

Abstract
MICU1 is a mitochondrial inner membrane protein that inhibits mitochondrial calcium entry; elevated MICU1 expression is characteristic of many cancers, including ovarian cancer. MICU1 induces both glycolysis and chemoresistance and is associated with poor clinical outcomes. However, there are currently no available interventions to normalize aberrant MICU1 expression. Here, we demonstrate that microRNA‐195‐5p (miR‐195) directly targets the 3′ UTR of the MICU1 mRNA and represses MICU1 expression. Additionally, miR‐195 is under‐expressed in ovarian cancer cell lines, and restoring miR‐195 expression reestablishes native MICU1 levels and the associated phenotypes. Stable expression of miR‐195 in a human xenograft model of ovarian cancer significantly reduces tumor growth, increases tumor doubling times, and enhances overall survival. In conclusion, miR‐195 controls MICU1 levels in ovarian cancer and could be exploited to normalize aberrant MICU1 expression, thus reversing both glycolysis and chemoresistance and consequently improving patient outcomes.
Keywords: chemoresistance, glycolysis, MICU1/CBARA1, miR‐195, Ovarian cancer
Subject Categories: Cancer, RNA Biology
miR‐195 represses MICU1 expression in ovarian cancer, thus reversing both glycolysis and chemoresistance and consequently improving patient survival. miR‐195 might thus be a potential tool for ovarian cancer therapy.
Introduction
Metabolic reprogramming is characteristic of cancer cells, which generally shift toward aerobic glycolysis in order to meet the high energy demands of their uncontrolled proliferation (Lunt & Vander Heiden, 2011; Ward & Thompson, 2012). This shift to glycolysis is recognized as an important determinant of both tumor growth and chemoresistance in various malignancies (Ganapathy‐Kanniappan & Geschwind, 2013; Morandi & Indraccolo, 2017). As a result, glycolytic pathways represent viable targets for therapeutic intervention, and several anti‐glycolytic agents, targeting different enzymes, have been used to sensitize tumor cells to therapy (Fanciulli et al, 2000; Xu et al, 2005; Hsu & Sabatini, 2008; Dang et al, 2011; Birsoy et al, 2012; Dang, 2012; Wagner et al, 2015). Unfortunately, however, the ubiquity of enzymes involved in glycolysis results in systemic toxicity when broad‐spectrum anti‐glycolytic agents are used, which limits their utility (Ganapathy‐Kanniappan & Geschwind, 2013). Thus, while inhibition of glycolysis remains a promising strategy for anti‐cancer interventions, better‐targeted treatment options are required before it can have a significant clinical impact.
Mitochondrial calcium uptake 1 (MICU1) is a protein on the inner mitochondrial membrane that interacts with the mitochondrial calcium uniporter (MCU), a pore‐forming unit to modulate mitochondrial Ca2+ (mCa2+) uptake in response to cytosolic Ca2+ concentration [Ca2+]c (Mishra et al, 2017; Paupe & Prudent, 2018). Disulfide‐linked hetero‐oligomerization of MICU1 and MICU2 regulates MCU activity depending on the cytosolic Ca2+ levels. MICU1 acts both as an activator or inhibitor of m(Ca2+) uptake (Patron et al, 2014; Matesanz‐Isabel et al, 2016). Mallilankaraman et al (2012b) have demonstrated that the mitochondrial protein MICU1 is required to maintain normal mCa2+ levels under basal conditions (Nemani et al, 2020). In its absence, mitochondria become constitutively loaded with Ca2+, triggering excessive reactive oxygen species (ROS) generation, and sensitivity to apoptotic stress. MICU1 interacts with the uniporter pore‐forming subunit MCU and sets a Ca2+ threshold for mCa2+ uptake without affecting the kinetic properties of MCU‐mediated Ca2+ uptake (Mallilankaraman et al, 2012b). Thus, MICU1 is a gatekeeper of MCU‐mediated mCa2+ uptake that is essential to prevent mCa2+ overload and associated stress. We recently demonstrated that overexpression of MICU1 in ovarian cancer cells induced both glycolysis and chemoresistance and also showed that overexpression of MICU1 in ovarian cancer patients correlated with poor overall survival (Chakraborty et al, 2017). Recent reports show that both intracellular Ca2+ homeostasis and MICU1 expression are altered in a range of cancer cells and also contribute to poor prognosis in cancer patients (Mallilankaraman et al, 2012b; Hall et al, 2014; Chakraborty et al, 2017; Monteith et al, 2017). Thus, Ca2+ homeostasis and, more specifically, MICU1 represent potential targets for interventional strategies to address the glycolytic status of cancer cells. Targeting of MICU1 would overcome the systemic toxicity associated with the use of broad‐spectrum inhibitors by exploiting its overexpression in cancer cells compared to normal cells. Understanding how the expression of MICU1 is regulated in cancer cells is an important step in developing strategies to suppress its activities and thus reverse glycolysis and sensitize drug‐resistant cells to therapy. However, apart from regulation by FOXD1 during development and AKT‐mediated phosphorylation in tumor models (Shanmughapriya et al, 2018; Marchi et al, 2019a), regulation of MICU1 is not well understood.
MicroRNAs (miRNAs) are small evolutionarily conserved single‐stranded noncoding RNAs (19–25 nucleotides in length) that post‐transcriptionally regulate the translation and stability of mRNAs. miRNAs are implicated in many cellular processes including cell cycle, apoptosis, autophagy, stemness, differentiation, inflammation, drug resistance, transformation, and migration, as well as Ca2+ homeostasis and oxidative phosphorylation (Ambros, 2004; Iorio & Croce, 2012; Hayes et al, 2014). Additionally, miRNAs are frequently deregulated in cancer and play a key role in the regulation of cancer‐associated glycolytic pathways (Dang, 2010; Gao et al, 2012; Gomez de Cedron & Ramirez de Molina, 2016). miR‐195 has been shown to be a tumor suppressor in several cancer models including breast (Singh et al, 2015), hepatocellular (Yang et al, 2014), gastric (Deng et al, 2013), and lung cancers (Yu et al, 2018). Additionally, serum levels of miR‐195 are a diagnostic and prognostic marker for osteosarcoma (Cai et al, 2015), and downregulation of miR‐195 is associated with both lymph node metastasis and poor prognosis in colorectal cancer patients (Wang et al, 2012). However, little is known about the role of miR‐195 in regulating MICU1 expression in ovarian cancer.
Herein, we show that miR‐195 regulates expression of MICU1 in ovarian cancer. Normalizing MICU1 expression by ectopic expression of miR‐195 increases mCa2+ uptake, reverses glycolysis, inhibits tumor growth, and increases overall survival in a human xenograft model of ovarian cancer. With the advent of therapeutic miRNAs, some of which are advancing from bench to clinic (Rupaimoole & Slack, 2017), our findings provide an opportunity to exploit miR‐195 as a potential therapeutic option to normalize MICU1 expression thereby reversing glycolysis and enhancing therapeutic sensitivity, and ultimately leading to improved patient outcomes.
Results
miR‐195 regulates expression of MICU1
As an initial step in identifying miRNAs as potential regulators of MICU1, we used the web‐interface of miRWalk2 (Dweep & Gretz, 2015). miRWalk2 is a software tool that predicts miRNA targets utilizing twelve different algorithms (miRWalk, Microt4, miRanda, mirbridge, miRDB, miRMap, miRNAMap, Pictar2, PITA, RNA22, RNAhybrid, and Targetscan) and predicted a significant number of miRNAs which could potentially target MICU1. Those miRNAs predicted to target MICU1 by more than nine prediction methods are shown in Table 1 and those predicted to bind the 3′ UTR of MICU1 with a minimum seed length of 7 bp and P < 0.05 by 12 different programs are shown in Table EV1. miRNAs of the miR‐15 family (namely miR‐15a/b, miR‐16, miR‐195, and miR‐497) predominated among those predicted to interact at the MICU1 3′ UTR positions (Table 1). Several members of this miR‐15 family, including miR‐15a/b, miR‐16, miR‐195, and miR‐497, have demonstrable associations with ovarian cancer (Xu et al, 2015; Dwivedi et al, 2016). Based on bioinformatics analysis, we selected two representative members of the miR‐15 family, miR‐15a‐5p (miR‐15a), and miR‐195‐5p (miR‐195) and evaluated their effect on MICU1 expression. Since miRNAs modulate gene expression by either modulating mRNA stability or by inhibiting translation, we determined target protein abundance by immunoblotting. We assayed two ovarian cancer cell lines transfected with either a non‐target miR‐control (miR‐CTL) or one of the target miRNAs, miR‐15a, or miR‐195. While miR‐195 significantly decreased the expression of MICU1 in both cell lines, miR‐15a had no effect (Figs 1A and EV1A). However, miR‐15a did reduce expression of its established targets BMI1 and BCL2 (Cimmino et al, 2005; Dwivedi et al, 2016), thereby demonstrating that it was ectopically expressed and active in the studied cell lines (Figs 1A and EV1A). Similarly, miR‐195 decreased the expression of its known target BCL2 (Singh & Saini, 2012) indicating the specificity of targeting in these cells (Figs 1A and EV1A). miR‐195‐mediated regulation of MICU1 was further confirmed in OVCAR4, CP20, and OVSAHO cell lines; efficient transfection of miR‐195 and reduced expression of MICU1 in these cells were, respectively, demonstrated by RT–qPCR and immunoblotting (Fig EV1B and C). These data conclusively showed that the MICU1 mRNA was targeted by miR‐195 but not by miR‐15a.
