

Abstract
Autotaxin (ATX) converts lysophosphatidylcholine and sphingosyl‐phosphorylcholine into lysophosphatidic acid and sphingosine 1‐phosphate, respectively. Despite the pivotal function of ATX in lipid metabolism, mechanisms by which ATX regulates immune and inflammatory disorders remain elusive. Here, using myeloid cell lineage‐restricted Atx knockout mice, we show that Atx deficiency disrupts membrane microdomains and lipid rafts, resulting in the inhibition of Toll‐like receptor 4 (TLR4) complex formation and the suppression of adaptor recruitment, thereby inhibiting TLR4‐mediated responses in macrophages. Accordingly, TLR4‐induced innate immune functions, including phagocytosis and iNOS expression, are attenuated in Atx‐deficient macrophages. Consequently, Atx−/− mice exhibit a higher bacterial prevalence in the intestinal mucosa compared to controls. When combined with global Il10−/− mice, which show spontaneous colitis due to the translocation of luminal commensal microbes into the mucosa, myeloid cell lineage‐restricted Atx knockout accelerates colitis development compared to control littermates. Collectively, our data reveal that Atx deficiency compromises innate immune responses, thereby promoting microbe‐associated gut inflammation.
Keywords: ectonucleotide pyrophosphatase/phosphodiesterase family member 2, ENPP2, inflammatory bowel diseases, lipid raft, toll‐like receptor 4
Subject Categories: Immunology; Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
Autotaxin is a secreted lysophospholipase that hydrolyzes lysophosphatidylcholine. Atx loss disrupts plasma membrane lipid rafts in macrophages, impairing TLR4‐induced innate immune responses, thereby accelerating gut inflammation.
Introduction
Myeloid lineage cells, including neutrophils, monocytes/macrophages, and dendritic cells (DCs), express a group of pattern recognition receptors (PRRs) which recognize a variety of microbial pattern molecules. One such PRR is Toll‐like receptor 4 (TLR4), which specifically recognizes lipopolysaccharide (LPS) (Poltorak et al, 1998; Rhee & Hwang, 2000). Upon recognizing LPS in macrophages, TLR4 relocates to lipid rafts of the plasma membrane where it forms a complex with the co‐receptor CD14 and then proceeds to recruit a combination of adaptor molecules (MYD88, MAL/TIRAP, TRIF, and TRAM) to its cytoplasmic TIR domain. MYD88 then associates with the serine/threonine protein kinases Irak‐1/4 to activate the transcription factors NFκB and AP‐1, leading to the production of cytokines and chemokines involved in inflammatory and immune responses. Additionally, the TLR4‐CD14 complex can be internalized to cytosolic endosomes, leading to the activation of the transcription factor IRF‐3 and subsequent induction of interferon (IFN)‐α/β production for anti‐viral defense (Rajaiah et al, 2015). By these two pathways, macrophages that recognize LPS produce an array of cytokines and chemokines, which either kill invading microorganisms directly or assist effector T and B cells in eliminating them (Iwasaki & Medzhitov, 2015). Therefore, macrophages have a critical role at the first line of immune defense against invading microbes.
Autotaxin (ATX, also known as ENPP2) is a secreted lysophospholipase D that hydrolyzes lysophosphatidylcholine (LPC) into lysophosphatidic acid (LPA) (Stracke et al, 1992). Relatively high concentrations of ATX protein have been identified in a variety of biological fluids including blood, urine, seminal fluids, and cerebrospinal fluids (Nakamura et al, 2008). ATX expression has also been detected in melanoma cells, endothelial cells, adipocytes, lung epithelial cells and macrophages, and breast cancer cells (Stracke et al, 1992; Nakamura et al, 2008; David et al, 2010; Oikonomou et al, 2012). Increased protein levels of ATX have also been measured in the serum samples of patients with liver injury and in the lung tissues of patients with idiopathic pulmonary fibrosis (Kremer et al, 2010; Oikonomou et al, 2012), suggesting the potential involvement of ATX in the pathogenesis or pathophysiology of certain diseases.
Inflammatory bowel diseases (IBD) is a chronic inflammatory disorder in the gut, which is caused by uncontrolled immune responses against commensal microbes in genetically susceptible individuals (Mitchell et al, 2018a). Previous studies have identified increased Atx mRNA expression in the inflamed colons of mice in which colitis was induced either by oral administration of dextran sulfate sodium (DSS) (Hozumi et al, 2013; Lin et al, 2019) or by adoptive transfer of CD4+;CD25− T cells. Increased Atx mRNA levels have similarly been observed in the inflamed mucosa of Crohn's disease (CD) and ulcerative colitis (UC) patients (Hozumi et al, 2013). A recent study also suggested that Atx deletion reduced the severity of DSS‐induced colitis in mice (Lin et al, 2019). However, because residual DSS contamination in RNA samples from DSS‐treated mice inhibits quantitative real‐time PCR (Kerr et al, 2012; Viennois et al, 2013), these observations ought to be tested by an alternative method. Moreover, the previous studies heavily depend on the model of DSS‐induced colitis, which is primarily a model for epithelial injury; likewise, the T‐cell transfer colitis model relies on T‐cell migration in immunocompromised SCID mice. In both cases, these models do not accurately reflect the underlying pathology of human IBD. Thus, the functional role of ATX deficiency in chronic gut inflammation has yet to be conclusively determined.
In this study, we examine the association of ATX with the development of chronic intestinal inflammation by evaluating the serum level of ATX protein in IBD patients and then further dissecting innate and adaptive immune mechanisms in myeloid cell‐restricted Atx knockout (ko) mice in order to uncover the mechanism by which Atx deficiency modulates TLR4‐mediated immune responses.
Results
ATX serum protein levels are lower in IBD patients than in normal subjects
To examine the association of ATX with intestinal inflammation, we evaluated the ATX protein content in serum samples of IBD patients classified as UC and CD, along with healthy control subjects. Surprisingly, we observed that ATX protein levels were markedly lower in the serum of UC and CD patients than in normal controls (Fig 1A), suggesting that reduced expression of ATX may be associated with the development or perpetuation of the chronic inflammatory disorder in the intestine.
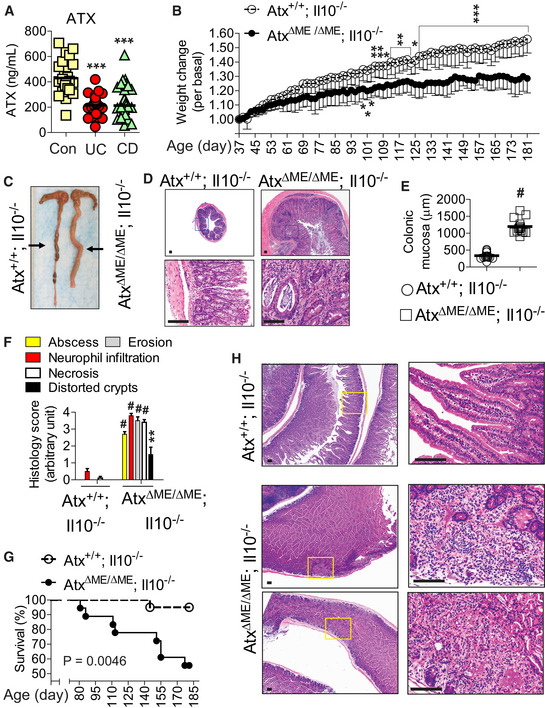
Figure 1. Myeloid cell lineage‐restricted Atx gene deletion accelerated the development of spontaneous gut inflammation in an Il10‐deficient condition.
-
AATX serum protein levels were measured in blood serum samples from IBD patients and healthy controls using ELISA: active UC (n = 26), active CD (n = 34), control (Con) (n = 26).
-
BAge‐ and sex‐matched AtxΔΜΕ/ΔΜΕ;Il10−/− mice (n = 9) and Atx+/+;Il10−/− littermates (n = 9) were examined for the development of spontaneous colitis. Body weight changes were monitored every other day, starting at the age of 37 days and ending at the age of 6 months. Results are means ± SD. Data were compared by two‐way ANOVA (with treatment and times), followed by the multiple‐comparison Bonferroni t‐test to assess differences between groups.
-
CGross appearance of the full‐length mouse colon. Each line on a ruler represents a millimeter (mm).
-
D, EH&E‐stained section of the mouse colon. Insets are enlarged in the lower panel. Colonic mucosa thickness was measured using ZEISS Axio Imager Z1 microscope (n = 14/group) (E).
-
FHistological parameters were quantified with H&E colon sections (n = 10/group) (D) and shown as mean ± SEM.
-
GSurvival of AtxΔΜΕ/ΔΜΕ;Il10−/− (n = 17) and Atx+/+;Il10−/− littermates (n = 18) was analyzed by the Kaplan–Meier method (Log‐rank P = 0.0046).
-
HPresented are H&E‐stained sections of the small intestine in a “Swiss‐Roll” form. Inset areas are enlarged in the right panel.
It is worth noting that gut microbes are capable of translocating from the lumen into the intestinal mucosa in normal conditions. However, when immune function is impaired these microbes can induce intestinal inflammation in an immune compromised condition if not eliminated by innate and adaptive mucosal immune mechanisms. This notion prompted us to hypothesize that Atx deficiency may alter the innate and adaptive immune responses to gut microbes, thereby promoting the development of intestinal inflammation. Therefore, we generated myeloid cell lineage‐restricted Atx‐ko (AtxΔΜΕ/ΔΜΕ) mice and Atx+/+ littermates on a C57BL/6 background by crossing LysM‐Cre mice (Clausen et al, 1999) with Atx‐floxed mice (van Meeteren et al, 2006). We confirmed that these mice are otherwise healthy without any noticeable phenotypic changes.
Myeloid cell lineage‐specific Atx‐ko accelerates the development of spontaneous colitis in an interleukin‐10 (Il10)‐deficient condition
The translocation of indigenous gut microbes to the mucosa can result in pathology when the immune system is severely compromised (Casanova & Abel, 2009). For instance, in Il10−/− mice bacterial translocation can spontaneously elicit deleterious intestinal inflammation (Kuhn et al, 1993; Sellon et al, 1998). For this reason, we harnessed Il10−/− mice to examine the potential role of the Atx deficiency in chronic intestinal inflammation. We hypothesized that Atx deficiency in myeloid cells would accelerate the spontaneous induction of colitis in Il10−/− mice. To test this hypothesis, we generated AtxΔΜΕ/ΔΜΕ; Il10−/− mice on a C57BL/6 background which harbored a myeloid cell lineage‐specific Atx‐ko and a global interleukin‐10 gene deletion. Over the 6‐month experimental period following birth, AtxΔΜΕ/ΔΜΕ; Il10−/− mice exhibited a noticeably slowed weight‐gaining pattern reflecting the development of intestinal inflammation, whereas Atx+/+; Il10−/− littermates demonstrated a gradual and consistent pattern of weight gain (Fig 1B). Indeed, AtxΔΜΕ/ΔΜΕ; Il10−/− mice exhibited gross inflammation throughout the colon, characterized by a pale and enlarged colon with reduced colon contents; meanwhile, Atx+/+;Il10−/− mice had normal colons (Fig 1C). Because they were suffering from chronic colitis, the colonic mucosa of AtxΔΜΕ/ΔΜΕ; Il10−/− mice was thicker than that of Atx+/+;Il10−/− mice, indicative of chronic colitis (Fig 1D and E). Through microscopic examination, the colonic mucosa of AtxΔΜΕ/ΔΜΕ;Il10−/− mice were characterized by prominent abscesses, massive neutrophil infiltration, increased epithelial erosion, marked architectural distortion of crypts, and enhanced necrosis, all of which were almost negligible in the colons of Atx+/+; Il10−/− mice (Fig 1F). In accordance with the histological manifestations of colitis, the expression of pro‐inflammatory factors was dramatically increased in the colons of AtxΔΜΕ/ΔΜΕ; Il10−/− mice compared to Atx+/+; Il10−/− mice (Fig EV1). Reduced levels of serum albumin and total protein were observed in AtxΔΜΕ/ΔΜΕ;Il10−/− mice relative to Atx+/+; Il10−/− mice (Fig EV2A) indicating protein‐losing enteropathy, which is a typical manifestation of severe intestinal inflammation. Because they were experiencing massive inflammation, substantially increased mortality was observed in AtxΔΜΕ/ΔΜΕ;Il10−/− mice compared to Atx+/+;Il10−/− mice during the experimental period (Fig 1G).
Figure EV1. Pro‐inflammatory cytokine gene expression is up‐regulated in the colon of AtxΔΜΕ/ΔΜΕ;Il10−/− mice compared to Atx+/+;Il10−/− mice.
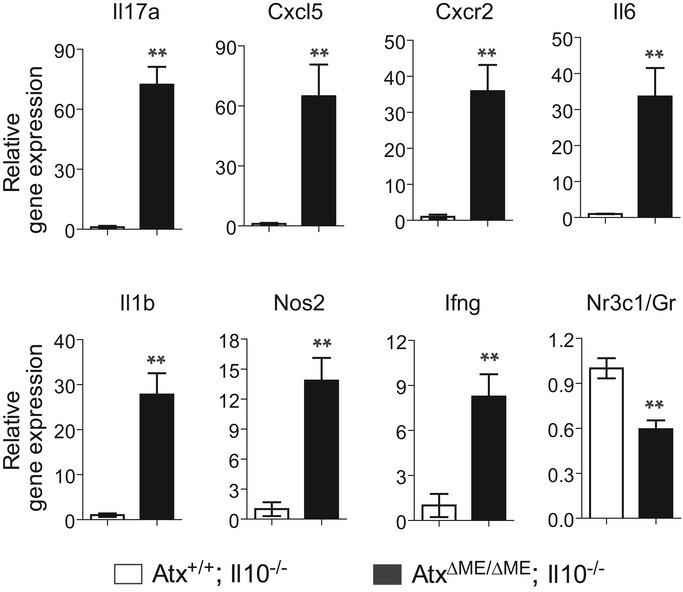
The mRNA level of pro‐inflammatory cytokine genes was evaluated through qPCR with the full‐thickness colon of AtxΔΜΕ/ΔΜΕ;Il10−/− and Atx+/+;Il10−/− mice. n = 6/group. The data are shown as mean ± SEM. **P < 0.01 (Mann–Whitney U‐test).
