

Abstract
Senescent cells display senescence‐associated (SA) phenotypic programs such as stable proliferation arrest (SAPA) and a secretory phenotype (SASP). Senescence‐inducing persistent DNA double‐strand breaks (pDSBs) cause an immediate DNA damage response (DDR) and SAPA, but the SASP requires days to develop. Here, we show that following the immediate canonical DDR, a delayed chromatin accumulation of the ATM and MRN complexes coincides with the expression of SASP factors. Importantly, histone deacetylase inhibitors (HDACi) trigger SAPA and SASP in the absence of DNA damage. However, HDACi‐induced SASP also requires ATM/MRN activities and causes their accumulation on chromatin, revealing a DNA damage‐independent, non‐canonical DDR activity that underlies SASP maturation. This non‐canonical DDR is required for the recruitment of the transcription factor NF‐κB on chromatin but not for its nuclear translocation. Non‐canonical DDR further does not require ATM kinase activity, suggesting structural ATM functions. We propose that delayed chromatin recruitment of SASP modulators is the result of non‐canonical DDR signaling that ensures SASP activation only in the context of senescence and not in response to transient DNA damage‐induced proliferation arrest.
Keywords: chromatin, DNA damage response, MRN complex, NF‐κB, senescence secretome
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Expression of SASP factors requires a non‐canonical function of the DNA damage response (DDR) activated in senescent cells, which is characterized by an accumulation of ATM and MRN complexes on chromatin, independent of the presence of DNA DSBs.
Introduction
Cellular senescence is a tumor suppressor mechanism that relies on a stable senescence‐associated (SA) proliferation arrest (SAPA) to limit the multiplication of cells at risk for neoplastic transformation (Lowe et al, 2004; Rodier & Campisi, 2011; van Deursen, 2014; Gonzalez et al, 2016; Lee & Schmitt, 2019). Unlike apoptotic cells, which are rapidly eliminated (Christophorou et al, 2005, 2006; Roos & Kaina, 2013), senescent cells remain viable and have multifaceted biological functions. These include roles in embryonic development (Munoz‐Espin et al, 2013; Storer et al, 2013), viral infection (Appay et al, 2007), placental biology (Chuprin et al, 2013), wound healing (Demaria et al, 2014), and tissue remodeling that occurs during cancer treatment (Gonzalez et al, 2016; Demaria et al, 2017; Milanovic et al, 2018; Fleury et al, 2019). Senescent cells are found with increased frequency in aging tissues and at sites of age‐associated pathologies, including cancer, while their targeted elimination can restore naturally or prematurely aged tissue functions and extend lifespan in mice (Dimri et al, 1995; Herbig et al, 2006; Baker et al, 2011, 2016; Burd et al, 2013; van Deursen, 2014; Chang et al, 2016; Demaria et al, 2017; Fuhrmann‐Stroissnigg et al, 2017; Jeon et al, 2017). Although SAPA largely accounts for tumor suppression aspects of senescence, most beneficial and detrimental microenvironmental functions of senescent cells are mediated via their paracrine pro‐inflammatory SASP (Krtolica et al, 2001; Parrinello et al, 2005; Coppe et al, 2008; Alspach et al, 2014; Laberge et al, 2015; Malaquin et al, 2016).
Establishing the SASP following senescence induction requires a complex multiday genetic program that involves temporally interlaced molecular pathways converging toward the activation of NF‐κB, the major transcription factor regulating the SASP (Acosta et al, 2008; Rodier & Campisi, 2011; Baker & Sedivy, 2013; Ito et al, 2017). Cellular senescence is almost always a consequence of genotoxic stresses (i.e., telomere shortening, oncogene‐induced mitotic stress, irradiation) that cause DNA double‐strand breaks (DSBs) and a DNA damage response (DDR) signaling cascade (d'Adda di Fagagna et al, 2003; Bartkova et al, 2005; Di Micco et al, 2006; Mallette et al, 2007). In senescent cell nuclei, irreparable DSBs generate continuous DDR signaling from persistent DNA damage foci (DDF) or ‘DNA segments with chromatin alterations reinforcing senescence’ (DNA‐SCARS) (Rodier et al, 2011; Fumagalli et al, 2012; Hewitt et al, 2012; Fuhrmann‐Stroissnigg et al, 2017). In addition to DNA‐SCARS and reinforcing the link between DNA damage and senescence, TREX1‐, PARP‐, GATA4‐, p38MAPK‐, or cGAS/STING‐related damage responses have been linked to both SAPA and SASP (Freund et al, 2011; Ohanna et al, 2011; Kang et al, 2015; Dou et al, 2017; Ito et al, 2018; Takahashi et al, 2018; De Cecco et al, 2019). The DDR is necessary for both SAPA and SASP, but important differences in the temporal kinetics of each SA phenotype can be noted (Rodier et al, 2009; Coppe et al, 2011; Pazolli et al, 2012; Kang et al, 2015; Malaquin et al, 2015, 2016). For example, DSBs trigger immediate local recruitment of the MRE11‐Rad50‐NBS1 (MRN) complex on nearby chromatin. This promotes near‐simultaneous ATM kinase recruitment and activation (phosphorylation at serine 1981 [S1981‐ATM]) that leads to DDR signaling and cell cycle checkpoints activation, including the CHK2 and p53/p21Cip1 pathways controlling SAPA (Chen et al, 1995; Serrano et al, 1997; d'Adda di Fagagna et al, 2003; Beausejour et al, 2003; Herbig et al, 2004; Rodier et al, 2009). Alternatively, the DDR‐dependent expression of many SASP factors develop over a period of many days after the formation of senescence‐inducing DNA lesions, long after the initial canonical DDR signal has emerged from DSBs. In this context, persistent DDF have been proposed as reservoirs of active DDR signaling (Coppe et al, 2008, 2011; Rodier et al, 2009, 2011; Freund et al, 2011; Baar et al, 2017). Thus, despite its absolute requirement for the initiation and maintenance of SASP, the canonical early activation of ATM and the DDR at DSBs are not sufficient to trigger the SASP program, suggesting that delayed partnered ATM activity, perhaps at persistent DDF could be involved (Rodier et al, 2009, 2011). Some evidence has shown that histone deacetylase inhibitors (HDACis) can trigger a p53/p21‐ and p16‐dependent SAPA and SA‐β‐galactosidase activity (SAβGAL), another key senescence hallmark (Dimri et al, 1995; Ogryzko et al, 1996; Munro et al, 2004). HDACi‐induced senescence was also shown to trigger the expression of IL‐6 and IL‐8 (Orjalo et al, 2009; Pazolli et al, 2012), in the absence of global γH2AX phosphorylation (Pazolli et al, 2012), suggesting that part of the DDR‐associated SASP may occur independently of DSBs (Bakkenist & Kastan, 2003; Pazolli et al, 2012).
Here, we compared SA secretory programs triggered by HDACi (sodium butyrate [NaB]) or DNA damage (X‐Ray irradiation [XRA]). We found that NaB triggered a full SASP, similar to the XRA‐induced SASP, but in the absence of persistent DDF or classical DDR activation. Strikingly, despite an apparent lack of DDR, the genetic depletion of ATM, MRE11 or NBS1 completely abrogated the NaB‐induced SASP as previously observed for the XRA‐SASP (Rodier et al, 2009, 2011). Accompanying this discordance, we observed that while classical DDR activity appeared within minutes of DNA damage exposure, the NaB‐induced SASP occurred within days and the XRA‐SASP required up to a week to manifest. In line with a late role for the DDR in SASP maturation, for both XRA and NaB, we observed a delayed global accumulation of MRN complex on chromatin, which coincided with NF‐κB chromatin recruitment and SASP development. Finally, we observed that the role of ATM in SASP is independent of its kinase activity while the nuclease activities of MRE11 are partially involved. Overall, we suggest that the SASP is regulated by delayed non‐canonical, chromatin‐associated DDR signaling outside of persistent DDF essential for NF‐κB activity, providing additional novel pharmaceutical targets to regulate SA phenotypes.
Results
HDAC inhibition triggers a rapid SASP in the absence of DNA damage and canonical DDR activation
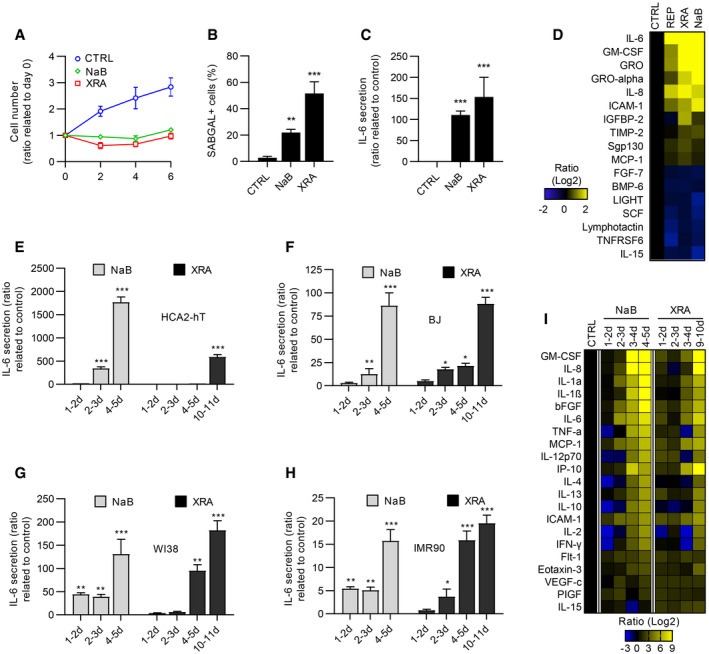
To investigate whether a canonical DDR is both essential and directly responsible for the SASP, we exposed normal immortalized human fibroblasts (HCA2‐hT) to the HDACi NaB. Alternatively, we exposed these cells to a single dose of 10 Gray (Gy) XRA, which is known to trigger a full senescence program associated with persistent DDR signaling and DNA‐SCARS (Coppe et al, 2008; Rodier et al, 2009, 2011). As expected, we observed stable proliferation arrest using either NaB or XRA (Fig 1A), and long‐term treatment (9 days) generated SAβGAL and IL‐6 secretion as previously observed (Ogryzko et al, 1996; Pazolli et al, 2012) (Figs 1B and C, and EV1A). Using non‐immortalized HCA2 cells, we then probed whether sustained NaB treatment generated a SASP profile similar to that from two typical models of DNA damage‐induced senescence (replicative senescence [REP] or irradiation). Quantitative antibody arrays revealed that the six most secreted proteins in REP or irradiated (XRA) cells were also significantly increased by NaB (IL‐6, GM‐CSF, GRO, GRO‐alpha, IL‐8, and ICAM‐1; P < 0.05), and overall, 17 proteins were differentially secreted at similar levels in both XRA‐ and NaB‐treated cells (P < 0.05; Fig 1D). Importantly, factors that were secreted at lower levels after REP or XRA senescence also decreased after NaB treatment, suggesting that HDACi does not trigger a global upregulation of secreted factors. Accordingly, the secretory profile (120 proteins) of NaB‐treated cells positively correlated with the profiles of cells that senesced by replicative exhaustion and irradiation, supporting common NaB‐XRA SASP programs (Appendix Fig S1A).