Table 1.
miRNAs predicted to bind the 3′ end of MICU1 with a minimum seed length of 7, by different prediction methods
| miRNA | miRWalk | Microt4 | miRanda | mirbridge | miRDB | miRMap | Pictar2 | PITA | RNA22 | RNAhybrid | Targetscan | SUM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hsa‐miR‐195‐5p | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | 9 |
| hsa‐miR‐15a‐5p | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | 9 |
| hsa‐miR‐15b‐5p | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | 9 |
| hsa‐miR‐424‐5p | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | 9 |
| hsa‐miR‐497‐5p | Y | Y | Y | N | N | Y | Y | Y | Y | Y | Y | 9 |
| hsa‐miR‐1206 | Y | Y | Y | N | Y | Y | N | Y | Y | Y | Y | 9 |
| hsa‐miR‐589‐5p | Y | Y | Y | N | Y | Y | N | Y | Y | Y | Y | 9 |
| #of predictions for MICU1 | 2,794 | 429 | 1,649 | 2 | 69 | 4,032 | 48 | 222 | 409 | 14,632 | 2,571 |
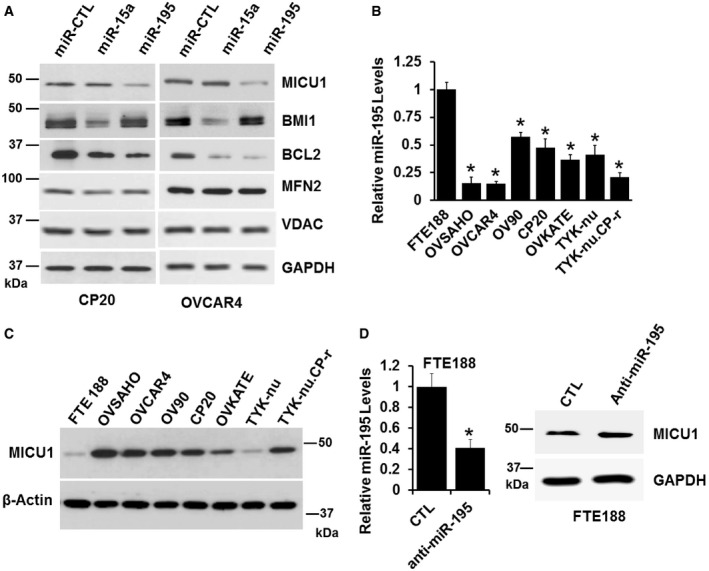
Figure 1. miR‐195 and MICU1 levels are inversely related in ovarian cancer cell lines.
-
ACP20 and OVCAR4 cells were transfected with either non‐target miRNA control (miR‐CTL), miR‐15a, or miR‐195. Seventy‐two hours following transfection, cells were lysed and immunoblotted for detection of MICU1, BMI1, BCL2, and MFN2. GAPDH and VDAC were used as loading control.
-
BmiR‐195 expression in FTE188 and ovarian cell lines as determined by RT–qPCR normalized with U6. Data are mean ± SD, n = 3 (biological repeats). *P < 0.05, Student's t‐test.
-
CExpression of MICU1 in FTE188 and various ovarian cell lines as determined by immunoblotting. Actin is used as the loading control.
-
DAnti‐miR-195 was transfected in the FTE188 cell line, level of miR‐195 was measured using RT–qPCR (left) and MICU1 levels were measured using immunoblotting (right). Data are mean ± SD, n = 3 (biological repeats). *P < 0.05, Student's t‐test.
Figure EV1. miR‐195 regulates MICU1 expression.

-
AGraph showing densitometric quantification of Western blot bands for CP20 and OVCAR4 cells in Fig 1A (n = 3, biological repeats mean ± SE) normalized with GAPDH. *P < 0.05, Student's t‐test.
-
BOvarian cancer cells were transfected with either non‐target microRNA (miR‐CTL) or miR‐195. Total RNA was isolated and miR‐195 levels normalized with U6 were plotted. Data are mean ± SD, n = 3 biological repeats, *P < 0.05, Student's t‐test.
-
CMICU1 expression in cells transfected with miR‐CTL and miR‐195. β‐Actin was used as the loading control.
-
DGraph showing densitometric quantification of Western blot bands for ovarian cancer cells in Fig 1C. Values are represented as mean fold change ± SD, n = 3 biological repeats, *P < 0.05, Student's t‐test.
Based on these findings, we next determined the relationship between the endogenous cellular expression of miR‐195 and MICU1 levels. First, we used RT–qPCR to determine miR‐195 levels; compared to non‐malignant fallopian tube epithelial cells (FTE188), the expression of miR‐195 was significantly lower in seven different ovarian cancer cell lines (Fig 1B). We then determined MICU1 protein levels in the same cell lines; in six of the ovarian cancer cell lines, MICU1 levels were significantly higher than in the non‐malignant FTE188 cells (Figs 1C and EV1D). In TYK‐nu cells, MICU1 levels were similar to those in FTE188 cells, suggesting complexity in MICU1 regulation in addition to that mediated by miR‐195 in these particular cells (Figs 1C and EV1D). However, in general, MICU1 protein levels were inversely related to miR‐195 expression in ovarian cancer cells. Targeting of endogenous MICU1 by miR‐195 was confirmed by ectopic expression of anti‐miR‐195 in FTE188 cells, which resulted in increased MICU1 levels. The targeting of miR‐195 through anti‐miR‐195 in FTE188 cells was confirmed using RT–qPCR (Fig 1D). Taken together these data indicate that miR‐195 targets the MICU1 mRNA thereby reducing expression of MICU1.
Expression of miR‐195 regulates cancer‐associated cellular phenotypes: clonal growth, invasion, and migration
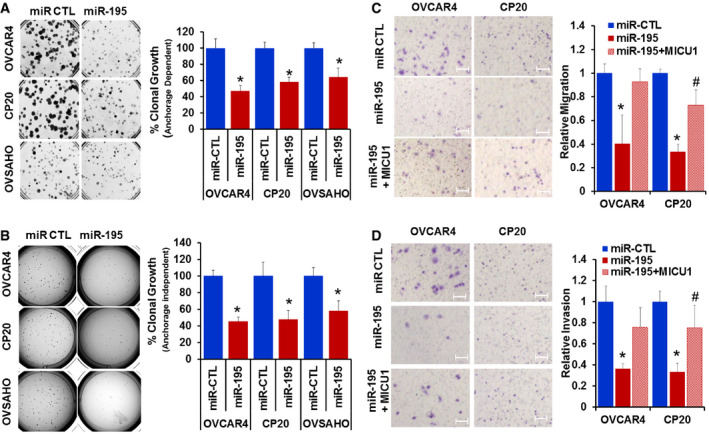
Having demonstrated that miR‐195 regulates expression of MICU1, we next sought to determine the role of miR‐195 in regulating several cellular phenotypes responsible for aggressiveness in cancer, specifically clonal growth, invasion, and migration. Three cell lines were selected for these experiments: OVCAR4 and OVSAHO for their characteristic miR‐195 and MICU1 expression levels, as well as CP20 for its cisplatin‐resistant phenotype. OVCAR4, OVSAHO, and CP20 cells were transfected with miR‐195; efficient transfection was confirmed using RTqPCR (Fig EV2). Compared to the controls, a significant decrease in clonal growth occurred in all the miR‐195‐transfected cells. The anchorage‐dependent clonal growth of OVCAR4, CP20, and OVSAHO was decreased, respectively, by 53, 42, and 36% and in the anchorage‐independent assay, the clonal growth of these cells was respectively reduced by of 55, 52, and 42%. (Fig 2A and B). Having demonstrated a role for miR‐195 in regulating clonal growth, we next assessed the significance of miR‐195 ectopic expression on the migratory and invasive potential of CP20 and OVCAR4 cells. In a Boyden chamber‐based migration and invasion assay, miR‐195‐transfected OVCAR4 and CP20 cells had decreased migration by 60 and 67%, (Fig 2C) and invasion by 64 and 67%, respectively (Fig 2D), when compared to control. To confirm that the decrease in the migratory and invasive potential of ovarian cancer cells was mediated through MICU1, we ectopically expressed a miR‐195 non‐responsive MICU1 construct (i.e., pLYS1‐MICU1‐Flag, with only the MICU1 coding sequence and thus not a target for miR‐195) in miR‐195‐transfected cells. Ectopic expression of MICU1 in miR‐195‐transfected cells rescued the migratory and invasive potential of these cells to the levels of non‐targeted miR‐control‐transfected cells (Fig 2C and D). Thus, re‐expression of miR‐195 inhibits clonal growth as well as cellular migration and invasion, all of which are hallmarks of cancer progression (Hanahan & Weinberg, 2011), and this is mediated through MICU1.
Figure EV2. Quantitation of microRNA expression.
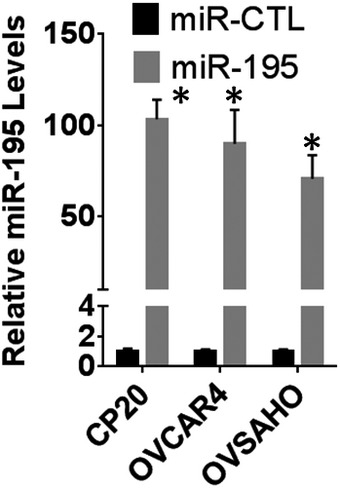
Ovarian cancer cells were transfected with either non‐target microRNA (miR‐CTL) or miR‐195. Total RNA was isolated and miR‐195 levels normalized with U6 were plotted. Data are mean ± SD, n = 3 biological repeats, *P < 0.05, Student's t‐test.
Figure 2. Ectopic expression of miR‐195 suppresses ovarian cancer clonal growth, migration, and invasion.
-
A, BCP20, OVCAR4, and OVSAHO cells were transfected with non‐target microRNA control (miR‐CTL) or miR‐195; 24 h post‐transfection, cells were recounted and plated as single cells for anchorage‐dependent (A) or anchorage‐independent (B) clonal growth. After 10 (CP20) or 14 (OVCAR4 and OVSAHO) days, colonies were quantified using an Optronix GelCount colony counter. The left panel shows representative images of the colonies and the right panel depicts a graphical representation of data presented as percent clonal growth relative to the control (miR‐CTL).
-
CMigration of miR‐CTL, miR‐195, and miR‐195 + MICU1 (pLYS1‐MICU1‐Flag) transfected cells, toward serum gradient was examined and the number of cells per field was counted. The left panel shows representative images and the right panel depicts a graphical representation of relative migration compared to the control (miR‐CTL).
-
DInvasion of miR‐CTL, miR‐195, and miR‐195 + MICU1 cells through fibronectin‐coated filters was examined using Boyden chamber and number of cells per field was counted. Left panel shows representative images and the right panel depicts a graphical representation of relative invasion compared to the control (miR‐CTL).
miR‐195 targets the 3′ UTR of MICU1
Having demonstrated that miR‐195 regulates expression of MICU1, we next sought to determine whether this regulation was mediated by a direct interaction between miR‐195 and the MICU1 mRNA. The putative interaction site of miR‐195 on the MICU1 3′ UTR was mapped using TargetScan (Fig 3A), and site‐directed mutagenesis was used to delete this putative binding site from the luciferase construct. The MICU1 3′ UTR luciferase construct was used to perform reporter assays and showed that re‐expression of miR‐195 dose‐dependently decreased MICU1 3′ UTR luciferase activity while no change was noted with a non‐target miRNA control (Fig 3B). Deletion of the proposed miR‐195 interaction site on the MICU1 3′ UTR almost completely reversed miR‐195‐mediated inhibition of luciferase activity (Fig 3B). Collectively, these results show that miR‐195 directly targets the MICU1 3′ UTR.
Figure 3. MICU1 is a direct target of miR‐195.