Figure EV2. AtxΔΜΕ/ΔΜΕ;Il10−/− mice exhibit no pathologic phenotype in the liver and kidney.
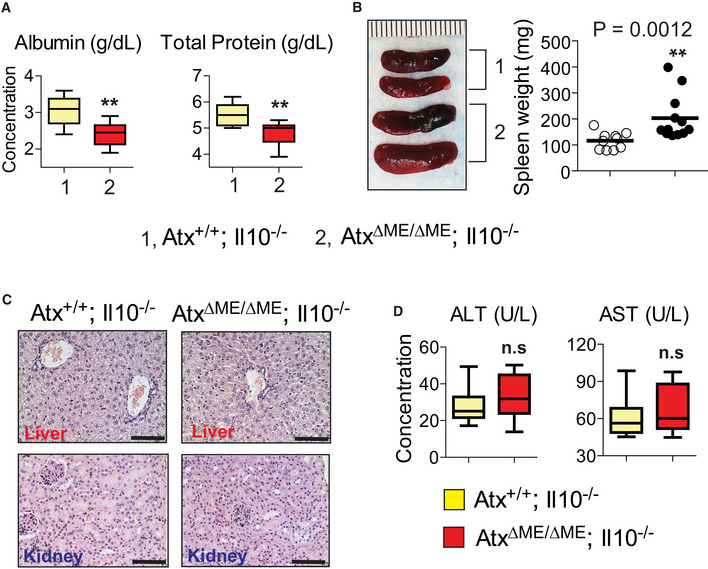
- Albumin and total protein were measured in the blood serum of the mice (n = 12/group).
- Gross images of the spleen (left) and spleen weight (right) were presented. Each line on a ruler represents a millimeter (mm).
- Presented are representative photographs of H&E‐stained sections of the liver and kidney. Scale bar indicates 100 μm.
- The level of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) protein was measured from the mouse blood serum. **P < 0.01 (Mann–Whitney U‐test). not significant (n.s.).
Crohn's disease (CD)‐like enteritis is observed in the small intestines of AtxΔΜΕ/ΔΜΕ;Il10−/− mice
CD is a regional enteritis that frequently affects the small intestine, but can develop in any part of the gastrointestinal tract. CD is characterized by inflammation that is transmural from mucosa to serosa and discontinuous, exhibiting patchy and regional inflammation. Therefore, neutrophils can be focally present and infiltration can be prominently observed in the submucosa and serosa. Surprisingly, AtxΔΜΕ/ΔΜΕ;Il10−/− mice exhibited CD‐like enteritis in the small intestine in addition to colitis, while the small intestines of age‐matched Atx+/+;Il10−/− mice were normal (Fig 1H). Similarly to the pathology of human CD, the small intestines of AtxΔΜΕ/ΔΜΕ;Il10−/− mice were characterized by focal inflammation with regional infiltration and discontinuous mucosal necrosis. These data indicate that, in addition to colitis, AtxΔΜΕ/ΔΜΕ;Il10−/− mice develop CD‐like enteritis.
Because they were suffering from chronic intestinal inflammation, AtxΔΜΕ/ΔΜΕ; Il10−/− mice had larger spleens than age‐matched Atx+/+;Il10−/− mice (Fig EV2B). However, other extra‐intestinal organs, including the liver and kidney, were normal in both AtxΔΜΕ/ΔΜΕ; Il10−/− and Atx+/+; Il10−/− mice. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST), which signify pathology in the liver, were similar between the groups (Fig EV2C and D). Therefore, these data indicate that the development of inflammation was confined to the small intestine and colon of AtxΔΜΕ/ΔΜΕ;Il10−/− mice.
Elevated levels of the Bacteroides genus in the fecal microbiome are a commonly observed microbial manifestation of colitis in IL‐10 signaling‐defective mice (Bloom et al, 2011; Im et al, 2014; Mitchell et al, 2018a). In line with these studies, we observed through 16S rRNA gene sequencing that age‐ (8 weeks old) and sex‐matched AtxΔΜΕ/ΔΜΕ; Il10−/− and Atx+/+; Il10−/− mice had distinct fecal microbiomes with elevated levels of the Bacteroides genus in AtxΔΜΕ/ΔΜΕ; Il10−/− (37.2%) mice compared to Atx+/+; Il10−/− (14.3%, P = 0.0152) mice (Fig EV3A and B). Within the Bacteroides genus, we further identified sharp increases in Bacteroides spp. and Bacteroides acidifaciens in the fecal samples of AtxΔΜΕ/ΔΜΕ; Il10−/− mice compared to Atx+/+; Il10−/− mice (Fig EV3C). These data demonstrate that the myeloid cell‐restricted Atx‐ko condition is capable of promoting the development of colitis in an IL‐10‐deficient condition.
Figure EV3. Altered fecal microbiome of AtxΔΜΕ/ΔΜΕ;Il10−/− mice signifies the microbial manifestation of mouse colitis.
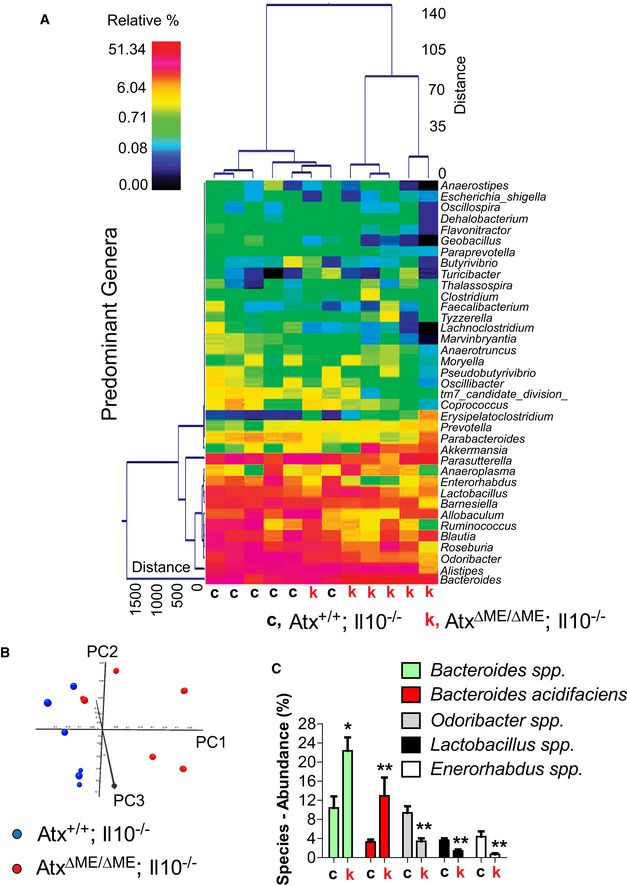
- A dual hierarchal dendrogram was generated based on the predominant genera using Ward's minimum variance clustering and Manhattan distances. More similar microbial populations between AtxΔΜΕ/ΔΜΕ;Il10−/− and Atx+/+;Il10−/− mice were mathematically clustered closer together. The samples with a more similar consortium of genera cluster closer together with the length of connecting lines (top of heatmap) related to the similarity; shorter lines between two samples indicate closely matched microbial consortia. The heatmap represents the relative percentages of each genus. The legend for the heatmap is provided in the upper left corner. The predominant genera are represented along the right Y‐axis. AtxΔΜΕ/ΔΜΕ;Il10−/− mouse is indicated by “k”, while “c” stands for Atx+/+;Il10−/− mice.
- Principal coordinates analysis (PCoA) plot of weighted UniFrac metrics was generated. PCoA plots represent the three (PC1, PC2, and PC3) highest discriminating axes, explaining 87.3 percent of the variation between the groups (P = 0.003).
- The major species identified in the fecal samples of AtxΔΜΕ/ΔΜΕ;Il10−/− mice (k) and Atx+/+;Il10−/− littermates (c) were analyzed to compare the abundance at the species level. The abundance of Bacteroides was dramatically increased in the fecal samples of AtxΔΜΕ/ΔΜΕ;Il10−/− mice compared to Atx+/+;Il10−/− mice. Results are means ± SD, *P < 0.05, **P < 0.01 (Mann–Whitney U‐test).
Furthermore, through microarray analysis we identified that the genes associated with anti‐bacterial responses were differentially expressed in the intestines of AtxΔΜΕ/ΔΜΕ; Il10−/− mice compared to Atx+/+;Il10−/− mice (Fig EV4). For instance, the mRNA level of the genes encoding Tlr5, Naip1, Xiap, Il18, and Sugt1 was substantially reduced in the intestines of AtxΔΜΕ/ΔΜΕ; Il10−/− mice compared to controls; however, the mRNA levels of Tnf, Nlrp3, Lyz2, Lbp, Cd14, and Mefv levels were markedly increased. These data indicate that the myeloid cell‐restricted Atx‐ko condition results in altered immune responses in the intestine, which is home to approximately 70–80% of host immune cells.
Figure EV4. Altered anti‐bacterial responses in the colon of AtxΔΜΕ/ΔΜΕ;Il10−/− mice compared to Atx+/+;Il10−/− littermates.
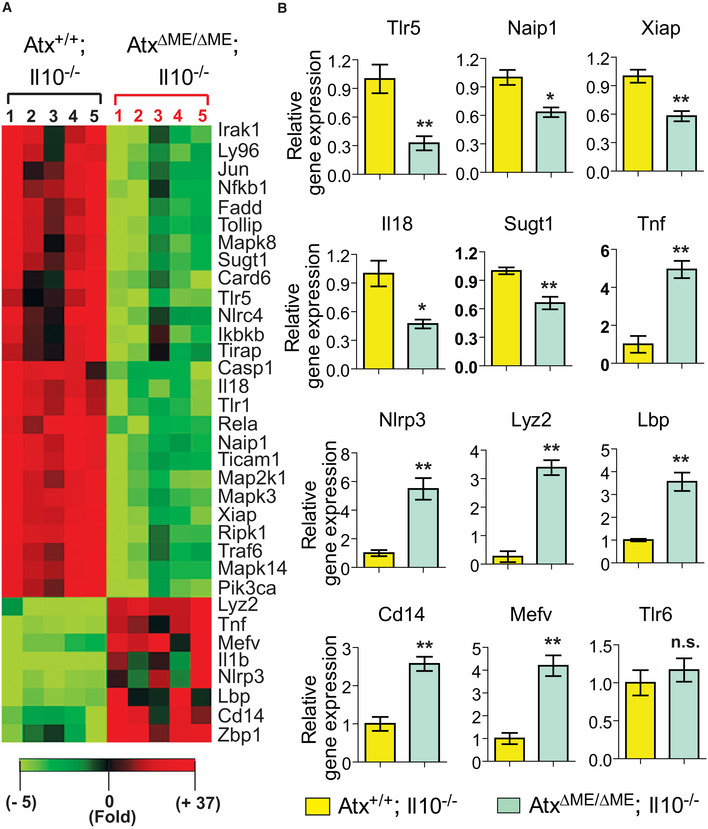
- The expression of anti‐bacterial response‐focused genes was evaluated using the full‐thickness colon tissues of AtxΔΜΕ/ΔΜΕ; Il10−/− mice and Atx+/+;Il10−/− littermates. An array of gene expression revealing a significant difference was visualized in the heatmap.
- Representative gene expression exhibiting a significant difference was independently confirmed by qPCR. n = 5/group. Error bars indicate ± SEM. *P < 0.05, **P < 0.01 (Mann–Whitney U‐test). not significant (n.s.).
Together, these data strongly indicate that Atx gene deletion in myeloid cells may inhibit immune mechanisms against commensal microbes of the intestine, thereby accelerating microbe‐associated intestinal inflammation in an Il10‐deficient condition.
Atx deficiency disrupts plasma membrane lipid rafts in macrophages
While primarily known for its ability to convert LPC into LPA (Stracke et al, 1992), ATX is also capable of hydrolyzing sphingosyl‐phosphorylcholine (SPC) to produce sphingosine 1‐phosphate (S1P) (Clair et al, 2003). LPC interacts with cholesterol to decrease cell membrane fluidity (Mio et al, 1985). Large amounts of cholesterol and sphingolipids are tightly packed alongside saturated phospholipids in certain regions of the plasma membrane, giving rise to phase separations of the membrane microdomain called “lipid rafts” (Simons & Ikonen, 1997). Lipid rafts are characterized by the tight packing of lipids, which allows them to accommodate only a limited subset of membrane proteins, including TLR4 and the glycosylphosphatidylinositol (GPI)‐anchored receptor CD14 (Triantafilou et al, 2002). Because ATX hydrolyzes LPS and SPC, both of which participate in maintaining lipid raft structure, it is likely that ATX plays a significant role in maintaining the integrity of lipid rafts.
Interestingly, much like the enhanced development of colitis that we observed in AtxΔΜΕ/ΔΜΕ; Il10−/− mice, it has been demonstrated that Tlr4−/−; Il10−/− mice exhibit accelerated development of colitis compared to Tlr4+/+; Il10−/−, suggesting that the disruption of TLR4‐mediated responses promotes the development of colitis in an Il10‐deficient condition (Matharu et al, 2009).