Figure 1. Characterization of SASP induced by HDACi.
-
AHCA2‐hT cells that were untreated, irradiated (10 Gy of XRA), or treated with 5 mM of sodium butyrate (NaB) were fixed at 2, 4, and 6 days following treatment. Cell proliferation was evaluated using DRAQ5 staining. Fluorescence intensity was reported on day 0. Data are means ± SD of triplicates and are representative of two independent experiments.
-
BSA‐ß‐galactosidase assays of HCA2‐hT cells fixed 9 days post‐XRA (10 Gy) or NaB treatment (2 mM). Data are means ± SD of triplicates and are representative of two independent experiments.
-
CIL‐6 secretion was analyzed by ELISA in conditioned media (CM) of HCA2‐hT cells that were collected 9 days after XRA (10 Gy) or NaB (2 mM). Data are the means ± SD of triplicates and are representative of three independent experiments.
-
DSoluble factors secreted by untreated or senescence‐induced HCA2 cells: replicative senescence (REP), 10 days post‐XRA(10 Gy) or treated 10 days with 2 mM of NaB were analyzed by antibody arrays.
-
E–H(E) HCA2‐hT, (F) BJ, (G) WI‐38, or (H) IMR‐90 cells were treated with NaB (5 mM) or XRA (10 Gy). CM were collected for the indicated period and IL‐6 secretion was assessed by ELISA. Data are means ± SD of triplicates and are representative of three independent experiments.
-
IHCA2‐h cells were treated with NaB (5 mM) or XRA (10 Gy) and CM were collected for the indicated period. Secretion of 40 soluble factors was measured by a multiplex immunoassay (40‐Plex MSD®). Data represent the means of two independent experiments.
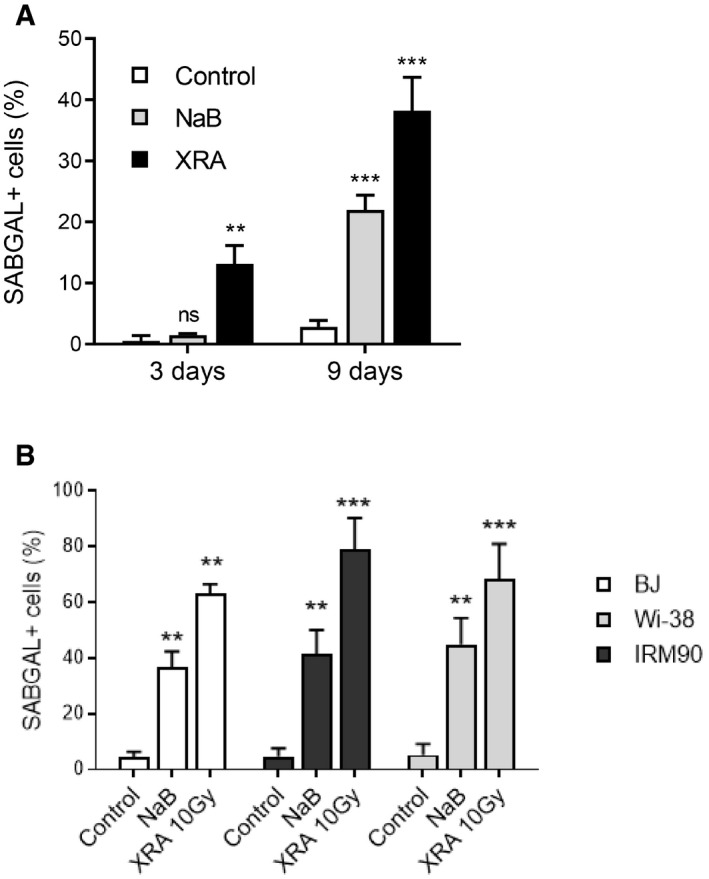
Figure EV1. Quantification of SA‐β‐galactosidase assays in NaB‐ or XRA‐ treated fibroblasts.
-
AHCA2‐hTert fibroblasts were irradiated (XRA) with 10 Gy or treated with 2 mM of NaB. Three or 9 days later, cells were fixed and SA‐ß‐galactosidase assays were performed. Data are means ± SD of triplicates and are representative of two independent experiments.
-
BBJ, WI‐38, or IMR90 fibroblasts were irradiated (XRA) with 10 Gy or treated with 5 mM of NaB. Nine days later, cells were fixed and SA‐ß‐galactosidase assays were performed. Data are means ± SD of triplicates and are representative of two independent experiments. Unpaired t‐test: **P < 0.01; ***P < 0.001, ns, non‐significant.
Interestingly, we noticed that the IL‐6 secretion induced by NaB occurred much earlier than the XRA‐induced Il‐6 secretion in HCA2‐hT cells (Fig 1E). Indeed, IL‐6 secretion significantly increased 2–3 days following the NaB treatment compared to 8–9 days for XRA (Fig 1E). This pattern of IL‐6 secretion was observed with another HDACi (Trichostatin A [TSA]; Appendix Fig S1B) and was similar for independent normal human fibroblasts cultures (BJ, IMR90 and WI38 cells; Fig 1F–H), which was also accompanied by increased SAβGAL (Fig EV1B). NaB‐induced senescence and SASP were indistinguishable from XRA‐induced phenotypes and were also detected in aggressive PC‐3 prostate cancer cells, overall suggesting that NaB‐induced senescence phenotypes are broadly conserved in cells that can undergo senescence (Appendix Fig S2). We further characterized the temporal kinetics of SASP factors induced by HDACi using sensitive multiplex immunoassays and compared the secretory profiles of 40 cytokines and chemokines from NaB‐exposed cells (NaB‐SASP) and irradiated cells (XRA‐SASP). As expected, irradiation gradually increased the secretion of pro‐inflammatory factors including IL‐6, IL‐8, and GM‐CSF, reaching maximal SASP levels in 10 days (Fig 1I, right panel). In contrast, NaB triggered secretion of SASP levels or higher in only 3 days (Fig 1I, compare left and right panels). Levels of SASP factors that were elevated in the NaB‐SASP at 24–48 or 48–72 h were comparably lower or absent in corresponding XRA‐SASP. Thus, HDACi strongly accelerated the expression of a mature SASP when compared to DNA damage.
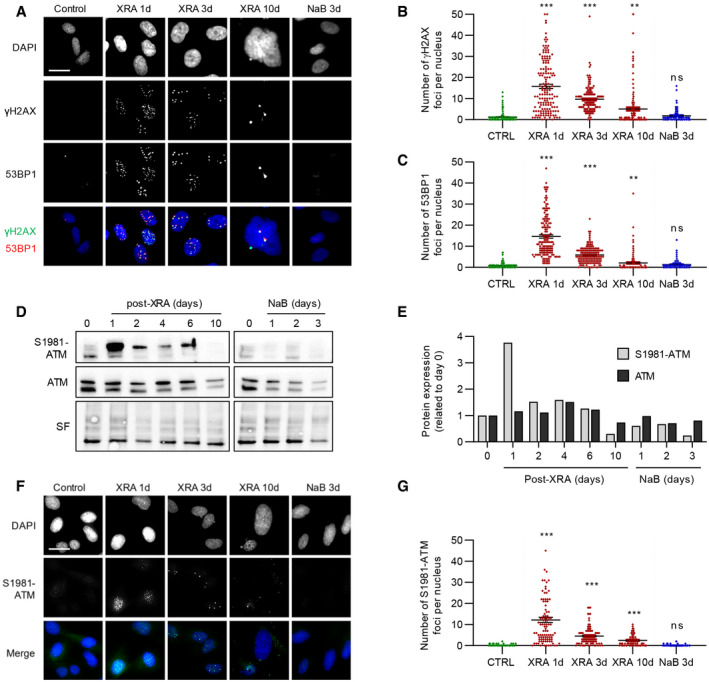
Since the SASP has been previously linked to persistent DDR signaling (Rodier et al, 2009, 2011), we evaluated the presence of DDF characterized by the colocalization of 53BP1 and γH2AX. As expected, 10 Gy of XRA triggered a large number of DSBs foci (1 day), which were mostly repaired within a few days, leaving behind residual persistent DDF in senescent cells (10 days; Fig 2A–C) (Rodier et al, 2011). In contrast, NaB‐treated cells did not display any γH2AX or 53BP1 nuclear foci even after 72 h of treatment (Fig 2A–C). We further probed DDR activation via detection of the phosphorylated form of ATM at serine S1918 (S1981‐ATM) in whole cell extracts (Fig 2D). As expected, the total level of S1981‐ATM rapidly increased following exposure to XRA and gradually decayed to normal levels over 10 days as cells repaired part of their DNA lesions and entered senescence (Fig 2D and E). In contrast, S1981‐ATM remained undetectable in NaB‐treated cells even several days after treatment when cells have already developed a mature SASP profile (Fig 2D and E). Alternatively, phosphorylation of the ATM target and cell cycle regulator CHK2 (phospho T68‐CHK2; (Ahn et al, 2000)) was rapidly observed and was maintained after irradiation for up to 10 days, but remained undetected following NaB exposure, suggesting that canonical DDR signaling was not activated in this context (Appendix Fig S3A). To probe ATM activity following XRA or NaB treatment at the single‐cell level, we analyzed nuclear foci of S1981‐ATM by immunofluorescence (Fig 2F). Similar to DDF, a large number of S1981‐ATM foci were present early following XRA (24 h), and a few residual foci persisted until senescence (Fig 2F and G). Analysis of integrated single‐cell mean fluorescence intensity (MFI) of nuclear S1981‐ATM was consistent with whole cell extracts, revealing low ATM activity in either XRA‐ or NaB‐induced senescent cells (Appendix Fig S3B). Taken together, these results suggest that the SASP does not always correlate with the formation of persistent DSBs or with the degree of canonical ATM‐dependent DDR activity detected, particularly in HDACi‐induced senescent cells.