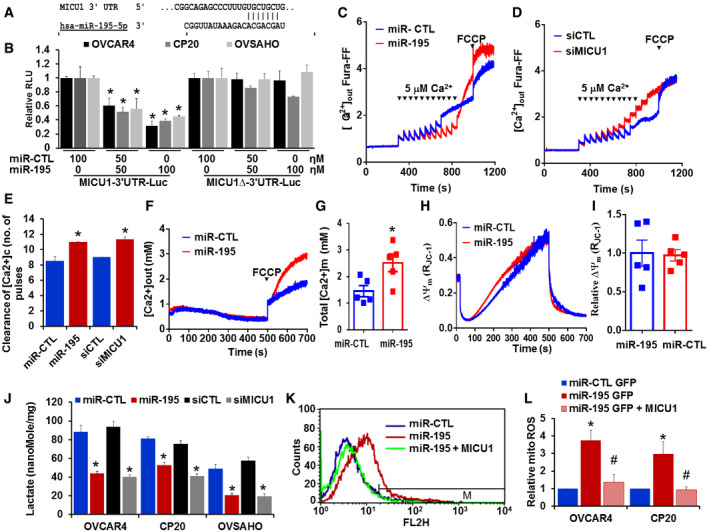
-
AThe miR‐195 binding site as predicted by TargetScan.
-
BMICU1 3′ UTR in LightSwitch™ 3′ UTR Reporter Vector (wild type or miR‐195 binding site deleted) was co‐transfected with either non‐target miR‐control (miR‐CTL) or different concentrations of miR‐195. After 24‐h transfection, luciferase (Renilla) activity was measured. Relative light units (RLU) compared to miR‐CTL were plotted. Data are mean ± SD n = 3 (biological repeats). *P < 0.05, Student's t‐test.
-
C, DCP20 cells were transfected as indicated, and 48 h post‐transfection, 4 × 106 cells were permeabilized, and extra‐mitochondrial calcium ([Ca2+]out) clearance was measured, representative traces of [Ca2+]out‐ clearance in miR‐CTL and miR‐195 or control siRNA (siCTL) and siMICU1 transfected cells. [Ca2+]out pulses and FCCP were delivered as indicated.
-
EBar graph illustrating the number of Ca2+ pulses handled by control miR, miR‐195, siCTL, and siMICU1 transfected cells. Mean ± SEM; n = 3–4 (biological repeats). *P < 0.05, Student's t‐test.
-
FRepresentative [Ca2+](m) traces after addition of FCCP (10 μM) in permeabilized CP20 cells transfected with non‐target miR‐CTL or miR‐195.
-
GQuantification of resting matrix [Ca2+](m) after addition of FCCP. n = 5 (biological repeats) *P < 0.05, Student's t‐test.
-
HRepresentative traces of mitochondrial membrane potential (∆Ψμ) in CP20 cells transfected with miR‐CTL or miR‐195.
-
IQuantification of relative ∆Ψμ (n = 5, biological repeats).
-
JIntracellular lactate was measured in miR‐CTL, miR‐195, siCTL, and siMICU1 expressing OVCAR4, CP20, and OVSAHO cells. n = 3, biological repeats *P < 0.05, Student's t‐test.
-
K, LmiR‐CTL, miR‐195, or miR‐195 + pLYS1‐MICU1‐Flag transfected cells were stained with MitoSOX Red, and mitochondrial ROS levels were determined by flow cytometry. The histogram shows representative staining and bar graph (right) shows results of three independent experiments. Mean ± SEM; n = 3, biological repeats. *P < 0.05 when comparing with miR‐CTL, # P < 0.05 when comparing with miR‐195 by Student's t‐test.
MICU1 mediates the effects of miR‐195
As discussed above, MICU1 is an important regulator of intracellular Ca2+ homeostasis. Mallilankaraman et al (2012b) and Csordas et al (2013) revealed the importance of MICU1 in preserving normal mCa2+ under basal conditions. When MICU1 levels are high, as in cancer, mitochondrial uptake of Ca2+ is inhibited (Chakraborty et al, 2017; Marchi et al, 2019a,b). Since miR‐195 targets the 3′ UTR of the MICU1 gene thus reducing MICU1 levels, we sought to determine the effect of the miR‐195 expression on mCa2+ homeostasis. In theory, re‐expression of miR‐195 will normalize MICU1 expression in ovarian cancer cells and thus will promote Ca2+ entry into the mitochondria (Mallilankaraman et al, 2012b). We determined mCa2+ uptake capacity in miR‐195 re‐expressing CP‐20 cells that were digitonin‐permeabilized. The cells were bathed in an intracellular‐like medium containing Fura‐FF to monitor the Ca2+ levels in the medium (Mallilankaraman et al, 2012a). Starting at 300 s, boluses of Ca2+ were added at 50‐s intervals; in response to repeated Ca2+ administration, cytosolic Ca2+ was cleared and mitochondrial uptake was increased. Compared to the control, CP20 cells expressing miR‐195 demonstrated increased mCa2+ (Fig 3C and E). Having shown that miR‐195 increased mCa2+ uptake in ovarian cancer cells, we next sought to determine if the silencing of MICU1 achieved the same result. We used siRNA to silence MICU1 in CP20 cells and showed that lack of MICU1 increased mCa2+ uptake (Fig 3D and E). Efficacy of MICU1 inhibition by miR‐195 and siMICU1 is shown in Fig EV3. It is noteworthy that both miR‐195 overexpression and siRNA‐mediated knockdown of MICU1 enhanced mitochondrial Ca2+ retention capacity (Fig 3C–E). Although the extra‐mitochondrial Ca2+ pulses in MICU1 siRNA‐treated condition were stacking after 9 pulses, the mitochondrial Ca2+ uptake rate (1/τ) was measurable (Control = −0.193; MICU1 siRNA = 0.03171), but it was absent in control cells suggesting that MICU1 silencing enhances mitochondrial Ca2+ retention capacity. These data further reinforce that miR‐195 acts via its effect on MICU1 to regulate mCa2+. The effect of miR‐195 on mitochondrial membrane potential and basal mitochondrial matrix Ca2+ concentration (Total [Ca2+]m) was evaluated in miR‐195‐transfected CP20 cells. We found that there was a significant increase in total [Ca2+]m (Fig 3F and G) without any effect on ∆Ψm (Fig 3H and 3I). To confirm that the change in [Ca2+]m is mediated through MICU1 and not BCL‐2, cCa2+ was evaluated. When miR‐195 was over‐expressed, there was no appreciable change in cCa2+ in CP20 cells (Fig EV4A and B). These data indicate that miR‐195 controls MICU1 abundance that modulates mCa2+ uptake without any effect on cCa2+ dynamics. Next, to determine whether miR‐195 overexpression has any effect on expression of the other MCU complex proteins, the MCU complex protein expression was evaluated by immunoblotting. As shown in Fig EV4C, no change in the protein abundance of MCU, MICU2, or EMRE was seen in CP20, OVCAR4, and OVSAHO cell lines. Moreover, neither the level of MFN2 protein nor the mitochondrial morphology was altered by miR‐195 transfection in CP20 or OVCAR4 cells (Fig EV4C and D), suggesting that ovarian cancer cells behave differently than breast cancer cell lines where miR‐195 has been shown to target MFN2 and control mitochondrial fission (Purohit et al, 2019). In addition to the association with Ca2+ homeostasis, high MICU1 levels are known to favor glycolysis in cancer cells. We have previously shown that the silencing of MICU1 in ovarian cancer cells significantly decreases intracellular lactate levels, which is an indicator of glycolysis (Chakraborty et al, 2017). Thus, in order to determine the impact of miR‐195 on glycolysis in ovarian cancer cells, we determined the intracellular lactate levels in cancer cells after re‐expressing miR‐195. In all three ovarian cancer cell lines, intracellular lactate levels were significantly decreased when either miR‐195 was expressed or MICU1 was silenced (Fig 3J). The effect of miR‐195 overexpression on mitochondrial oxidative stress was measured by using MitoSOX dye staining and flow cytometry‐based analysis. While a moderate but significant increase in mitochondrial ROS was observed in the miR‐195‐transfected cells, the co‐transfection of miR‐195 non‐responsive MICU1 decreased the miR‐195‐induced mitochondrial ROS to the levels of miR non‐target‐transfected cells (Fig 3K and L). These data both corroborate our previous findings and demonstrate that the effect of miR‐195 on glycolysis is likely mediated through MICU1.
Figure EV3. Efficacy of MICU1 inhibition by miR‐195 and siRNA .
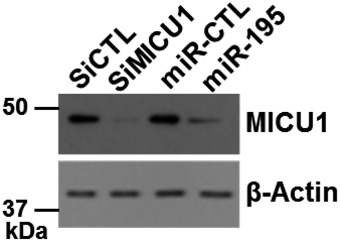
After siRNA and miR transfection as shown in the image, efficacy of MICU1 inhibition was evaluated using immunoblotting. Similarly, transfected cells were used to evaluate [Ca2+]out shown in Fig 3. β‐Actin was used as loading control.
Figure EV4. miR‐195 overexpression does not alter cytosolic Ca2+, MCU complex protein expression, and mitochondrial morphology in ovarian cancer.
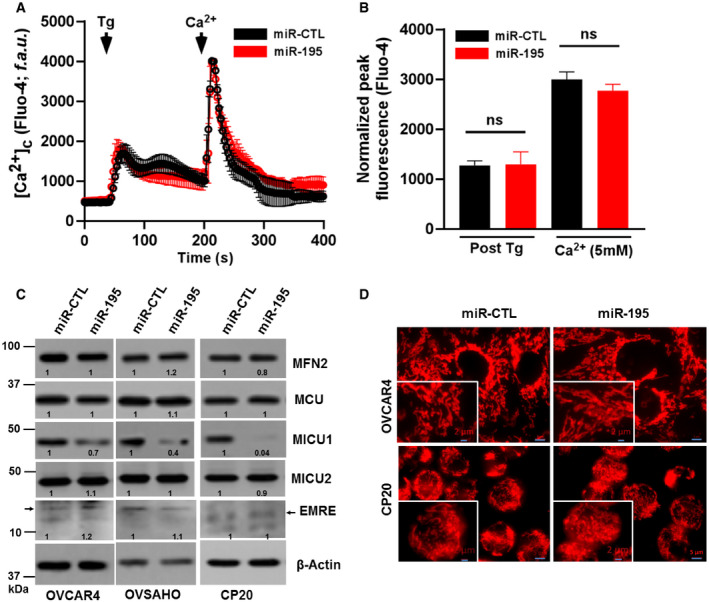
-
A, BAfter miR‐CTL or miR‐195 transfection in CP20 cells cytosolic Ca2+ was measured (A) Fluorescence traces (Fluo‐4 AM) vs time (s) showing Tg (4 μM) induced characteristic increase in cytosolic Ca2+ (5 mM) and uptake and retention of SOC activity following Ca2+ addition in both miR‐CTL and miR 195 transfected cells. Tg and Ca2+ were added as indicated. (B) Quantification of normalized peak fluorescence (Fluo‐4 AM) after Tg stimulation and Ca2+ addition. Non‐parametric t‐test determined the level of significance of fluorescence between miR‐CTL and miR‐195-treated cells (n = 3, biological repeats), (ns = not significant, Student's t‐test).
