverse was observed for the proportion of TFH cells in cluster 0 (Figures 3C and S3 A and Table S2B). This pattern was maintained irrespective of whether the patients’ samples were analyzed early (< 3 weeks from symptom onset) or later (> 3 weeks) in the course of illness (Figure S3B). Notably, the proportion of TFH cells in cluster 7 was not significantly different between hospitalized and non-hospitalized COVID-19 patients (Figure S3C).
Figure 3.

SARS-CoV-2-Reactive CD4+ T Cell Subsets Associated with Disease Severity
(A) Unsupervised clustering of COVID-19 patients based on the proportions of SARS-CoV-2-reactive CD4+ T cells in different clusters following 6 h peptide stimulation. Clusters with fewer than 5% of the total dataset are not depicted. Gender and hospitalization status per patient are indicated by different color schemes above the heatmap.
(B) Percentage of TFH cells (clusters 0, 5, and 7) in the total SARS-CoV-2-reactive CD4+ T cell pool for non-hospitalized and hospitalized COVID-19 patients; dots indicate data from a single subject. Data are mean ± SEM; significance for comparisons was computed using Mann-Whitney U test; ns, non-significant P value.
(C) Proportion of clusters 5 and 0 cells in SARS-CoV-2-reactive TFH cells (clusters 0, 5, and 7) in non-hospitalized and hospitalized COVID-19 patients. Data are mean ± SEM; significance for comparisons was computed using Mann-Whitney U test; ∗∗∗∗p < 0.0001.
(D) Violin plots showing normalized expression level (log2(CPM+1)) of ZBTB32 and ZBED2 transcripts in SARS-CoV-2-reactive cells from clusters 0, 5, and 7 (top); color indicates percentage of cells expressing indicated transcript. Plots below show average expression and percent of cells expressing selected transcripts in indicated clusters.
(E) Scatterplot displaying normalized co-expression level (log2(CPM+1)) between PRF1 and GZMB transcripts in SARS-CoV-2-reactive cells present in clusters 5 (left) and 0 (right). Numbers indicate percentage of cells in each quadrant.
(F) Correlation between percentage of SARS-CoV-2-reactive CD4+ TFH cells and S1/S2 antibody titers in 15 non-hospitalized (left) and 20 hospitalized (right) COVID-19 patients. Correlation coefficient r and the related P value were computed using Spearman correlation; ∗p < 0.05.
(G) Correlation between percentage of SARS-CoV-2-reactive CD4+ TFH cells form cluster 5 as a frequency of total CD4+ TFH and S1/S2 antibody titers (left two plots) and interval between symptom onset and blood draw (right two plots) in 15 non-hospitalized and 20 hospitalized (left) COVID-19 patients. Correlation coefficient r and the related P value were computed using Spearman correlation; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, non-significant P value.
(H) Single-cell trajectory analysis of cells in cluster 5 and 0 showing pseudotime, expression of indicated genes, and IFN response signature score.
Figure S3.

SARS-CoV-2-Reactive CD4+ T Cell Subsets Associated with Disease Severity, Related to Figure 3
(A) Average frequency of cells per cluster from hospitalized and non-hospitalized COVID-19 patients.
(B) Proportion of cluster 5 cells in SARS-CoV-2-reactive cytotoxic TFH cells (cluster 0, 5, and 7) in non-hospitalized and hospitalized COVID-19 patients who provided blood samples under 21 days (left) and over 21 days (right) after onset of symptoms. Data are mean ± S.E.M; significance for comparisons was computed using Mann-Whitney U test; ∗∗p < 0.01; ∗∗∗p < 0.001.
(C) Proportion of cluster 7 cells in SARS-CoV-2-reactive TFH cells in non-hospitalized and hospitalized COVID-19 patients. Data are mean ± SEM. Significance for comparisons was computed using Mann-Whitney U test; ns identifies non-significant P value.
(D) Volcano plot showing differentially expressed genes between SARS-CoV-2-reactive CD4+ T cells in cluster 5 versus cluster 0.
(E) Violin plots showing expression level (log2(CPM+1)) of PRF1 and GZMB transcripts in cells from clusters 0, 5 and 7.
(F) Scatterplot displaying co-expression level (log2(CPM+1)) of PRF1 and GZMB transcripts in SARS-CoV-2-reactive cells present in cluster 7. Numbers indicate percentage of cells in each quadrant.
(G) Concentration of S1/S2 antibodies in the circulation of 22 hospitalized and 16 hospitalized non-hospitalized COVID-19 patients. Data are mean ± S.E.M; significance for comparisons was computed using Mann-Whitney U test; ∗p < 0.05.
(H) Correlation between percentage of SARS-CoV-2-reactive CD4+ TFH cells form cluster 0 as a frequency of total CD4+ TFH cells and S1/S2 antibody titers (left two plots) and interval between symptom onset and blood draw (right two plots) in 15 non-hospitalized and 20 hospitalized (left) COVID-19 patients. Correlation coefficient r and the related P value were computed using Spearman correlation; ∗∗∗p < 0.001.
(I) FACS plots showing S1/S2-specific B cells in 9 COVID-19 patients. Patient ID and proportion of SARS-CoV-2-reactive TFH cells in cluster 5 is specified.
(J) Ingenuity pathway analysis (IPA) of genes with increased expression (adjusted p < 0.05 and log2 fold change > 1) between cells from cluster 5 versus cluster 0. Upstream regulatory network analysis of genes in IFN alpha pathway.
(K) GSEA for IFN response signature genes in cluster 5 versus cluster 0; ∗∗∗p < 0.001.
To determine the transcriptional features that differentiated SARS-CoV-2-reactive TFH cells present in cluster 5 from those in cluster 0, we performed single-cell differential gene expression analysis (Figure S3D and Table S3A). Transcripts encoding for transcription factors zinc finger BED-type-containing 2 (ZBED2) and zinc finger and BTB domain-containing protein 32 (ZBTB32) were enriched in TFH cells in cluster 5 and were also expressed at significantly higher levels in hospitalized COVID-19 patients (Figures 3D and S3D and Tables S3A and S3B). ZBTB32, also known as PLZP, belongs to a broad-complex, tramtrack and bric-à-brac zinc finger (BTB-ZF) family of transcriptional repressors like PLZF, B-cell lymphoma 6 (BCL6), and T-helper-inducing POZ-Kruppel-like factor (ThPOK) and has been shown to play a role in impairing anti-viral immune responses by negatively regulating T cell proliferation, cytokine production, and development of long-term memory cells (Piazza et al., 2004; Shin et al., 2017). ZBED2, a novel zinc finger transcription factor without a mouse ortholog, has been linked to T cell dysfunction in the context of anti-tumor immune response (Li et al., 2019) and more recently shown to repress expression of IFN target genes (Somerville et al., 2020). In support of potential dysfunctional properties of the cells in the TFH cluster 5, we found increased expression of several transcripts encoding for molecules linked to inhibitory function, like TIGIT, LAG3, TIM3, and PD1 (Thommen and Schumacher, 2018), and to negative regulation of T cell activation and proliferation, like DUSP4 and CD70 (Huang et al., 2012; O’Neill et al., 2017) (Figures 3D and S3D and Table S3A).
Most strikingly, TFH cells in cluster 5 expressed high levels of cytotoxicity-associated transcripts (PRF1, GZMB) (Figures 3E, S3D, and S3E), reminiscent of the recently described cytotoxic TFH cells, which were shown to directly kill B cells and associated with the pathogenesis of recurrent tonsillitis in children (Dan et al., 2019). Of relevance, recent studies reported a striking loss of germinal center B cells in the thoracic lymph nodes and spleen of patients who died of SARS-CoV-2 infection (Kaneko et al., 2020), as well as slightly lower SARS-CoV-2 spike protein (S)-specific immunoglobulin M (IgM) antibodies in deceased COVID-19 patients (Atyeo et al., 2020). On the basis of these findings, we hypothesized that the cytotoxic TFH cells (cluster 5) observed in hospitalized COVID-19 patients may impair humoral (B cell) immune responses to SARS-CoV-2. To test this association, we assessed the correlation between the proportions of SARS-CoV-2-reactive TFH cell subsets and immunoglobulin G (IgG) antibody titers against the SARS-CoV-2 S1/S2 (S1 and S2 subunits), which was higher in hospitalized patients (Figures 3F, 3G, and S3G). Although the total frequency of SARS-CoV-2-reactive TFH cells (clusters 0, 5, and 7) showed a positive correlation with antibody levels in hospitalized COVID-19 patients, but not in non-hospitalized COVID-19 patients (Figure 3F), the relative proportions of cytotoxic TFH cells (TFH cells in cluster 5) showed a strong negative correlation with anti-S1/S2 antibody levels in hospitalized COVID-19 patients (Figure 3G and Table S3C). Conversely, the proportions of TFH cells in cluster 0 (non-cytotoxic) were positively correlated with antibody concentrations in hospitalized COVID-19 patients (Figure S3H). We noted that the magnitude of cytotoxic TFH response (cluster 5) also showed a significant negative correlation with the time interval between onset of illness and sample collection, suggesting that their association with antibody levels could be confounded by the timing of analysis of patients’ samples (Figure 3G and Table S3C). Furthermore, we did not observe this negative association between cytotoxic TFH cells and anti-S1/S2 antibody levels in non-hospitalized patients, which suggested that other mechanisms such as lower viral titers may explain the low levels of anti-S1/S2 antibodies in non-hospitalized patients. To further assess effects on B cell function, we analyzed B cells specific for SARS-CoV-2 spike protein (S1 and S2 subunits) from nine patients with varying proportion of cytotoxic TFH cells. Notably, in the hospitalized patients with high proportions of cytotoxic TFH cells (patients 08, 09, and 16), we observed a much smaller number of S1/S2-specific B cells compared to those with lower proportions of these cytotoxic TFH cells (Figure S3I). Future longitudinal studies that examine the kinetics of T and B cell responses to SARS-CoV-2 are likely to provide more definitive and time-resolved associations between cytotoxic TFH cell and antibody responses.