Figure 2. HDACi does not trigger DSBs or canonical DDR activation.
-
AFluorescence images of 53BP1 and γH2AX stained by immunofluorescence in cells fixed at the indicated times. DAPI was used for nuclear counterstain. Bar scale = 25 μM.
-
B, CQuantification of the number of (B) γH2AX or (C) 53BP1 foci per nucleus (n = 150) using ImageJ. Means ± SEM of foci per nucleus were represented.
-
DWestern blot analysis of phosphorylated ATM (S1981‐ATM) or total ATM expression in BJ cells untreated (time 0), treated with NaB or XRA. Stain‐free (SF) imaging of the membrane was used as loading control.
-
EQuantification of S1981‐ATM and ATM expression detected in D using Image Lab (Bio‐Rad). Bars represent the ratio of expression related to the control.
-
FImages of S1981‐ATM immunofluorescence (green) and DAPI staining in control, irradiated or NaB‐treated BJ cells. Bar scale = 25μM
-
GQuantification of S1981‐ATM foci per nucleus (n = 150) using ImageJ. Means ± SEM of foci per nucleus were represented.
The MRN complex and ATM are required for NaB‐SASP
Because ATM is crucial for the production of oncogene‐, XRA‐, and REP‐induced SASPs (Rodier et al, 2009, 2011; Freund et al, 2011; Pazolli et al, 2012; Kang et al, 2015), we examined whether ATM and the ATM‐recruiting DNA damage sensor MRN complex were involved in NaB‐SASP, despite the absence of persistent DDF. We infected normal human fibroblasts with lentiviruses expressing short‐hairpin RNA against green fluorescent protein (shGFP; control), ATM (shATM), NBS1 (shNBS1), and MRE11 (shMRE11) and validated the protein depletion by Western blot (Fig 3A–C). As previously observed for the depletion of ATM and NBS1 (Rodier et al, 2009), shMRE11 prevented the secretion of Il‐6 (Fig EV2A) and of most SASP factors at 10 days post‐XRA (Fig EV2B), reinforcing the importance of the MRN complex in this context. Strikingly, the genetic depletion of ATM, MRE11, or NBS1 also prevented the IL‐6 secretion triggered by NaB (Fig 3D), a finding validated by multiplex immunoassay profiling that revealed a systematic reduction in SASP factors following MRE11 depletion (Fig 3E). To note that the depletion of MRE11 can also reduce the basal secretion of several cytokines (i.e., IL‐1α and β, IP‐10…; Figs 3E and EV2B). These results suggest that NaB‐induced SASP depends on the ATM/MRN DDR pathway, yet possibly act in a non‐canonical way uncoupled from persistent DSBs‐type DNA lesions. Indeed, we confirmed that SASP‐negative senescent ATM‐deficient cells, as well as the MRE11‐ and NBS1‐depleted cells, still displayed DDF in response to XRA probably via ATR/DNAPK compensation mechanisms following the loss of ATM, as previously reported (Appendix Fig S4) (Katyal et al, 2014). Overall, these results demonstrate that SASP correlates with non‐canonical DDR ATM activity rather than with DSBs‐type DNA lesions.
Figure 3. Genetic ablation of MRN and ATM impairs NaB‐induced SASP .
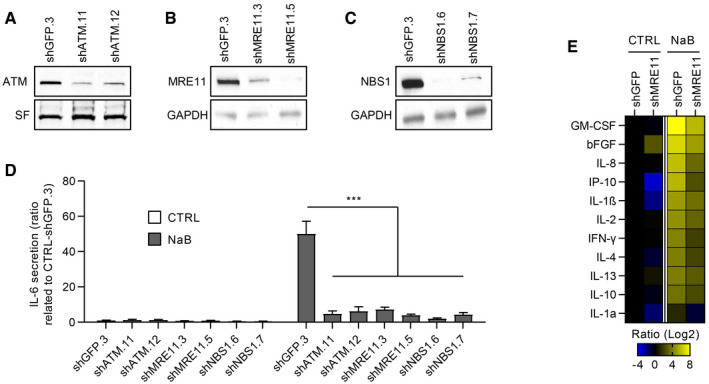
-
A–CDepletion of (A) ATM, (B) MRE11, or (C) NBS1 proteins was validated by Western blot. Stain‐free (SF) imaging of the membrane or GAPDH was used as loading control.
-
DCells were treated with 5 mM of NaB for 3 days and the conditioned media (CM) of the last 16 h were collected and assessed for IL‐6 secretion by ELISA. Data are the means ± SD of triplicates and represent at least three independent experiments. Unpaired t‐test: ***P < 0.001.
-
EBJ‐shGFP.3 and BJ‐shMRE11.5 cells were either untreated or exposed to 5 mM NaB for 2 days, and CM were collected for the next 24 h. Secreted soluble factors were evaluated using a multiplex immunoassay (40‐Plex MSD®). Average secretion of untreated BJ‐shGFP.3 cells was used as baseline. Heat map key indicates log2‐fold changes from control. Data represent two independent experiments.
Figure EV2. Impact of MRE11 depletion on XRA‐SASP.
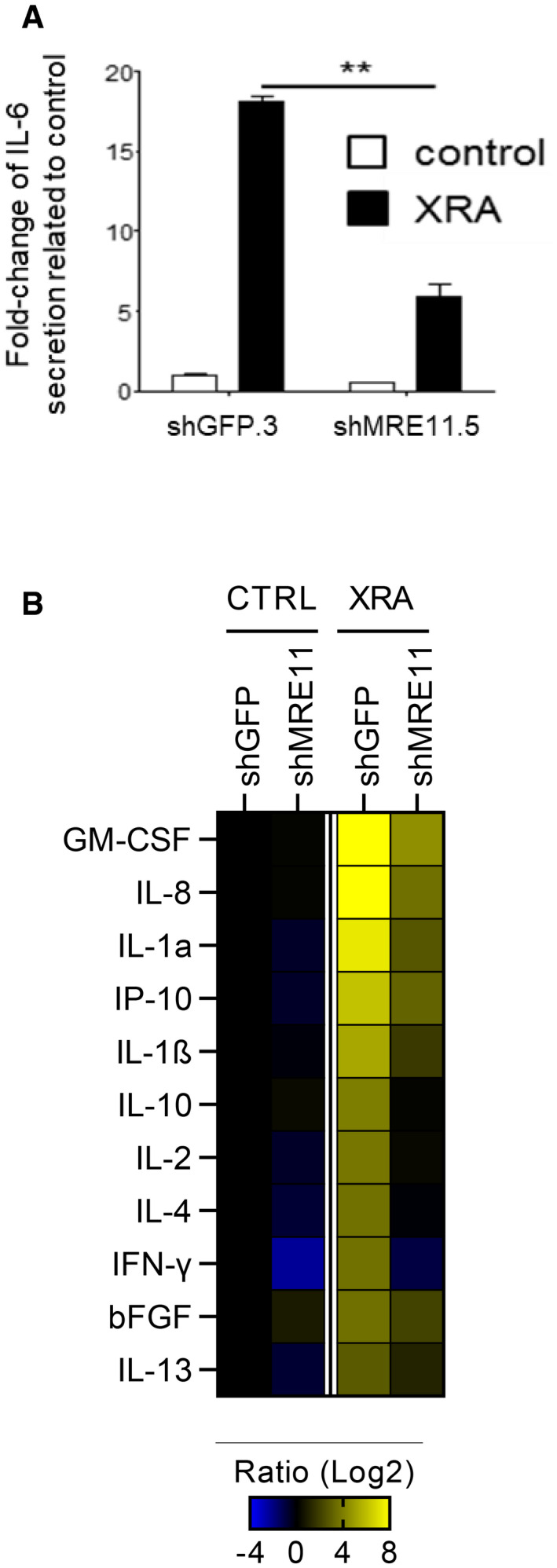
-
ABJ fibroblasts were infected with lentiviruses expressing shGFP.3 or shMRE11.5, selected with puromycin and allowed to recover for 7 days. BJ‐shGFP.3 and BJ‐shMRE11.5 cells were untreated (control) or irradiated with 10 Gy of XRA. After 9 days, serum‐free conditioned medium (SF‐CM) was collected over 24 h. IL‐6 secretion in SF‐CM was analyzed by ELISA. Data are the means ± SD of triplicates and representative of two independent experiments.
-
BSecreted soluble factors were evaluated using a multiplex immunoassay (40‐Plex MSD®). Average secretion of untreated BJ‐shGFP.3 cells was used as baseline. Heat map key indicates log2‐fold changes from control. Unpaired t‐test: **P < 0.01.