-
CAfter miR‐CTL or miR‐195 transfection in OVCAR4, OVSAHO, and CP20 cells, cell lysate was evaluated for the expression of MFN2, MCU, MICU1, MICU2, and EMRE. β‐Actin is used as loading control.
-
DImmunofluorescence of CP20 and OVCAR4 cells labeled with Mitotracker red (Scale bar 5 μM), inset shows enlarged image portion (Scale bar 2 μM).
Finally, we sought to confirm that the effects of miR‐195 were primarily mediated via MICU1. In order to do so, we generated stable CP20 and OVCAR4 cell lines that stably expressed either miR‐195‐GFP or non‐target miR‐GFP. These cells exhibited a near 100% stable GFP fluorescence (Fig 4A and B). miR‐195 overexpression in these cells was confirmed using RT–qPCR. (Fig 4C). pLYS1‐MICU1‐Flag (CDS only, hence non‐target for miR‐195) was ectopically expressed in the stable miR‐195‐GFP cells; MICU1 protein abundance was confirmed by immunoblotting (Fig 4D). miR‐195‐GFP cells and the same cells expressing the non‐responsive MICU1 were compared to miR non‐target control GFP cells in both anchorage‐dependent and anchorage‐independent clonal growth assays. The stable miR‐195‐GFP cells exhibited significantly reduced clonal growth as compared to the control, and re‐expression of MICU1 restored clonal growth comparable to the control levels (Fig 4E and F). Taken together, these results show that miR‐195 mediates its effects through MICU1 expression.
Figure 4. miR‐195 regulates clonal growth by targeting MICU1.
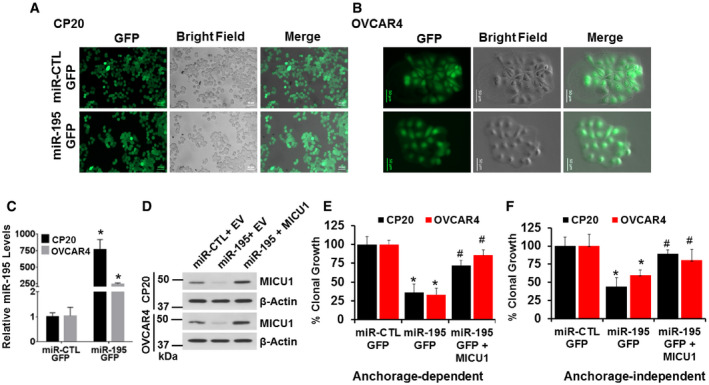
-
A, BStable cell lines for CP20 and OVCAR4 expressing GFP‐miR-195 or non‐target miR‐GFP were generated using lentiviral‐mediated transduction and were selected using puromycin; representative images are shown. Scale bars, 50 μm.
-
CRelative miR‐195 expression was measured using RT–qPCR in stable cell lines, normalized with U6. Mean ± SEM; n = 3, biological repeats. *P < 0.05, Student's t‐test.
-
DCP20 and OVCAR4 stable cell lines were transfected as either empty vector (EV) or pLYS1‐MICU1‐Flag (MICU1) and relative MICU1 levels were evaluated using immunoblotting.
-
E, FCP20 and OVCAR4 stable cell lines transfected as in (D) were counted and re‐plated for anchorage‐dependent (E) and independent clonal growth (F). Quantification compared to the miR‐CTL stable cells transfected with EV is shown. Mean ± SD; n = 3, Biological repeats. *P < 0.05 when comparing with miR‐CTL, # P < 0.05 when comparing with miR‐195 by Student's t‐test.
miR‐195 reduces tumor growth in vivo
Knowing that MICU1 impacts tumor progression and that MICU1 expression is regulated by miR‐195, we sought to determine the impact of miR‐195 on tumor growth in vivo. In order to determine the consequences of miR‐195 re‐expression in vivo, we compared CP20 ovarian cancer cells stably expressing either miR‐195‐GFP or miR‐GFP‐CTL in a xenograft model. The miR‐195 re‐expressing group had significantly lower tumor volumes than the control group (Fig 5A). To compare tumor growth rate over the entire experimental period, the tumor doubling time was calculated according to Mehrara et al (2007). Interestingly, the miR‐195 group had significantly greater (~ 9.3 days) tumor doubling time as compared to the control group (~ 4.3 days; Fig 5B). Similarly, based on the humane endpoint criteria, overall survival was greater in the miR‐195 group than in the control group (Fig 5C). These data demonstrate that miR‐195 reduces tumor burden and prolongs survival in in vivo models of ovarian cancer. Having shown the gross impacts of miR‐195 expression on tumor development in vivo, we sought to determine its impact on in vivo expression of MICU1 and aerobic glycolysis. We achieved this by immunoblotting tumor tissue samples for MICU1, pyruvate dehydrogenase (PDH), and phosphorylated PDH (pPDH), and pPDH is characteristically increased in cancer cells and is an indicator of enhanced glycolysis (Chakraborty et al, 2017). Additionally, we have previously shown that pPDH levels mirror MICU1 levels in ovarian cancer cells (Chakraborty et al, 2017). Tumors from the miR‐195 group had reduced expression of both MICU1 and pPDH when compared to the miR‐control group (Fig 5D). However, expression of PDH did not change, suggesting that miR‐195 normalizes MICU1 expression and reverses glycolysis as evidenced by decreased pPDH levels. Lysates from these groups were also immunoblotted for PARP and BCL2 levels and microscopic slides were evaluated for the expression of Ki67, CD31, and TUNEL using immunohistochemistry. Decreased Ki67 and CD31 in the miR‐195 group indicated significantly lower indices of proliferation and angiogenesis in this group compared to the control group. Similarly, increased PARP cleavage and TUNEL staining along with decreased BCL2 expression indicated greater apoptosis in the miR‐195 group than in the control group (Fig 5E and F). To confirm that the reduced tumor growth was mediated through MICU1, we repeated in vivo tumor growth experiments by generating cells stably overexpressing MICU1 in CP20‐miR‐195 GFP (miR‐195‐GFP + MICU1) stable cells (Fig EV5A and B). We implanted these cells subcutaneously as before along with miR‐GFP‐CTL, miR‐195‐GFP, and monitored tumor growth. The miR‐195 re‐expressing group had significantly lower tumor volumes than the control group while re‐expression of MICU1 in miR‐195‐GFP cells rescued the tumor growth inhibition phenotype due to miR‐195 overexpression and tumor volume was comparable to the miR‐CTL group (Fig 5G). These results confirmed that the observed phenotype of reduced tumor growth in ovarian cancer by miR‐195 overexpression was predominantly mediated through MICU1. We observed no signs of toxicity in any of the animals during the experiment, as assessed by changes in behavior, feeding habits, mobility, body weight, and H&E staining of vital organs (Fig EV5C and D). Together these results suggest that miR‐195, by targeting MICU1, reduces tumor burden and increases overall survival.
Figure 5. miR‐195 expression determines in vivo tumor growth.
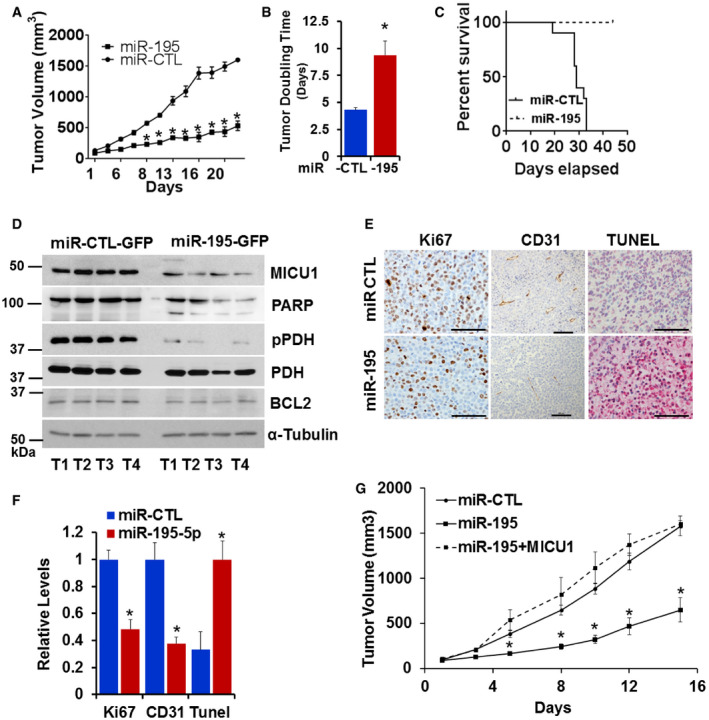
-
AFemale athymic mice were injected subcutaneously with either CP20 GFP‐miR-CTL or CP20‐ GFP-miR‐195 cells. Mice injected with the miR‐195 expressing cells (miR‐195) had significantly smaller tumor volumes than control mice (miR‐CTL). Data are mean ± SD, n = 10. *P < 0.05, Student's t‐test.
-
BTumor doubling time was significantly longer in the mice injected with miR‐195 expressing cells than in control mice. Data are mean ± SD, n = 10. *P < 0.05, Student's t‐test.
-
CPercent survival based on humane endpoint criteria was calculated by the Kaplan–Meier method and P values determined by log rank test.
-
DFour tumor samples from each group were analyzed for the expression of MICU1, pPDH, PDH, PARP, and BCL2 using immunoblotting. α tubulin was used as the loading control.
-
E, FRepresentative histology images of tumors from mice xenografts of CP20‐GPF-miR‐CTL (miR‐CTL) or CP20‐GPF-miR‐195 (miR‐195) cells with Ki67 expression (Scale bar = 50 μm) (E), CD31 positive vessels (Scale bar = 100 μm) and TUNEL staining (Scale bar = 50 μm) and (F) graph showing quantification of each marker in tumors from the experimental mice; values are mean ± SD, n = 10, *P < 0.05, Student's t‐test.
-
GFemale athymic mice were injected subcutaneously with either CP20 GFP‐miR-CTL or CP20‐ GFP-miR‐195 or CP20‐ GFP-miR‐195 + MICU1 cells and tumor volume was measured, values are mean ± SE, n = 10. *P < 0.05, Student's t‐test.