Next, to characterize upstream regulators that may induce the differentiation and maintenance of the cytotoxic TFH cells, we performed Ingenuity Pathway analysis (IPA) of the transcripts increased in SARS-CoV-2-reactive TFH cells in cluster 5 (cytotoxic) when compared to those in cluster 0 (Tables S3D and S3E). Surprisingly, we found that type 1 and 2 IFNs emerged as the top upstream activators of genes enriched in the cytotoxic TFH cluster (Figure S3J and Tables S3D and S3E). GSEA confirmed that IFN response signatures were also significantly enriched in the cytotoxic TFH cluster (cluster 5) (Figure S3K). Single-cell trajectory analysis showed that a large fraction of cytotoxic TFH cells (cluster 5) followed a separate trajectory from cluster 0 cells (Figure 3H), and cells in this track were enriched for the IFN response signature. In addition, we found that transcripts encoding perforin (PRF1) and the transcription factor ZBED2 were also enriched in the cytotoxic TFH cell trajectory, which suggested the hypothesis that ZBED2 may contribute to the differentiation or function of cytotoxic TFH cells, although further studies will be needed to verify this.
Massive Clonal Expansion of CD4-CTLs
While T cells with cytotoxic function are thought to predominantly consist of conventional MHC class I-restricted CD8+ T cells, MHC class II-restricted CD4+ T cells with cytotoxic potential (CD4-CTLs) have also been reported in several viral infections in humans and are associated with better clinical outcomes (Cheroutre and Husain, 2013; Juno et al., 2017; Meckiff et al., 2019; Weiskopf et al., 2015a). Paradoxically, in SARS-CoV-2 infection, we find that cells in the CD4-CTL clusters (Figure 4 A; cluster 6 and 9) were present at higher frequencies in some hospitalized COVID-19 patients compared to non-hospitalized patients, potentially contributing to disease severity, although we observed substantial heterogeneity in responses among patients (Figures 4B and 3A and Table S2B).
Figure 4.

SARS-CoV-2-Reactive CD4-CTLs and Single-Cell TCR Sequence Analysis
(A) UMAPs showing Seurat-normalized expression level of PRF1, GZMB, GNLY, and NKG7 transcripts in each virus-reactive cell.
(B) Percentage of CD4-CTLs (clusters 6 and 9) in the total SARS-CoV-2-reactive CD4+ T cell pool for non-hospitalized and hospitalized COVID-19 patients; dots indicate data from a single subject. Data are mean ± SEM; significance for comparisons was computed using Mann-Whitney U test; ns, non-significant P value..
(C) Violin plots showing normalized expression level (log2(CPM+1)) of transcription factors HOPX and ZEB2 and effector molecules CD72, GPR18, and SLAMF7 transcripts in virus-reactive cells from designated clusters (6 and 9) compared to an aggregation of remaining cells (Rest).
(D) UMAPs showing Seurat-normalized expression of CCL3, CCL4, CCL5, XCL1, and XCL2 transcripts in each virus-reactive cell.
(E) UMAP showing TCR clone size (log2, color scale) of SARS-CoV-2-reactive cells from COVID-19 patients (6 h stimulation condition).
(F) Histogram bar graph (top) displaying single-cell TCR sequence analysis of SARS-CoV-2-reactive cells. Each bar shows the number of TCRs shared between cells from individual clusters (rows, connected by lines). Connected lines (bottom) indicates what clusters are sharing TCRs. Clusters 6 (green), 9 (blue), and 11 (pink), i.e., CD4-CTLs, are highlighted.
(G) Single-cell trajectory analysis showing relationship between cells in different clusters (line), constructed using Monocle 3. Only SARS-CoV-2-reactive cells from COVID-19 patients (6 h stimulation condition) are shown.
Interrogation of the transcripts enriched in the CD4-CTL subsets pointed to several interesting molecules and transcription factors that are likely to play an important role in their maintenance and effector function. These include molecules like CD72 and GPR18 that are known to enhance T cell proliferation and maintenance of mucosal T cell subsets, respectively (Jiang et al., 2017; Wang et al., 2014) (Figures 4C and S4 A). Additional examples include transcription factors HOPX and ZEB2 (Figures 4C and S4A) that have been shown to positively regulate effector differentiation, function, persistence, and survival of T cells (Albrecht et al., 2010; Omilusik et al., 2015). Besides cytotoxicity-associated transcripts, the CD4-CTL subsets (clusters 6 and 9) and cytotoxic TFH cells (cluster 5) were highly enriched for transcripts encoding for a number of chemokines like CCL3 (also known as macrophage inflammatory protein [MIP]-1α), CCL4 (MIP-1β), and CCL5 (Figures 4D and S2F); these chemokines play an important role in the recruitment of myeloid cells (neutrophils, monocytes, macrophages), NK cells, and T cells expressing C-C type chemokine receptors (CCR)1, CCR3, and CCR5 (Hughes and Nibbs, 2018). The CD4-CTL subset in cluster 6 and cytotoxic TFH cells (cluster 5) also expressed high levels of transcripts encoding for chemokines XCL1 and XCL2 (Figures 4D, S4B, and S4C) that specifically recruit XCR1-expressing conventional type 1 dendritic cells (cDC1) to sites of immune responses where they play a key role in promoting the CD8+ T cell responses by antigen cross-presentation (Lei and Takahama, 2012). Overall, the transcriptomic features of SARS-CoV-2-reactive CD4-CTLs and cytotoxic TFH cells suggest that they are likely to play an important role in orchestrating immune responses by recruiting innate immune cells to enhance CD8+ T cell responses, while also directly mediating cytotoxic death of MHC class II-expressing virally infected cells.
Figure S4.

Single-Cell TCR Sequence Analysis and Analysis of SARS-CoV-2-Reactive CD4+ T Cells from 24 h Stimulation and Ex Vivo Conditions, Related to Figure 4
(A) Average expression and percent expression of selected transcripts in indicated clusters.
(B) Violin plots showing normalized expression level (log2(CPM+1)) of CCL3, CCL4, CCL5, XCL1, and XCL2 transcripts in designated clusters (6 and 9) compared to an aggregation of remaining cells (Rest).
(C) Scatterplots displaying co-expression level (log2(CPM+1)) of XCL1 and XCL2 transcripts in SARS-CoV-2-reactive cells present in designated clusters. Numbers indicate percentage of cells in each quadrant.
(D) Proportion of expanded SARS-CoV-2-reactive CD4+ T cells (clone size >2) in hospitalized and non-hospitalized COVID-19 patients (6 h stimulation condition). Data are mean ± S.E.M; significance for comparisons were computed using Mann-Whitney U test; ∗p < 0.05.
(E) Single-cell transcriptomes of memory CD4+ T cells expressing activation markers (CD38, HLA-DR, PD-1) ex vivo (0 h; blue) and sorted CD154+ CD69+ memory CD4+ T cells following 6 h stimulation with virus-specific peptide megapools (6 h; red) are displayed by UMAP. Seurat-based clustering of 122,292 cells.
(F) UMAP showing activation, TFH, and CD4-CTL signature scores for each cell.
(G) Violin plots showing expression level (log2(CPM+1)) of TNFRSF4, TNFRSF18, MIR155HG, CD200, IFNG, IL2, TNF, and POU2AF1 transcripts in 0- and 6 h time points.
(H) Number of cells from matched patients with shared (yellow) and unique (blue) TCRs between activation marker-positive cells sorted ex vivo (0 h) and 6 h peptide stimulated populations (left). Venn diagram illustrating the number of shared clones between activation marker-positive CD4+ T cells sorted ex vivo (0 h) and 6 h peptide stimulated populations.
The recovery of paired TCR sequences from individual single cells enabled us to link transcriptome data to clonotype information and evaluate the clonal relationship between different CD4+ T cell subsets as well as determine the nature of subsets that display greatest clonal expansion (Tables S4A and S4B). In SARS-CoV-2 infection, hospitalized patients were characterized by large clonal expansion of the virus-reactive CD4+ T cells (mean of 55.8%); in contrast, in non-hospitalized patients, recovered TCRs were less clonally expanded (mean of 38.0%) (Figure S4D). Among SARS-CoV-2-reactive CD4+ T cells, CD4-CTL subsets (clusters 6 and 9) displayed the greatest clonal expansion (> 75% of cells were clonally expanded), indicating preferential expansion and persistence of CD4-CTLs in some patients with COVID-19 illness (Figure 4E and Tables S4A and S4B). Analysis of clonally expanded SARS-CoV-2-reactive CD4+ T cells from COVID-19 patients showed extensive sharing of TCRs between cells in clusters 6 and 9, as well as those in cluster 11 (Figure 4F), which, notably, was enriched for the expression of XCL1 and XCL2 transcripts and also for cytotoxicity-associated transcripts, albeit at lower levels compared to the established CD4-CTL clusters (Figures 4D and S4C and Table S2F). Thus, cells in cluster 11 are likely to be an intermediate transition population, a hypothesis supported by single-cell trajectory analysis that showed potential temporal connection and transcriptional similarity between these subsets (Figure 4G).