ATM kinase activity is not required for the SASP
The kinase activity of ATM is required for canonical DDR immediately following the generation of DSBs (Malaquin et al, 2015; Blackford & Jackson, 2017). However, in the context of SASP, the levels of phospho‐ATM (S1981‐ATM) are very low or absent (Fig 2D–G). To determine whether the kinase activity of ATM was involved in the expression of SASP factors we used chemical inhibitors against ATM kinase activity (ATMi, Ku55933). Using a dose of XRA (1 Gy) to allow for easy visualization of individual DDF, we validated that ATMi completely prevented the formation of S1981‐ATM nuclear foci (Fig 4A) and negatively modulated the intensity of γH2AX DDF (Fig 4B). In the context of XRA, the early canonical DDR is key for effective proliferation arrest (Abraham, 2001). Indeed, if we blocked ATM kinase activity early simultaneously with irradiation and maintained inhibition for 10 days (ATMi D0), we observed increased genomic instability and increased γH2AX‐positive persistent DDF in SAβGAL‐positive senescent cells (Fig EV3A–C and Appendix Fig S5A and B). ATMi D0 also increased IL‐6 secretion by about twofold, consistent with an increased DNA damage load (Appendix Fig S5C, ATMi D0). To avoid altering canonical DDR signaling early after XRA, we treated cells with ATMi starting 8 days post‐XRA (ATMI D8; Fig 4C) and we did not observe any significant increase in the number of γH2AX‐positive persistent DDF (Figs 4D and EV3A–C) nor in the percentage of SAβGAL‐positive cells (Fig 4E), suggesting a more limited impact on proliferation and DNA damage phenotypes. Again, we observed that the inhibition of ATM kinase activity at day 8 post‐XRA did not have any significant impact on Il‐6 secretion or more globally, on the pro‐inflammatory XRA‐SASP (Fig 4F–H). Similarly, ATMi did not prevent the secretion of Il‐6 induced by NaB or the NaB‐SASP (Fig 4F–H). In summary, although ATM is essential for the SASP, as validated using genetic depletion approaches, the canonical kinase activity of ATM is apparently not required.
Figure 4. ATM kinase activity is not involved in NaB‐ and XRA‐SASPs.
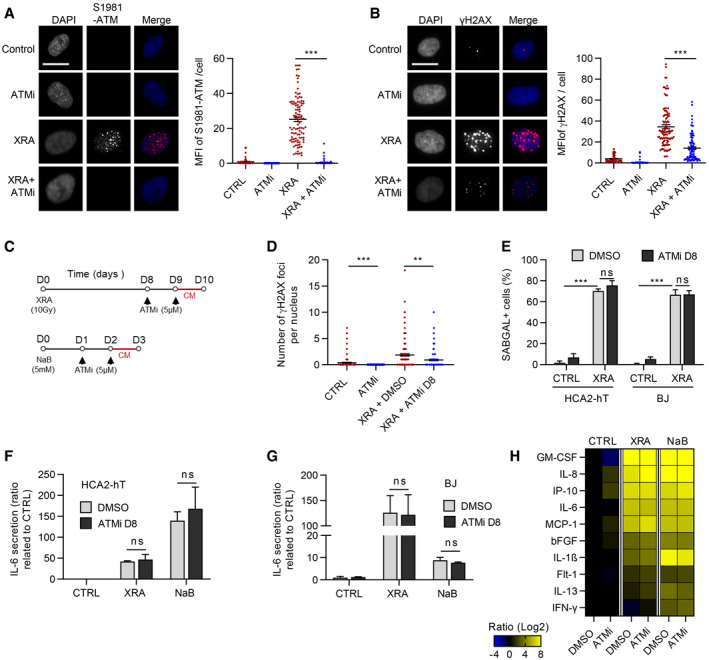
-
A, BHCA2‐hT cells were treated with 5 μM of Ku‐55933, an ATM inhibitor (ATMi), for 2 days and irradiated with 1 Gy of XRA. Two hours post‐XRA, cells were fixed and analyzed for S1981‐ATM or γH2AX immunofluorescence. (A) Fluorescence images of S1981‐ATM (red) and DAPI staining (left panel), and quantification of the S1981‐ATM mean fluorescence intensity (MFI) per nucleus (n = 150; right panel). (B) Fluorescence images of γH2AX (red) and DAPI staining (left panel), and quantification of the γH2AX foci number per nucleus (n = 150; right panel). Means of foci or MFI per nucleus ± SEM are shown, representative of two independent experiments. Bar scale = 10μM
-
CTimeline summarizing the sequence of XRA (10 Gy), NaB (5 mM), and Ku‐55933 (ATMi, 5 μM) treatments and the collection of the conditioned media (CM) used for figures D–H.
-
DQuantification of the γH2AX foci per nucleus (n = 150) of HCA2‐hT cells untreated (Control), treated with ATMi for 2 days (ATMi), 10 days post‐XRA with DMSO (XRA) or ATMi (XRA + ATMi D8) added for the last 2 days. Representative images of the immunofluorescence are given in Fig EV3A. Means of foci or MFI per nucleus ± SEM are representative of two independent experiments.
-
EBJ and HCA2‐hT cells were treated with the same conditions as in (D) and assessed for SA‐ß‐galactosidase assay. Data are mean ± SD of triplicates and are representative of two independent experiments.
-
F, GIL‐6 secretion assessed by ELISA in CM of (F) HCA2‐hT or (G) BJ cells untreated, irradiated or treated with NaB +/− ATMi. Data are the means ± SD of triplicates and are representative of three independent experiments.
-
HSecreted soluble factors by HCA2‐hT cells untreated (Control), irradiated, or treated with NaB +/− ATMi were quantified using multiplex immunoassay (40‐Plex MSD®). Average secretion of control cells was used as baseline. Heat map key indicates log2‐fold changes from control. Data represent two independent experiments.
Figure EV3. Impact of ATM inhibitor on persistent DNA damage foci.
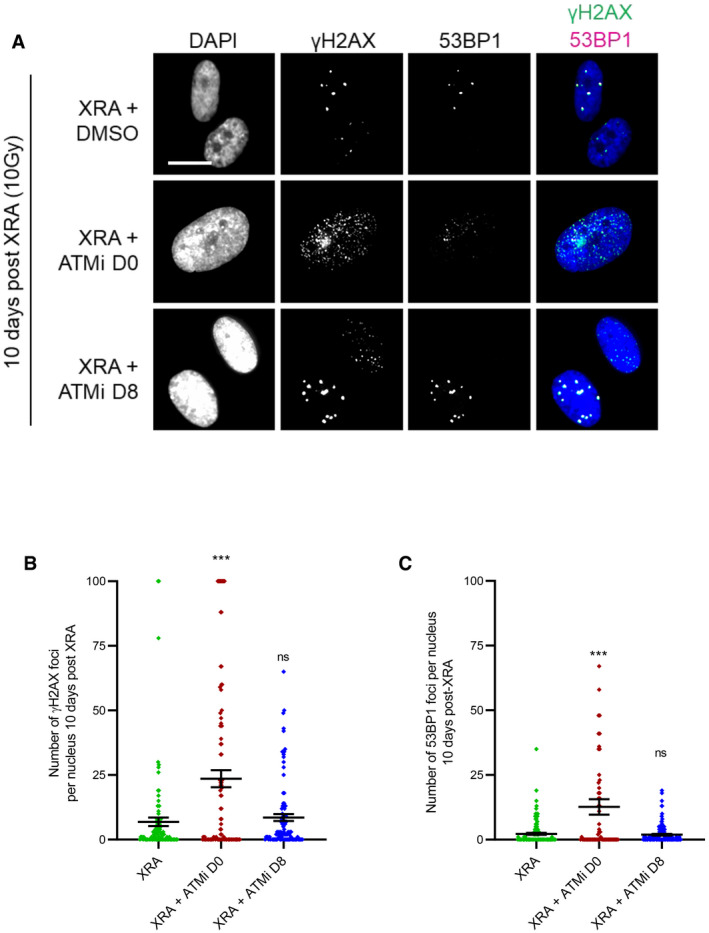
-
A–CHCA2‐hT or BJ cells were irradiated with 10 Gy of XRA. Ku‐55933 (ATMi, 5 μM) was added 1 h before XRA (ATMi D0) or 8 days post‐XRA (ATMi D8). (A) BJ cells were fixed 10 days post‐XRA and analyzed for γH2AX or 53BP1 immunofluorescence. Images of γH2AX (green) or 53BP1 (red) and DAPI staining. (B) Quantification of the number of γH2AX foci per nucleus (n = 150). (C) Quantification of the number of 53BP1 foci per nucleus (n = 150). Means ± SEM of foci per nucleus are representative of three independent experiments. Unpaired t‐test: ***P < 0.001, ns: non‐significant.
MRE11 nuclease activities contribute to the NaB‐ and XRA‐SASPs
We next investigated whether the MRE11 exo‐ and/or endonuclease activities were involved with the non‐canonical DDR activity and the expression of SASP factors. We used two different MRE11 inhibitors which have demonstrated inhibitory effects on exonuclease activity (Mirin) or endonuclease activity (PFM01) (Shibata et al, 2014). As expected, inhibition of the nuclease activities of MRE11 by Mirin or PFM01 altered the canonical DDR as illustrated by decreased Rad51 nuclear DDF at 2 h post‐XRA (Fig 5A). As for ATMi, cells were treated at 8 days post‐XRA in order to specifically target the MRE11 nuclease activities associated with the non‐canonical DDR, which did not alter the number of persistent DDF and the percentage of the SAβGAL‐positive cells (Fig 5B–D). Unlike for ATMi, usage of Mirin or PFM01 from day 0 (concomitantly with irradiation) did not have a measurable impact on persistent DDF or on the percentage of SAβGAL‐positive senescent cells 10 days later (Fig EV4A–C and Appendix Fig S6A and B). Importantly, inhibition of MRE11 nuclease activities from day 8 during the non‐canonical DDR was enough to reduce about 40% the secretion of Il‐6 (Fig 5E) and decrease the secretion of most pro‐inflammatory SASP factors (Fig 5G). Similarly, a strong reduction of Il‐6 secretion was observed if Mirin was added from day 0 concomitantly with XRA (Appendix Fig S6C). Mirin and PFM01 were also able to partially reduce the Il‐6 secretion induced by NaB and the NaB‐SASP (Fig 5F and G). We then compared the effect of MRE11 nuclease inhibition by Mirin with the genetic depletion of MRE11 on Il‐6 secretion induced by NaB (Fig 5H). Interestingly, the genetic depletion of MRE11 by shRNA reduced the NaB‐induced Il‐6 secretion more efficiently than exonuclease inhibition by Mirin (Fig 5H), suggesting that similar to ATM, the canonical enzymatic nuclease activities of MRE11 are only partially involved in the SASP.