Given the probable influence of ATX upon lipid raft integrity, along with the localization of the TLR4:CD14 complex to lipid rafts, we hypothesized that Atx deficiency would disrupt the integrity of the membrane microdomain to suppress TLR4‐mediated immune responses in myeloid cells.
To test this hypothesis, we examined the peritoneal macrophages from AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermates to confirm that macrophages from AtxΔΜΕ/ΔΜΕ mice did not produce ATX protein and that ATX production was conserved in macrophages from Atx+/+ littermates (Fig 2A). We further observed that the amount of total LPA and certain molecular species of LPA were reduced in the macrophages from AtxΔΜΕ/ΔΜΕ mice relative to controls (Fig 2B). Next, macrophages and DCs were visualized using transmission electron microscopy (TEM), which revealed that these cells had a similar microscopic appearance between AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermates (Fig 2C and D).
Figure 2. The integrity of lipid rafts was disrupted in Atx‐ko macrophages.
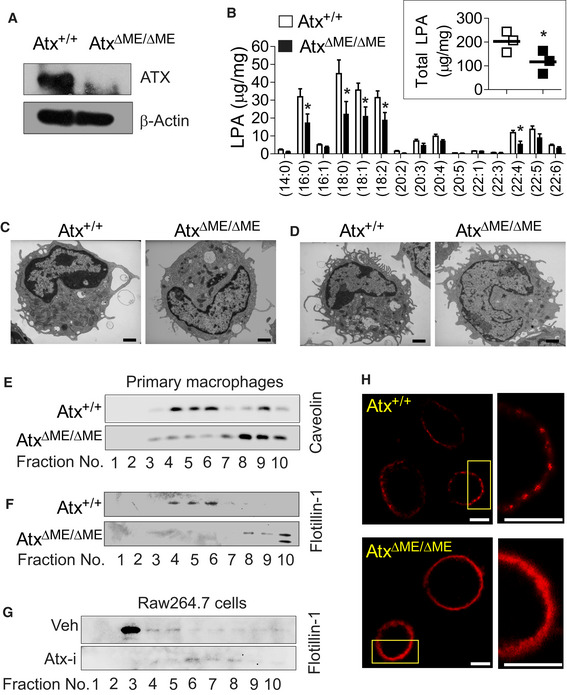
-
ATotal cell lysates of peritoneal macrophages from AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermates were subjected to immunoblotting analysis to confirm Atx deletion.
-
BPeritoneal macrophages from the mice were subjected to the ESI‐MS/MS method to quantify endogenous LPA. Individual molecular species of LPA and total LPA (inset graph) were compared. The data are shown as mean ± SEM (n = 3/group) *P < 0.05 (one‐tailed unpaired t‐test).
-
C, DThioglycolate‐elicited peritoneal immune cells harvested from the mice were immediately fixed for transmission electron microscopy to visualize the appearance of macrophages (C) and DCs (D). Scale bar indicates 1 μm.
-
E–GLipid rafts from peritoneal macrophages (E, F) or from mouse macrophage Raw264.7 cells treated with Atx inhibitor (Atx‐i) PF8380 (50 μM, 30 min) or vehicle (Veh., DMSO 0.1%) (G) were fractionated into 10 fractions though sucrose density‐gradient ultracentrifugation at 100,000 g for 17 h. Protein was purified and concentrated with the 3K cutoff filter, followed by Western blot analysis with antibody recognizing the lipid raft marker proteins Caveolin or Flotillin‐1.
-
HPeritoneal macrophages were stained with Alexa Fluor 594‐cholera toxin subunit B (Red). The plasma membrane was examined with FV10i confocal scanning microscopy. Inset areas were magnified at the right panel. Each scale bar indicates 10 μm.
To determine whether Atx deficiency might alter the integrity of lipid rafts, we carried out sucrose density‐gradient ultracentrifugation which is a powerful technique for fractionating lipid rafts. We found that the distribution of the lipid raft marker proteins Flotillin‐1 and Caveolin‐1 (Lu et al, 2016) was changed in peritoneal macrophages from AtxΔΜΕ/ΔΜΕ mice compared to those from Atx+/+ littermates. In controls, the presence of Flotillin‐1 and Caveolin‐1 was observed mainly in lipid raft fractions (fraction 4–6). However, in AtxΔΜΕ/ΔΜΕ mice the localization of these proteins was shifted into non‐lipid raft fractions (fraction 8–10) (Fig 2E and F). We further performed the lipid raft fraction assay with a mouse macrophage cell line (Raw264.7 cells) treated with Atx inhibitor PF8380 (Atx‐i) (Gierse et al, 2010) or vehicle. In vehicle‐treated Raw264.7 cells, Flotillin‐1 was identified in the lipid raft fraction (fraction 3–5). However, Atx inhibitor treatment shifted the distribution of Flotillin‐1 into non‐lipid raft fractions (fraction 6–8) of Raw264.7 cells (Fig 2G). These data suggest that Atx deficiency disrupts the integrity of lipid rafts in macrophages.
Lipid rafts are cell membrane microdomains composed of cholesterol and sphingolipids such as ganglioside GM1, which form a separate liquid‐ordered phase in the cell membrane lipid bilayer. To further examine the effect of Atx on lipid raft structure and provide a visual representation, macrophages were stained with cholera toxin B (CTXB) labeled with Alexa594 (Red). Because CTXB binds to ganglioside GM1, it can be used as a marker for the identification of lipid rafts in the plasma membrane (Blank et al, 2007). ATX expressing macrophages exhibited an aggregation of GM1, indicated as red dots or patches in the confocal micrographs (Fig 2H upper panel), representing the clustering of lipid rafts in the plasma membrane. However, Atx‐ko macrophages exhibited disrupted discontinuous phase separation and domain clustering, resulting in diffuse GM1‐staining in the plasma membrane (Fig 2H lower panel).
Taken together with the results of the sucrose density‐gradient fractionation assay, these data demonstrate that Atx deficiency disrupts the integrity of lipid rafts at least in macrophages.
Atx deficiency inhibits the interaction of TLR4 with its co‐receptor CD14
Lipid rafts are characterized by the tight packing of lipids, which allows them to accommodate only a limited subset of proteins, including TLR4 and CD14 (Triantafilou et al, 2002). To elicit LPS‐stimulated responses in macrophages, TLR4 not only forms a homodimer, but also complexes with CD14 at the membrane lipid raft. Given our finding that lipid rafts are disrupted in AtxΔΜΕ/ΔΜΕ macrophages, we hypothesized that this would disturb the interaction between TLR4 and CD14. Therefore, we performed a co‐immunoprecipitation assay which revealed that the LPS‐induced interaction between TLR4 and CD14 was abolished in AtxΔΜΕ/ΔΜΕ macrophages, whereas LPS stimulation clearly induced this interaction in control cells (Fig 3A). We similarly found that the treatment of Raw264.7 cells with ATX inhibitor suppressed the interaction of TLR4 and CD14 (Fig 3B).
Figure 3. Atx deficiency disturbed the formation of the LPS‐sensing‐TLR4 receptor complex.
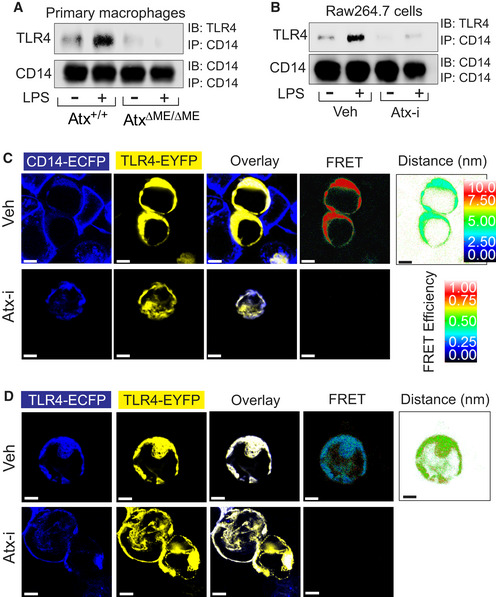
-
A, BPeritoneal macrophages (A) or Raw264.7 cells (B) were stimulated with LPS (20 ng/ml, 20 min) in the presence/absence of ATX inhibitor (50 μM, 30 min) or vehicle (Veh, DMSO 0.1%). Cell lysates were subjected to co‐immunoprecipitation (IP) with CD14 antibody, followed by immunoblotting (IB) with TLR4 antibody.
-
C, DThe physical interaction of TLR4‐CD14 or TLR4‐TLR4 was examined by FRET analysis. HEK293 cells were transfected with TLR4‐EYFP (yellow) and CD14‐ECFP (blue) (C) or TLR4‐EYFP and TLR4‐ECFP (D), followed by ATX inhibitor (50 μM, 30 min) or vehicle treatment (Veh, DMSO 0.1%). Cells were washed with PBS, fixed, and then visualized with filter sets for CFP and YFP. FRET images were visualized with the FRET filter set, expressed as corrected FRET efficiency and displayed in quantitative pseudocolor indicating the distance between proteins (arbitrary linear units of fluorescence intensity). Presented are the representative images from at least three independent experiments. Scale bar indicates 5 μm.
Source data are available online for this figure.
Next, we harnessed the technique of fluorescence resonance energy transfer (FRET), which allows for the examination of a physical protein–protein interaction inside living cells (Hernanz‐Falcon et al, 2004; Kawai et al, 2004). In FRET analysis, enhanced cyan fluorescent protein (ECFP) is used as the donor fluorophore and enhanced yellow fluorescent protein (EYFP) as the acceptor (Rizzo et al, 2004). Therefore, we generated CD14‐ECFP and TLR4‐EYFP encoding constructs to perform FRET analysis. HEK293 cells were co‐transfected with CD14‐ECFP (donor) and TLR4‐EYFP (acceptor) expression constructs, followed by treatment with ATX inhibitor or vehicle. As expected, CD14 and TLR4 were primarily observed at the plasma membrane (Fig 3C). While a strong FRET signal was measured in control cells, this signal was ablated in ATX inhibitor‐treated cells, indicating that blocking ATX suppresses the association between TLR4 and CD14. To examine whether ATX inhibition also affects the homodimerization of TLR4, HEK293 cells were co‐transfected with TLR4‐ECFP and TLR4‐EYFP encoding constructs, followed by treatment with inhibitor or vehicle. We observed a FRET signal between TLR4‐ECFP and TLR4‐EYFP in vehicle‐treated cells, but not in inhibitor‐treated cells (Fig 3D), indicating that TLR4‐TLR4 interaction is also disrupted by ATX inhibition.
Together with the immunoprecipitation data obtained from AtxΔΜΕ/ΔΜΕ macrophages and ATX inhibitor‐treated Raw264.7 cells, the results obtained from FRET analysis demonstrate that formation of the TLR4 and CD14 is disrupted in an ATX‐deficient condition.
Atx deficiency inhibits LPS‐induced internalization of TLR4 in macrophages
LPS stimulation induces the recruitment of TLR4 to lipid rafts where it cooperates with other signaling molecules and begins internalization to an early endosome, where it can mediate LPS‐stimulated intracellular signaling (Kagan et al, 2008; Rajaiah et al, 2015; Tsukamoto et al, 2018). Lipid rafts are critical to this process, in which they function as a signaling platform for TLR4‐mediated intracellular signaling. Thus, based on our finding of compromised lipid raft integrity in AtxΔΜΕ/ΔΜΕ macrophages, we next investigated whether TLR4 internalization is also disturbed by Atx deficiency. Atx+/+ macrophages demonstrated normal LPS‐induced TLR4 internalization (Fig 4A); however, AtxΔΜΕ/ΔΜΕ macrophages did not exhibit TLR4 internalization in response to LPS stimulation. In these macrophages, TLR4 was instead mostly confined to the plasma membrane even after LPS stimulation (Fig 4B). Because CD14 is essential for the internalization of TLR4, our findings suggest that the formation and subsequent internalization of the TLR4‐CD14 receptor complex are inhibited in AtxΔΜΕ/ΔΜΕ macrophages.
Figure 4. The LPS‐induced internalization of TLR4 was inhibited in Atx‐ko macrophages.
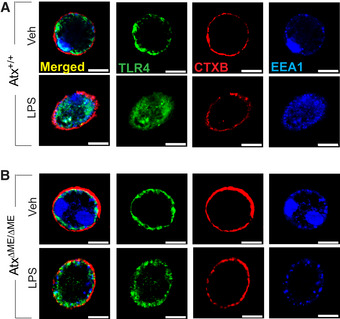
-
A, BPeritoneal macrophages from Atx+/+ (A) and AtxΔΜΕ/ΔΜΕ (B) mice were stimulated with LPS (20 ng/ml, 20 min), followed by fixation in 4% paraformaldehyde for 15 min at room temperature and staining with TLR4‐anti-rabbit FITC (Green), early endosomal marker EEA1‐anti-mouse Dylight405 (Blue), and Alexa Fluor 594‐CTXB (Red). TLR4 internalization was examined with confocal microscopy. Presented is the representative from 3 independent experiments in which more than 95% of the cells exhibited similar results. Scale bar, 10 μm.
