

Mutations in GRN, which encodes progranulin, are a common cause of familial frontotemporal dementia (FTD). FTD is a devastating disease characterised by neuronal loss in the frontal and temporal lobes that leads to changes in personality, behaviour and language. There are no effective treatments for this complex condition. TMEM106B is a well‐recognised risk factor for FTD caused by GRN mutation. While the specific relationship between progranulin and TMEM106B is unclear, it is well established that they are both required for correct lysosome function and trafficking. Elegant experiments have suggested that increased risk for FTD is due to elevated levels of TMEM106B (Nicholson et al, 2013; Gallagher et al, 2017). Therefore, recent work has explored the therapeutic potential of reducing TMEM106B levels, with initial results looking encouraging, as crossing a Grn‐deficient mouse to a Tmem106b knockout showed a rescue in FTD‐related behavioural defects and specific aspects of lysosome dysfunction (Klein et al, 2017). However, three independent studies in this issue report that completely removing Tmem106b from Grn knockout mice leads to clear exacerbation of phenotypes, causing severe motor deficits, neurodegeneration and enhanced lysosome abnormalities and gliosis. Remarkably, the double knockout mice also develop TDP‐43 pathology—a hallmark of FTD patients with GRN mutations that have not been consistently observed in either of the single knockouts. These concurrent publications that all reach the same surprising but definitive conclusion are a cautionary tale in the control of TMEM106B levels as a potential therapeutic for FTD. They also re‐ignite the debate as to whether loss or gain of TMEM106B function is critical for altering FTD risk.
Subject Categories: Molecular Biology of Disease, Neuroscience
Loss of TMEM106B has been suggested as therapeutic treatment of FTLD‐GRN. 3 studies in this issue instead show that loss of Tmem106b exacerbates FTLD pathologies causing motor deficits, neuronal loss, autophagy‐lysosome defects, glial activation, TDP‐43 aggregation and early death in Grn deficient mice.
Feng et al (2020) used CRISPR/Cas9 to remove Tmem106b in their double‐cross. The resultant ataxia and hindlimb phenotypes are severe and reached experiment endpoint by 5.5 months. Defects are most pronounced in the spinal cord, with a decrease in neurons and marked gliosis. At 5 months old, these mice exhibit enlarged lysosomal vacuoles at the axon initial segment of motor neurons. Werner et al (2020) report strikingly similar results, with severe motor defects at 4–5 months of age in their double knockouts. Zhou et al (2020) report progressive motor defects in their mouse models at the slightly later age of 8 months. However, their initial model retained 5–10% Tmem106b protein; when they used CRISPR to completely ablate Tmem106b, the onset of motor deficits was significantly advanced. Like Feng et al (2020), Zhou et al (2020) report significant loss of motor neurons at 12 months, and in addition a reduction in myelin density, with disorganised myelin fibres and degenerated myelin debris. All papers report significantly enhanced gliosis in the double knockouts, as well as the presence of phosphorylated TDP‐43. These reports clearly show a significant exacerbation of phenotype in Grn −/− mice upon removal of Tmem106b (Fig 1).
Figure 1. Removing Tmem106b from progranulin null mice exacerbates disease phenotype.
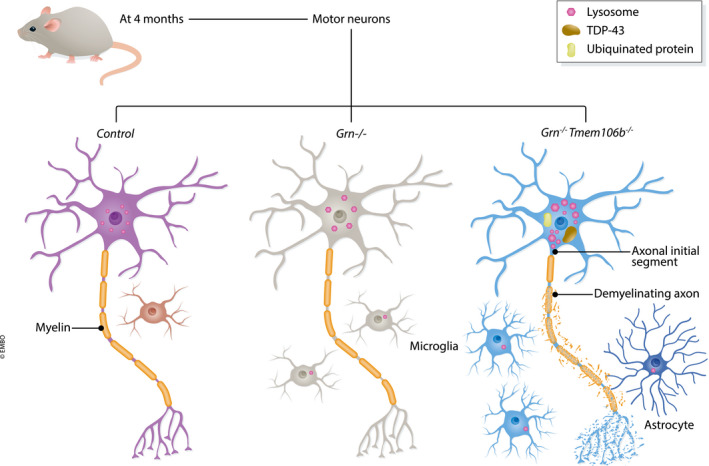
Three publications show that removing the FTD risk factor Tmem106b from FTD model granulin knockout mice severely worsens phenotype. By 4 months, compared with the single knockouts, motor neurons from double knockout mice show exacerbated lysosomal abnormalities, with enlarged lysosomes clustering at the axon initial segment, ubiquitinated protein deposits, phosphorylated TDP‐43 accumulation and enhanced gliosis. This pathology results in severe motor deficits in the double knockouts.