Figure 5. Inhibition of MRE11 nuclease activities partially reduce NaB‐ and XRA‐SASPs.
-
AHCA2‐hT cells infected with mAG‐Geminin lentivirus were treated with 5 μM of Mirin or PFM01 for 2 days and irradiated with 1 Gy of XRA. Two hours post‐XRA, cells were fixed and assessed for Rad51 immunofluorescence. Left panel: Fluorescence images of Rad51 (red), Geminin (Green), and DAPI staining Bar scale = 10 μM. Right panel: quantification of the Rad51 foci number per nucleus in Geminin‐positive nuclei (cells in G2‐phase; n = 50). Means of foci or MFI per nucleus ± SEM are shown and are representative of two independent experiments.
-
BTimeline summarizing the sequence of XRA (10 Gy), NaB (5 mM), and Mirin (5 μM) and PFM01 (5 μM) treatments and collection of conditioned media (CM) used for panels (D–G).
-
CQuantification of the γH2AX foci per nucleus (n = 150) of HCA2‐hT cells treated with DMSO, with Mirin for 2 days (Mirin), 10 days post‐XRA (XRA) or 10 days post‐XRA with Mirin added for the last 2 days (XRA + Mirin D8). Representative images of the immunofluorescence are given in Figure EV4A. Means of foci or MFI per nucleus ± SEM are representative of three independent experiments.
-
DSA‐ß‐galactosidase assay in BJ and HCA2‐hT cells that were irradiated and treated with DMSO or treated with Mirin or PFM01. Data are mean ± SD of triplicate and are representative of two independent experiments.
-
E, FIL‐6 secretion assessed by ELISA in CM of HCA2‐hT and BJ cells that were (E) irradiated or (F) NaB‐treated and treated with DMSO or treated with Mirin or PFM01. Data are the means ± SD of triplicates and are representative of four independent experiments.
-
GSecreted soluble factors by HCA2‐hT cells untreated (CTRL), irradiated, or treated with NaB +/− Mirin were quantified using a multiplex immunoassay (40‐Plex MSD®). Average secretion of control cells was used as baseline. Heat map key indicates log2‐fold changes from control. Data represent two independent experiments.
-
HBJ cells infected with lentiviruses expressing shGFP.3 or shMRE11.5 were treated with NaB (5 mM) for 3 days with DMSO or Mirin (5 μM). CM of the last 16 h were collected and assessed for Il‐6 secretion. Secretion of BJ‐shGFP.3 treated with DMSO was used as baseline. Data are the means ± SD of triplicates and represent two independent experiments.
Figure EV4. Impact of MRE11 inhibitor on persistent DNA damage foci.
-
A–CHCA2‐hT or BJ cells were irradiated with 10 Gy of XRA. Five μM of Mirin was added 1 h before XRA (Mirin D0) or 8 days post‐XRA (Mirin D8). (A) BJ cells were fixed 10 days post‐XRA and analyzed for γH2AX or 53BP1 immunofluorescence. Left panel: images of γH2AX (green) or 53BP1 (red) and DAPI staining. Middle panel: quantification of the number of γH2AX foci per nucleus (n = 150). Right panel: quantification of the number of 53BP1 foci per nucleus (n = 150). Means ± SEM of foci per nucleus are representative of three independent experiments. Unpaired t‐test: *P < 0.05; **P < 0.01, ns: non‐significant.
The SASP is correlated with an accumulation of ATM, MRE11, and NF‐κB on the chromatin
Canonical DDR activity requires the association of the MRN complex and ATM with the chromatin in the vicinity of DSBs (Blackford & Jackson, 2017) as illustrated by the formation of DDF in Fig 2. Despite the absence of obvious DDF, we investigated whether ATM and the MRN complex still bound chromatin during the SASP‐associated non‐canonical DDR. We isolated proteins from the nucleus and from the chromatin in XRA‐ or NaB‐treated fibroblasts using subcellular fractionation. As expected for the canonical DDR, a strong increase of S1981‐ATM was observed 1 day following XRA in both the nuclear soluble and chromatin fractions (Fig 6A and B), confirming the rapid canonical DDR activation induced by DSBs. A strong increase of S1981‐ATM 1 day after XRA was also observed in the cytosolic fraction but rapidly returned to basal levels (Appendix Fig S7). Over time nuclear soluble S1981‐ATM gradually decreased similarly returning to basal levels (or lower) a few days post‐XRA (Fig 6A and B). Alternatively, a large proportion of S1981‐ATM persisted on the chromatin even 10 days after XRA (Fig 6A and B). Unlike S1981‐ATM, chromatin‐associated MRE11 was not enriched during early canonical DDR, but like S1981‐ATM, MRE11 gradually accumulated in the chromatin fraction a few days following XRA (Figs 6A and B, and EV5A). Thus, the persistence of almost all residual S1981‐ATM nuclear activity in the chromatin fraction and more importantly the unique enrichment of chromatin‐associated MRE11 could characterize the non‐canonical DDR. In line with this suggestion, subcellular fractionations of NaB‐treated cells that did not display any apparent DDF or focal accumulation of S1981‐ATM (Fig 2F and G) revealed an accumulation of MRE11, ATM, and S1981‐ATM proteins on chromatin a few days following treatment initiation while no noticeable difference was observed in the nuclear soluble fraction (Figs 6C and D, and EV5A). Altogether these results suggest that senescence is associated with delayed non‐canonical DDR functions independent of DSBs.
Figure 6. Chromatin localization of DDR components and p65 in NaB‐ and XRA‐induced senescent cells.
-
A–HHCA2‐hT cells were untreated (time 0), irradiated with 10 Gy of XRA or treated with 5 mM of NaB. At the indicated times, cells were collected and protein from the nuclear soluble and chromatin fractions were isolated by subcellular fractionation. Expression of S1981‐ATM, ATM, MRE11, or p65 was assessed by Western blot. (A) Western blot images and (B) quantification of S1981‐ATM, ATM, and MRE11 protein expression from irradiated HCA2‐hT cells. (C) Western blot images and (D) quantification of S1981‐ATM, ATM, and MRE11 protein expression from HCA2‐hT cells treated with NaB. (E) Western blot images and (F) quantification of p65 protein expression from irradiated HCA2‐hT cells. (G) Western blot images and (H) quantification of p65 protein expression from HCA2‐hT cells treated with NaB.
Figure EV5. Chromatin localization of p65 and MRE11 in senescent BJ cells.
-
A, BCells were untreated (time 0), irradiated with 10 Gy of XRA, or treated with 5 mM of NaB. At the indicated time, BJ cells were collected and proteins from the (A) nuclear soluble or (B) chromatin fractions were isolated by subcellular fractionation. Expression of MRE11 or p65 was assessed by Western blot. Stain‐free (SF) imaging of the membrane was used as loading control.
-
CBJ cells infected with lentiviruses expressing shGFP.3 or shATM.12 were treated with 5 mM of NaB for 3 days. Cells were collected and protein from chromatin fraction was isolated by subcellular fractionation. Expression of p65, MRE11, and ATM proteins in chromatin fractions was analyzed by Western blot. Stain‐free (SF) imaging of the membrane was used as loading control.
-
DBJ cells infected with lentiviruses expressing shGFP.3 or shNBS1.6 were treated with 5 mM of NaB. At the indicated times, cells were collected protein from chromatin fraction were isolated by subcellular fractionation. Expression of p65, MRE11, and ATM proteins in chromatin fractions was analyzed by Western blot. Stain‐free (SF) imaging of the membrane was used as loading control.
Source data are available online for this figure.
The non‐canonical DDR contributes to the binding of NF‐κB on chromatin
The expression of SASP factors requires the activation of the NF‐κB pathway, involving NF‐κB translocation into the nucleus and binding on the chromatin at SASP promoter sites (Chien et al, 2011). Consistent with this idea, we observed that the p65 subunit of NF‐κB was increased first in the nuclear soluble fraction and then in the chromatin fractions > 4 days after XRA (Figs 6E and F, and EV5B) or 2–3 days following NaB treatment (Figs 6G and H, and EV5B), which correlated with the expression of SASP factors. To determine whether the delayed non‐canonical DDR can impact the nuclear or chromatin localization of NF‐κB, we performed cell fractionation of fibroblasts depleted of MRE11 and treated with NaB or XRA. Interestingly, the depletion of MRE11 in combination with NaB treatment did not impact p65 kinetics in the nuclear soluble fraction (Appendix Fig S8A and B, but completely prevented the accumulation of chromatin‐associated p65 (Fig 7A). Similar results were obtained following exposure to XRA (Fig 7B and Appendix Fig S8C and D). Alteration of p65 accumulation on chromatin was also observed with the depletion of ATM (Fig EV5C) or NBS1 (Fig EV5D), suggesting that non‐canonical functions of MRN/ATM are essential for p65 recruitment on the chromatin, but not for nuclear accumulation of the transcription factor. Notably, the depletion of ATM altered the recruitment of MRE11 on chromatin (Fig EV5C), while the depletion of MRE11 did not impact ATM binding (Fig 7A and B). Finally, to determine whether the exo‐ and/or endonuclease activities of MRE11 were involved in the accumulation of p65 on the chromatin, we performed cell fractionation on fibroblasts treated with Mirin and PFM01 in similar conditions described in Fig 5B. Consistent with the effects observed on the SASP, the addition of Mirin or PFM01 at 8 days post‐XRA partially reduced the accumulation of both p65 and MRE11 in the chromatin fraction, while no significant change was observed in the nuclear soluble fraction (Fig 7C and Appendix Fig S9A and B). We observed a similar partial reduction of p65 and MRE11 accumulation on the chromatin when the cells were treated with NaB (Fig 7D and Appendix Fig S9C and D). Overall, the genetic depletion of MRE11 was more efficient than the inhibition of MRE11 nuclease activities, which was consistent with their impact on IL‐6 secretion (Fig 5H). These results reveal that the non‐canonical DDR is required for the recruitment of NF‐κB on chromatin, a key transcription factor that establishes the SASP.