Initial reports in patients with acute COVID-19 have suggested that circulating T cells that express activation markers such as CD38, HLA-DR, and PD-1 ex vivo (without in vitro peptide stimulation) are enriched for SARS-CoV-2-reactive T cells (Braun et al., 2020; Thevarajan et al., 2020). However, a recent study indicated that bystander T cells reactive to other antigens (e.g., CMV and EBV) can also express these activation markers, likely to be non-specifically activated without TCR engagement (Sekine et al., 2020). Thus, studies in active SARS-CoV-2 infection that just examine T cells expressing activation markers are not likely to reveal the full potential effector function of SARS-CoV-2-reactive T cells. To determine the specificity and molecular features of such T cells expressing activation markers ex vivo, we isolated CD38high HLA-DRhigh PD-1+ memory CD4+ T cells from hospitalized COVID-19 patients and performed single-cell transcriptome and TCR sequence analysis of >20,000 cells. CD4+ T cells expressing activation markers ex vivo clustered distinctly from the SARS-CoV-2-reactive CD4+ T cells, which were isolated following in vitro stimulation with SARS-CoV-2 peptides for 6 h (Figure S4E and Tables S2C–S2E, S4C, and S4D). The CD4+ T cells expressing activation markers ex vivo displayed reduced activation and TFH signature scores and had lower expression of transcripts encoding effector cytokines (IFN-γ, IL-2, TNFα), activation markers (OX40), and TFH associated genes (CD200, POU2AF1) (Figures S4F and S4G). Furthermore, by comparison of single-cell TCR sequences, we found that 33.8% of SARS-CoV-2-reactive CD4+ T cells shared clonotypes with CD4+ T cells expressing activation markers ex vivo, and 12.2% of CD4+ T cells expressing activation markers ex vivo shared their TCRs with SARS-CoV-2-reactive CD4+ T cells (Figure S4H and Tables S4E and S4F). Our findings indicate that using surface activation markers as a strategy to enrich for SARS-CoV-2-reactive T cells without SARS-CoV-2 peptide stimulation (ARTE assay) may not capture the full spectrum of SARS-CoV-2-reactive T cells, like TFH biology and their cytokine profiles, although the transcriptomic features of such in vitro activated cells may be affected by antigen-presenting cells present in the cultures.
SARS-CoV-2-Reactive TREG Cells Are Reduced in Hospitalized COVID-19 Patients
In order to capture SARS-CoV-2-reactive CD4+ T cells that may not upregulate the activation markers (CD154 and CD69) after 6 h of in vitro stimulation with SARS-CoV-2 peptide pools, we stimulated PMBCs from the same cultures for a total of 24 h (see STAR Methods) and captured cells based on co-expression of activation markers CD137 (4-1BB) and CD69, a strategy that allowed us to additionally capture antigen-specific regulatory T cells (TREG) (Bacher et al., 2016) (Figures 5 A and S5 A). Our analysis of a total of 38,519 single-cell CD4+ T cell transcriptomes revealed 6 distinct clusters (Figures 5A–5C and Tables S5A–S5C). The TFH subset (cluster D) was detectable at relatively lower frequencies in the 24 h condition, though they represented the major CD4+ T cell subsets in the 6 h stimulation condition (Figures 2A and 5A). Consistent with delayed kinetics of activation of central memory T (TCM) cells, we identified a higher proportion of CD4+ T cells expressing transcripts linked to central memory cells (CCR7, IL7R, and TCF7) (cluster C) (Figures 2A, 5A, and 5C).
Figure 5.

Analysis of SARS-CoV-2-Reactive CD4+ T Cells from 24 h Stimulation Condition
(A) Single-cell transcriptomes of sorted CD137+ CD69+ memory CD4+ T cells following 24 h stimulation with SARS-CoV-2-specific peptide megapools are displayed by UMAP. Seurat-based clustering of 38,519 cells colored based on cluster type.
(B) Heatmap showing expression of the most significantly enriched transcripts in each cluster (see Table S5C). Seurat marker gene analysis—comparison of cluster of interest versus all other cells—shown are top 200 transcripts with adjusted P value < 0.05, log2 fold change > 0.25, and > 10% difference in the percentage of cells expressing differentially expressed transcript between two groups compared.
(C) Plot showing average expression (color scale) and percent of expression (size scale) of selected marker gene transcripts in each cluster.
(D) UMAP showing Seurat-normalized expression level of FOXP3 transcripts (left). Percentage of TREG cells (cluster A) in the total SARS-CoV-2-reactive CD4+ T cell pool for non-hospitalized and hospitalized COVID-19 patients; dots indicate data from a single subject (right plot). Data are mean ± SEM; significance for comparisons was computed using Mann-Whitney U test; ∗∗∗p < 0.001.
(E) Average frequency of cells per cluster from hospitalized and non-hospitalized COVID-19 patients.
(F) UMAP showing CD4-CTL signature score for each cell (left) and percentage of CD4-CTLs (clusters B and F) in the total SARS-CoV-2-reactive CD4+ T cell pool for non-hospitalized and hospitalized COVID-19 patients; dots indicate data from a single subject (left plot). Data are mean ± SEM. Significance for comparisons was computed using Mann-Whitney U test; ns, non-significant P value.
(G) Correlation between percentage of SARS-CoV-2-reactive CD4+ TREG and percentage of SARS-CoV-2-reactive CD4-CTLs in 13 non-hospitalized and 17 hospitalized (left) COVID-19 patients. Correlation coefficient r and the related P value were computed using Spearman correlation; ∗∗∗∗p < 0.0001.
(H) UMAP showing Seurat-normalized expression level of IL1R2 transcripts (left) and percentage of TFR cells (IL1R2-expressing cells in cluster A) in the total SARS-CoV-2-reactive CD4+ T cell pool for non-hospitalized and hospitalized COVID-19 patients; dots indicate data from a single subject (left plot). Data are mean ± SEM; significance for comparisons were computed using Mann-Whitney U test; ∗∗∗p < 0.001.
(I) Correlation between percentage of SARS-CoV-2-reactive cytotoxic TFH cells (proportion of TFH cells in cluster 5, from 6 h stimulation dataset as in Figure 3C) and percentage of TFR cells (IL1R2-expressing cells in cluster A) in 25 COVID-19 patients (left). Correlation coefficient r was computed using Spearman correlation; ns, non-significant P value..
Figure S5.

Analysis of SARS-CoV-2-Reactive CD4+ T Cells from 24 h Stimulation Condition, Related to Figure 5
(A) Representative FACS plots showing surface staining of CD137 and CD69 in memory CD4+ T cells stimulated for 24 h with SARS-CoV-2 peptide pools, post-enrichment (CD137-based), in hospitalized and non-hospitalized COVID-19 patients (left). Summary of number of cells sorted in 14 hospitalized and 17 non-hospitalized COVID-19 patients (right); data are mean ± SEM.
(B) GSEA for TREG, cytotoxicity, TFH and TH17 signature genes in a given cluster compared to the rest of the cells; ∗∗p < 0.01; ∗∗∗p < 0.001.
(C) Unsupervised clustering of 17 hospitalized and 13 non-hospitalized COVID-19 patients based on the proportions of SARS-CoV-2-reactive CD4+ T cells in different clusters following 24 h peptide stimulation. Clusters with fewer than 5% of the total dataset are not depicted. Hospitalization status (red versus green) and sex (pink versus blue) are indicated in the annotation rows immediately below the dendrogram.
(D) UMAP showing TCR clone size (log2, color scale) of SARS-CoV-2-reactive cells from COVID-19 patients (24 h stimulation condition).
(E) Proportion of clonally expanded (clone size >2) and non-expanded cells in each cluster (24 h stimulation condition).
(F) GSEA for TFH and TFR signature genes in IL1R2+ cells compared to IL1R2– cells in cluster A; ∗p < 0.05; ∗∗∗p < 0.001.
The largest cluster (cluster A) was characterized by high expression of FOXP3 transcripts, which encodes for the TREG master transcription factor forkhead box P3 (FOXP3) (Rudensky, 2011) (Figures 5A–5D and Table S5C). Independent GSEA analysis showed significant positive enrichment of TREG signature genes in this cluster, suggesting that cells in this cluster represented SARS-CoV-2-reactive TREG cells (Figure S5B and Table S2H). Notably, the proportion of cells in the TREG cluster was significantly lower in hospitalized COVID-19 patients compared to non-hospitalized patients (Figures 5D, 5E, and S5C and Tables S5A and S5B), suggesting a potential defect in the generation of immunosuppressive SARS-CoV-2-reactive TREG cells in hospitalized patients. Consistent with our data from 6 h stimulation condition, we found that cells in the CD4-CTL clusters (clusters B and F) were present at higher frequencies in some hospitalized COVID-19 patients (Figures 5E, 5F, and S5C and Tables S5A and S5B). They also showed the greatest clonal expansion compared to other clusters (Figures S5D an S5E and Table S4B), suggesting potential importance of the CD4-CTL subset in driving immune responses to SARS-CoV-2 infection.