Why these seemingly disparate results between the original double knockout data from Klein et al and these new papers? On closer inspection, these conclusions are not entirely different. Klein et al report that accumulation of autofluorescent material—a sign of impaired lysosome function—is not rescued, and neither is gliosis. However, they do show correction of the increased levels and activity of lysosomal proteins in Grn −/− single knockouts, phenotypes that were exacerbated in the three new reports. An early rescue, at 4 months of age, of two behavioural phenotypes (hyperactivity and disinhibition) is also reported. Interestingly, the Zhou et al (2020) paper used the same Tmem106b knockout model as Klein et al. A key piece of information in the Zhou paper may help explain the different phenotypic reports from the different laboratories. Zhou et al (2020) show here that this model is an imperfect knockout of Tmem106b, with 5–10% protein levels remaining in the mice. It is clear from these new papers that neurons are delicately tuned to the correct levels of Tmem106b. The residual levels of Tmem106b in the cortex may have been enough to correct the behavioural phenotype at 4 months of age, prior to the age‐related motor deficits first observed at 6 months in the Zhou paper; although undoubtedly the long‐term consequence of loss of the majority of Tmem106b is detrimental.
Does this mean that Tmem106b manipulation no longer harbours viable therapeutic potential for disease? Not necessarily. There are several caveats that preclude drawing this conclusion. Although the homozygous Grn knockout mouse model is commonly used as a model of FTD, it is not a completely accurate genetic model. In humans, haploinsufficiency due to heterozygous loss of function of GRN drives disease, while rare homozygous mutations instead result in lysosomal storage disorder. Therefore, effects may be exaggerated in Grn homozygous knockout mice. A more genetically accurate model of partial reduction of Tmem106b in a heterozygous Grn knockout mouse has already been reported and appeared neutral, as neither rescue nor exacerbation of social deficits or lysosomal abnormalities was observed (Arrant et al, 2018). This highlights that partial loss of Tmem106b is likely to have distinct effects to its complete ablation. Partial reduction of Tmem106b using antisense oligonucleotides in primary cortical neuronal cultures rescued in vitro lysosomal trafficking defects caused by a rare FTD‐causing mutation in CHMP2B (Clayton et al, 2018), indicating partial reduction can be beneficial in some contexts.
A striking result from all three reports is that complete loss of Tmem106b leads to the accumulation of phosphorylated TDP‐43, as it has been a long‐standing puzzle as to why even homozygous Grn knockout mice do not reliably develop TDP‐43 inclusions, even though they are present in all Grn mutation patients. This raises the possibility that loss of function of TMEM106B could contribute to FTD risk, rather than the gain of function previously suggested. However, opposing this argument, overexpression of TMEM106B in Grn−/− mice also enhances lysosomal abnormalities in aged mice (Zhou et al, 2017), indicating that either too little or too much TMEM106B is detrimental. The effects of the risk allele may then be cell‐type specific or modify specific lysosomal functions of TMEM106B in addition to changing its levels. Intriguingly, another just‐published paper reports the accumulation of cytoplasmic TDP‐43 in Grn homozyogus knockout mouse thalamic neurons from 12 months of age, that can be reduced by blocking microglial‐derived complement (Zhang et al, 2020). The role of TMEM106B was not investigated. Given the enhanced microgliosis in Grn−/−;Tmem106b−/− double knockouts, the exacerbated TDP‐43 pathology that is also observed could be driven at least in part by this mechanism.