Figure 7. Impact of MRE protein depletion or MRE nuclease activity inhibition on p65 chromatin localization.
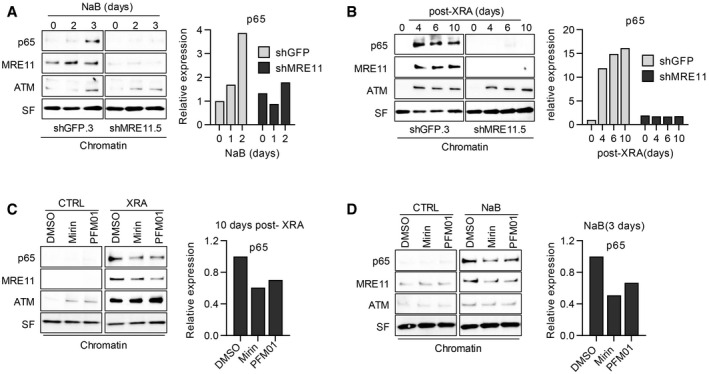
-
A, BBJ cells infected with lentiviruses expressing shGFP.3 or shMRE11.5 were treated with (A) NaB (5 mM) or (B) 10 Gy of XRA. At the indicated times, cells were collected and protein from chromatin fractions was isolated by subcellular fractionation.
-
C, DHCA2‐hT cells were (C) irradiated with 10 Gy and at 8 days post‐XRA, Mirin (5 μM).or PMF01 (5 μM) were added for 2 days or (D) cells were treated with NaB (5 mM) +/− Mirin or PFM01 added 24 h after NaB. Alternatively, non‐irradiated HCA2‐hT cells were treated with DMSO, Mirin or PFM01 for 2 days. Ten days post‐XRA or after 3 days of NaB treatment, cells were collected and protein from the chromatin fraction were isolated.
Discussion
The SASP shapes the tissue microenvironment of senescent cells in physiological and pathological contexts and represents a potential target to improve the manipulation of senescence in age‐related pathologies or cancer (Baker et al, 2011, 2016; Zhu et al, 2015, 2017; Chang et al, 2016; Oubaha et al, 2016; Fuhrmann‐Stroissnigg et al, 2017; Jeon et al, 2017). Following exposure to senescence‐inducing stimuli, the SASP evolves over time and at least two specific secretomes are now distinguished (Acosta et al, 2008; Kuilman et al, 2008; Rodier et al, 2009; Kang et al, 2015; Hoare et al, 2016; Malaquin et al, 2016; Ito et al, 2017; De Cecco et al, 2019): (i) an early TGF‐β‐rich anti‐inflammatory secretome dependent on NOTCH‐1 is first expressed; and (ii) a late pro‐inflammatory SASP follows, containing IL‐6 and IL‐8 and dependent on the NF‐κB and C/EBPβ transcription factors (Acosta et al, 2008; Rodier et al, 2009; Hoare et al, 2016). NOTCH‐1 is essential to coordinate the switch between these two secretomes by promoting secretome (i) and repressing secretome (ii), while many senescence‐activated signaling pathways including the DDR, p38MAPK, or the inflammasome apparently converge to collectively promote NF‐κB activity and secretome (ii). However, it remains unclear how secretome (ii) or the late pro‐inflammatory SASP is established (Rodier et al, 2009; Freund et al, 2011; Laberge et al, 2015; Malaquin et al, 2016).
Although we and others have previously shown that ATM is required for the establishment of both SAPA and the late SASP (d'Adda di Fagagna et al, 2003; Herbig et al, 2004; Rodier et al, 2009), we noted an unexplained disconnection between the kinetics of rapid canonical DDR activity and the delayed expression of SASP factors (Rodier et al, 2009). Indeed, the canonical DDR is activated within minutes following DSBs‐type DNA lesions resulting in immediate cell cycle checkpoints and proliferation arrest, but the DDR‐dependent SASP is expressed much later and is perhaps related to unresolved persistent DSBs or DNA‐SCARS (Rodier et al, 2009, 2011). Here, we highlighted that the expression of SASP factors depends on delayed non‐canonical DDR activity independent of rapid canonical DSBs‐type DDR signals. Unlike the canonical DDR, the non‐canonical DDR is characterized by a persistent binding of almost all available active ATM on the chromatin as well as a gradual accumulation of MRE11 on the senescent cell's chromatin in the absence of obvious DSBs. Importantly, this enrichment of ATM and MRN complex on chromatin was not directly linked to unresolved persistent DSBs nor to the rapid canonical DDR activity where chromatin‐associated MRE11 enrichment was undetectable by cell fractionation.
The specific role of non‐canonical DDR complexes in the regulation of specific SASP factors expression remains unclear and will require further exploration, but nonetheless appears directly related to the regulation of NF‐κB chromatin binding. This is consistent with NF‐κB being the major transcription factor regulating the SASP in multiple senescence contexts. For example, the genetic inhibition of the NF‐κB subunit (p65) or other components of the NF‐kB pathway (IkBα) prevents the SASP in XRA‐, RAS‐, and even HDACi‐induced senescent fibroblasts (Chien et al, 2011; Freund et al, 2011; Pazolli et al, 2012). The non‐canonical DDR triggered by HDACi generates a SASP that is indistinguishable from the pro‐inflammatory SASP produced by DNA damage in fibroblasts. Along those lines, HDACi‐induced senescent cells co‐injected with cancer cells in xenografts can stimulate cancer cell growth (Pazolli et al, 2012). Overall, given our findings and the fact that HDACi induce a stable SAPA and SASP in the absence of DNA damage or classical DDR activity in normal cells (Ogryzko et al, 1996; Pospelova et al, 2009; Pazolli et al, 2012), it appears likely that the non‐canonical activities of ATM/MRN described here are more important for the SASP than the immediate DDR activated in response to DSBs.
Previous studies have shown that in response to DNA damage, ATM can be translocated into the cytosol, contributing to the activation of NF‐κB (Hinz et al, 2010; Wu et al, 2010; Wang et al, 2017). In this condition, ATM interacts with NEMO, a regulator of the IKK complex (Miyamoto, 2011). The translocation of the ATM/NEMO complex into the cytoplasm results in the activation of IKKα/β, leading to the degradation of IκB proteins, which initiate nuclear translocation of NF‐κB (Wu et al, 2006; Miyamoto, 2011). However, we observed only a transient increase of S1981‐ATM in the cytosolic fraction the first day following the XRA (Appendix Fig S7), which is not consistent with the timing of SASP factors expression. Moreover, the non‐canonical DDR was essential for NF‐κB binding on the chromatin, but not for nuclear translocation, highlighting a new layer of NF‐κB regulation in this context. Only the chromatin fraction of S1981‐ATM, and MRE11, persisted over time suggesting that the role of non‐canonical DDR could be directly associated with the final steps of transcriptional regulation for SASP factors. Recent evidence demonstrates that DDR signaling can be involved in transcriptional initiation and elongation (Bunch et al, 2015; Puc et al, 2015; Singh et al, 2015; Capell et al, 2016). For example, in oncogene‐induced senescence, an enrichment of γH2AX at the promoters and gene bodies of the most highly up‐regulated SASP genes has been observed (Capell et al, 2016). Another study showed an interplay between HMGA2 (HMG AT‐hook 2 protein), ATM, and H2AX as a novel mechanism of transcription initiation for TGFβ1‐induced gene expression (Singh et al, 2015). Finally, it has been demonstrated in prostate cancer cells that androgen‐regulated gene expression involves the recruitment of MRE11 and the DDR machinery on enhancers of actively transcribed genes (Puc et al, 2015). In NaB‐ and XRA‐induced senescence, we demonstrated that the MRN complex and ATM are required for both the chromatin binding of NF‐κB and the presence of SASP factors. Surprisingly, as demonstrated via a combination of genetic and chemical approaches, this non‐canonical role of MRN/ATM was independent of the classical kinase activity of ATM and only partially dependent on the nuclease activities of MRE11, suggesting a structural function. We also found that ATM appeared first on the chromatin during non‐canonical DDR signaling and was required for the delayed chromatin binding of MRE11, which itself is not required for ATM recruitment. This suggests that unlike canonical DDR, ATM precedes MRE11 during non‐canonical DDR signaling. In support of non‐canonical functions for the MRN/ATM duo, recent mouse models expressing kinase‐dead (KD) ATM have also revealed unexpected structural functions of ATM since these mice displayed more severe genomic instability compared to a simple ATM deletion (Yamamoto et al, 2012; Menolfi & Zha, 2020).
Overall, we propose that the delayed chromatin recruitment of the MRN/ATM complex during the senescence program is the result of non‐canonical DDR signaling essential for the chromatin recruitment of the NF‐κB transcription factor. This delayed non‐canonical DDR cascade ensures SASP activation only in the context of cellular senescence, and not in response to a transient DNA damage‐induced proliferation arrest regulated by rapid canonical DDR signaling. Recognizing the tissue consequences of paracrine senescence activation, this multistep process may provide time for cell repair before initiating broader tissue repair. We believe that this knowledge may provide new ways to pharmacologically exploit the DDR without necessarily impacting its key functions in DNA repair and cell cycle checkpoints. In the future, further understanding of the crosstalk between low‐level, chromatin‐associated, non‐canonical DDR signaling and well‐established SASP regulators, including p38MAPK, mTOR, cytokine loops, and possibly the epigenetic regulation network, will help further decipher SA extracellular communication programs. This may serve as a template to understand how senescent cells shape their microenvironment, which will be key to exploit senescence in an intelligent way during the development of new therapies for SA human diseases.