Correlation analysis of the proportion of CD4-CTLs and TREG in our 24 h dataset revealed a significant negative correlation, which indicated that patients with an impaired TREG response to SARS-CoV-2 mounted a stronger CD4-CTL response (Figure 5G and Table S5D). A recent study in a murine model showed that cytotoxic TFH responses are curtailed by a subset of TREG cells called follicular regulatory T (TFR) cells (Xie et al., 2019). To determine if such association is observed in our datasets, we first quantified TFR cells based on the expression of IL1R2 (Eschweiler et al., 2020) from cells in the TREG cluster A (Figure 5H). Independent GSEA confirmed that IL1R2-expressing cells were significantly enriched for follicular and TFR signature genes (Figure S5F), which indicated they represent TFR cells. Over 40% of the cells in the TREG cluster expressed IL1R2; this indicates that a strong circulating TFR response is generated in SARS-CoV-2 infection. Importantly, the proportion of TFR cells was significantly lower in hospitalized COVID-19 patients (Figure 5H) and showed a modest negative correlation with the proportion of cytotoxic TFH cells (Figure 5I and Table S5E). On the basis of these findings and the known function of these TREG subsets, we hypothesize that the magnitude of TREG and TFR responses to SARS-CoV-2 are likely to modulate cytotoxic CD4+ T and B cell responses in COVID-19 illness, although further studies are required to confirm this hypothesis.
Discussion
There is an urgent need to better understand the molecular determinants of protective and pathogenic immune responses in COVID-19. Given the importance of CD4+ T cells in anti-viral immunity, studying this adaptive immune cell population is likely to provide insights into the nature of host responses observed in patients with COVID-19. Current studies on antigen-specific CD4+ T cells are limited to flow-cytometry-based phenotyping of SARS-CoV-2-responding cells using limited sets of markers (Braun et al., 2020; Grifoni et al., 2020; Thieme et al., 2020), which thus fail to comprehensively capture the breadth of CD4+ T cells that respond to SARS-CoV-2. Unbiased approaches employing single-cell RNA-seq assays can provide these insights; however, to our knowledge, single-cell studies to date have only examined total CD4+ T cells in blood or bronchoalveolar lavage specimens from patients with COVID-19 illness (Vabret et al., 2020). Due to the rarity of SARS-CoV-2-specific cells in the total CD4+ T cell populations, signals from these cells are likely to be masked by the relative abundance of other non-antigen-specific CD4+ T cells. Furthermore, despite a profusion of single-cell transcriptomic studies, the analysis of virus-specific or any antigen-specific T cells, as such in humans, has lagged behind, partly due to the challenges imposed by methods to isolate antigen-specific T cells in sufficient numbers. Here, we have overcame these issues and performed single-cell transcriptomic study of >100,000 virus-reactive CD4+ T cells, focused on SARS-CoV-2-reactive cells from 40 COVID-19 patients with varying disease severity, and compared their molecular profile to CD4+ T cells reactive to other common respiratory viruses.
We find remarkable heterogeneity in the nature of CD4+ T cell subsets that are reactive to SARS-CoV-2 and other respiratory viruses and across individual patients and with differing severity of COVID-19. Polyfunctional TH1 cells, which are abundant among FLU-reactive CD4+ T cells and considered to be protective (Seder et al., 2008), were present in lower frequencies among SARS-CoV-2-reactive CD4+ T cells. Lower frequencies of TH17 cells were also observed among SARS-CoV-2-reactive CD4+ T cells. In contrast, we find increased proportions of SARS-CoV-2-reactive cytotoxic TFH cells in hospitalized COVID-19 patients. Cytotoxic TFH cells can kill B cells and dampen germinal center responses (Xie et al., 2019), and to our knowledge this is the first description of circulating cytotoxic TFH cells in humans. Importantly, the magnitude of the cytotoxic TFH response to SARS-CoV-2 was stronger early in the course of illness and negatively correlated with antibody levels to SARS-CoV-2 S. Recent reports have found that patients with fatal COVID-19 infections have abrogated germinal center B cell responses (Kaneko et al., 2020) and very slightly reduced levels of S-specific IgM antibodies (Atyeo et al., 2020), the mechanistic basis of which is not known. Our findings of strong cytotoxic TFH responses early in the illness may provide the link to defects in B cells responses in some patients with severe and fatal COVID-19 illness.
Another striking observation is the abundance in SARS-CoV-2-reactive CD4+ T cells of CD4-CTLs that express high levels of transcripts encoding for multiple chemokines (XCL1, XCL2, CCL3, CCL4, and CCL5), particularly from some hospitalized COVID-19 patients. This suggests that the CD4-CTL responses in COVID-19 illness may be linked to pathogenesis, although further studies in animal models and large-scale association studies in COVID-19 patients are required to verify or refute this hypothesis. Notably, some hospitalized COVID-19 patients showed impaired TREG response to SARS-CoV-2, and such patients mounted a strong CD4-CTL response, raising another interesting association that warrants testing in larger studies.
Limitations and Future Directions
The limitation of this study is the relatively small sample size considering the heterogeneity observed in the nature of CD4+ T cell responses to SARS-CoV-2. Analysis of patients in the acute and convalescent phase of illness fails to discriminate effector and long-term memory CD4+ T cell responses. Serial sampling of the same patients in the recovered phase is likely to provide insights into the nature and persistence of memory CD4+ T cell responses to SARS-CoV-2.
Because the negative association between cytotoxic TFH cell responses and anti-spike antibody levels was not observed in non-hospitalized patients, the potential role of cytotoxic TFH cells in antibody responses cannot be generalized. Furthermore, the higher proportions of cytotoxic TFH cells in hospitalized patients may merely reflect higher viral titers and IFN production. Longitudinal studies are required to clarify the association between aberrant cytotoxic TFH responses and their impact on modulating the magnitude and duration of protective antibody responses to SARS-CoV-2. The role of CD4-CTLs in protective or pathogenic immune responses to SARS-CoV-2 needs to be clarified in pre-clinical models. Future studies in COVID-19 patients should also examine the relationships between the subsets of SARS-CoV-2-reactive CD4+ T cells in the blood and those observed in the mucosal tissues where control of SARS-CoV-2 infection is critical.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 | Biolegend | SK7; RRID: AB_10640737 |
| CD3 | Biolegend | UCHT1; RRID: AB_314060 |
| CD4 | Biolegend | OKT4; RRID: AB_2561866 |
| CD8a | Biolegend | RPA-T8; RRID: AB_314134 |
| CD8b | eBioscience | SIDI8BEE; RRID: AB_2762625 |
| CD14 | Biolegend | HCD14; RRID: AB_830693 |
| CD14 | Biolegend | M5E2; RRID: AB_493695 |
| CD19 | Biolegend | HIB19; RRID: AB_314248 |
| CD27 | Biolegend | M-T271; RRID: AB_2561825 |
| CD38 | Biolegend | HIT-2; RRID: AB_2072782 |
| CD38 | Biolegend | SK1; RRID: AB_2564510 |
| CD40 (blocking) | Miltenyi Biotec | HB14; RRID: AB_10839704 |
| CD45 | BD Bioscience | HI30; RRID: AB_2744399 |
| CD45RA | Biolegend | HI100; RRID: AB_493763 |
| CD56 | Biolegend | HCD56; RRID: AB_10896424 |
| CD69 | Biolegend | FN50; RRID: AB_2563696 |
| CD137 (4-1BB) | Biolegend | 4B4-1; RRID: AB_2566258 |
| CD137 (4-1BB) | Miltenyi Biotec | REA765; RRID: AB_2654994 |
| CD138 | Biolegend | MI15; RRID: AB_2562899 |
| CD154 (CD40L) | Biolegend | 24-31; RRID: AB_314829 |
| CD154 (CD40L) | Miltenyi Biotec | 5C8; RRID: AB_2751206 |
| CD197 (CCR7) | BD Bioscience | 3D12; RRID: AB_2744306 |
| CD279 (PD-1) | BD Bioscience | EH12.1; RRID: AB_2739399 |
| CD298 (β2M; TotalSeq-C) | Biolegend | LNH-94; 2M2; RRID: AB_2801031, AB_2801032, AB_2801033, AB_2820042, AB_2820043, AB_2820044, AB_2820045, AB_2820046 |
| HLA-DR | BD Bioscience | G46-6; RRID: AB_2732846 |
| IgD | Biolegend | IA6-2; RRID: AB_2563269 |
| IgG | Biolegend | M1310G05; RRID: AB_2565788 |
| IgM | Biolegend | MHM-88; RRID: AB_2562916 |
| Ki-67 | BD Bioscience | B56; RRID: AB_2732007 |
| Nucleoprotein | Sino Biological | |
| S1/S2 Protein | Sino Biological | |
| Biological Samples | ||
| Cryopreserved PBMCs from hospitalized and non-hospitalized COVID-19 patients | Southampton University Hospital | |
| Cryopreserved PBMCs from healthy non-exposed subjects | San Diego Blood Bank | |
| Cryopreserved PBMCs from subjects before and/or after receiving flu vaccination | La Jolla Institute for Immunology | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Peptivator SARS-CoV-2 Prot M (membrane glycoprotein) | Miltenyi Biotec | 130-126-703 |
| Peptivator SARS-CoV-2 Prot S (spike glycoprotein) | Miltenyi Biotec | 130-126-701 |
| SARS-CoV-2 megapools (CD4-R and CD4-S) | La Jolla Institute for Immunology - Sette | |
| Human Parainfluenza (HPIV) megapool | La Jolla Institute for Immunology - Sette | |
| Human Metapneumovirus (HMPV) megapool | La Jolla Institute for Immunology - Sette | |
| Human Influenza (HA) megapool | La Jolla Institute for Immunology - Sette | |
| Fixable Viability Dye eFluor 780 | eBioscience | C34557 |
| Deposited Data | ||
| Sequencing Data | Gene Expression Omnibus | GSE152522 |
| Software and Algorithms | ||
| Flowjo v10 | Flowjo | https://www.flowjo.com/ |
| Prism 8 | Graphpad | https://www.graphpad.com |
| Cellranger v3.1.0 | 10x Genomics | https://www.10xgenomics.com |
| Seurat v3.1.5 | (Stuart et al., 2019) | https://www.satijalab.org/seurat |
| R v3.6.1 | R Core team | www.R-project.org |
| UpSetR v1.4.0 | (Conway et al., 2017) | https://github.com/hms-dbmi/UpSetR |
| Monocle3 v0.2.1 | (Trapnell et al., 2014) | https://cole-trapnell-lab.github.io/monocle3/ |
| MAST v1.10.0 | (Finak et al., 2015) | https://www.bioconductor.org/packages/release/bioc/html/MAST.html |
| FGSEA | (Korotkevich et al., 2019) | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Other | ||
| Chromium Single Cell 5¢ Feature Barcode Library | 10x Genomics | 1000080 |
| Chromium Single Cell 5¢ Library & Gel Bead Kit | 10x Genomics | 1000006 |
| Chromium Single Cell 5¢ Library Construction Kit | 10x Genomics | 1000020 |
| Chromium Single Cell C(D)J Enrichment Kit, Human T Cell | 10x Genomics | 1000005 |
| Chromium Single Cell Chip A Kit | 10x Genomics | 1000151 |
| Chromium i7 Multiplex Kit | 10x Genomics | 120262 |
| Chromium i7 Multiplex Kit N, Set A | 1000084 | |
| TexMACS | Miltenyi Biotec | 130-097-19 |
Resource Availability
Lead contact
Further information and requests for reagents may be directed to the lead contact, Pandurangan Vijayanand (vijay@lji.org).