The most striking neuronal defects reported in the double knockouts were observed in motor neurons—thus more closely resembling a motor neuron disease phenotype, even though GRN mutations cause FTD and not ALS. Motor neurons contain extraordinarily long axons, which may render them more susceptible to subtle perturbation in Tmem106b levels, given the known involvement of Tmem106b in endolysosomal trafficking (Schwenk et al, 2014). Indeed, Tmem106b‐deficient mice were recently reported to accumulate LAMP‐1‐positive vacuoles at the axon initial segment (Lüningschrör et al, 2020), while rare human mutations in TMEM106B cause a hypomyelination disorder (Simons et al, 2017).
These new papers prove conclusively that TMEM106B levels need tight spatiotemporal regulation. The robust defects in motor neurons indicate that specific subsets of neurons in the brain may be differentially sensitive to loss of GRN and TMEM106B. While cortical neurons are potentially susceptible to excess TMEM106B (leading to FTD), it appears motor neurons are more susceptible to too little of the protein (leading to motor deficits). Further studies addressing these issues are needed to tease apart the cost–benefit analysis of regional modulation of TMEM106B levels in FTD.
EMBO Reports (2020) 21: e51668
See also: X Zhou et al, T Feng et al and G Werner et al (October 2020)
References
- Arrant AE, Nicholson AM, Zhou X, Rademakers R, Roberson ED (2018) Partial Tmem106b reduction does not correct abnormalities due to progranulin haploinsufficiency. Mol Neurodegener 13: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, Milioto C, Muralidharan B, Norona FE, Edgar JR, Soriano A, Jafar‐Nejad P, Rigo F, Collinge J, Isaacs AM (2018)Frontotemporal dementia causative CHMP2B impairs neuronal endolysosomal traffic‐rescue by TMEM106B knockdown. Brain 141: 3428–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Mai S, Marie Roscoe J, Sheng RR, Ullah M, Zhang J, Iscol Katz I, Yu H, Xiong W, Hu F (2020)Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep 21: e50219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL, Chesi A, Manduchi E, Wells AD, Grant SFA et al (2017)A Dementia‐Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am J Hum Genet 101: 643–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, Strittmatter SM (2017) Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia‐Related Phenotypes in Progranulin‐Deficient Mice. Neuron 95: 281–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüningschrör P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D, Wanner R, Lüllmann‐Rauch R, Wefers B, Wurst W et al (2020)The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep 30: 3506–3519 [DOI] [PubMed] [Google Scholar]
- Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB III, Castanedes‐Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R et al (2013)TMEM106B p. T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 126: 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, Lichtenthaler SF, Hoogenraad CC, Capell A, Haass C et al (2014)The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J 33: 450–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons C, Dyment D, Bent SJ, Crawford J, D'Hooghe M, Kohlschütter A, Venkateswaran S, Helman G, Poll‐The BT, Makowski CC et al (2017)A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain, 140: 3105–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner G, Damme M, Schludi M, Gnörich J, Wind K, Fellerer K, Wefers B, Wurst W, Edbauer D, Brendel M et al (2020)Loss of TMEM106B potentiates lysosomal and FTLD‐like pathology in progranulin deficient mice. EMBO Rep: 21: e50241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Velmeshev D, Hashimoto K, Huang YH, Hofmann JW, Shi X, Chen J, Leidal AM, Dishart JG, Cahill MK et al (2020)Neurotoxic microglia promote TDP‐43 proteinopathy in progranulin deficiency. Nature. 10.1038/s41586-020-2709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Brady OA, Murphy KA, Hu F (2017)Elevated TMEM106B levels exaggerate lipofuscin accumulation and lysosomal dysfunction in aged mice with progranulin deficiency. Acta Neuropathol Commun 5: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC, Parsons TM, Castanedes‐Casey M, Phillips V, Librero AL et al (2020)Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin‐deficient mice. EMBO Rep: 21: e50197 [DOI] [PMC free article] [PubMed] [Google Scholar]