Atx deficiency blocks the recruitment of TIR domain‐containing adaptors to TLR4
To investigate the effect of Atx deficiency on the recruitment of TLR4‐associated signaling molecules, we performed a series of immunoprecipitation assays in AtxΔΜΕ/ΔΜΕ and Atx+/+ macrophages and in ATX inhibitor‐treated Raw264.7 cells. We found that the LPS‐induced interaction of TLR4 with MAL/TIRAP was eliminated in AtxΔΜΕ/ΔΜΕ macrophages, but remained intact in LPS‐stimulated Atx+/+ macrophages (Fig 5A). Similarly, in Raw264.7 cells the LPS‐induced interaction between TLR4 and MAL/TIRAP was disrupted by ATX inhibitor treatment (Fig 5B). The LPS‐induced interaction of TLR4 with MYD88 was also inhibited in AtxΔΜΕ/ΔΜΕ macrophages but preserved in Atx+/+ macrophages (Fig 5C). The interaction of TLR4 and MYD88 following LPS treatment was likewise ablated in ATX inhibitor‐treated Raw264.7 cells (Fig 5D). Similarly, we observed that the LPS‐induced association of MAL/TIRAP with MYD88 was inhibited in AtxΔΜΕ/ΔΜΕ macrophages (Fig 5E).
Figure 5. Recruitment of TLR4‐associated adaptor molecules was inhibited in Atx‐ko macrophages.
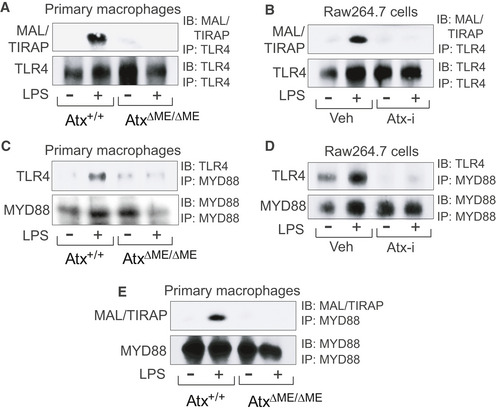
-
A–EThe macrophages from AtxΔΜΕ/ΔΜΕ and Atx+/+ mice were stimulated with LPS (20 ng/ml, 20 min) (A, C, E). Raw264.7 cells were stimulated by LPS with the ATX inhibitor (50 μM, 30 min) or vehicle (Veh, DMSO 0.1%) (B, D). The cell lysates were subjected to immunoprecipitation (IP), followed by immunoblotting (IB) analysis with the antibodies indicated. Presented is the representative from three independent experiments.
Source data are available online for this figure.
Together, these results demonstrate that when Atx is defective, TLR4 is unable of recruit TIR domain‐containing adaptor molecules.
TLR4‐mediated signaling pathways are inhibited in Atx‐ko macrophages, while TLR2‐ and non‐TLR‐mediated signaling are preserved
We next evaluated TLR4‐mediated signaling pathways in primary macrophages. The LPS‐induced activation of NFκB (p65), MAPKs (p38, JNK1/2, ERK1/2), and IRF‐3 was dramatically reduced in AtxΔΜΕ/ΔΜΕ macrophages, but preserved in Atx+/+ macrophages (Fig 6A). It has previously been suggested that LPS stimulation upregulates ATX protein by enhancing the stability of its mRNA transcript in macrophages (Awada et al, 2014; Sun et al, 2016). In agreement with these earlier findings, we observed that LPS stimulation enhanced the ATX protein level in Atx+/+ macrophages. We further confirmed that the major signaling molecules essential for LPS‐induced responses were expressed at similar levels in AtxΔΜΕ/ΔΜΕ and Atx+/+ macrophages (Fig 6B). Similarly to AtxΔΜΕ/ΔΜΕ mice, the macrophages obtained from Atx‐heterozygous mice also demonstrated suppression of LPS‐induced signaling pathways relative to wild‐type mice (Fig 6C). We additionally confirmed that ATX inhibitor treatment in Raw264.7 cells decreased LPS‐induced NFκB (p65) and ERK1/2 activation (Fig 6D). In contrast to the inhibition of TLR4 signaling observed in AtxΔΜΕ/ΔΜΕ macrophages, the intracellular signaling pathways mediated by TLR2 (stimulated by peptidoglycan or Pam3CSK4), IL‐1R (stimulated by IL‐1β), and TNF receptor (stimulated by TNFα) were preserved in AtxΔΜΕ/ΔΜΕ and Atx+/+ macrophages (Fig 6E–H).
Figure 6. TLR4‐induced signaling pathways were inhibited in Atx‐ko macrophages.
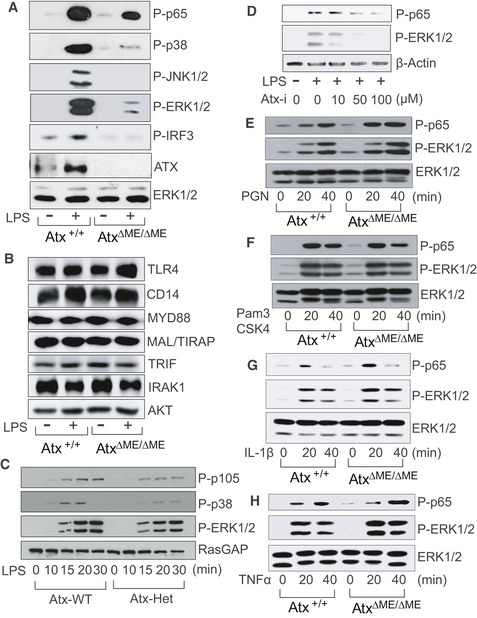
-
A–CThe macrophages from AtxΔΜΕ/ΔΜΕ and Atx+/+ mice (A, B) or Atx‐heterozygous (Atx‐Het) mice and wild‐type littermates (C) were stimulated by LPS [20 ng/ml, 20 min (A, B) or indicated time point (C)].
-
DRaw264.7 cells were stimulated by LPS with the ATX inhibitor.
-
E–HThe macrophages from AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermates were activated with TLR2 ligand Peptidoglycan (PGN, 30 ng/ml) (E), Pam3CSK4 (1 μg/ml) (F), IL‐1β (100 ng/ml) (G), or TNFα (25 ng/ml) (H) for the indicated time points.
Taken together, our results demonstrate that Atx deficiency specifically inhibits TLR4‐mediated responses in macrophages.
Atx deletion suppresses LPS‐induced cytokine production
To further investigate the inhibition of TLR4‐induced signaling in AtxΔΜΕ/ΔΜΕ macrophages, we next evaluated LPS‐induced cytokine production. The LPS‐stimulated secretion of TNFα, IL‐6, and IL‐1β was reduced in AtxΔΜΕ/ΔΜΕ macrophages compared to controls (Fig 7A). Similarly, ATX inhibitor treatment suppressed LPS‐induced cytokine secretion in Raw264.7 cells (Fig 7B). Upon analyzing single cytokine‐producing cells by fluorescence‐activated cell sorting (FACS), we likewise identified that the production of LPS‐stimulated cytokines was substantially reduced in AtxΔΜΕ/ΔΜΕ macrophages compared to Atx+/+ cells (Fig 7C–E).
Figure 7. LPS‐induced cytokine production was reduced in Atx‐ko macrophages.
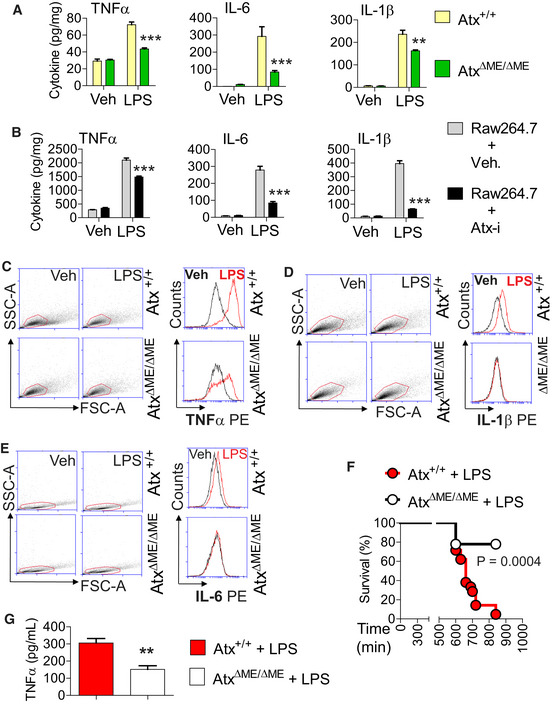
-
A, BPeritoneal macrophages from AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermates (A) or Raw264.7 cells (B) were stimulated with LPS (50 ng/ml, 8 h) in the presence/absence of the ATX inhibitor (50 μM, 30 min). The culture medium was collected for ELISA to measure the level of secreted cytokines. All assays were performed in triplicate, and data are shown as mean ± SEM.
-
C–EPeritoneal macrophages were stimulated with LPS or vehicle (endotoxin‐free water), after which intracellular cytokine production was measured through FACS analysis. With the scatter dot plot, the gate was set so that the viable cells were selected to analyze the intensity of the fluorescence signal by flow cytometry. The overlay plot represents the cytometry data.
-
FTo induce sepsis via LPS injection (Shirey et al, 2013; Voss et al, 2016), age (9–10 weeks)‐ and sex‐matched AtxΔΜΕ/ΔΜΕ mice (n = 9) and Atx+/+ littermates (n = 21) were injected with LPS (i.p. 25 mg/kg). Survival was monitored for up to 16 h, and analyzed by the Kaplan‐Meier method (Log‐rank P = 0.0004).
-
GBlood samples were collected from AtxΔΜΕ/ΔΜΕ (n = 7) and Atx+/+ (n = 6) mice 10 h after LPS injection, after which the serum TNFα protein level was measured. The data are shown as mean ± SEM.
To further characterize the inhibitory effect of ATX deficiency in LPS‐induced cytokine production, we investigated the in vivo effect of Atx deficiency in an LPS‐induced mouse sepsis model (Shirey et al, 2013) in which a cytokine storm induced by LPS injection causes severe systemic inflammation. We challenged AtxΔΜΕ/ΔΜΕ and Atx+/+ littermates with an intraperitoneal (i.p.) injection of LPS. After LPS administration, AtxΔΜΕ/ΔΜΕ mice had a markedly higher survival rate than LPS‐treated Atx+/+ mice (Fig 7F) and a correspondingly lower serum level of TNFα (Fig 7G).
Taken together, these results demonstrate that Atx deficiency leads to reduced cytokine production in response to LPS in both in vitro and in vivo settings.
Innate immune effector functions are impaired in AtxΔΜΕ/ΔΜΕ macrophages
Phagocytosis is a crucial innate immune effector mechanism of macrophages (Blander & Medzhitov, 2004). Therefore, we investigated whether Atx deficiency alters LPS‐induced immune effector functions of macrophages. Fc gamma receptor (FcgR)‐mediated phagocytic activity of macrophages was enhanced in Atx+/+ macrophages in response to LPS, but was substantially reduced in macrophages from AtxΔΜΕ/ΔΜΕ mice (Fig 8A and B). Intriguingly, AtxΔΜΕ/ΔΜΕ macrophages were not observed to form the phagocytic cup at the plasma membrane in response to LPS, whereas LPS‐treated Atx+/+ macrophages were able to successfully form the phagocytic cup formation (Fig 8C).
Figure 8. Innate immune effector function was suppressed in Atx‐ko macrophages compare to controls.
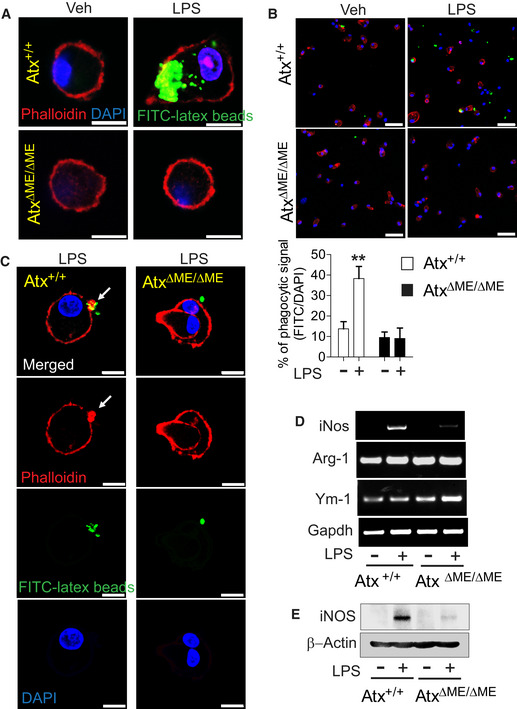
-
A, BConfocal laser scanning micrographs of the macrophages from AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermates. Cells were incubated with IgG‐opsonized latex beads in the absence/presence of LPS (20 ng/ml, 2 h) and stained with Phalloidin (F‐actin) and DAPI. Internalized beads were examined under a higher(A) and a lower magnification (B) to quantify % phagocytosis by dividing latex bead‐positive cell numbers by the total number of DAPI‐positive cells (n = 11–16 per group). The data are analyzed with results accumulated from three independent experiments and shown as mean ± SEM (B). **P < 0.01 (Mann–Whitney U‐test). Scale bars are 5 μm (A) and 50 μm (B), respectively.
-
CLPS‐stimulated macrophages were co‐incubated with IgG‐opsonized latex beads for 40 min. Phagocytic cups were visualized by F‐actin staining with Phalloidin‐iFluor 647. The arrow indicates the phagocytic cup formed in the plasma membrane. Scale bar is 10 μm.
-
D, EAfter LPS (20 ng/ml, 4 h) treatment, LPS‐induced iNos, Arginase‐1 (Arg‐1), and Ym‐1 mRNA expression were evaluated by semi‐quantitative PCR (D). With LPS stimulation (20 ng/ml, 8 h), iNOS protein production was examined by immunoblotting analysis (E).
We next evaluated the expression of inducible nitric oxide synthase (iNOS), which produces the effector molecule nitric oxide in macrophages. We found that LPS‐induced iNos expression was substantially decreased in AtxΔΜΕ/ΔΜΕ macrophages compared to controls, whereas Arg‐1 and Ym‐1 expression were similar between groups (Fig 8D and E).