Materials availability
SARS-CoV-2, Human Influenza (FLU), Parainfluenza (HPIV) and Metapneumovirus (HMPV) epitope pools utilized in this paper will be made available to the scientific community upon request and execution of a material transfer agreement (MTA) directed to Dr. Alessandro Sette (alex@lji.org). There might be restrictions on the availability of the peptide reagents due to cost and limited quantity
Data and Code availability
Scripts are available in our repository on GitHub (https://github.com/vijaybioinfo/COVID19_2020). Sequencing data for this study has been deposited onto the Gene Expression Omnibus with the accession number GSE152522.
Experimental Model and Subject Details
COVID-19 patients and samples
Ethical approval for this study from the Berkshire Research Ethics Committee 20/SC/0155 and the Ethics Committee of La Jolla Institute for Immunology (LJI) was in place. Written consent was obtained from all subjects. 22 hospitalized patients in a large teaching hospital in the south of England with SARS-CoV-2 infection, confirmed by reverse transcriptase polymerase chain reaction (RT-PCR) assay for detecting SARS-CoV-2, between April-May 2020 were recruited to the study. A further cohort of 18 participants consisting of healthcare workers who were not hospitalized with COVID-19 illness, confirmed based on RT-PCR assay or serological evidence of SARS-CoV-2 antibodies, were also recruited over the same period. All subjects provided up to 80 mL of blood for research studies. Clinical and demographic data were collected from patient records for hospitalized patients including comorbidities, blood results, drug intervention, radiological involvement, thrombotic events, microbiology, and virology results (Table S1A). The 22 hospitalized patients had a median age of 60 (33-82), 17 of these patients (77%) were men and this cohort consisted of 16 (73%) White British/White Other, 4 (18%) Indian, and 2 (9%) Black British patients. All hospitalized patients survived to discharge from hospital. All hospitalized patients were still symptomatic at time of blood collection, whereas some of the non-hospitalized patients (4/18) were symptom free (Table S1A). The 18 non-hospitalized participants had a median age of 39 (22-50), 8 (44%) of these participants were men and this cohort consisted of 15 (83%) White British/White Other, 2 (11%) Arab, and 1 (6%) Chinese participant. We noted that the median age of the non-hospitalized patients was lower than the hospitalized COVID-19 patients.
Healthy controls
To study HPIV, HMPV, and SARS-CoV-2-reactive CD4+ T cells from healthy non-exposed subjects (pre-COVID-19 pandemic), we utilized de-identified buffy coat samples from 5 healthy adult donors who donated blood at the San Diego Blood Bank before 2019, prior to the Covid-19 pandemic. Donors were considered to be in good health, free of cold or flu-like symptoms and with no history of Hepatitis B or Hepatitis C infection. The median age was 50 (32-71) and 4 of these patients (80%) were men. To study FLU-reactive cells, we obtained de-identified blood samples from 8 donors enrolled in the LJI Normal Blood Donor Program before and/or after (12 - 14 days) receiving the FLUCELVAX vaccine (September and October 2019). The median age was 37 (26-57) and 5 of these patients (63%) were women. Approval for the use of this material was obtained from the LJI Ethics Committee.
Method Details
PBMC processing
Peripheral blood mononuclear cells (PBMCs) were isolated from up to 80ml of anti-coagulated blood by density centrifugation over Lymphoprep (Axis-Shield PoC AS, Oslo, Norway) and cryopreserved in 50% decomplemented human antibody serum, 40% complete RMPI 1640 medium and 10% DMSO.
SARS-CoV-2 peptide pools
Pools of lyophilized peptides covering the immunodominant sequence of the spike glycoprotein and the complete sequence of the membrane glycoprotein of SARS-CoV-2 (15-mer sequences with 11 amino acids overlap) were obtained from Miltenyi Biotec (Thieme et al., 2020) resuspended and stored according to the manufacturer’s instructions.
SARS-CoV-2 antibody testing
The LIAISON SARS-CoV-2 S1/S2 IgG (DiaSorin S.p.A., Saluggia, Italy) was utilized as per the manufacturer’s instructions to obtain quantitative antibody results from plasma samples via an indirect chemiluminescence immunoassay (CLIA) in a United Kingdom Accreditation Service (UKAS) diagnostic laboratory at University Hospital Southampton. Sample results were interpreted as positive (≥ 15 AU/mL), Equivocal (≥ 12.0 and < 15.0 AU/mL) and negative (< 12 AU/mL).
SARS-CoV-2 spike protein-specific B cell responses
To assess the level of SARS-CoV-2 S1/S2-specific B cells, cells were prepared in staining buffer (PBS with 2% FBS and 2 mM EDTA), FcγR blocked (clone 2.4G2, BD Biosciences), stained with indicated primary antibodies and biotinylated S1/S2 proteins (Sino Biological) for 30 min at 4°C; washed, and subsequently stained with streptavidin-BV421. Patients 10, 24 and 49 were analyzed on a different day with a lower intensity violet laser and required different gating.
Epitope Megapool of peptide (MP) design
The Human Parainfluenza (HPIV) and Metapneumovirus (HMPV) CD4+ T cell peptide megapools (MPs) were produced by sequential lyophilization of viral-specific epitopes as previously described (Carrasco Pro et al., 2015, Weiskopf et al., 2015b). Full lists of the viral protein sequences derived from the uniprot database and used for the HPIV and HMPV MP designs are available in Table S1F. T cell prediction was performed using TepiTool tool, available in identification epitope database analysis resources (IEDB-AR, LJI), applying the 7-allele prediction method and a median cutoff ≤20 (Dhanda et al., 2019, Paul et al., 2015, Paul et al., 2016). For the HA-influenza MP, we selected 177 experimentally defined epitopes, retrieved by querying the IEDB database (www.IEDB.org) on 07/12/19 with search parameters “positive assay only, No B cell assays, No MHC ligand assay, Host: Homo Sapiens and MHC restriction class II.” The list of epitopes was enriched with predicted peptides derived from the HA sequences of the vaccine strains available in 2017-2018 and 2018-2019 (A/Michigan/45/2015(H1N1), B/Brisbane/60/2008, A/Hong_Kong/4801/2014(H3N2), A/Michigan/45/2015(H1N1), A/Alaska/06/2016(H3N2), B/Iowa/06/2017, and B/Phuket/3073/2013). The resulting peptides were then clustered using the IEDB cluster 2.0 tool and the IEDB recommended method (cluster-break method) with a 70% cut off for sequence identity applied (Dhanda et al., 2019, Dhanda et al., 2018) (Table S1E). Peptides were synthesized as crude material (A&A, San Diego, CA), resuspended in DMSO, pooled according to each MP composition and finally sequentially lyophilized (Carrasco Pro et al., 2015). For screening healthy non-exposed subjects (samples provided before the current pandemic) who cross-react to SARS-CoV-2, we screened 20 healthy non-exposed subjects using SARS-CoV-2 peptide CD4-R and CD4-S pools, as described (Grifoni et al., 2020).