Figure EV5. MICU1 stable cell line and toxicity assessment in miR‐CTL and miR‐195 tumor model.
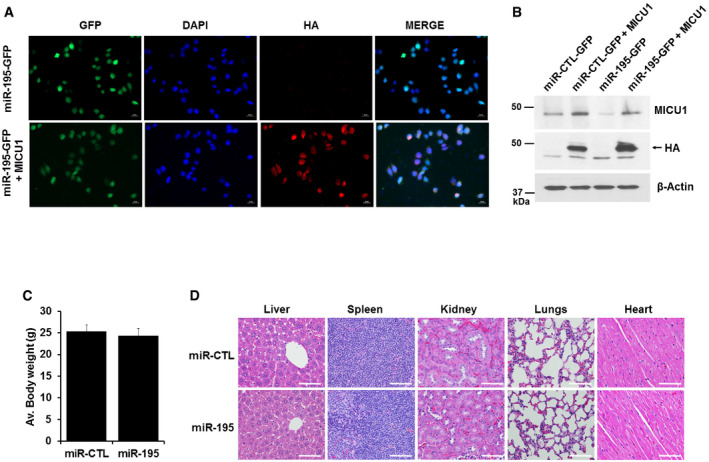
-
A, BMICU1 was stably expressed in miR stable transfected CP20 cells by using pLenti‐C-HA-IRES‐BSD plasmid (OriGene Technologies); MICU1 stable cells were selected using Blasticidin. MICU1 expression in stable cells is shown by IFC (A) and immunoblotting (B).
-
C, DToxicity in experimental mice caused by miR‐195 overexpressing CP20 cells was assessed by changes in body weight and histology of vital organs. (C) Average body weight of mice from miR‐CTL and miR‐195 experimental groups immediately before euthanasia. Mean ± SD, n = 10 is shown. (D) Representative H&E‐stained sections of tissues (liver, spleen, kidneys, lungs, and heart) from miR‐CTL and miR‐195 mice visualized using an upright epi‐fluorescent microscope (Nikon NI‐U, Plan Apochromatic). Scale bar 25 μM.
Discussion
In this report, we used in silico and in vitro techniques to demonstrate that miR‐195 is an important negative regulator of MICU1 expression in ovarian cancer. Via its inhibition of MICU1, miR‐195 further regulates the delicate balance of mitochondrial Ca2+ that ultimately shifts cancer cells toward glycolysis, the latter being a major determinant of tumor growth and resistance to therapy (Morisaki & Katano, 2003; Guerra et al, 2017). Moreover, tumors from the miR‐195 group had significantly lower indices of proliferation, angiogenesis, and increased apoptosis. Increased PARP cleavage and decreased BCL‐2 are likely to be responsible for increased apoptosis in the miR‐195 group. Recently, Singh et al (2015) reported that overexpression of miR‐195 in breast cancer cells inhibits cell proliferation, migration, and invasion, decreases the expression of mesenchymal markers, and enhances the expression of epithelial markers. Although they did not examined MICU1 expression, our data would suggest that miR‐195 acts through MICU1 to also regulate Ca2+ in this case. In addition, when miR‐195 was re‐expressed in ovarian cancer cells, decreased clonal growth, invasion, and migration in vitro, as well as reduced tumor development in vivo, was observed. That this reduced tumor vigor is mediated through MICU1 was further confirmed in in vitro and in vivo conditions. miR‐195 is an important regulator of cancer progression is supported by the reports that it is implicated in both the epithelial‐to‐mesenchymal transition as well as migration and invasion in prostate cancer (Liu et al, 2015; Wu et al, 2015).
In addition to the role in cancer progression, it is likely that low miR‐195 levels are associated with resistance to chemotherapy. Previously, we reported that high levels of MICU1 impact both chemoresistance and cancer progression (Chakraborty et al, 2017). This indicates that the miR‐195 and MICU1 interaction is also likely to impact chemoresistance. Further, the relationship between miR‐195, MICU1, and chemoresistance is supported by the observation that taxol‐resistant ovarian cancer cells significantly under‐express miR‐195 when compared to taxol‐sensitive cells (Kim et al, 2014). Additionally, others have reported that miR‐195 regulates chemotherapy sensitivity and multidrug resistance in gastric cancer (Ye et al, 2017; Nie et al, 2018), epithelial‐to‐mesenchymal transition (Liu et al, 2015) and, migration and invasion (Wu et al, 2015) in prostate cancer (Nie et al, 2018).
Because miR‐195 acts as a tumor suppressor, is associated with chemoresistance, and is under‐expressed in multiple cancers (Soon et al, 2009; Xu et al, 2009; Heneghan et al, 2010; Liu et al, 2010; Brenner et al, 2011; Li et al, 2011; Sekiya et al, 2011; Bai et al, 2012; Chen et al, 2012; Fei et al, 2012; Lin et al, 2012; Mao et al, 2012; Wang et al, 2012, 2013, 2014, 2019; Zhang et al, 2012; Bhattacharya et al, 2013; Deng et al, 2013; Fu et al, 2013; Furuta et al, 2013; Hui et al, 2013; Guo et al, 2014), restoration of miR‐195 represents a potentially important anti‐cancer strategy. Restoring miR‐195 expression in cancer cells would inhibit clonal growth, as well as migration and invasion, which is mediated via MICU1 regulated Ca2+ homeostasis and the associated inhibition of glycolysis. Cancer cells undergo metabolic reprogramming and preferentially utilize glucose via aerobic glycolysis, the so‐called Warburg effect (Popgeorgiev et al, 2018). This metabolic reprogramming is implicated in cancer progression and therapy resistance in numerous malignancies (Sun et al, 2018). Numerous reports show that inhibition of glycolysis sensitizes tumor cells to treatment. For example, treatment with 2‐DG, a hexokinase II inhibitor rendered tumor cells more sensitive to both chemotherapy (adriamycin or paclitaxel) and radiation therapy (Maschek et al, 2004; Dwarakanath & Jain, 2009). Similarly, the glycolytic inhibitors 3‐bromopyruvate, targeting GAPDH, and oxamate, a lactate dehydrogenase inhibitor, sensitize tumor cells to chemotherapeutic agents (Ganapathy‐Kanniappan et al, 2010; Zhou et al, 2010). However, despite these promising results, targeting the glycolytic pathway for therapy has been met with limited success largely due to undesirable side effects arising from the ubiquity of the enzymes involved in glycolysis (Ganapathy‐Kanniappan & Geschwind, 2013). Thus, the use of broad‐spectrum glycolysis inhibitors in cancer treatment appears untenable. However, exploiting differential MICU1 overexpression in cancer may mitigate issues related to systemic toxicity and represents a potentially valid approach to target glycolysis in cancer. While MICU1 is clearly a good target for cancer treatment, there are currently no chemical inhibitors of MICU1 identified. However, since we have shown that MICU1 is directly regulated by miR‐195, miRNA delivery should be considered as a viable alternative therapy. This is especially true in light of the increasingly successful transition of miRNA therapy from the bench to the clinic in recent years (Kaczmarek et al, 2017). Several miR‐targeted therapies have reached clinical development, including a mimic of the tumor suppressor miR‐34, which reached phase I clinical trials for treating cancer (Bader, 2012), and an anti‐miRNA targeted at miR‐122, which reached phase II trials for treating hepatitis (Rupaimoole & Slack, 2017). RNA‐based therapeutics combined with conventional chemotherapy agents offer a new approach to treat cancer, and it is likely that in the near future RNA‐based combination therapies will move into the clinic for this purpose. In this context validation of miR‐195 as a potential therapeutic target has great potential to translate MICU1‐targeted therapy into clinical care.
Materials and Methods
Cell culture
The human ovarian cancer cell lines OVCAR4 (NIH, Repository of Tumors and Tumor Cell Lines, USA), A2780‐CP20 (developed by sequential exposure of the A2780 parental cell line to increasing concentrations of cisplatin, henceforth termed CP20 were a kind gift from Dr. Anil Sood, MD Anderson Cancer Center) (Sood et al, 2001). TYK‐nu (JCRB0234.0), TYK‐nu.CPr (JCRB0234.1), and OVSAHO (JCRB1046) were from the Japanese Collection of Research Bioresources Cell Bank., and OV90 cells (ATCC® CRL‐11732™) were purchased from ATCC. Immortalized normal fallopian tube epithelial cells (FTE188) were a kind gift from Dr. Jinsong Liu, MD Anderson Cancer Center. All the cell lines were routinely cultured in RPMI (OV90, CP20, OVCAR4, OVSAHO, and OVKATE); ATCC‐EMEM (TYK‐nu and TYK‐nu.CPr) and 1:1 Media 199: MCDB 105 (FTE188). Media was supplemented with 10% heat‐inactivated FBS (Gibco, Grand Island, NY, USA) and 100 units penicillin and 100 μg streptomycin/ml (Invitrogen, Rockford, IL, USA) in a 5% CO2 humidified atmosphere.
RNA isolation, reverse transcription, and quantitation of microRNA expression levels
For quantification of endogenous or transfected miR‐195 expression levels, total RNA was isolated using the mirVana™ miRNA Isolation Kit (Thermo Fisher Scientific), and following quantification, 1 μg RNA was reverse transcribed (miRNA cDNA Synthesis Kit, with Poly(A) Polymerase Tailing, from Applied Biological Materials Inc., Richmond, BC, Canada). Quantitative polymerase chain reaction was performed using SYBR Green (iQ™ SYBR® Green Supermix from Bio‐Rad) based RT–qPCR. Relative microRNA expression levels were calculated using the comparative Ct method with U6 as the normalizer (Dwivedi et al, 2016). Primers for miR‐15a and miR‐195 and U6 were purchased from Applied Biological Materials Inc., Richmond, BC, Canada.
microRNA transfection
microRNA transfection was performed in CP20, OVCAR4, and OVSAHO cell lines in 6 cm culture dishes containing 5 × 105 cells using HiPerFect Transfection Reagent (Qiagen, Germantown, MD) following the manufacturers protocol and using 100 nM miR‐IDIAN Mimic Negative Control (Dharmacon, cat No‐CN‐001000‐01, Fisher Scientific, Pittsburgh PA, 15275, USA) or 100 nM Pre‐miR‐precursor hsa‐miR‐195‐5p or hsa‐miR‐15a (Ambion®, Grand Island, NY 14072 USA).