Taken together, these results suggest that Atx deficiency is capable of disrupting innate immune effector mechanisms in macrophages, thereby leading to compromised innate immunity.
AtxΔΜΕ/ΔΜΕ mice have increased bacterial infiltration into the intestinal mucosa
Indigenous gut microbes are capable of translocating across the single layer of epithelial cells that forms a critical barrier between the lumen and mucosa of the intestine. The translocation of indigenous gut microbes across the intestinal barrier is a normal phenomenon that occurs in healthy subjects without deleterious effects, due to the intervention of an adequate clearing immune system employing residential intestinal macrophages and recruited neutrophils (Van Leeuwen et al, 1994). However, when the immune system is severely compromised, this same phenomenon can result in pathology (Casanova & Abel, 2009). Given the reduced immune responses in the macrophages of AtxΔΜΕ/ΔΜΕ mice relative to Atx+/+ mice, we further hypothesized that AtxΔΜΕ/ΔΜΕ mice would have elevated levels of bacteria in the intestinal mucosa. To test this hypothesis, we performed fluorescence in situ hybridization (FISH) with mouse intestinal sections to detect the presence of bacteria with FITC‐labeled pan‐bacterial EUB338 (Amann et al, 1990) and F27 (Weisburg et al, 1991) probes. We found that the prevalence of bacteria in the mucosa of the colon and small intestine was increased in AtxΔΜΕ/ΔΜΕ mice compared to Atx+/+ littermates (Fig EV5A and B). However, myeloid cell lineage‐restricted Atx gene deletion did not appear to affect colonic barrier integrity, as both Atx+/+ and AtxΔΜΕ/ΔΜΕ mice have an intact colonic epithelial cell barrier (Fig EV5C).
Figure EV5. AtxΔΜΕ/ΔΜΕ mice have elevated levels of bacteria in the intestinal mucosa.
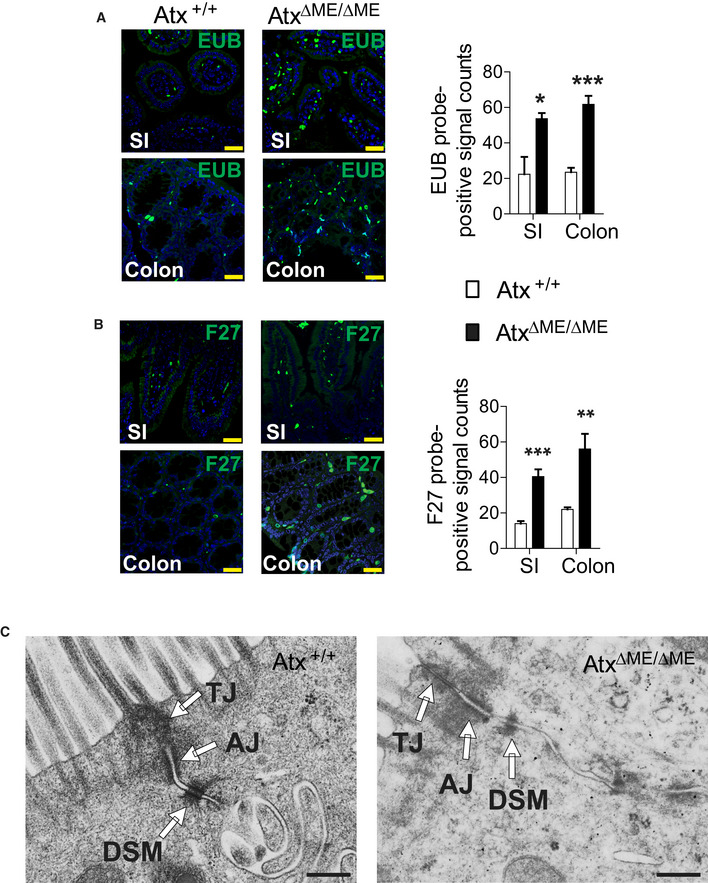
-
A, BThe bacterial load in the intestinal mucosa of AtxΔΜΕ/ΔΜΕ and Atx+/+ mice was evaluated by Fluorescent In Situ Hybridization (FISH) using FITC‐labeled pan‐bacterial EUB probe (A) and FITC‐labeled pan‐bacterial F27 probe (B). Scale bar is 30 μm. Graph indicates the count of FITC‐positive signals quantified from three or more independent experiments (n = 3–4 per group). The data are shown as mean ± SEM. Small Intestine (SI).
-
CElectron micrographs of intact cell‐to-cell adhesion in the colonic epithelium from age (8 weeks)‐matched AtxΔΜΕ/ΔΜΕ mice and Atx+/+ littermate. Tight junction (TJ), Adherens junction (AJ), Desmosome (DSM). Scale bar indicates 500 nm.
Discussion
After being generated by ATX, LPA(Stracke et al, 1992) and S1P (Clair et al, 2003) have been suggested to interact with their specific receptors to mediate an array of downstream effects, including Ca2+ entry, cell migration, cytokine expression, angiogenesis, fibrosis, and cancer development (Itagaki et al, 2005; Cao et al, 2017). The overexpression of exogenous ATX in mouse microglia cells has been suggested to suppress TNFα and IL‐6 expression (Awada et al, 2014). Exogenous treatment with LPC, a substrate of ATX, could activate TLR2 or TLR4 in TLR2‐ or TLR4‐transfected HEK293 cells in the absence of stimulation by their specific ligands, but LPC treatment inhibited LPS‐induced responses in mouse macrophages (Carneiro et al, 2013). However, treating cells with exogenous LPC may cause a variety of indirect effects, thus resulting in off‐target responses which may differ from the effects mediated solely by endogenous ATX. Therefore, the interplay between ATX and TLR4‐mediated responses has yet to be clearly investigated, particularly as concerns the direct impact of Atx deficiency on TLR4 engagement and subsequent immune effector functions.
In this study, we demonstrate that Atx deficiency disturbs the integrity of the plasma membrane lipid rafts, thereby disrupting the formation of the LPS‐sensing receptor complex composed of TLR4 and CD14. Consequently, Atx deficiency inhibits LPS/TLR4‐mediated intracellular signaling pathways. In addition to TLR4, other membrane receptors such as IL‐1R and TLR2 may also reside in lipid rafts, where they regulate receptor‐mediated intracellular signaling pathways (Oakley et al, 2009; Hellwing et al, 2018). However, it is worth noting that the disruption of lipid rafts could not abort IL‐1R‐mediated signaling events, as IL‐1R can also be activated in non‐lipid raft regions of the plasma membrane (Windheim, 2016). Likewise, it has been suggested that the lipid composition of lipid rafts does not affect TLR2 engagement (Hellwing et al, 2018). In contrast, the activation of TLR4 by LPS is highly dependent upon the lipid raft integrity because CD14 specifically resides in lipid rafts where it interacts with LPS. After this initial recognition, TLR4 migrates to lipid rafts where the CD14:TLR4 receptor complex is then formed in myeloid cells (Triantafilou et al, 2002). In this way, TLR4 forms a heteromeric complex with CD14 at the plasma membrane lipid raft to elicit LPS‐induced responses. Therefore, the integrity of lipid rafts is required for TLR4 activation. In this context, we believe that Atx deficiency alters TLR4‐mediated signaling pathways, whereas TLR2‐, IL‐1R‐, and TNF‐R‐mediated responses are preserved.
Regarding the association of ATX with intestinal inflammation, previous studies have generally suggested that blockade of ATX may have a protective effect in experimental mouse colitis. For instance, increased Atx mRNA levels have been observed in the inflamed colons of DSS‐induced (Hozumi et al, 2013; Lin et al, 2019) and T‐cell‐transferred colitis mice (Hozumi et al, 2013). Increased ATX protein levels have been observed in the inflamed colons of DSS‐treated Alkaline‐SMase‐ko mice (Zhang et al, 2018). ATX inhibitor treatment can ameliorate experimental mouse colitis, such as DSS‐induced (Hozumi et al, 2013; Thirunavukkarasu et al, 2016; Dong et al, 2019), T‐cell‐transferred (Hozumi et al, 2013), and SAMP1/Fc mouse colitis (Dhillon et al, 2019). Similarly, Atx deletion reduced the severity of DSS‐induced colitis in mice (Lin et al, 2019). However, a recent study demonstrated that ATX can be produced as a pro‐inflammatory factor, as the NFκB signaling pathway induces the expression of ATX (Wu et al, 2010). According to this study, the TNFα‐NFκB axis substantially upregulates the expression of ATX, meaning that inflammatory factors are capable of inducing Atx gene expression. Indeed, increased levels of ATX protein were found in patients with chronic liver diseases such as chronic hepatitis C infection (Ando et al, 2018), primary biliary cholangitis (Joshita et al, 2018), and non‐alcoholic fatty liver disease (Fujimori et al, 2018). Accordingly, in the inflamed intestine, it is reasonable to believe that a plethora of inflammatory factors therein are able to induce the expression of ATX; in this case, an elevated ATX level should be an outcome of the inflammation. Therefore, regarding Atx expression in IBD patients or in experimental mouse models of IBD, it should be underscored that the inflammatory response itself could elicit the expression of Atx. Therefore, we speculate that an ATX blockade might be able to confer a protective effect in experimental mouse colitis.
When it comes to the interaction between gut microbes and host immunity, it is of importance to note that in normal conditions, gut luminal bacteria translocate into the submucosa where they help to shape host immunity; and subsequently and subsequently eliminated by innate and adaptive immune mechanisms. During this elimination process, the involvement of anti‐inflammatory mediators such as IL‐10 plays a critical role in suppressing the microbe‐induced inflammatory response. Due to potent expression of IL‐10, intestinal macrophages can produce limited levels of pro‐inflammatory cytokines in response to microbes, while retaining their highly phagocytic activity. In this way, intestinal homeostasis can be maintained even during active immune responses against invading microbes in the intestinal mucosa. Much in line with the role of IL‐10 as an essential anti‐inflammatory factor in gut mucosal immune responses, mice deficient in IL‐10 spontaneously develop colitis in a gut microbe‐dependent manner. Indeed, genetic defects of the IL‐10 encoding gene cause aggressive intestinal inflammation in humans; therefore, mutation of the Il10 gene is a well‐known genetic factor for IBD in humans (Glocker et al, 2009; Mitchell et al, 2018a). Accordingly, IL‐10 mice are one of the most clinically relevant animal models of human IBD.
It is worth noting that TLR4 activation initiates and fortifies macrophage‐associated immune mechanisms necessary for the eradication of invading microbes. Therefore, TLR4 and IL‐10 double knockout mice exhibit accelerated colitis development compared to IL‐10‐ko mice (Matharu et al, 2009). Considering the premise of the prior studies about Atx and IBD and the nature of gut mucosal immune responses, in this we harness Il10−/− mice to investigate the impact of Atx in intestinal inflammation. In light of our finding that Atx‐ko impairs TLR4‐mediated immune mechanisms of macrophages and also given the aforementioned study finding that TLR4‐ko worsens the development of colitis in IL‐10 mice (Matharu et al, 2009), we theorized that Atx deficiency may accelerate gut inflammation in an IL‐10‐deficient condition. Indeed, we observed accelerated colitis development in AtxΔΜΕ/ΔΜΕ;Il10−/− mice compared to Atx+/+;Il10−/− littermates. Recent studies have recast IBD as being characterized by abnormal immune responses against commensal gut microbes in genetically susceptible individuals (Yan et al, 2017). In agreement with this notion, our study provides an important component of the mechanism by which gut microbes can cause the onset and perpetuation of inflammatory disorders in the gut.
In this study, we identify an increased abundance of Bacteroides in the feces of AtxΔΜΕ/ΔΜΕ;Il10−/− mice compared to that of Atx+/+;Il10−/− littermates. The abundance of the Bacteroides genus is commonly increased in the feces of IL‐10‐ko mice with spontaneous colitis (Im et al, 2014; Mitchell et al, 2018a). Therefore, the elevated level of Bacteroides in AtxΔΜΕ/ΔΜΕ;Il10−/− mice suggests that these mice are similarly experiencing chronic colitis as IL‐10‐ko mice do in a commensal microbe‐dependent manner. Moreover, Bacteroides can adhere to and invade intestinal epithelium to fulfill its pathogenic role (Nakano et al, 2008) and commensal Bacteroides are capable of inducing colitis in genetically susceptible mouse models of colitis (Bloom et al, 2011). Accordingly, Bacteroides can be considered pathobionts, at least in mice. In this context, we speculate that elevated levels of Bacteroides may excel at invading the intestinal epithelium of Atx‐ko mice, in which Atx deficiency dampens mucosal immune defense mechanisms. Consequently, these bacteria can accelerate the onset of and subsequently perpetuate gut inflammation in an IL‐10‐deficient condition.
Macrophages exist at the frontline of innate immunity against invading microbes and are extremely prevalent in the intestine, which contains the body's largest population of macrophages (Bain et al, 2014). Thus, the immune effector function of macrophages plays a pivotal role in maintaining gut immunological homeostasis. Given the keystone role of macrophages in the anti‐microbial immune defense of the intestinal mucosa, it is likely that compromised immune effector functioning of macrophages is correlated with the onset and perpetuation of inflammatory disorders in the gut (Casanova & Abel, 2009). One recent study suggested that LPA stimulation can induce the development of macrophages through induction of Akt/mTOR signaling pathway and PPARγ activation in mice and humans, indicating that LPA is capable of regulating immune defense mechanisms (Ray & Rai, 2017). Accordingly, it is possible that reduced LPA levels in the Atx‐deficient mice may dampen the macrophage development and relevant inflammatory responses, leading to compromised immunity. Nonetheless, our data clearly show that Atx deficiency suppresses TLR4‐mediated responses by inhibiting CD14:TLR4 receptor complex formation, resulting in compromised innate and adaptive immune responses. Together with this biochemical evidence, our findings of reduced ATX protein levels in IBD patients and accelerated colitis development in AtxΔΜΕ/ΔΜΕ;Il10−/− mice signify the considerable physiological relevance of ATX in the onset and perpetuation of bacteria‐associated chronic inflammation in the gut.