Materials and Methods
Cells and culture conditions
Primary fibroblasts (HCA2 and BJ foreskin fibroblasts, WI‐38 and IMR‐90) were obtained from J. Smith (University of Texas, San Antonio) and cultured under ambient oxygen levels in Dulbecco's modified Eagle's media (DMEM) supplemented with 10% fetal bovine serum (FBS), 2.5 μg/ml fungizone, and 100 U/ml penicillin/streptomycin. Unless specified, early passage fibroblasts were used and defined by < 35 cumulative population doublings (PD) and a 24‐h BrdU labeling index of > 75%. PDs of primary cells were determined as follows: current PD = last PD + log2(cell number/cells seeded). Cell populations were considered replicatively senescent when cells achieved 24‐h labeling indices of < 5%. The 293FT packaging cells (Invitrogen) were used to generate lentiviruses as previously described (Rodier et al, 2011). When indicated, the media was supplemented with sodium butyrate (Sigma‐Aldrich) at 5 mM, Trichostatin A (Sigma‐Aldrich) at 100 ng/ml, Mirin (Sigma‐Aldrich) at 5 μM, PFM01 (Sigma‐Aldrich) at 5 μM, or KU‐55933 (Sigma‐Aldrich) at 5 μM.
Irradiation
Cells were X‐irradiated with a total dose of 10 Gy at rates equal to or above 0.75 Gy/min using Gammacell® 3000 irradiator Elan.
Viruses and infections
Viruses were produced as described previously (Campeau et al, 2009; Rodier et al, 2011). Briefly, mAG‐hGeminin(1/110)/pCSII‐EF‐MCS, given by Dr Jean‐yves Masson (Université de Laval, Quebec, Canada), was cloned into a Gateway entry vector and transferred to a lentiviral destination vector under the control of the CMV promoter (Campeau et al, 2009). The plasmid containing TRC lentiviral human shRNA against GFP (control), ATM, MRE11, and NBS1 were purchased from Open Biosystems and had the following target sequences: shATM.11, TRCN0000039951, 5′ ‐ AAACCCAGGGCTGCCTTGGAAAAG ‐ 3′; shATM.12, TRCN0000039952, 5′ ‐ AAACCCAGGGCTGCCTTGGAAAAG ‐ 3′; shATM.13, TRCN0000010299, 5′ ‐ AAACCCAGGGCTGCCTTGGAAAAG ‐ 3′; shMRE11.3, TRCN0000039869, 5′ ‐ AAACCCAGGGCTGCCTTGGAAAAG ‐ 3′; shMRE11.5, TRCN0000039871, 5′ ‐ AAACCCAGGGCTGCCTTGGAAAAG ‐ 3; shNBS1.6,TRCN0000010393, 5′‐AAACCCAGGGCTGCCTTGGAAAAG‐ 3′;shNBS1.7, TRCN0000010392, 5′ ‐ AAACCCAGGGCTGCCTTGGAAAAG – 3.
Cells were infected with virus titered to achieve 90% infectivity with 4 μg/ml of polybrene in a final volume of 1 ml in six‐well plates. Infections were followed 48 h later by puromycin or hygromycin selection (1 and 100 μg/ml, respectively).
Preparation of conditioned media and analysis of secreted SASP factors
Cells were seeded in 96‐well plates (1,500 cells per well), treated, or irradiated 24 h after seeding and maintained in culture before the preparation of conditioned media (CM). CM were prepared as previously described with minor modifications (Rodier, 2013). Briefly, cells were washed three times with serum‐free medium, followed by incubation in 1% FBS DMEM containing antibiotics and when applicable NaB and/or inhibitors (Ku55933, Mirin, PFM01) for 16 h. CM were collected and stored at −80°C until assayed. CM were assessed by ELISAs (Il‐6) using kits and procedures from R&D (IL‐6 #D06050, IL8 #DY208). Data were normalized to the cell number determined by DRAQ5™ staining of the nucleus and analyzed by the live‐imaging system Incucyte®. CM were also analyzed using antibody arrays (Chemicon, Human Arrays VI and VII, cat #AA1001CH‐8) according to the manufacturer's instructions with modifications as previously described (Coppe et al, 2008). Finally, CM were also analyzed using the multispot electrochemiluminescence immunoassay system for 40 secreted factors using the V‐Plex human kit from Meso Scale Discovery (MSD; #K15209D) following the manufacturer's instructions.
DNA content‐based cell counting with DRAQ5™
Cells were seeded in 96‐well plates, treated, or irradiated 24 h after the seeding and grown for the determined period. At the end, floating cells were removed with a PBS wash and the attached cells were fixed 10 min in formalin (Sigma) followed by three quick washes using PBS. Cells were stained with DRAQ5™ (1:10,000; Thermo Scientific) for 1 h and washed three times with PBS + 1% Triton. The far‐red fluorescence intensity from each well was captured using an Odyssey® imaging system (Li‐Cor). Fluorescence intensity was analyzed by Image Studio™ Software (Li‐Cor) and used to determine cell proliferation.
Senescence‐associated β‐galactosidase detection
Senescence‐associated β‐galactosidase assays were performed as previously described (Dimri et al, 1995). Briefly, cells grown in six‐well plates were washed once with 1× PBS and fixed with 10% formalin for 5 min, then washed again with PBS, and finally incubated at 37°C for 16 h in a staining solution composed of 1 mg/ml 5‐bromo‐4‐chloro‐3‐indolyl‐β‐galactosidase in dimethylformamide (20 mg/ml stock), 5 mM potassium ferricyanide, 150 mM NaCl, 40 mM citric acid/sodium phosphate, and 2 mM MgCl2, at pH 6.0. Afterward, cells were washed twice with PBS and pictures were taken for quantification.
Immunofluorescence
Cells seeded onto coverslips in 12‐well plates or Falcon™ chambered cell culture slides were fixed in formalin for 10 min at room temperature (RT), washed once in PBS, and permeabilized in PBS‐0.25% Triton for 10 min. Slides were washed once in PBS and blocked for 1 h in PBS containing 1% bovine serum albumin (BSA) and 4% normal donkey serum. For Rad51, fixed cells were permeabilized in PBS‐0.25% Triton + 0.2N HCl for 10 min. Slides were washed once in PBS and blocked for 30‐min PBS containing 8% of BSA and 1% FBS. Primary antibodies (listed in Appendix Table S1) diluted in blocking buffer were added, and slides were incubated overnight at 4°C. Cells were washed three times and incubated with secondary antibodies for 1 h at RT, then washed again. Coverslips were mounted onto slides using Prolong® Gold anti‐fade reagent with DAPI (Life Technologies Inc.). Images (400× magnification) were obtained using a Zeiss microscope (Zeiss Axio Observer Z1, Carl Zeiss, Jena, Germany). Number of foci or MFI per nucleus were counted using ImageJ software in > 150 nuclei of each condition.
Protein preparation and Western blot analysis
Cells were seeded in six‐well plates for total protein extraction or 100‐mm petri dishes for protein fractionation and immunoprecipitation, treated, or irradiated 24 h after the seeding and grown for the determined period. Total protein cell lysates were prepared by scraping cells with mammalian protein extraction reagent (MPER, Thermo Fisher Scientific) containing a protease and phosphatase inhibitor cocktail (Sigma‐Aldrich Inc). Subcellular fractionation was performed using the subcellular protein fractionation kit (Thermo Fisher Scientific; # 78840) according to the manufacturer's instructions. Briefly, cells were collected by trypsinization, centrifuged for 5 min at 1,000 g, and proteins were isolated from cytoplasmic, membrane, soluble nuclear, and chromatin‐bound fractions. All collected proteins were stored at −80°C. Protein concentration was measured using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific). Proteins were separated in stain‐free, 4–15% gradient Tris‐glycine SDS–polyacrylamide gels (Mini PROTEAN® TGX Stain‐Free™ Gels, Bio‐Rad Laboratories). Gels were activated by UV exposure for 45 s using ChemiDoc MP imaging system (Bio‐Rad Laboratories). After protein transfer onto PVDF membranes (Hybond‐C Extra, GE Healthcare Life Sciences), membranes were imaged for stain‐free staining and total protein was quantified using ImageLab 6 software (Bio‐Rad Laboratories). Membranes were blocked with 2% BSA in PBS for 1 h and probed with primary antibodies (see Appendix Table S1) overnight at 4°C. Bound primary antibodies were detected with peroxidase‐conjugated secondary antibodies (Cell Signaling Technology) and enhanced chemiluminescence (Bio‐Rad Laboratories). Chemiluminescence was detected using the ChemiDoc MP. The control for protein loading was evaluated using the stain‐free technology (Bio‐Rad Laboratories) or by specific antibodies (GAPDH). Bands were quantified using ImageLab 6 software and normalized with the stain‐free staining of the total protein.
Author contributions
NM and FR designed the study. NM, M‐AO, AM, SN, CS, and J‐PC performed experiments and collected/analyzed data. GC provided technical assistance. FAM and JC.provided technical support, expertise, and conceptual advice. FR provided financial support. NM and FR wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 6
Acknowledgements
We thank members of the Rodier laboratory as well as Jacqueline Chung for valuable comments and discussions. We thank the Institut du cancer de Montréal (ICM) Imaging and Live imaging platform. This work was supported by the ICM (F.R.) and by the Canadian Institute for Health Research (CIHR MOP‐114962 to F.R.; and MOP‐133442 to F.A.M.) and the Terry Fox Research Institute (TFRI 1030 to F.R.). F.R. is supported by a Fonds de Recherche Québec Santé (FRQS) junior I‐II career award (22624, 33070). F.A.M. holds the Canada Research Chair in Epigenetics of Aging and Cancer. F.R. is a researcher of CRCHUM/ICM, which receive support from the FRQS. N.M was supported by Mitacs acceleration postdoctoral fellowship. A.M., M‐A.O., and S.N. are recipients of ICM Canderel fellowships. S.N. is also an FRQS PhD fellowship recipient. C.S. obtained PhD fellowships from the Cole Foundation and from Hydro‐Québec.