Cell lysis and western blotting
Immunoblotting was performed on cell lysates in RIPA buffer (Boston Bioproducts, Ashland, MA, USA) supplemented with the protease–phosphatase mix (Thermo Fisher Scientific, Grand Island, NY 14072 USA). Briefly, the cell lysate was separated on 10–15% Tris‐glycine SDS–PAGE gel and transferred to Immun‐Blot® PVDF membrane (Bio‐Rad, Hercules, California 94547USA). Membranes were blocked in 5% non‐fat dry milk in TBS with 0.1% TWEEN‐20 (TBST) for 1 h at room temperature followed by overnight incubation with indicated primary antibodies (MICU1 (CST, 1:1,000), BMI1(Invitrogen, 1:1,000), BCL2(CST, 1:1,000), MFN2 (CST, 1:1,000), VDAC (CST, 1:1,000), PARP (CST, 1:1,000), pPDH (CST, 1:1,000), PDH (CST, 1:1,000), MCU (CST, 1:1,000), MICU2 (CST, 1:1,000), EMRE (CST, 1:1,000), β Actin (Sigma at 1:10,000 dilutions), GAPDH (Sigma, 1:10,000), and alpha‐Tubulin (CST, 1:10,000) in TBST with 5% BSA. Membranes were washed and incubated with secondary antibody for 1 h at room temperature. Equal loading was verified by immunoblotting with GAPDH, β‐actin, or α‐tubulin.
Soft agar assay
Forty‐eight hours post‐transfection 1 × 103 cells (OVCAR4, CP20 or OVSAHO) in RPMI medium containing 10% FBS with 0.3% agar (SeaPlaque™ GTG™ Agarose, Lonza Allendale, NJ 07401, USA) were seeded on top of 0.6% agar in the same medium in each well of 12‐well plates. After 10 (CP20) or 14 (OVCAR4/OVSAHO) days, the colonies were imaged and counted using an Optronix GelCount colony counter (Abingdon OX14 4SA, United Kingdom).
MICU1 3′ UTR reporter assay
For reporter assays, 104 cells were plated in 96‐well white polystyrene plates. MICU1 3′ UTR reporter plasmid (100 ng, Switchgear Genomics product id 806421) in LightSwitch™ 3′UTR Reporter Vector was co‐transfected with either non‐target microRNA control or different concentrations of miR‐195. Twenty‐four hours following transfection, luciferase (Renilla) activity was measured using LightSwitch™ assay reagent. Site‐directed mutagenesis was performed using the QuikChange Lightning Site‐Directed Mutagenesis Kit (Agilent) to delete the miR‐195 binding site within the MICU1 3′ UTR.
Plasmids
LightSwitch™ MICU1‐3′UTR (S806421) was purchased from switchgear genomics (CA, USA). pLenti‐III‐miR‐GFP Control and LentimiR‐a‐GFP‐hsa‐miR‐195‐5p. Vector was purchased from Applied Biological Materials Inc (Richmond, BC Canada). pLYS1‐MICU1‐Flag (Plasmid #50058) was obtained from Addgene. For overexpression studies, MICU1 gene was cloned in plenti‐C‐HA‐IRES‐BSD plasmid (by OriGene Technologies).
Generation of stable cell lines
For generation of miR‐195 stable cell line, lentiviral particles were made using either LentimiR‐a‐non‐target miR‐GFP or LentimiR‐a‐GFP‐hsa‐miR‐195‐5p along with GAG packaging plasmid and VSVG envelope plasmid in HEK‐293T cell line and were used to transduce OVCAR4 and CP20 cell lines. Resultant cell lines were selected with 2 μg/ml puromycin for 2 weeks. Selection efficiency was verified using GFP and overexpression was verified using RT–qPCR for miR‐195. For MICU1 over expression stable CP20 cells, lentiviral particles were generated using pLenti‐ MICU1‐C‐HA‐IRES‐BSD plasmid along with GAG packaging plasmid and VSVG envelope plasmid in HEK‐293T cell line and were used to transduce CP20‐miR‐CTL‐GFP or CP20‐GFP‐miR‐195 stable cells. MICU1 stable cells were selected using blasticidin. MICU1 expression in stable cells is shown by IFC and immunoblotting.
Lactate assay
Lactate concentration was determined by using the Lactate Assay Kit (#MAK064) from Sigma as per the manufacturer's instructions. Briefly, 5 × 105 cells, were seeded in a 60‐mm plate, transfecting as indicated and then lysed in lactate assay buffer. Aliquots of lysate (5–10 μl) were distributed in 96‐well plates suitable for absorbance measurement. Lactate concentration was determined by an enzymatic assay, which results in a colorimetric (570 nm) product and normalized with the protein concentration.
Boyden chamber migration and invasion Assay
After 48 h transfection with scrambled non‐target miRNA or miR‐195 or miR‐195 + pLYS1‐MICU1‐Flag (a gift from Vamsi Mootha, Addgene plasmid # 50058) (Sancak et al, 2013), OVCAR4 or CP20 cells were serum‐starved overnight, detached from culture plates by trypsinization and 1 × 105 cells were plated into 8 μm transwell chambers in 200 μl of serum‐free RPMI1640 medium. The lower chambers of the plate were supplied with 650 μl of RPMI1640 medium with 10% FBS. The cells were allowed to migrate for 12 h after which cells were processed with Three‐Step Stain Set. Cells in the upper chamber were removed using a cotton swab, and cells migrating through the membrane were counted. Cell invasion studies were performed using a Boyden chamber equipped with membranes pre‐coated with 100 μg/ml fibronectin (#F1141, Sigma) for 16 h.
Ca2+ uptake and ∆Ψμ measurement in permeabilized cell system
The measurement of extra‐mitochondrial calcium [Ca2+]out clearance as an indicator of [Ca2+](m) retention was achieved as described previously (Mallilankaraman et al, 2012a). Briefly, 4 × 106 cells were washed in an extracellular‐like Ca2+‐free buffer (in mM: 120 NaCl, 5 KCl, 1 KH2PO4, 0.2 MgCl2, 0.1 EGTA, and 20 HEPES‐NaOH, pH 7.4) and stored on ice for 10 min. Following centrifugation, cells were transferred to an intracellular‐like medium (permeabilization buffer, in mM: 120 KCl, 10 NaCl, 1 KH2PO4, 20 HEPES‐Tris, pH 7.2, and digitonin (40 μg/ml). The cell suspension supplemented with succinate (5 mM) was placed in a fluorimeter and permeabilized by gentle stirring. Fura‐FF (0.5 μM; Cayman Chemical) was added at 0 s, and JC‐1 (Thermo Fisher Scientific, 800 nM) at 20 s. Fluorescence was monitored in a temperature‐controlled (37°C) multiwavelength‐excitation dual wavelength‐emission spectrofluorometer (Delta RAM, Photon Technology International) using 340‐nm/380‐nm for Fura‐FF. A series of Ca2+ boluses (5 μM) and mitochondrial uncoupler FCCP (2 μM) were added at the indicated time points and the Ca2+ uptake was monitored simultaneously. Traces indicative of the Fura‐FF ratio are from three independent experiments.
Assessment of mitochondrial matrix Ca2+ and ∆Ψm
Briefly, 4 × 106 cells were washed in an extracellular‐like Ca2+ free buffer (in mM: 120 NaCl, 5 KCl, 1 KH2PO4, 0.2 MgCl2, 0.1 EGTA, and 20 HEPES‐NaOH, pH 7.4) and stored on ice for 10 min. Following centrifugation, cells (4 × 106) were transferred to an intracellular‐like medium (permeabilization buffer, in mM: 120 KCl, 10 NaCl, 1 KH2PO4, 20 HEPES‐Tris, pH 7.2, and digitonin [40 μg/ml]). The cell suspension supplemented with succinate (5 mM) was placed in a fluorimeter and permeabilized by gentle stirring. Fura‐FF (Cayman Chemical, 0.5 μM) was added at 0 s, and JC‐1 (Thermo Fisher Scientific, 800 nm) at 20 s. Fluorescence was monitored in a temperature‐controlled (37°C) multiwavelength‐excitation dual wavelength‐emission spectrofluorometer (Delta RAM, Photon Technology International) using 490‐nm excitation/535‐nm emission for the monomer, 570‐nm excitation/595‐nm emission for the J‐aggregate of JC1 and 340‐nm/380‐nm for Fura‐FF. After baseline recording, mitochondrial uncoupler FCCP (10 μM) were added at the indicated time point and Ca2+ released from the mitochondria was calculated as resting matrix [Ca2+]m.
Measurements of Cytosolic Ca2+ transients
CP20 cells were grown on collagen‐coated coverslips in RPMI medium containing 10% FBS. The cultured cells were stained with Fluo‐4 AM (5 μM; Thermo Fisher Scientific, Calcium‐bound; Ex‐494 nm, Em‐506 nm) for 30 min at room temperature. The stained cells were imaged on a SP8, Leica DMi8 Confocal Microscope, coupled with an environmental chamber. Time‐lapse imaging was performed using 63× oil objective under Ca2+ and Mg2+ free condition. Cells were first stimulated with Thapsigargin (Millipore Sigma, Tg., 4 μM) at 45 s and then with a bolus of Ca2+ (5 mM) at 210 s. ER Ca2+ release and store‐operated Ca2+ entry (SOCE) changes were recorded and analyzed using Leica Application Suite X.
Mitochondrial morphology
To assess mitochondrial morphology, cells were stained with MitoTracker™ Red CMXRos (M7512, Invitrogen, USA). Approximately 2 × 104 cells were plated on 12‐mm coverslips and 72‐h post‐transfection cells were treated with Mitotracker Red (100 nM) for 15 min at 37°C; after three times washing with PBS, the cells were supplemented with warm medium and observed under microscope. Cell images were acquired with a 63× objective using a Zeiss Axio Observer. Z1 (Göttingen, Germany).
Mitochondrial ROS
After 72‐h miR transfection, mitochondrial ROS was measured using MitoSOX (Invitrogen) staining (2.5 μM for 10 min at 37°C). Data were acquired with a FACS Calibur (BD Biosciences) and analyzed with Flow Jo analytical software.
Preclinical model of ovarian cancer
Female athymic nude mice (NCrnu; 5–6 weeks old) were purchased from ENVIGO. All mice were housed and maintained under specific pathogen‐free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US Department of Agriculture, US Department of Health and Human Services and National Institutes of Health. All studies were approved and supervised by the OUHSC Institutional Animal Care and Use Committee. A total of 20 animals were randomly divided into two groups (N = 10 per group). Animals received 1 × 106 cells in 100 μl HBBS by subcutaneous injection: group 1 received miR‐CTL‐GFP‐CP20 cells and the second group received miR‐195‐GFP‐CP20 cells. For the rescue experiment, female athymic nude mice were similarly injected with either miR‐CTL‐GFP‐CP20, miR‐195‐GFP‐CP20, or miR‐195‐GFP + MICU1‐CP20 cells (n = 10 per group). Once the tumor reached ~ 100 mm3 the mice were monitored for three additional weeks or until their tumor volume attained the pre‐determined IACUC limits, at which point they were euthanized.