Materials and Methods
Human serum samples
Serum samples were provided by the UCLA IBD Biobank in the Division of Digestive Diseases, David Geffen School of Medicine, UCLA. Board‐certified gastroenterologists at UCLA determined the diagnosis of UC and CD on the basis of clinical, endoscopic, radiological, and histological criteria. Venous blood samples (5 ml) were obtained from each IBD (CD, UC) patient and control subject. After being centrifuged, the serum was separated in aliquots to avoid multiple freeze–thaw cycles and stored at −80°C until use. Patients with cancer or previous chemotherapy or radiation therapy were excluded. Patients with any infectious disease were also excluded. All human blood samples were collected and analyzed with the approval of the UCLA Institutional Review Board (IRB number: 12‐000420). All participants were provided with complete information about the study and gave written informed consent to the study protocol. The patients examined in this study have never been included in any of the previous studies.
Animals
Atx‐floxed mice (van Meeteren et al, 2006) and Atx‐heterozygous knockout mice (van Meeteren et al, 2006) were kindly provided by Dr. Moolenaar (The Netherlands Cancer Institute, Amsterdam, the Netherlands). Their genotypes were determined by a genotyping PCR protocol (van Meeteren et al, 2006; Dusaulcy et al, 2011). Macrophage‐specific Cre‐expressing LysM‐Cre mice (Clausen et al, 1999) and Il10−/− (Kuhn et al, 1993) mice on a C57BL/6 background were purchased from the Jackson laboratory (Bar Harbor, ME). Genotyping PCR was performed in accordance with the protocol provided by the Jackson laboratory. Atx‐floxed mice were crossed with LysM‐Cre mice to generate macrophage‐specific Atx‐ko (AtxΔΜΕ/ΔΜΕ) and littermate control mice (Atx+/+) mice. An AtxΔΜΕ/ΔΜΕ mouse was crossed with an Il10−/− mouse to generate AtxΔΜΕ/ΔΜΕ;Il10−/− mice. AtxΔΜΕ/ΔΜΕ mice and AtxΔΜΕ/ΔΜΕ;Il10−/− mice were backcrossed into a C57BL/6 background for at least 8 generations prior to performing the experiments. All animal experiments were approved by the Institutional Animal Care and Use Committees of Oakland University and Pusan National University. Mice were bred and maintained in a specific pathogen‐free condition with normal drinking water ad libitum at the AAALAC accredited animal facility of the Biomedical Research Support Facility, Oakland University (IACUC no. 16122), and Pusan National University (IACUC No. PNU‐2018‐1843) under the approval of the IACUC.
Reagents and antibodies
ATX antibody (van Meeteren et al, 2006) was kindly provided by Dr. Moolenaar (The Netherlands Cancer Institute, Amsterdam, the Netherlands). The ATX‐specific inhibitor (PF8380) (Gierse et al, 2010) was obtained from Echelon Biosciences Incorporated (Salt Lake City, UT). Ultrapure LPS (E. coli 0111:B4), Peptidoglycan, Pam3CSK4, and endotoxin‐free water were obtained from InvivoGen (San Diego, CA). Mouse recombinant TNFα was from BioLegend (575202, San Diego, CA). Mouse IL‐1β and mouse interferon gamma protein (IFNγ) were from Abcam (Cambridge, MA). Antibodies recognizing P‐p65 (3033S), P‐p105 (4806S), P‐p38 (9211S), P‐JNK1/2 (9251S), P‐ERK1/2 (9101S), P‐IRF‐3 (4947S), ERK1/2 (9102S), Flotillin‐1(3253), Caveolin‐1 (3238), and AKT (9272S) were obtained from Cell Signaling Technology (Danvers, MA). The early endosomal marker EEA1 monoclonal antibody (Cat. No. MAB8047) was purchased from R&D Systems (Minneapolis, MN), and the secondary antibody DyLight™ 405 AffiniPure Goat Anti‐Mouse IgG (H+L) (Code No. 115‐475‐166) was obtained from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). CD14 (M‐305), MYD88 (HFL‐296), and TLR4 (25) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). Alexa Fluor 594‐labeled‐CTXB (C22842) and Brefeldin A (00‐4506‐51) were obtained from Thermo Fisher Scientific (Waltham, MA). Phalloidin‐iFluor 647 (ab176759) was from Abcam (Cambridge, MA). FACS staining buffer was prepared in DPBS containing 1% FBS and 0.09% sodium azide, followed by filtration through 0.2 μm pore membrane. Goat anti‐Rabbit IgG‐heavy and light chain antibody FITC conjugated (A120‐101F) was purchased from Bethyl Laboratories, Inc. (Montgomery, TX). Mouse IL‐1β/IL‐1F2 PE‐conjugated antibody (Cat. No. IC4013P) was obtained from R&D Systems (Minneapolis, MN). PE Rat anti‐mouse TNFα (Cat. No. 554419), PE Rat anti‐mouse IL‐6 (Cat. No. 554401), and PE Rat anti‐mouse MHC class II (IA‐/I‐E) (Cat. No. 557000) antibodies were purchased from BD Biosciences (San Jose, CA). Monensin (554724) and dispase (BD354235) were obtained from BD Bioscience. HBSS without Ca2+ and Mg+ (14175) and with Ca2+ and Mg+ (14025) were from Thermo Fisher Scientific. Percoll (P1644), HEPES (H4034), Phorbol 12‐myristate 13‐acetate (PMA, P8139), A23187 (C7522), DNase I (DN25), and Collagenase VIII (C9697) were from Sigma‐Aldrich (St. Louis, MO). The CD14 antibody used in immune fluorescence staining (17000‐1‐AP) was purchased by ProteinTech Group (Rosemont, IL).
Primary peritoneal macrophages were prepared from mice using thioglycolate elicitation and cultivated as described previously (Rhee et al, 2003). The mouse monocyte/macrophage Raw264.7 cell line (ATC® TBI‐71) and human embryonic kidney (HEK293) cell line (ATC® CRL‐1573) were purchased from ATCC (Manassas, VA) and cultivated as previously described at 37°C in a 5% CO2 air environment.(Mitchell et al, 2018b). For the inhibitor treatment, cells were pre‐treated with 50 μM of ATX inhibitor (PF8380) for 30 min at 37°C.
DNA expression constructs
pTLR4‐ECFP and pTLR4‐EYFP expression constructs
The murine TLR4 encoding construct (pcDNA3.1‐TLR4) (Rhee & Hwang, 2000) was treated with XhoI restriction enzyme to obtain the DNA fragment spanning the start codon to the transmembrane region, but excluding the cytoplasmic TIR domain. The DNA fragment was inserted into the SalI site of pECFP‐N1 and pEYFP‐N1 plasmids to generate pTLR4‐ECFP and pTLR4‐EYFP, respectively. The pTLR4‐ECFP and pTLR4‐EYFP constructs do not have a cytoplasmic TIR domain, which was replaced with the ECFP or EYFP encoding region.
pcDNA3.1‐CD14‐ECFP expression construct
The cloning scheme is described in Appendix Fig S1.
At each step of subcloning, we performed DNA sequencing to confirm the integrity of the complete cDNA sequence and to verify the sequence in frame. The expression of each construct was also confirmed by Western blotting after transfecting them into HEK293 cells.
Sucrose density‐gradient ultracentrifugation
Sucrose density‐gradient ultracentrifugation was carried out to fractionate lipid rafts. Peritoneal macrophages or Raw264.7 cells were seeded at 1 × 107 cells/plate. Raw264.7 cells were treated with ATX inhibitor PF8380 (50 μM, 30 min) or vehicle (0.1% DMSO). Cells were washed with cold PBS and harvested. Cell pellets were resuspended in 2 ml TNE/Triton X‐100 buffer [25 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, and 1% Triton X‐100]. Lysates were sheared by a 22G needle and incubated for 20 min on ice. An equal amount of 80% sucrose in TNE buffer was added to the lysate to make 40% sucrose. 40% sucrose with lysates were overlaid with 7 ml 35% sucrose and 3 ml 5% sucrose solution. Samples were centrifuged for 18 h at 100,000 g at 4°C. The gradient was divided into 10 fractions. Protein was purified and concentrated with 3K cutoff filter (Cat. No. UFC200324, Sigma‐Aldrich, St. Louis, MO). Protein concentration was then determined, followed by the immunoblotting procedure.
Measurement of endogenous lysophosphatidic acid (LPA) by LC‐MS/MS analysis
Peritoneal macrophages were harvested from AtxΔΜΕ/ΔΜΕ and Atx+/+ littermates and stabilized for 4 days without any stimulation. Cells were collected by scraping, and the same number of cells were collected and stored at −80°C until the lipid extraction. Lysophosphatidic acid (LPA) was analyzed by the Wayne State University Lipidomics Core Facility in accordance with the method separating lysophosphatidylcholine (LPC) and lysophosphatidylserine (LPS) from LPA for the accurate detection of LPA in biological samples (Zhao & Xu, 2009). C17‐LPA from Avanti Polar lipids (Birmingham, AL) was used as an internal standard for quantitation. Lysophospholipids (LPLs) extraction was performed as described previously (Zhao & Xu, 2009). HPLC column was used for the separation of LPC and LPS from LPLs. The LC‐MS analysis was performed on a QTRAP 5500 mass spectrometer (SCIEX). Samples (10 μl) were directly injected into the MS system; the flow rate was 0.2 ml/min, and the duration was 1.5 min/sample. The concentration of LPA was normalized by the protein concentration of cell lysates.
Transmission electron microscopy
Peritoneal macrophages were harvested from AtxΔΜΕ/ΔΜΕ and littermate Atx+/+ mice, followed by immediate fixation for transmission electron microscopy, as previously described (Im et al, 2014; Howe et al, 2018).
Lipid raft staining of macrophages
Peritoneal macrophages from mice were cultivated for 3 days in chamber slides and stained with the lipid raft marker (Blank et al, 2007) Alexa Fluor 594‐labeled cholera toxin subunit B (CTXB). Cells were then washed twice with PBS and incubated for 10 min at 4°C with Alexa Fluor 594‐labeled‐CTXB (1 μg/ml), which binds to the pentasaccharides of the plasma membrane ganglioside GM1 as a lipid raft marker. Cells were washed twice with PBS and fixed in 4% paraformaldehyde for 15 min at room temperature. The cell membrane was examined by the FV10i confocal laser scanning microscope (Olympus Inc, Center Valley, PA). Images were analyzed by FV10i FluoView software.
FRET measurement
HEK293 cells were co‐transfected with a combination of CD14‐ECFP, TLR4‐ECFP, and TLR4‐EYFP encoding plasmid constructs. Transfected cells were treated with the ATX inhibitor PF8380 (50 μM, 30 min, 37°C) or vehicle (DMSO 0.1%) and fixed prior to FRET measurement. To calculate FRET, Z‐stacks of images were acquired; FRET between CD14‐ECFP and TLR4‐EYFP or between TLR4‐ECFP and TLR4‐EYFP was evaluated from the whole image on a pixel‐by‐pixel basis.(Zal et al, 2002; Hernanz‐Falcon et al, 2004; Kawai et al, 2004).
FRET was measured by using FV1000 (Olympus). Phase 1 was set with CFP/YFP/FRET channel, and phase 2 was added by EYFP. In order to analyze FRET results from samples, we set donor or acceptor only controls. Images were acquired as phase 1 and phase 2 filter set. After acquisition, donor only control images in phase 1 (channel 1 or 2) and acceptor only control images in phase 1 (channel 2) and phase 2 (channel 3) were loaded in FRET setting section. This was applied to ECFP/EYFP FRET samples. The background signal was also normalized with control samples. To calculate FRET efficiency and distance, Förster distance (i.e., the donor‐acceptor distance at which FRET efficiency is 50%) was set with 5.2767 nm for CFP‐YFP. For FRET efficiency, we set the range as 0–100%. After data acquisition, the average intensities of CFP, FRET, and YFP were measured, and fluorescence was calculated through the FRET filter set consisting of a FRET component, precision FRET (PFRET). The non‐FRET components were subtracted by the following equation: PFRET = acceptor channel image of donor excited, with donor and acceptor dyed sample image – DSBT (donor spectral bleed‐through) – ASBT (acceptor spectral bleed‐through). The final PFRET image is presented as a quantitative pseudocolor image. For quantification, normalized FRET was calculated on the co‐localization areas of CD14‐ECFP and TLR4‐EYFP or TLR4‐ECFP and TLR4‐EYFP on a pixel‐by‐pixel basis. FRET was calculated for at least 20 cells each in five independent experiments.
Confocal microscopy with immunofluorescence staining
Peritoneal macrophages from the mice were plated in chamber slides. After overnight stabilization, cells were stimulated with LPS (20 ng/ml) or vehicle for 20 min. Cells were then washed twice with PBS and incubated for 10 min at 4°C with Alexa Fluor 594‐labeled cholera toxin subunit B (CTXB) (1 μg/ml), which binds to the pentasaccharides of the plasma membrane ganglioside GM1 as a lipid raft marker. Cells were washed twice with PBS and fixed in 4% paraformaldehyde for 15 min at room temperature (RT) and permeabilized with 0.3% Triton X‐100 in PBS for 10 min at RT. Cells were washed with PBS and blocked with 1% normal goat serum and 0.3% Triton X‐100 in PBS for 1 h at RT. Cells were then incubated overnight at 4°C with TLR4 antibody and EEA1 antibody in blocking buffer. Samples were washed with PBS and incubated with the secondary antibodies (FITC‐conjugated anti‐rabbit IgG‐heavy and light chain secondary antibody, 1:200 dilution, for TLR4; and DyLight™ 405 AffiniPure goat anti‐mouse IgG H+L secondary antibody, 1:200 dilution, for EEA1) for 1 h at room temperature. Cells were washed three times with PBS and mounted. Images were visualized with FV10i confocal scanning microscopy, and images were analyzed by FV10i FluoView software.