Antigen-reactive T cell enrichment (ARTE) assay
Enrichment and FACS sorting of virus-reactive CD154+ CD4+ memory T cells following peptide pool stimulation was adapted from Bacher et al. 2016 (Bacher et al., 2016). Briefly, PBMCs from each donor, were thawed, washed, plated in 24-well culture plates at a concentration of 5 × 106 cells/mL in 1 mL of serum-free TexMACS medium (Miltenyi Biotec) and left overnight (5% CO2, 37 °C). Cells were stimulated by the addition of individual virus-specific peptide pools (1 μg/mL) for 6 h in the presence of a blocking CD40 antibody (1 μg/mL; Miltenyi Biotec). For subsequent MACS-based enrichment of CD154+, cells were sequentially stained with fluorescence-labeled surface antibodies (antibody list in Table S1G), Cell-hashtag TotalSeq-C antibody (0.5 μg/condition), and a biotin-conjugated CD154 antibody (clone 5C8; Miltenyi Biotec) followed by anti-biotin microbeads (Miltenyi Biotec). Labeled cells were added to MS columns (Miltenyi Biotec) and positively selected cells (CD154+) were eluted and used for FACS sorting of CD154+ memory CD4+ T cells. The flow-through from the column was collected and re-plated to harvest cells responding 24 h after peptide stimulation. Analogous to enrichment for CD154+, CD137-expressing CD4+ memory T cells were positively selected by staining with biotin-conjugated CD137 antibody (clone REA765; Miltenyi Biotec) followed by anti-biotin MicroBeads and applied to a new MS column. Following elution, enriched populations were immediately sorted using a FACSAria Fusion Cell Sorter (Becton Dickinson) based on dual expression of CD154 and CD69 for the 6 h stimulation condition, and CD137 and CD69 for the 24 h stimulation condition. The gating strategy used for sorting is shown in Figures S1A and S4B. All flow cytometry data were analyzed using FlowJo software (version 10).
Cell isolation and single-cell RNA-seq assay (10x platform)
For combined single-cell RNA-seq and TCR-seq assays (10x Genomics), a maximum of 60,000 virus-reactive memory CD4+ T cells from up to 8 donors were pooled by sorting into low retention 1.5 mL collection tubes, containing 500 μl of a 1:1 solution of PBS:FBS supplemented with recombinant RNase inhibitor (1:100, Takara). For healthy donors, when possible, equal numbers of cells were isolated from each donor and pooled before 10x Genomics single-cell RNA-seq experiments. For analysis of FLU-reactive CD4+ T cell responses, we sequenced paired pre- and post-vaccination samples from 4 donors and supplemented this with 2 non-paired samples for both pre- and post-vaccination. Samples from both pre- and post-vaccination were pooled for analysis of FLU-reactive CD4+ T cells. Following sorting, ice-cold PBS was added to make up to a volume of 1400 μl. Cells were then centrifuged for 5 min (600 g at 4 °C) and the supernatant was carefully removed leaving 5 to 10 μl. 25 μl of resuspension buffer (0.22 μm filtered ice-cold PBS supplemented with ultra-pure bovine serum albumin; 0.04%, Sigma-Aldrich) was added to the tube and the pellet was gently but thoroughly resuspended. Following careful mixing, 33 μl of the cell suspension was transferred to a PCR-tube for processing as per the manufacturer’s instructions (10x Genomics).
Briefly, single-cell RNA-sequencing library preparation was performed as per the manufacturer’s recommendations for the 10x Genomics 5' TAG v1.0 chemistry with immune profiling and cell surface protein technology. Both initial amplification of cDNA and library preparation were carried out with 13 cycles of amplification; V(D)J and cell surface protein libraries were generated corresponding to each 5'' TAG gene expression library using 9 cycles and 8 cycles of amplification, respectively. Libraries were quantified and pooled according to equivalent molar concentrations and sequenced on Illumina NovaSeq6000 sequencing platform with the following read lengths: read 1 – 101 cycles; read 2 – 101 cycles; and i7 index - 8 cycles.
Single-cell transcriptome analysis
Reads from single-cell RNA-seq were aligned and collapsed into Unique Molecular Identifiers (UMI) counts using 10x Genomics’ Cell Ranger software (v3.1.0) and mapped to GRCh37 reference (v3.0.0) genome. Hashtag UMI counts for each TotalSeq-C antibody capture library were generated with the Feature Barcoding Analysis pipeline from Cell Ranger. To demultiplex donors, UMI counts of cell barcodes were first obtained from the raw data output, and only cells with at least 100 UMI for the hashtag with the highest UMI counts were considered for donor assignment. Donor identities were inferred by MULTIseqDemux (autoThresh = TRUE and maxiter = 10) from Seurat (v3.1.5) using the UMI counts. Each cell barcode was assigned a donor ID, marked as a Doublet or having a Negative enrichment. Cells were re-classified as doublets if the ratio of UMI counts between the top 2 barcodes was less than 3. Cells labeled as Doublet or Negative were removed from downstream analyses. Raw 10x data were independently aggregated using Cell Ranger’s aggr function (v3.1.0). Donors P28 and P48 were not stained with hashtag antibodies and therefore did not contribute to any donor-specific data. The merged data was transferred to the R statistical environment for analysis using the package Seurat (v3.1.5) (Stuart et al., 2019). To further minimize doublets and to eliminate cells with low quality transcriptomes, cells expressing < 800 and > 4400 unique genes, < 1500 and > 20,000 total UMI content, and > 10% of mitochondrial UMIs were excluded. The summary statistics for all the single-cell transcriptome libraries are provided in Table S2C-E and indicate good quality data with no major differences in quality control metrics across multiple batches, where batches are groups of donors whose libraries were sequenced together (Figure S2A). This procedure was independently applied for data from CD4+ T cells stimulated for 0 and 6 h, 6 and 24 h.
For single-cell transcriptome analysis only genes expressed in at least 0.1% of the cells were included. The transcriptome data was then log-transformed and normalized (by a factor of 10,000) per cell, using default settings in Seurat software (Stuart et al., 2019). Variable genes with a mean UMI expression greater than 0.01 and explaining 25% of the total variance were selected using the Variance Stabilizing Transformation method, as described (Stuart et al., 2019). Transcriptomic data from each cell was then further scaled by regressing the number of UMI-detected and percentage of mitochondrial counts. For data from CD4+ T cells stimulated for 6 h, principal component analysis was performed using the variable genes, and based on the standard deviation of PCs in the “elbow plot,” the first 38 principal components (PCs) were selected for further analyses. Cells were clustered using the FindNeighbors and FindClusters functions in Seurat with a resolution of 0.6. The robustness of clustering was independently verified by other clustering methods and by modifying the number of PCs and variable genes utilized for clustering. Analysis of clustering patterns across multiple batches revealed no evidence of strong batch effects (Figure S2A). For data from CD4+ T cells stimulated for 24 h, the first 16 PCs were selected for further analyses. Cluster 6 (G) in the 24 h dataset was merged with cluster 0 (A) after being identified as TREG. For 0 and 6 h aggregation analysis, 30 PCs were taken. Finally, cells were clustered using the FindNeighbors and FindClusters functions in Seurat with a resolution of 0.6 and 0.2 for 6 and 0 h aggregation and 24 h, respectively. Further visualizations of exported normalized data such as UMAP or “violin” plots were generated using the Seurat package and custom R scripts. Violin shape represents the distribution of cell expressing transcript of interest (based on a Gaussian Kernel density estimation model) and are colored according to the percentage of cells expressing the transcript of interest.
Single-cell differential gene expression analysis
Pairwise single-cell differential gene expression analysis was performed using the MAST package in R (v1.8.2) (Finak et al., 2015) after conversion of data to log2 counts per million (log2(CPM + 1)). A gene was considered differentially expressed when Benjamini-Hochberg adjusted P-value was < 0.05 and a log2 fold change was more than 0.25. For finding cluster markers (transcripts enriched in a given cluster) the function FindAllMarkers from Seurat was used.
Gene Set Enrichment Analysis and Signature Module Scores
GSEA scores were calculated with the package fgsea in R using the signal-to-noise ratio (or the log2 fold change for cluster 5 versus cluster 0 comparison) as a metric. Gene sets were limited by minSize = 3 and maxSize = 500. Normalized enrichment scores were presented as GSEA plots. Signature module scores were calculated with AddModuleScore function, using default settings in Seurat. Briefly, for each cell, the score is defined by the mean of the signature gene list after the mean expression of an aggregate of control gene lists is subtracted. Control gene lists were sampled (same size as the signature list) from bins created based on the level of expression of the signature gene list. Gene lists used for analysis are provided in Table S2H.