Immunohistochemistry
Immunohistochemistry was performed according to the manufacturer's protocol using the automated BONDRX Leica platform and Leica Bond‐III™ Polymer Refine Detection system (DS 9800). In brief, FFPE tissues were sectioned at 4 μm and were mounted on positively charged slides. The slides were dried overnight at room temperature and incubated at 60°C for 45 min followed by deparaffinization and rehydration in an automated Multistainer (Leica ST5020). Subsequently, these slides were transferred to the Leica Bond‐III™, treated for target retrieval at 100°C for 20 min in a retrieval solution, at pH 6.0. The sections were incubated with 5% goat serum (01‐6201; Thermo Fisher Scientific) for 30 min. Endogenous peroxidase was blocked using peroxidase‐blocking reagent, followed by the selected primary antibody (Ki67 1:1,500 (Abcam, AB15580), CD31 1:50 (Abcam ab28364) incubation for 60 min. For the secondary antibody, post‐primary IgG‐linker and/or Poly‐HRP IgG reagents was used. Detection was done using 3,3′‐diaminobenzidine tetrahydrochloride (DAB), as chromogen and counter stained with hematoxylin. Completed slides were dehydrated (Leica ST5020) and mounted (Leica MM24). Tunnel staining was performed after deparaffinization of 4 μM sections using In situ Cell Death Detection Kit, AP (Roche Diagnostics GmbH, Manheim, Germany) following the manufacturer's protocol. Images were taken using a Nikon Eclipse Ni microscope.
Statistical analysis
All the experiments were performed in triplicate and repeated independently three times. Data are expressed as means ± standard deviation (SD) or ± standard error. Student's t‐test was used for statistical analysis with significance set at P < 0.05. For animal experiments, 10 mice were assigned per treatment group. This sample size gave 80% power to detect a 50% reduction in tumor weight with 95% confidence. Mouse and tumor weights for each group were compared using Student's t‐test (for comparisons of two groups). Statistical analyses were done using GraphPad Prism. A two‐tailed P ≤ 0.05 was deemed statistically significant.
Author contributions
GR, SKDD, RB, MM, and PM designed research; GR and SKDD developed methodology; GR, SKDD, YZ, AD, RK, SS, and MNH acquired data; SK, JTD, and JDW did the bioinformatics analysis; GR, SKDD, RB, MM, PM, YZ, AD, RK, SS, and MNH analyzed and interpreted data; SKDD, GR, RB, and PM wrote and/or revised the manuscript; PM and RB provided administrative, technical, material support, and supervised the study. All authors edited the manuscript.
Conflict of interest
JTD has received consulting fees or honoraria from Janssen Pharmaceuticals, GlaxoSmithKline, AstraZeneca, and Hoffman‐La Roche. JTD is a scientific advisor to LAM Therapeutics and holds equity in NuMedii, Ayasdi, and Ontomics. KS has received consulting fees or honoraria from Kencor Health, OccamzRazor, Philips Healthcare, Alphabet, McKinsey and Company, LEK Consulting and Parthenon‐EY.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
This work was supported by National Institutes of Health Grants 2CA136494, CA213278. We thank the Peggy and Charles Stephenson Cancer Center at the University of Oklahoma Health Sciences Center for a seed grant and also acknowledge an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20 GM103639) for the use of Histology and Immunohistochemistry Core, which provided immunohistochemistry and image analysis service. This research was also supported by Peggy and Charles Stephenson Endowed Chair fund to PM. We thank the Laboratory for Molecular Biology and Cytometry Research at OUHSC for the use of the Core Facility which provided DNA sequencing and flow cytometry service; use of the core facility was supported in part by the National Cancer Institute Cancer Center Support Grant P30CA225520 awarded to the University of Oklahoma Stephenson Cancer Center. KS and JTD acknowledge support from the National Institutes of Health: National Center for Advancing Translational Sciences (NCATS, grant number UL1TR000067); and Clinical and Translational Science Awards (CTSA) grant. Research reported in this publication was supported in part by the Oklahoma Tobacco Settlement Endowment Trust awarded to the University of Oklahoma//Stephenson Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Oklahoma Tobacco Settlement Endowment Trust.
EMBO Reports (2020) 21: e48483
Contributor Information
Resham Bhattacharya, Email: resham-bhattacharya@ouhsc.edu.
Priyabrata Mukherjee, Email: priyabrata-mukherjee@ouhsc.edu.
Data availability
This study includes no data deposited in external repositories.
References
- Ambros V (2004) The functions of animal microRNAs. Nature 431: 350–355 [DOI] [PubMed] [Google Scholar]
- Bader AG (2012) miR‐34 ‐ a microRNA replacement therapy is headed to the clinic. Front Genet 3: 120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Yang W, Yang HX, Liao Q, Ye G, Fu G, Ji L, Xu P, Wang H, Li YX et al (2012) Downregulated miR‐195 detected in preeclamptic placenta affects trophoblast cell invasion via modulating ActRIIA expression. PLoS ONE 7: e38875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Schmitz U, Wolkenhauer O, Schonherr M, Raatz Y, Kunz M (2013) Regulation of cell cycle checkpoint kinase WEE1 by miR‐195 in malignant melanoma. Oncogene 32: 3175–3183 [DOI] [PubMed] [Google Scholar]
- Birsoy K, Sabatini DM, Possemato R (2012) Untuning the tumor metabolic machine: targeting cancer metabolism: a bedside lesson. Nat Med 18: 1022–1023 [DOI] [PubMed] [Google Scholar]
- Brenner B, Hoshen MB, Purim O, David MB, Ashkenazi K, Marshak G, Kundel Y, Brenner R, Morgenstern S, Halpern M et al (2011) MicroRNAs as a potential prognostic factor in gastric cancer. World J Gastroenterol 17: 3976–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Zhao H, Tang J, Wu H (2015) Serum miR‐195 is a diagnostic and prognostic marker for osteosarcoma. J Surg Res 194: 505–510 [DOI] [PubMed] [Google Scholar]
- Chakraborty PK, Mustafi SB, Xiong X, Dwivedi SKD, Nesin V, Saha S, Zhang M, Dhanasekaran D, Jayaraman M, Mannel R et al (2017) MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun 8: 14634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Untiveros GM, McKee LA, Perez J, Li J, Antin PB, Konhilas JP (2012) Micro‐RNA‐195 and ‐451 regulate the LKB1/AMPK signaling axis by targeting MO25. PLoS ONE 7: e41574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M et al (2005) miR‐15 and miR‐16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102: 13944–13949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R et al (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17: 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2010) Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res 70: 859–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, Hamaker M, Sun P, Le A, Gao P (2011) Therapeutic targeting of cancer cell metabolism. J Mol Med (Berl) 89: 205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012) Links between metabolism and cancer. Genes Dev 26: 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Guo Y, Song H, Xiao B, Sun W, Liu Z, Yu X, Xia T, Cui L, Guo J (2013) MicroRNA‐195 and microRNA‐378 mediate tumor growth suppression by epigenetical regulation in gastric cancer. Gene 518: 351–359 [DOI] [PubMed] [Google Scholar]
- Dwarakanath B, Jain V (2009) Targeting glucose metabolism with 2‐deoxy‐D‐glucose for improving cancer therapy. Future Oncol 5: 581–585 [DOI] [PubMed] [Google Scholar]
- Dweep H, Gretz N (2015) miRWalk2.0: a comprehensive atlas of microRNA‐target interactions. Nat Methods 12: 697 [DOI] [PubMed] [Google Scholar]
- Dwivedi SK, Mustafi SB, Mangala LS, Jiang D, Pradeep S, Rodriguez‐Aguayo C, Ling H, Ivan C, Mukherjee P, Calin GA et al (2016) Therapeutic evaluation of microRNA‐15a and microRNA‐16 in ovarian cancer. Oncotarget 7: 15093–15104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanciulli M, Bruno T, Giovannelli A, Gentile FP, Di Padova M, Rubiu O, Floridi A (2000) Energy metabolism of human LoVo colon carcinoma cells: correlation to drug resistance and influence of lonidamine. Clin Cancer Res 6: 1590–1597 [PubMed] [Google Scholar]
- Fei X, Qi M, Wu B, Song Y, Wang Y, Li T (2012) MicroRNA‐195‐5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett 586: 392–397 [DOI] [PubMed] [Google Scholar]
- Fu MG, Li S, Yu TT, Qian LJ, Cao RS, Zhu H, Xiao B, Jiao CH, Tang NN, Ma JJ et al (2013) Differential expression of miR‐195 in esophageal squamous cell carcinoma and miR‐195 expression inhibits tumor cell proliferation and invasion by targeting of Cdc42. FEBS Lett 587: 3471–3479 [DOI] [PubMed] [Google Scholar]
- Furuta M, Kozaki K, Tanimoto K, Tanaka S, Arii S, Shimamura T, Niida A, Miyano S, Inazawa J (2013) The tumor‐suppressive miR‐497‐195 cluster targets multiple cell‐cycle regulators in hepatocellular carcinoma. PLoS ONE 8: e60155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy‐Kanniappan S, Vali M, Kunjithapatham R, Buijs M, Syed LH, Rao PP, Ota S, Kwak BK, Loffroy R, Geschwind JF (2010) 3‐bromopyruvate: a new targeted antiglycolytic agent and a promise for cancer therapy. Curr Pharm Biotechnol 11: 510–517 [DOI] [PubMed] [Google Scholar]
- Ganapathy‐Kanniappan S, Geschwind JF (2013) Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 12: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Sun L, He X, Cao Y, Zhang H (2012) MicroRNAs and the Warburg Effect: new players in an old arena. Curr Gene Ther 12: 285–291 [DOI] [PubMed] [Google Scholar]
- Gomez de Cedron M, Ramirez de Molina A (2016) Microtargeting cancer metabolism: opening new therapeutic windows based on lipid metabolism. J Lipid Res 57: 193–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra F, Arbini AA, Moro L (2017) Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg 1858: 686–699 [DOI] [PubMed] [Google Scholar]
- Guo H, Li W, Zheng T, Liu Z (2014) MiR‐195 targets HDGF to inhibit proliferation and invasion of NSCLC cells. Tumour Biol 35: 8861–8866 [DOI] [PubMed] [Google Scholar]
- Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME (2014) Mitochondrial calcium uniporter activity is dispensable for MDA‐MB‐231 breast carcinoma cell survival. PLoS ONE 9: e96866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hayes J, Peruzzi PP, Lawler S (2014) MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med 20: 460–469 [DOI] [PubMed] [Google Scholar]