Flow cytometry analysis of intracellular cytokine production
Peritoneal macrophage cells were seeded in six‐well plates (1 × 106 cells/well). Twenty‐four h after seeding, cells were co‐stimulated with Brefeldin A (1:1,000) and LPS (50 ng/ml, 6 h) for IL‐6, LPS (1 ng/ml, 4 h) for TNFα, or LPS (20 ng/ml, 4 h) for IL‐1β. Cells were harvested and washed with PBS, followed by fixation and permeabilization with Cytofix/Cytoperm (554714, BD Biosciences, San Jose, CA) for 30 min at 4°C. Cells were stained intracellularly with PE‐conjugated rat anti‐mouse TNFα (BD Biosciences) or PE‐conjugated anti‐mouse IL‐1β/IL‐1F2 (R&D Systems) or PE‐conjugated rat anti‐mouse IL‐6 (BD Biosciences), followed by washing with 1× washing buffer. Flow cytometry analysis was performed with Accuri C6 (BD Biosciences).
LPS‐induced mouse sepsis model
To induce endotoxic shock (Shirey et al, 2013; Voss et al, 2016), age‐ (9–10 weeks old) and sex‐matched AtxΔΜΕ/ΔΜΕ and Atx+/+ mice were injected (i.p. 25 mg/kg) with Ultrapure LPS (E. coli 0111:B4) (InvivoGen, San Diego, CA). Survival was monitored over 14 h after LPS injection. Blood samples were collected from the mice by cardiac puncture.
Phagocytosis measurement and visualization of phagocytic cup formation
Peritoneal macrophage cells (5 × 104 cells/well) were seeded in 4 well‐chamber slides and treated with vehicle (endotoxin‐free water) or LPS (20 ng/ml), followed by a 2‐h incubation. Phagocytosis was examined with a Phagocytosis Assay Kit (IgG‐FITC) (Cat. No. 500290, Cayman Chemical, Ann Arbor, MI). To evaluate phagocytosis with confocal microscopy, latex beads‐rabbit IgG‐FITC complex was added directly to the medium at a 1:200 dilution. Cells were incubated with FITC‐conjugated latex beads at 37°C for 2 h or 40 min, followed by cell fixation with 4% (w/v) paraformaldehyde solution. Cells were washed with PBS to remove fixation solution, permeabilized with PBS containing 0.1% (w/v) Triton X‐100 for 5 min, and then washed with PBS. Permeabilized cells were stained with 1× Phalloidin‐iFluor 647 (Abcam, ab176759, Cambridge, MA) for 1 h at RT. Cells were washed twice with assay buffer and sealed with VECTASHIELD® Mounting Medium with 4′,6‐diamidino‐2‐phenylindole.. Images were visualized with a LSM‐800 confocal laser scanning microscope. Acquired images were then analyzed by ZEN blue software.
Peritoneal macrophages (5 × 104 cells/well) were used to examine phagocytic cup formation. After vehicle or LPS (20 ng/ml) treatment for 2 h, peritoneal macrophages were incubated with latex beads‐rabbit IgG‐FITC complex (1:200) for 30 min. Without PBS washing, the phagocytic cup formation was stopped by fixation in 4% (w/v) paraformaldehyde for 30 min at RT, and cells were permeabilized by incubating for 5 min in PBS containing 0.1% (w/v) Triton X‐100. After removing permeabilizing solution, phagocytic cups were visualized by F‐actin staining with 1× Phalloidin‐iFluor 647 solution for 1 h. VECTASHIELD® Mounting Medium with 4′,6‐diamidino‐2‐phenylindole containing solution was followed by PBS washing. Phagocytic cups were examined under LSM‐800 confocal laser scanning microscope (ZEISS, Oberkochen, Germany) and analyzed by ZEISS ZEN blue software.
Fluorescent in situ hybridization (FISH)
For FISH experiments, we used the FITC‐labeled pan‐bacterial F27 probe (5′‐AGA GTT TGA TCM TGG CTC AG‐3′) (Weisburg et al, 1991), and FITC‐labeled pan‐bacterial EUB338 probe (5′‐GCT GCC TCC CGT AGG AGT‐3′) (Amann et al, 1990), as F27 and EUB338 probes specifically target the bacterial 16S rRNA gene V1 region (Amann et al, 1990; Weisburg et al, 1991). In accordance with literature protocols (Wallner et al, 1993; Salzman et al, 2010; Dishaw et al, 2016), we took the colon and small intestine from the sex‐ and age (3 months old)‐matched mice, followed by fixation in 10% neutral‐buffered formalin solution and preparation of paraffin‐embedded sections. The sections were dewaxed with xylene for 7 min and repeated twice. The slide sections were rehydrated by soaking in 100, 95, and 70 ethanol sequentially for 2 min each, twice. The sections were then rinsed with distilled water and placed in 95–99°C sodium citrate buffer for 10 min for antigen retrieval. After washing three times, slides were washed in PBS for 5 min. For staining, the tissue sections were placed in 1% Triton X‐100 and washed in PBS three times. The sections were incubated in the hybridization solution [20 mM Tris–Cl (pH 7.4) + 0.9 M NaCl + 0.1% SDS + 30% Formamide (for EUB338)] containing 250 μg of either FITC‐labeled pan‐bacterial F27 probe or FITC‐labeled pan‐bacterial EUB338 probe at 46°C overnight (Wallner et al, 1993; Dishaw et al, 2016). The sections were then rinsed in 48°C pre‐warmed washing buffer for 15 min. The slides were rinsed by placing them in a petri dish with distilled water for several seconds and were then allowed to air‐dry. The slides were mounted with VECTASHIELD® Mounting Medium with 4′,6‐diamidino‐2‐phenylindole and examined by FV10i (Olympus Inc.) with image analysis by FV10i FluoView software.
Spontaneous colitis development in mice
A breeding pair composed of a male and a female AtxΔΜΕ/+;Il10−/− mouse was prepared for breeding. After being born, male and female pups were separated at 4 weeks of age from the parents, followed by determination of the genotype using mouse tail biopsies. AtxΔΜΕ/ΔΜΕ;Il10−/− and Atx+/+;Il10−/− mice were co‐housed in separate cages with < 4 mice per cage. From 37 days of age, body weight change and mortality were monitored every other day until the mouse reached an age of 6 months, as described previously (Im et al, 2014; Mitchell et al, 2018a).
Semi‐quantitative RT–PCR
Primary macrophages were seeded in six‐well plates (1 × 106 cells/well). Cells were stimulated by 20 ng/ml LPS for 4 h. After stimulation, cells were rinsed with cold DPBS. For RNA extraction, Ribo EX was added, and cells were harvested. Homogenized cells in Ribo EX were incubated for 5 min at RT to dissociate nucleoproteins. Chloroform was added, and samples were vigorously mixed. The samples were centrifuged at 12,000 g for 15 min 4°C. Following centrifugation, the mixture separated into lower red, phenol chloroform phase, an interphase, and a colorless upper aqueous phase. The upper aqueous phase containing RNA was carefully transferred without touching the interphase into a fresh tube. To precipitate the RNA from the aqueous phase, isopropyl alcohol was mixed and at RT for 10 min. Samples were centrifuged at 12,000 g for 10 min at 4°C. Pellets were washed with 75% ethanol twice. After removing supernatant, the precipitated RNA was left at RT for 5 min to dry the pellet. RNA pellets were dissolved in RNase‐free water, and RNA concentration was quantified. cDNA was synthesized by using RT‐&GO Mastermix (MP Biomedicals, 11RTRAG001) and 500 ng of RNAs. An equal amount of RNA (2 ng) was used for semi‐quantitative RT–PCR. Primers for murine iNos, 5′‐CCCTTCCGAAGTTTCTGGCAGCAGC‐3′ (forward), 5′‐GGCTGTCAGAGCCTCGTCGTGGCTTTGG‐3′ (reverse); murine arginase‐I, 5′‐AAGAAAAGGCCGATTCACCT‐3′ (forward), 5′‐CACCTCCTCTGCTGTCTTCC‐3′ (reverse); murine Ym‐1, 5′‐GGGCATACCTTTATCCTGAG‐3′ (forward), 5′‐CCACTGAAGTCATCCATGTC‐3′ (reverse); murine Gapdh, 5′‐CTCACTGGCATGGCCTTCCG‐3′ (forward), 5′‐ACCACCCTGTTGCTGTAG‐3′ (reverse). PCR products were analyzed by DNA electrophoresis to visualize mRNA expression.
PCR‐based gene microarray analysis and Quantitative real‐time PCR
PCR‐based gene microarray analysis (Qiagen, Valencia, CA) focused on anti‐bacterial response (PAMM‐148Z) and innate and adaptive immunity pathways (PAMM‐052Z) were studied in accordance with the manufacturer's instructions (Im et al, 2014). Quantitative real‐time PCR was done as described (Im et al, 2014; Howe et al, 2018).
Fecal microbiome analysis
Age‐ and sex‐matched and co‐fostered AtxΔΜΕ/ΔΜΕ;Il10−/− and littermate Atx+/+;Il10−/− mice were co‐housed in a SPF condition without any experimental intervention. When the mice reached the age of 8 weeks old, the feces were harvested from the colon and snap‐frozen in liquid nitrogen before sequencing. Microbiome was analyzed as we previously described (Im et al, 2014; Howe et al, 2018).
Immunoblot, immunoprecipitation, and analysis were carried out as described previously (Mitchell et al, 2018b).
Statistics
Differences in survival were estimated by the Kaplan–Meier method. The log‐rank (Mantel–Cox) test was used to compare significant survival difference. Data of body weight change were compared by Two‐way ANOVA, followed by the multiple‐comparison Bonferroni t‐test. P values < 0.05 were considered significant. Unless stated otherwise, statistical analysis was conducted with GraphPad Prism (GraphPad Software, Inc., San Diego. CA).
Author contributions
SHR and EI conceived and designed all experiments. SJK, JM, CH, JC, EI, and SHR performed experiments. SJK, EI, and SHR analyzed the data. SJK, CH, AP, EI, and SHR contributed the mice. AO, CP, DWH, and SHR contributed the human samples. SHR generated the plasmid constructs. SJK, EI, and SHR wrote the paper. EI and SHR directed the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 8
Acknowledgements
We thank Dr. Wouter Moolenaar (The Netherlands Cancer Institute, Amsterdam, the Netherlands) for the Atx‐floxed mouse, Atx‐heterozygous mouse, and Atx antibody; Dr. Krishna Rao Maddipati at the Lipidomics Core Facility, Wayne State University for the LPA analysis by LC‐MS/MS. This research was supported by a grant from Oakland University and the National Institutes of Health (DK079015, S.H.R), and by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2019R1A2C1010536 to E.I.).
EMBO Reports (2020) 21: e49332
Contributor Information
Eunok Im, Email: eoim@pusan.ac.kr.
Sang Hoon Rhee, Email: srhee@oakland.edu.
Data availability
No primary datasets have been generated and deposited.