EMBO Reports (2020) 21: e50718
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N et al (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018 [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay‐Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere‐initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- Ahn JY, Schwarz JK, Piwnica‐Worms H, Canman CE (2000) Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res 60: 5934–5936 [PubMed] [Google Scholar]
- Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, Donlin MJ, Marsh T, Piwnica‐Worms D, Monahan J et al (2014) p38MAPK plays a crucial role in stromal‐mediated tumorigenesis. Cancer Discov 4: 716–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V, Almeida JR, Sauce D, Autran B, Papagno L (2007) Accelerated immune senescence and HIV‐1 infection. Exp Gerontol 42: 432–437 [DOI] [PubMed] [Google Scholar]
- Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA et al (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169: 132–147.e116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 479: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Sedivy JM (2013) Probing the depths of cellular senescence. J Cell Biol 202: 11–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A et al (2016) Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature 530: 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C et al (2005) DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 434: 864–870 [DOI] [PubMed] [Google Scholar]
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J (2003) Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22: 4212–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage Response. Mol Cell 66: 801–817 [DOI] [PubMed] [Google Scholar]
- Bunch H, Lawney BP, Lin YF, Asaithamby A, Murshid A, Wang YE, Chen BP, Calderwood SK (2015) Transcriptional elongation requires DNA break‐induced signalling. Nat Commun 6: 10191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE (2013) Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)‐luciferase model. Cell 152: 340–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4: e6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS et al (2016) MLL1 is essential for the senescence‐associated secretory phenotype. Genes Dev 30: 321–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W et al (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22: 78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN (1995) Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci USA 92: 4337–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS et al (2011) Control of the senescence‐associated secretory phenotype by NF‐kappaB promotes senescence and enhances chemosensitivity. Genes Dev 25: 2125–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou MA, Martin‐Zanca D, Soucek L, Lawlor ER, Brown‐Swigart L, Verschuren EW, Evan GI (2005) Temporal dissection of p53 function in vitro and in vivo . Nat Genet 37: 718–726 [DOI] [PubMed] [Google Scholar]
- Christophorou MA, Ringshausen I, Finch AJ, Swigart LB, Evan GI (2006) The pathological response to DNA damage does not contribute to p53‐mediated tumour suppression. Nature 443: 214–217 [DOI] [PubMed] [Google Scholar]
- Chuprin A, Gal H, Biron‐Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V (2013) Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 27: 2356–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J (2011) Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286: 36396–36403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD et al (2019) L1 drives IFN in senescent cells and promotes age‐associated inflammation. Nature 566: 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME et al (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell 31: 722–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM et al (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7: 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM (2014) The role of senescent cells in ageing. Nature 509: 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A et al (2006) Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature 444: 638–642 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira‐Smith O et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo . Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z et al (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury H, Malaquin N, Tu V, Gilbert S, Martinez A, Olivier MA, Sauriol A, Communal L, Leclerc‐Desaulniers K, Carmona E et al (2019) Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun 10: 2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J (2011) p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J 30: 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann‐Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, Grassi D, Gregg SQ, Stripay JL, Dorronsoro A et al (2017) Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun 8: 422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM et al (2012) Telomeric DNA damage is irreparable and causes persistent DNA‐damage‐response activation. Nat Cell Biol 14: 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez LC, Ghadaouia S, Martinez A, Rodier F (2016) Premature aging/senescence in cancer cells facing therapy: good or bad? Biogerontology 17: 71–87 [DOI] [PubMed] [Google Scholar]
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell 14: 501–513 [DOI] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM (2006) Cellular senescence in aging primates. Science 311: 1257 [DOI] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FD, Correia‐Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress‐induced senescence. Nat Commun 3: 708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C (2010) A cytoplasmic ATM‐TRAF6‐cIAP1 module links nuclear DNA damage signaling to ubiquitin‐mediated NF‐kappaB activation. Mol Cell 40: 63–74 [DOI] [PubMed] [Google Scholar]
- Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama R et al (2016) NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 18: 979–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Hoare M, Narita M (2017) Spatial and temporal control of senescence. Trends Cell Biol 27: 820–832 [DOI] [PubMed] [Google Scholar]
- Ito T, Teo YV, Evans SA, Neretti N, Sedivy JM (2018) Regulation of cellular senescence by polycomb chromatin modifiers through distinct DNA damage‐ and histone methylation‐dependent pathways. Cell Rep 22: 3480–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N et al (2017) Local clearance of senescent cells attenuates the development of post‐traumatic osteoarthritis and creates a pro‐regenerative environment. Nat Med 23: 775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ (2015) The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: aaa5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, Lee Y, Nitiss KC, Downing SM, Li Y, Shimada M, Zhao J, Russell HR, Petrini JH, Nitiss JL et al (2014) Aberrant topoisomerase‐1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat Neurosci 17: 813–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA 98: 12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell 133: 1019–1031 [DOI] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson‐Edell KA, Liu S et al (2015) MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Schmitt CA (2019) The dynamic nature of senescence in cancer. Nat Cell Biol 21: 94–101 [DOI] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432: 307–315 [DOI] [PubMed] [Google Scholar]
- Malaquin N, Carrier‐Leclerc A, Dessureault M, Rodier F (2015) DDR‐mediated crosstalk between DNA‐damaged cells and their microenvironment. Front Genet 6: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaquin N, Martinez A, Rodier F (2016) Keeping the senescence secretome under control: molecular reins on the senescence‐associated secretory phenotype. Exp Gerontol 82: 39–49 [DOI] [PubMed] [Google Scholar]
- Mallette FA, Gaumont‐Leclerc MF, Ferbeyre G (2007) The DNA damage signaling pathway is a critical mediator of oncogene‐induced senescence. Genes Dev 21: 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menolfi D, Zha S (2020) ATM, ATR and DNA‐PKcs kinases‐the lessons from the mouse models: inhibition not equal deletion. Cell Biosci 10: 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, Dorr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA et al (2018) Senescence‐associated reprogramming promotes cancer stemness. Nature 553: 96–100 [DOI] [PubMed] [Google Scholar]
- Miyamoto S (2011) Nuclear initiated NF‐kappaB signaling: NEMO and ATM take center stage. Cell Res 21: 116–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Espin D, Canamero M, Maraver A, Gomez‐Lopez G, Contreras J, Murillo‐Cuesta S, Rodriguez‐Baeza A, Varela‐Nieto I, Ruberte J, Collado M et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118 [DOI] [PubMed] [Google Scholar]
- Munro J, Barr NI, Ireland H, Morrison V, Parkinson EK (2004) Histone deacetylase inhibitors induce a senescence‐like state in human cells by a p16‐dependent mechanism that is independent of a mitotic clock. Exp Cell Res 295: 525–538 [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH (1996) Human fibroblast commitment to a senescence‐like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol 16: 5210–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanna M, Giuliano S, Bonet C, Imbert V, Hofman V, Zangari J, Bille K, Robert C, Bressac‐de Paillerets B, Hofman P et al (2011) Senescent cells develop a PARP‐1 and nuclear factor‐{kappa}B‐associated secretome (PNAS). Genes Dev 25: 1245–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J (2009) Cell surface‐bound IL‐1alpha is an upstream regulator of the senescence‐associated IL‐6/IL‐8 cytokine network. Proc Natl Acad Sci USA 106: 17031–17036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, Bourdel G, Popovic N, Rezende FA, Kaufman RJ et al (2016) Senescence‐associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med 8: 362ra144 [DOI] [PubMed] [Google Scholar]
- Parrinello S, Coppe JP, Krtolica A, Campisi J (2005) Stromal‐epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 118: 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazolli E, Alspach E, Milczarek A, Prior J, Piwnica‐Worms D, Stewart SA (2012) Chromatin remodeling underlies the senescence‐associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res 72: 2251–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV (2009) Pseudo‐DNA damage response in senescent cells. Cell Cycle 8: 4112–4118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puc J, Kozbial P, Li W, Tan Y, Liu Z, Suter T, Ohgi KA, Zhang J, Aggarwal AK, Rosenfeld MG (2015) Ligand‐dependent enhancer activation regulated by topoisomerase‐I activity. Cell 160: 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol 11: 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Campisi J (2011) Four faces of cellular senescence. J Cell Biol 192: 547–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Munoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppe JP, Campeau E, Beausejour CM, Kim SH et al (2011) DNA‐SCARS: distinct nuclear structures that sustain damage‐induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci 124: 68–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F (2013) Detection of the senescence‐associated secretory phenotype (SASP). Methods Mol Biol 965: 165–173 [DOI] [PubMed] [Google Scholar]
- Roos WP, Kaina B (2013) DNA damage‐induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 332: 237–248 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum‐Fikkert S, Kertokalio A, Romoli F et al (2014) DNA double‐strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53: 7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Ozturk N, Cordero J, Mehta A, Hasan D, Cosentino C, Sebastian C, Kruger M, Looso M, Carraro G et al (2015) High mobility group protein‐mediated transcription requires DNA damage marker gamma‐H2AX. Cell Res 25: 837–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert‐Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130 [DOI] [PubMed] [Google Scholar]
- Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, Watanabe S, Kawamoto S, Miyata K, Barber GN et al (2018) Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9: 1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Mani AM, Wu ZH (2017) DNA damage‐induced nuclear factor‐kappa B activation and its roles in cancer progression. J Cancer Metastasis Treat 3: 45–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZH, Shi Y, Tibbetts RS, Miyamoto S (2006) Molecular linkage between the kinase ATM and NF‐kappaB signaling in response to genotoxic stimuli. Science 311: 1141–1146 [DOI] [PubMed] [Google Scholar]
- Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V (2010) ATM‐ and NEMO‐dependent ELKS ubiquitination coordinates TAK1‐mediated IKK activation in response to genotoxic stress. Mol Cell 40: 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Wang Y, Jiang W, Liu X, Dubois RL, Lin CS, Ludwig T, Bakkenist CJ, Zha S (2012) Kinase‐dead ATM protein causes genomic instability and early embryonic lethality in mice. J Cell Biol 198: 305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M et al (2015) The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14: 644–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann‐Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, Kirkland JL (2017) New agents that target senescent cells: the flavone, fisetin, and the BCL‐XL inhibitors, A1331852 and A1155463. Aging (Albany NY) 9: 955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 6