Single-cell trajectory analysis
The “branched” trajectory was constructed using Monocle 3 (v0.2.1, default settings) (Trapnell et al., 2014) with the number of UMI, percentage of mitochondrial UMI as the model formula and including the highly variable genes from Seurat for consistency. After setting a single partition for all cells, the cell-trajectory was projected on the PCA and UMAP generated from Seurat analysis. The ‘root’ was selected by the get_earliest_principal_node function provided in the package. Monocle 3 alpha was used to analyze cluster 0 and 5 using the DDRTree algorithm for dimensional reduction after selecting the top 500 highly variable genes with Seurat.
T cell receptor (TCR) sequence analysis
Reads from single-cell V(D)J TCR sequence enriched libraries (Table S2D) were processed with the vdj pipeline from Cell Ranger (v3.1.0 and human annotations reference GRCh38, v3.1.0, as recommended). In brief, the V(D)J transcripts were assembled and their annotations were obtained for each independent library. In order to perform combined analysis of single-cell transcriptome and TCR sequence from the same cells, V(D)J libraries were first aggregated using a custom script. Then cell barcode suffixes from these libraries were revised according to the order of their gene expression libraries. Unique clonotypes, as defined by 10x Genomics as a set of productive Complementarity-Determining Region 3 (CDR3) sequences, were identified across all library files and their frequency and proportion (clone statistics) were calculated based on the aggregation result considering only the cells present in the gene expression libraries. This procedure was independently applied for data from CD4+ T cells stimulated for 6 and 24 h. Based on the vdj aggregation files, barcodes captured by our gene expression data and previously filtered to keep only good-quality cells, were annotated with a specific clonotype ID alongside their clone size (number of cells with the same clonotypes in either one or both the TCR alpha and beta chains) and other statistics (Table S4A,B,E and F). Cells that share clonotype with more than 1 cell were called as clonally expanded (clone size >2). Clone size for each cell was visualized on UMAP, depicting only SARS-CoV-2-reactive CD4+ T cells. Sharing of clonotype between cells in different clusters was depicted using the tool UpSetR (Conway et al., 2017). Finally, in order to assess the sharing between the 0- and 6 h datasets, the same aggregation process was applied for all of the vdj libraries from these data and only SARS-CoV-2-reactive CD4+ T cells specifically isolated from matched patients between sets were considered.
Quantification and Statistical Analysis
Processing of data, applied methods and codes are described in the respective section in the STAR Methods. The number of subjects, samples, replicates analyzed, and the statistical test performed are indicated in the figure legends or STAR methods. Statistical analysis for comparison between two groups were assessed with Mann Whitney U test and correlation assessed with spearman test with using GraphPad Prism.
Acknowledgments
We thank Luke Smith for patient recruitment and sample collection; Callum Dixon, Benjamin Johnson, Lydia Scarlett, and Silvia Austin for collection of clinical data; Céline Galloway, Oliver Wood, Katy McCann, and Lindsey Chudley for sample processing; and Sharon Gilchrist for the illustration. We thank the La Jolla Institute (LJI) Flow Cytometry Core for assisting with cell sorting and the LJI’s Clinical Studies Core for organizing sample collection. We thank Peter Friedmann and Anusha Preethi Ganesan for providing critical feedback on the manuscript. This work was funded by NIH grants U19AI14274, U19AI142742-0S1 (to P.V., A.S., and C.H.O.), U19AI118626 (to P.V., A.S., and G.S.), R01HL114093 (to P.V., F.A., and G.S.), R35-GM128938 (F.A), S10RR027366 (BD FACSAria-II), and S10OD025052 (Illumina Novaseq6000); the William K. Bowes, Jr. Foundation (P.V.); and Whittaker Foundation (C.H.O.). Supported by the Wessex Clinical Research Network and National Institute for Health Research UK.
Author Contributions
B.J.M., S.J.C., C.H.O., and P.V. conceived the work. B.J.M., S.J.C, C.H.O., and P.V. designed the study and wrote the manuscript. S.J.C. supervised patient identification, recruitment, sample collection, and processing. E.P. supervised the analysis of viral PCR and serology tests. A.G., D.W., and A.S. provided essential reagents for the isolation of viral-reactive CD4+ T cells. B.J.M. and A.K. performed ARTE assay and FACS sorting, S.E. performed B cell analyses, and H.S. performed single-cell RNA-sequencing under the supervision of G.S., C.H.O., and P.V. C.R.-S. and V.F. performed bioinformatic analyses under the supervision of G.S., F.A., C.H.O., and P.V.
Declaration of Interests
The authors declare no competing interests.
Published: October 5, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.10.001.
Supplemental Information
References
- Acharya D., Wang P., Paul A.M., Dai J., Gate D., Lowery J.E., Stokic D.S., Leis A.A., Flavell R.A., Town T., et al. Interleukin-17A Promotes CD8+ T Cell Cytotoxicity To Facilitate West Nile Virus Clearance. Journal of Virology. 2016;91 doi: 10.1128/JVI.01529-16. e01529–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht I., Niesner U., Janke M., Menning A., Loddenkemper C., Kühl A.A., Lepenies I., Lexberg M.H., Westendorf K., Hradilkova K., et al. Persistence of effector memory Th1 cells is regulated by Hopx. European Journal of Immunology. 2010;40:2993–3006. doi: 10.1002/eji.201040936. [DOI] [PubMed] [Google Scholar]
- Atyeo C., Fischinger S., Zohar T., Slein M.D., Burke J., Loos C., Mcculloch D.J., Newman K.L., Wolf C., Yu J., et al. Distinct Early Serological Signatures Track with Sars-Cov-2 Survival. Immunity. 2020;53:524–532.e4. doi: 10.1016/j.immuni.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher P., Schink C., Teutschbein J., Kniemeyer O., Assenmacher M., Brakhage A.A., Scheffold A. Antigen-reactive T cell enrichment for direct, high-resolution analysis of the human naive and memory Th cell repertoire. J Immunol. 2013;190:3967–3976. doi: 10.4049/jimmunol.1202221. [DOI] [PubMed] [Google Scholar]
- Bacher P., Heinrich F., Stervbo U., Nienen M., Vahldieck M., Iwert C., Vogt K., Kollet J., Babel N., Sawitzki B., et al. Regulatory T Cell Specificity Directs Tolerance Versus Allergy against Aeroantigens in Humans. Cell. 2016;167:1067–1078.e16. doi: 10.1016/j.cell.2016.09.050. [DOI] [PubMed] [Google Scholar]
- Bacher P., Hohnstein T., Beerbaum E., Rocker M., Blango M.G., Kaufmann S., Rohmel J., Eschenhagen P., Grehn C., Seidel K., et al. Human Anti-Fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida Albicans. Cell. 2019;176:1340–1355.e15. doi: 10.1016/j.cell.2019.01.041. [DOI] [PubMed] [Google Scholar]
- Bentebibel S.E., Lopez S., Obermoser G., Schmitt N., Mueller C., Harrod C., Flano E., Mejias A., Albrecht R.A., Blankenship D., et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5:176ra32. doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J., Loyal L., Frentsch M., Wendisch D., Georg P., Kurth F., Hippenstiel S., Dingeldey M., Kruse B., Fauchere F., et al. Sars-Cov-2-Reactive T Cells in Healthy Donors and Patients with Covid-19. Nature. 2020 doi: 10.1038/s41586-020-2598-9. [DOI] [PubMed] [Google Scholar]
- Carrasco Pro S., Sidney J., Paul S., Lindestam Arlehamn C., Weiskopf D., Peters B., Sette A. Automatic Generation of Validated Specific Epitope Sets. J Immunol Res. 2015;2015:763461. doi: 10.1155/2015/763461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroutre H., Husain M.M. CD4 CTL: living up to the challenge. Semin Immunol. 2013;25:273–281. doi: 10.1016/j.smim.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway J.R., Lex A., Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 2017;33:2938–2940. doi: 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan J.M., Havenar-Daughton C., Kendric K., Al-Kolla R., Kaushik K., Rosales S.L., Anderson E.L., Larock C.N., Vijayanand P., Seumois G., et al. Recurrent Group A Streptococcus Tonsillitis Is an Immunosusceptibility Disease Involving Antibody Deficiency and Aberrant Tfh Cells. Sci Transl Med. 2019;11:Eaau3776. doi: 10.1126/scitranslmed.aau3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda S.K., Vaughan K., Schulten V., Grifoni A., Weiskopf D., Sidney J., Peters B., Sette A. Development of a novel clustering tool for linear peptide sequences. Immunology. 2018;155:331–345. doi: 10.1111/imm.12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda S.K., Mahajan S., Paul S., Yan Z., Kim H., Jespersen M.C., Jurtz V., Andreatta M., Greenbaum J.A., Marcatili P., et al. IEDB-AR: immune epitope database-analysis resource in 2019. Nucleic Acids Res. 2019;47(W1):W502–W506. doi: 10.1093/nar/gkz452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschweiler S., Clarke J., Ramirez Suastegui C., Panwar B., Madrigal A., Chee S., Karydis I., Woo E., Alzetani A., Elsheikh S. T Fr Cells Inhibit Anti-Tumor Immunity and Are Responsive to Immune Checkpoint Blockade. Ssrn. 2020 doi: 10.2139/Ssrn.3596586. [DOI] [Google Scholar]
- Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi, et al. Targets of T Cell Responses to Sars-Cov-2 Coronavirus in Humans with Covid-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.Y., Lin Y.C., Hsiao W.Y., Liao F.H., Huang P.Y., Tan T.H. DUSP4 deficiency enhances CD25 expression and CD4+ T-cell proliferation without impeding T-cell development. Eur J Immunol. 2012;42:476–488. doi: 10.1002/eji.201041295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C.E., Nibbs R.J.B. A guide to chemokines and their receptors. FEBS J. 2018;285:2944–2971. doi: 10.1111/febs.14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Björkström N.K., Melum E. Intact CD100-CD72 Interaction Necessary for TCR-Induced T Cell Proliferation. Front Immunol. 2017;8:765. doi: 10.3389/fimmu.2017.00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juno J.A., van Bockel D., Kent S.J., Kelleher A.D., Zaunders J.J., Munier C.M. Cytotoxic CD4 T Cells-Friend or Foe during Viral Infection? Front Immunol. 2017;8:19. doi: 10.3389/fimmu.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko N., Kuo H.-H., Boucau J., Farmer J.R., Allard-Chamard H., Mahajan V.S., Piechocka-Trocha A., Lefteri K., Osborn M., Bals J., et al. The Loss of Bcl-6 Expressing T Follicular Helper Cells and the Absence of Germinal Centers in Covid-19. Cell. 2020;183:143–157. doi: 10.1016/j.cell.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G., Sukhov V., Sergushichev A. Fast gene set enrichment analysis. BioRxiv. 2019 doi: 10.1101/060012. [DOI] [Google Scholar]
- Koutsakos M., Wheatley A.K., Loh L., Clemens E.B., Sant S., Nussing S., Fox A., Chung A.W., Laurie K.L., Hurt et al Circulating Tfh Cells, Serological Memory, and Tissue Compartmentalization Shape Human Influenza-Specific B Cell Immunity. Sci Transl Med. 2018;10:Eaan8405. doi: 10.1126/scitranslmed.aan8405. [DOI] [PubMed] [Google Scholar]
- Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- Lei Y., Takahama Y. XCL1 and XCR1 in the immune system. Microbes Infect. 2012;14:262–267. doi: 10.1016/j.micinf.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Li H., Van Der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., Van Akkooi A.C.J., Van Den Braber M., Rozeman E.A., Haanen J., Blank C.U., et al. Dysfunctional Cd8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2019;176:775–789.e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locci M., Havenar-Daughton C., Landais E., Wu J., Kroenke M.A., Arlehamn C.L., Su L.F., Cubas R., Davis M.M., Sette A., et al. International AIDS Vaccine Initiative Protocol C Principal Investigators Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–769. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W.T., Yao X.T., Peng Q., Chen D.K. The protective and pathogenic roles of IL-17 in viral infections: friend or foe? Open Biol. 2019;9:190109. doi: 10.1098/rsob.190109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meckiff B.J., Ladell K., McLaren J.E., Ryan G.B., Leese A.M., James E.A., Price D.A., Long H.M. Primary EBV Infection Induces an Acute Wave of Activated Antigen-Specific Cytotoxic CD4+ T Cells. J Immunol. 2019;203:1276–1287. doi: 10.4049/jimmunol.1900377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill R.E., Du W., Mohammadpour H., Alqassim E., Qiu J., Chen G., McCarthy P.L., Lee K.P., Cao X. T Cell-Derived CD70 Delivers an Immune Checkpoint Function in Inflammatory T Cell Responses. J Immunol. 2017;199:3700–3710. doi: 10.4049/jimmunol.1700380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omilusik K.D., Best J.A., Yu B., Goossens S., Weidemann A., Nguyen J.V., Seuntjens E., Stryjewska A., Zweier C., Roychoudhuri R., et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med. 2015;212:2027–2039. doi: 10.1084/jem.20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil V.S., Madrigal A., Schmiedel B.J., Clarke J., O’rourke P., De Silva A.D., Harris E., Peters B., Seumois G., Weiskopf D., Sette A., Vijayanand P. Precursors of Human Cd4(+) Cytotoxic T Lymphocytes Identified by Single-Cell Transcriptome Analysis. Sci Immunol. 2018;3:Eaan8664. doi: 10.1126/sciimmunol.aan8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S., Lindestam Arlehamn C.S., Scriba T.J., Dillon M.B., Oseroff C., Hinz D., McKinney D.M., Carrasco Pro S., Sidney J., Peters B., Sette A. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 2015;422:28–34. doi: 10.1016/j.jim.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S., Sidney J., Sette A., Peters B. Tepitool: A Pipeline for Computational Prediction of T Cell Epitope Candidates. Curr Protoc Immunol. 2016;114:18.19.1–18.19.24. doi: 10.1002/cpim.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza F., Costoya J.A., Merghoub T., Hobbs R.M., Pandolfi P.P. Disruption of PLZP in mice leads to increased T-lymphocyte proliferation, cytokine production, and altered hematopoietic stem cell homeostasis. Mol Cell Biol. 2004;24:10456–10469. doi: 10.1128/MCB.24.23.10456-10469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensky A.Y. Regulatory T cells and Foxp3. Immunol Rev. 2011;241:260–268. doi: 10.1111/j.1600-065X.2011.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F. Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol. 2016;34:317–334. doi: 10.1146/annurev-immunol-032414-112056. [DOI] [PubMed] [Google Scholar]
- Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., Mcvicker G., Seumois G., Rao A., Kronenberg M., Peters B., Vijayanand P. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell. 2018;175:1701–1715.e16. doi: 10.1016/j.cell.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder R.A., Darrah P.A., Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.-B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild Covid-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H.M., Kapoor V.N., Kim G., Li P., Kim H.R., Suresh M., Kaech S.M., Wherry E.J., Selin L.K., Leonard W.J., et al. Transient expression of ZBTB32 in anti-viral CD8+ T cells limits the magnitude of the effector response and the generation of memory. PLoS Pathog. 2017;13:e1006544. doi: 10.1371/journal.ppat.1006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits M., Zoldan K., Ishaque N., Gu Z., Jechow K., Wieland D., Conrad C., Eils R., Fauvelle C., Baumert T.F., et al. Follicular T helper cells shape the HCV-specific CD4+ T cell repertoire after virus elimination. J Clin Invest. 2020;130:998–1009. doi: 10.1172/JCI129642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville T.D.D., Xu Y., Wu X.S., Maia-Silva D., Hur S.K., de Almeida L.M.N., Preall J.B., Koo P.K., Vakoc C.R. ZBED2 is an antagonist of interferon regulatory factor 1 and modifies cell identity in pancreatic cancer. Proc Natl Acad Sci USA. 2020;117:11471–11482. doi: 10.1073/pnas.1921484117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902 E21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevarajan I., Nguyen T.H.O., Koutsakos M., Druce J., Caly L., van de Sandt C.E., Jia X., Nicholson S., Catton M., Cowie B., et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med. 2020;26:453–455. doi: 10.1038/s41591-020-0819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieme C., Anft M., Paniskaki K., Blázquez Navarro A., Doevelaar A., Seibert F.S., Hoelzer B., Konik M.J., Brenner T., Tempfer C. The Sars-Cov-2 T-Cell Immunity Is Directed against the Spike, Membrane, and Nucleocapsid Protein and Associated with Covid 19 Severity. Ssrn. 2020 doi: 10.2139/Ssrn.3606763. [DOI] [Google Scholar]
- Thommen D.S., Schumacher T.N. T Cell Dysfunction in Cancer. Cancer Cell. 2018;33:547–562. doi: 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., et al. Sinai Immunology Review Project Immunology of COVID-19: Current State of the Science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Chan C.C., Yang M., Deng J., Poon V.K., Leung V.H., Ko K.H., Zhou J., Yuen K.Y., Zheng B.J., Lu L. A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cell Mol Immunol. 2011;8:462–468. doi: 10.1038/cmi.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Sumida H., Cyster J.G. GPR18 is required for a normal CD8αα intestinal intraepithelial lymphocyte compartment. J Exp Med. 2014;211:2351–2359. doi: 10.1084/jem.20140646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D., Bangs D.J., Sidney J., Kolla R.V., De Silva A.D., de Silva A.M., Crotty S., Peters B., Sette A. Dengue virus infection elicits highly polarized CX3CR1+ cytotoxic CD4+ T cells associated with protective immunity. Proc Natl Acad Sci USA. 2015;112:E4256–E4263. doi: 10.1073/pnas.1505956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D., Cerpas C., Angelo M.A., Bangs D.J., Sidney J., Paul S., Peters B., Sanches F.P., Silvera C.G., Costa P.R., et al. Human CD8+ T-Cell Responses Against the 4 Dengue Virus Serotypes Are Associated With Distinct Patterns of Protein Targets. J Infect Dis. 2015;212:1743–1751. doi: 10.1093/infdis/jiv289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M.M., Fang S., Chen Q., Liu H., Wan J., Dent A.L. Follicular regulatory T cells inhibit the development of granzyme B-expressing follicular helper T cells. JCI Insight. 2019;4:E128076. doi: 10.1172/jci.insight.128076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts are available in our repository on GitHub (https://github.com/vijaybioinfo/COVID19_2020). Sequencing data for this study has been deposited onto the Gene Expression Omnibus with the accession number GSE152522.