References
- Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA (1990) Combination of 16S rRNA‐targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56: 1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando W, Yokomori H, Kaneko F, Kaneko M, Igarashi K, Suzuki H (2018) Serum autotaxin concentrations reflect changes in liver stiffness and fibrosis after antiviral therapy in patients with chronic hepatitis C. Hepatol Commun 2: 1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awada R, Saulnier‐Blache JS, Gres S, Bourdon E, Rondeau P, Parimisetty A, Orihuela R, Harry GJ, d'Hellencourt CL (2014) Autotaxin downregulates LPS‐induced microglia activation and pro‐inflammatory cytokines production. J Cell Biochem 115: 2123–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Bravo‐Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, Malissen B, Osborne LC, Artis D, Mowat AM (2014) Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 15: 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R (2004) Regulation of phagosome maturation by signals from toll‐like receptors. Science 304: 1014–1018 [DOI] [PubMed] [Google Scholar]
- Blank N, Schiller M, Krienke S, Wabnitz G, Ho AD, Lorenz HM (2007) Cholera toxin binds to lipid rafts but has a limited specificity for ganglioside GM1. Immunol Cell Biol 85: 378–382 [DOI] [PubMed] [Google Scholar]
- Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, Dunne WM Jr, Allen PM, Stappenbeck TS (2011) Commensal bacteroides species induce colitis in host‐genotype‐specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 9: 390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Aoki Y, Badri L, Walker NM, Manning CM, Lagstein A, Fearon ER, Lama VN (2017) Autocrine lysophosphatidic acid signaling activates beta‐catenin and promotes lung allograft fibrosis. J Clin Invest 127: 1517–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro AB, Iaciura BM, Nohara LL, Lopes CD, Veas EM, Mariano VS, Bozza PT, Lopes UG, Atella GC, Almeida IC et al (2013) Lysophosphatidylcholine triggers TLR2‐ and TLR4‐mediated signaling pathways but counteracts LPS‐induced NO synthesis in peritoneal macrophages by inhibiting NF‐kappaB translocation and MAPK/ERK phosphorylation. PLoS ONE 8: e76233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL, Abel L (2009) Revisiting Crohn's disease as a primary immunodeficiency of macrophages. J Exp Med 206: 1839–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clair T, Aoki J, Koh E, Bandle RW, Nam SW, Ptaszynska MM, Mills GB, Schiffmann E, Liotta LA, Stracke ML (2003) Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine‐1‐phosphate. Cancer Res 63: 5446–5453 [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277 [DOI] [PubMed] [Google Scholar]
- David M, Wannecq E, Descotes F, Jansen S, Deux B, Ribeiro J, Serre CM, Gres S, Bendriss‐Vermare N, Bollen M et al (2010) Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid‐dependent activation of osteoclasts. PLoS ONE 5: e9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon AK, Kremer AE, Kummen M, Boberg KM, Elferink RPO, Karlsen TH, Beuers U, Vesterhus M, Hov JR (2019) Autotaxin activity predicts transplant‐free survival in primary sclerosing cholangitis. Sci Rep 9: 8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishaw LJ, Leigh B, Cannon JP, Liberti A, Mueller MG, Skapura DP, Karrer CR, Pinto MR, De Santis R, Litman GW (2016) Gut immunity in a protochordate involves a secreted immunoglobulin‐type mediator binding host chitin and bacteria. Nat Commun 7: 10617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YL, Duan XY, Liu YJ, Fan H, Xu M, Chen QY, Nan Z, Wu H, Deng SJ (2019) Autotaxin‐lysophosphatidic acid axis blockade improves inflammation by regulating Th17 cell differentiation in DSS‐induced chronic colitis mice. Inflammation 42: 1530–1541 [DOI] [PubMed] [Google Scholar]
- Dusaulcy R, Rancoule C, Gres S, Wanecq E, Colom A, Guigne C, van Meeteren LA, Moolenaar WH, Valet P, Saulnier‐Blache JS (2011) Adipose‐specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J Lipid Res 52: 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori N, Umemura T, Kimura T, Tanaka N, Sugiura A, Yamazaki T, Joshita S, Komatsu M, Usami Y, Sano K et al (2018) Serum autotaxin levels are correlated with hepatic fibrosis and ballooning in patients with non‐alcoholic fatty liver disease. World J Gastroenterol 24: 1239–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierse J, Thorarensen A, Beltey K, Bradshaw‐Pierce E, Cortes‐Burgos L, Hall T, Johnston A, Murphy M, Nemirovskiy O, Ogawa S et al (2010) A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J Pharmacol Exp Ther 334: 310–317 [DOI] [PubMed] [Google Scholar]
- Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D et al (2009) Inflammatory bowel disease and mutations affecting the interleukin‐10 receptor. N Engl J Med 361: 2033–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwing C, Schoeniger A, Roessler C, Leimert A, Schumann J (2018) Lipid raft localization of TLR2 and its co‐receptors is independent of membrane lipid composition. PeerJ 6: e4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernanz‐Falcon P, Rodriguez‐Frade JM, Serrano A, Juan D, del Sol A, Soriano SF, Roncal F, Gomez L, Valencia A, Martinez AC et al (2004) Identification of amino acid residues crucial for chemokine receptor dimerization. Nat Immunol 5: 216–223 [DOI] [PubMed] [Google Scholar]
- Howe C, Kim SJ, Mitchell J, Im E, Kim YS, Kim YS, Rhee SH (2018) Differential expression of tumor‐associated genes and altered gut microbiome with decreased Akkermansia muciniphila confer a tumor‐preventive microenvironment in intestinal epithelial Pten‐deficient mice. Biochim Biophys Acta Mol Basis Dis 1864: 3746–3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozumi H, Hokari R, Kurihara C, Narimatsu K, Sato H, Sato S, Ueda T, Higashiyama M, Okada Y, Watanabe C et al (2013) Involvement of autotaxin/lysophospholipase D expression in intestinal vessels in aggravation of intestinal damage through lymphocyte migration. Lab Invest 93: 508–519 [DOI] [PubMed] [Google Scholar]
- Im E, Jung J, Pothoulakis C, Rhee SH (2014) Disruption of pten speeds onset and increases severity of spontaneous colitis in il10(‐/‐) mice. Gastroenterology 147: 667–679 e610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki K, Kannan KB, Hauser CJ (2005) Lysophosphatidic acid triggers calcium entry through a non‐store‐operated pathway in human neutrophils. J Leukoc Biol 77: 181–189 [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R (2015) Control of adaptive immunity by the innate immune system. Nat Immunol 16: 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshita S, Umemura T, Usami Y, Yamashita Y, Norman GL, Sugiura A, Yamazaki T, Fujimori N, Kimura T, Matsumoto A et al (2018) Serum autotaxin is a useful disease progression marker in patients with primary biliary cholangitis. Sci Rep 8: 8159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R (2008) TRAM couples endocytosis of Toll‐like receptor 4 to the induction of interferon‐beta. Nat Immunol 9: 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S et al (2004) Interferon‐alpha induction through Toll‐like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5: 1061–1068 [DOI] [PubMed] [Google Scholar]
- Kerr TA, Ciorba MA, Matsumoto H, Davis VR, Luo J, Kennedy S, Xie Y, Shaker A, Dieckgraefe BK, Davidson NO (2012) Dextran sodium sulfate inhibition of real‐time polymerase chain reaction amplification: a poly‐A purification solution. Inflamm Bowel Dis 18: 344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer AE, Martens JJ, Kulik W, Rueff F, Kuiper EM, van Buuren HR, van Erpecum KJ, Kondrackiene J, Prieto J, Rust C et al (2010) Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 139: 1008–1018, 1018 e1001 [DOI] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W (1993) Interleukin‐10‐deficient mice develop chronic enterocolitis. Cell 75: 263–274 [DOI] [PubMed] [Google Scholar]
- Lin S, Haque A, Raeman R, Guo L, He P, Denning TL, El‐Rayes B, Moolenaar WH, Yun CC (2019) Autotaxin determines colitis severity in mice and is secreted by B cells in the colon. FASEB J 33: 3623–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JC, Chiang YT, Lin YC, Chang YT, Lu CY, Chen TY, Yeh CS (2016) Disruption of lipid raft function increases expression and secretion of monocyte chemoattractant protein‐1 in 3T3‐L1 adipocytes. PLoS ONE 11: e0169005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matharu KS, Mizoguchi E, Cotoner CA, Nguyen DD, Mingle B, Iweala OI, McBee ME, Stefka AT, Prioult G, Haigis KM et al (2009) Toll‐like receptor 4‐mediated regulation of spontaneous Helicobacter‐dependent colitis in IL‐10‐deficient mice. Gastroenterology 137: 1380–1390 e1381‐1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, Pettit TR, Wakelam MJ, Saulnier‐Blache JS, Mummery CL et al (2006) Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol 26: 5015–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio M, Ikeda A, Akagi M, Tasaka K (1985) Inhibitory effect of lysophosphatidylcholine on the histamine release from rat peritoneal mast cells. Agents Actions 16: 113–117 [DOI] [PubMed] [Google Scholar]
- Mitchell J, Kim SJ, Koukos G, Seelmann A, Veit B, Shepard B, Blumer‐Schuette S, Winter HS, Iliopoulos D, Pothoulakis C et al (2018a) Colonic inhibition of phosphatase and tensin homolog increases colitogenic bacteria, causing development of colitis in Il10‐/‐ MICE. Inflamm Bowel Dis 24: 1718–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J, Kim SJ, Seelmann A, Veit B, Shepard B, Im E, Rhee SH (2018b) Src family kinase tyrosine phosphorylates Toll‐like receptor 4 to dissociate MyD88 and Mal/Tirap, suppressing LPS‐induced inflammatory responses. Biochem Pharmacol 147: 119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nangaku M, Ohkawa R, Okubo S, Yokota H, Ikeda H, Aoki J, Yatomi Y (2008) Analysis of serum and urinary lysophospholipase D/autotaxin in nephrotic syndrome. Clin Chem Lab Med 46: 150–151 [DOI] [PubMed] [Google Scholar]
- Nakano V, Piazza RM, Cianciarullo AM, Bueris V, Santos MF, Menezes MA, Mendes‐Ledesma MR, Szulczewski V, Elias WP, Pumbwe L et al (2008) Adherence and invasion of Bacteroidales isolated from the human intestinal tract. Clin Microbiol Infect 14: 955–963 [DOI] [PubMed] [Google Scholar]
- Oakley FD, Smith RL, Engelhardt JF (2009) Lipid rafts and caveolin‐1 coordinate interleukin‐1beta (IL‐1beta)‐dependent activation of NFkappaB by controlling endocytosis of Nox2 and IL‐1beta receptor 1 from the plasma membrane. J Biol Chem 284: 33255–33264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikonomou N, Mouratis MA, Tzouvelekis A, Kaffe E, Valavanis C, Vilaras G, Karameris A, Prestwich GD, Bouros D, Aidinis V (2012) Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Cell Mol Biol 47: 566–574 [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C et al (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088 [DOI] [PubMed] [Google Scholar]
- Rajaiah R, Perkins DJ, Ireland DD, Vogel SN (2015) CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc Natl Acad Sci USA 112: 8391–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R, Rai V (2017) Lysophosphatidic acid converts monocytes into macrophages in both mice and humans. Blood 129: 1177–1183 [DOI] [PubMed] [Google Scholar]
- Rhee SH, Hwang D (2000) Murine TOLL‐like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase. J Biol Chem 275: 34035–34040 [DOI] [PubMed] [Google Scholar]
- Rhee SH, Jones BW, Toshchakov V, Vogel SN, Fenton MJ (2003) Toll‐like receptors 2 and 4 activate STAT1 serine phosphorylation by distinct mechanisms in macrophages. J Biol Chem 278: 22506–22512 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW (2004) An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol 22: 445–449 [DOI] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson‐Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D et al (2010) Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol 11: 76–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB (1998) Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin‐10‐deficient mice. Infect Immun 66: 5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, McAlees J, Gioannini TL, Weiss J et al (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497: 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387: 569–572 [DOI] [PubMed] [Google Scholar]
- Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, Liotta LA (1992) Identification, purification, and partial sequence analysis of autotaxin, a novel motility‐stimulating protein. J Biol Chem 267: 2524–2529 [PubMed] [Google Scholar]
- Sun S, Zhang X, Lyu L, Li X, Yao S, Zhang J (2016) Autotaxin expression is regulated at the post‐transcriptional level by the RNA‐binding proteins HuR and AUF1. J Biol Chem 291: 25823–25836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirunavukkarasu K, Tan B, Swearingen CA, Rocha G, Bui HH, McCann DJ, Jones SB, Norman BH, Pfeifer LA, Saha JK (2016) Pharmacological characterization of a potent inhibitor of autotaxin in animal models of inflammatory bowel disease and multiple sclerosis. J Pharmacol Exp Ther 359: 207–214 [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K (2002) Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide‐induced cell activation. J Cell Sci 115: 2603–2611 [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Takeuchi S, Kubota K, Kobayashi Y, Kozakai S, Ukai I, Shichiku A, Okubo M, Numasaki M, Kanemitsu Y et al (2018) Lipopolysaccharide (LPS)‐binding protein stimulates CD14‐dependent Toll‐like receptor 4 internalization and LPS‐induced TBK1‐IKK‐IRF3 axis activation. J Biol Chem 293: 10186–10201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen PA, Boermeester MA, Houdijk AP, Ferwerda CC, Cuesta MA, Meyer S, Wesdorp RI (1994) Clinical significance of translocation. Gut 35: S28–S34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viennois E, Chen F, Laroui H, Baker MT, Merlin D (2013) Dextran sodium sulfate inhibits the activities of both polymerase and reverse transcriptase: lithium chloride purification, a rapid and efficient technique to purify RNA. BMC Res Notes 6: 360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss OH, Murakami Y, Pena MY, Lee HN, Tian L, Margulies DH, Street JM, Yuen PS, Qi CF, Krzewski K et al (2016) Lipopolysaccharide‐induced CD300b receptor binding to toll‐like receptor 4 alters signaling to drive cytokine responses that enhance septic shock. Immunity 44: 1365–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner G, Amann R, Beisker W (1993) Optimizing fluorescent in situ hybridization with rRNA‐targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 14: 136–143 [DOI] [PubMed] [Google Scholar]
- Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173: 697–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windheim M (2016) Interleukin‐1‐induced gene expression requires the membrane‐raft‐dependent internalization of the interleukin‐1 receptor. Cell Signal 28: 1520–1529 [DOI] [PubMed] [Google Scholar]
- Wu JM, Xu Y, Skill NJ, Sheng H, Zhao Z, Yu M, Saxena R, Maluccio MA (2010) Autotaxin expression and its connection with the TNF‐alpha‐NF‐kappaB axis in human hepatocellular carcinoma. Mol Cancer 9: 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Hedl M, Abraham C (2017) An inflammatory bowel disease‐risk variant in INAVA decreases pattern recognition receptor‐induced outcomes. J Clin Invest 127: 2192–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zal T, Zal MA, Gascoigne NR (2002) Inhibition of T cell receptor‐coreceptor interactions by antagonist ligands visualized by live FRET imaging of the T‐hybridoma immunological synapse. Immunity 16: 521–534 [DOI] [PubMed] [Google Scholar]
- Zhang P, Chen Y, Zhang T, Zhu J, Zhao L, Li J, Wang G, Li Y, Xu S, Nilsson A et al (2018) Deficiency of alkaline SMase enhances dextran sulfate sodium‐induced colitis in mice with upregulation of autotaxin. J Lipid Res 59: 1841–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Xu Y (2009) Measurement of endogenous lysophosphatidic acid by ESI‐MS/MS in plasma samples requires pre‐separation of lysophosphatidylcholine. J Chromatogr B Analyt Technol Biomed Life Sci 877: 3739–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 8
Data Availability Statement
No primary datasets have been generated and deposited.