- Heneghan HM, Miller N, Kelly R, Newell J, Kerin MJ (2010) Systemic miRNA‐195 differentiates breast cancer from other malignancies and is a potential biomarker for detecting noninvasive and early stage disease. Oncologist 15: 673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134: 703–707 [DOI] [PubMed] [Google Scholar]
- Hui W, Yuntao L, Lun L, WenSheng L, ChaoFeng L, HaiYong H, Yueyang B (2013) MicroRNA‐195 inhibits the proliferation of human glioma cells by directly targeting cyclin D1 and cyclin E1. PLoS ONE 8: e54932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio MV, Croce CM (2012) microRNA involvement in human cancer. Carcinogenesis 33: 1126–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek JC, Kowalski PS, Anderson DG (2017) Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med 9: 60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YW, Kim EY, Jeon D, Liu JL, Kim HS, Choi JW, Ahn WS (2014) Differential microRNA expression signatures and cell type‐specific association with Taxol resistance in ovarian cancer cells. Drug Des Devel Ther 8: 293–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhao Y, Liu C, Chen X, Qi Y, Jiang Y, Zou C, Zhang X, Liu S, Wang X et al , et al (2011) Analysis of MiR‐195 and MiR‐497 expression, regulation and role in breast cancer. Clin Cancer Res 17: 1722–1730 [DOI] [PubMed] [Google Scholar]
- Lin Y, Wu J, Chen H, Mao Y, Liu Y, Mao Q, Yang K, Zheng X, Xie L (2012) Cyclin‐dependent kinase 4 is a novel target in micoRNA‐195‐mediated cell cycle arrest in bladder cancer cells. FEBS Lett 586: 442–447 [DOI] [PubMed] [Google Scholar]
- Liu L, Chen L, Xu Y, Li R, Du X (2010) microRNA‐195 promotes apoptosis and suppresses tumorigenicity of human colorectal cancer cells. Biochem Biophys Res Commun 400: 236–240 [DOI] [PubMed] [Google Scholar]
- Liu C, Guan H, Wang Y, Chen M, Xu B, Zhang L, Lu K, Tao T, Zhang X, Huang Y (2015) miR‐195 Inhibits EMT by targeting FGF2 in prostate cancer cells. PLoS ONE 10: e0144073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464 [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M et al (2012a) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14: 1336–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ et al (2012b) MICU1 is an essential gatekeeper for MCU‐mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151: 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao JH, Zhou RP, Peng AF, Liu ZL, Huang SH, Long XH, Shu Y (2012) microRNA‐195 suppresses osteosarcoma cell invasion and migration in vitro by targeting FASN. Oncol Lett 4: 1125–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Corricelli M, Branchini A, Vitto VAM, Missiroli S, Morciano G, Perrone M, Ferrarese M, Giorgi C, Pinotti M et al (2019a) Akt‐mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. EMBO J 38: e99435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Vitto VAM, Patergnani S, Pinton P (2019b) High mitochondrial Ca(2+) content increases cancer cell proliferation upon inhibition of mitochondrial permeability transition pore (mPTP). Cell Cycle 18: 914–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR, Lampidis TJ (2004) 2‐deoxy‐D‐glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non‐small cell lung cancers in vivo . Cancer Res 64: 31–34 [DOI] [PubMed] [Google Scholar]
- Matesanz‐Isabel J, Arias‐del-Val J, Alvarez‐Illera P, Fonteriz RI, Montero M, Alvarez J (2016) Functional roles of MICU1 and MICU2 in mitochondrial Ca(2+) uptake. Biochim Biophys Acta 1858: 1110–1117 [DOI] [PubMed] [Google Scholar]
- Mehrara E, Forssell‐Aronsson E, Ahlman H, Bernhardt P (2007) Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res 67: 3970–3975 [DOI] [PubMed] [Google Scholar]
- Mishra J, Jhun BS, Hurst S, O‐Uchi J, Csordas G Sheu SS (2017) The mitochondrial Ca(2+) uniporter: structure, function, and pharmacology. Handb Exp Pharmacol 240: 129–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteith GR, Prevarskaya N, Roberts‐Thomson SJ (2017) The calcium‐cancer signalling nexus. Nat Rev Cancer 17: 367–380 [DOI] [PubMed] [Google Scholar]
- Morandi A, Indraccolo S (2017) Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer 1868: 1–6 [DOI] [PubMed] [Google Scholar]
- Morisaki T, Katano M (2003) Mitochondria‐targeting therapeutic strategies for overcoming chemoresistance and progression of cancer. Curr Med Chem 10: 2517–2521 [DOI] [PubMed] [Google Scholar]
- Nemani N, Dong Z, Daw CC, Madaris TR, Ramachandran K, Enslow BT, Rubannelsonkumar CS, Shanmughapriya S, Mallireddigari V, Maity S et al , et al (2020) Mitochondrial pyruvate and fatty acid flux modulate MICU1‐dependent control of MCU activity. Sci Signal 13: eaaz6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H, Mu J, Wang J, Li Y (2018) miR1955p regulates multidrug resistance of gastric cancer cells via targeting ZNF139. Oncol Rep 40: 1370–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paupe V, Prudent J (2018) New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem Biophys Res Commun 500: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popgeorgiev N, Jabbour L, Gillet G (2018) Subcellular localization and dynamics of the Bcl‐2 family of proteins. Front Cell Dev Biol 6: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit PK, Edwards R, Tokatlidis K, Saini N (2019) MiR‐195 regulates mitochondrial function by targeting mitofusin‐2 in breast cancer cells. RNA Biol 16: 918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16: 203–222 [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovacs‐Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA et al (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342: 1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya Y, Ogawa T, Iizuka M, Yoshizato K, Ikeda K, Kawada N (2011) Down‐regulation of cyclin E1 expression by microRNA‐195 accounts for interferon‐beta‐induced inhibition of hepatic stellate cell proliferation. J Cell Physiol 226: 2535–2542 [DOI] [PubMed] [Google Scholar]
- Shanmughapriya S, Tomar D, Dong Z, Slovik KJ, Nemani N, Natarajaseenivasan K, Carvalho E, Lu C, Corrigan K, Garikipati VNS et al (2018) FOXD1‐dependent MICU1 expression regulates mitochondrial activity and cell differentiation. Nat Commun 9: 3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Saini N (2012) Downregulation of BCL2 by miRNAs augments drug‐induced apoptosis–a combined computational and experimental approach. J Cell Sci 125: 1568–1578 [DOI] [PubMed] [Google Scholar]
- Singh R, Yadav V, Kumar S, Saini N (2015) MicroRNA‐195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci Rep 5: 17454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood AK, Seftor EA, Fletcher MS, Gardner LM, Heidger PM, Buller RE, Seftor RE, Hendrix MJ (2001) Molecular determinants of ovarian cancer plasticity. Am J Pathol 158: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon PS, Tacon LJ, Gill AJ, Bambach CP, Sywak MS, Campbell PR, Yeh MW, Wong SG, Clifton‐Bligh RJ, Robinson BG et al (2009) miR‐195 and miR‐483‐5p identified as predictors of poor prognosis in adrenocortical cancer. Clin Cancer Res 15: 7684–7692 [DOI] [PubMed] [Google Scholar]
- Sun L, Suo C, Li ST, Zhang H, Gao P (2018) Metabolic reprogramming for cancer cells and their microenvironment: beyond the Warburg Effect. Biochim Biophys Acta Rev Cancer 1870: 51–66 [DOI] [PubMed] [Google Scholar]
- Wagner W, Ciszewski WM, Kania KD (2015) L‐ and D‐lactate enhance DNA repair and modulate the resistance of cervical carcinoma cells to anticancer drugs via histone deacetylase inhibition and hydroxycarboxylic acid receptor 1 activation. Cell Commun Signal 13: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang J, Ma H, Zhang J, Zhou X (2012) Downregulation of miR‐195 correlates with lymph node metastasis and poor prognosis in colorectal cancer. Med Oncol 29: 919–927 [DOI] [PubMed] [Google Scholar]
- Wang R, Zhao N, Li S, Fang JH, Chen MX, Yang J, Jia WH, Yuan Y, Zhuang SM (2013) MicroRNA‐195 suppresses angiogenesis and metastasis of hepatocellular carcinoma by inhibiting the expression of VEGF, VAV2, and CDC42. Hepatology 58: 642–653 [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Y, Lan H, Li J (2014) MiR‐195 inhibits the growth and metastasis of NSCLC cells by targeting IGF1R. Tumour Biol 35: 8765–8770 [DOI] [PubMed] [Google Scholar]
- Wang H, Ren S, Xu Y, Miao W, Huang X, Qu Z, Li J, Liu X, Kong P (2019) MicroRNA‐195 reverses the resistance to temozolomide through targeting cyclin E1 in glioma cells. Anticancer Drugs 30: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21: 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ji A, Wang X, Zhu Y, Yu Y, Lin Y, Liu Y, Li S, Liang Z, Xu X et al (2015) MicroRNA‐195‐5p, a new regulator of Fra‐1, suppresses the migration and invasion of prostate cancer cells. J Transl Med 13: 289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P (2005) Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res 65: 613–621 [PubMed] [Google Scholar]
- Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP, Zhuang SM (2009) MicroRNA‐195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology 50: 113–121 [DOI] [PubMed] [Google Scholar]
- Xu S, Fu GB, Tao Z, OuYang J, Kong F, Jiang BH, Wan X, Chen K (2015) MiR‐497 decreases cisplatin resistance in ovarian cancer cells by targeting mTOR/P70S6K1. Oncotarget 6: 26457–26471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li M, Chang S, Wang L, Song T, Gao L, Hu L, Li Z, Liu L, Yao J et al (2014) MicroRNA‐195 acts as a tumor suppressor by directly targeting Wnt3a in HepG2 hepatocellular carcinoma cells. Mol Med Rep 10: 2643–2648 [DOI] [PubMed] [Google Scholar]
- Ye R, Wei B, Li S, Liu W, Liu J, Qiu L, Wu X, Zhao Z, Li J (2017) Expression of miR‐195 is associated with chemotherapy sensitivity of cisplatin and clinical prognosis in gastric cancer. Oncotarget 8: 97260–97272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zhang Y, Cavazos D, Ma X, Zhao Z, Du L, Pertsemlidis A (2018) miR‐195 targets cyclin D3 and survivin to modulate the tumorigenesis of non‐small cell lung cancer. Cell Death Dis 9: 193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QQ, Xu H, Huang MB, Ma LM, Huang QJ, Yao Q, Zhou H, Qu LH (2012) MicroRNA‐195 plays a tumor‐suppressor role in human glioblastoma cells by targeting signaling pathways involved in cellular proliferation and invasion. Neuro Oncol 14: 278–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB et al (2010) Warburg effect in chemosensitivity: targeting lactate dehydrogenase‐A re‐sensitizes taxol‐resistant cancer cells to taxol. Mol Cancer 9: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.