

Summary
Repair of covalent DNA-protein crosslinks (DPCs) by DNA-dependent proteases has emerged as an essential genome maintenance mechanism required for cellular viability and tumor suppression. However, how proteolysis is restricted to the crosslinked protein while leaving surrounding chromatin proteins unharmed has remained unknown. Using defined DPC model substrates, we show that the DPC protease SPRTN displays strict DNA structure-specific activity. Strikingly, SPRTN cleaves DPCs at or in direct proximity to disruptions within double-stranded DNA. In contrast, proteins crosslinked to intact double- or single-stranded DNA are not cleaved by SPRTN. NMR spectroscopy data suggest that specificity is not merely affinity-driven but achieved through a flexible bipartite strategy based on two DNA binding interfaces recognizing distinct structural features. This couples DNA context to activation of the enzyme, tightly confining SPRTN’s action to biologically relevant scenarios.
Keywords: DNA-protein crosslink, DNA repair, DNA structure, formaldehyde, protease, SPRTN, Spartan, topoisomerases, Wss1
Graphical Abstract
Highlights
-
•
DNA-protein crosslink cleavage by SPRTN is coupled to recognition of DNA context
-
•
DNA-protein crosslinks are only cleaved in proximity to activating DNA structures
-
•
Two distinct interfaces recognize DNA with single- and double-stranded features
-
•
Activation of SPRTN depends on simultaneous engagement of both DNA binding interfaces
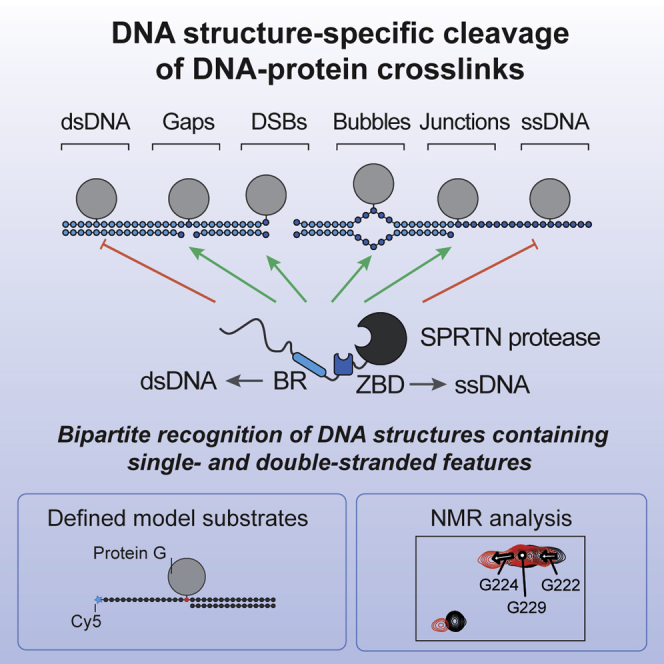
Reinking et al. show that the protease SPRTN degrades DNA-protein crosslinks in a DNA structure-specific manner, which restricts cleavage to biologically relevant scenarios. NMR analysis reveals that specificity is achieved by a bipartite strategy relying on two DNA binding interfaces that recognize single- and double-stranded features within the substrate.
Introduction
Genome stability is constantly challenged by various types of DNA damage (Lindahl, 1993). Efficient detection and repair of DNA lesions is crucially important to prevent premature aging and cancer development (Jackson and Bartek, 2009). A particular type of lesion, covalent DNA-protein crosslinks (DPCs), has recently become the focus of intense research efforts. DPCs are induced by various reactive metabolites and chemotherapeutic agents and can also be caused by entrapment of enzymatic reaction intermediates (Barker et al., 2005; Stingele et al., 2017). DPCs are highly toxic because they block chromatin transactions such as transcription and replication (Duxin et al., 2014; Fu et al., 2011; Nakano et al., 2012, 2013). DPCs pose an exceptional challenge for repair because they are very diverse in nature with respect to the identity of the crosslinked protein and depending on the DNA context in which they occur (Nakano et al., 2017). DPCs form within double-stranded DNA (dsDNA) (e.g., those induced by formaldehyde or acetaldehyde), at DNA nicks (trapped topoisomerase 1 [TOP1]), DNA gaps (polymerase β adducts), or at dsDNA ends/breaks (SPO11 adducts, trapped topoisomerase 2 [TOP2]) (Chen et al., 2013; Lu et al., 2010; Neale et al., 2005; Quiñones et al., 2015).
DPCs can be repaired through degradation of the protein component by proteases of the Wss1/SPRTN family, which is essential for maintaining genome stability, cellular viability, tumor suppression, and prevention of premature aging (Lessel et al., 2014; Lopez-Mosqueda et al., 2016; Maskey et al., 2014; Mórocz et al., 2017; Reinking et al., 2020; Stingele et al., 2014, 2016; Vaz et al., 2016). These proteases tackle the complexity of DPCs with an open and, thus, unselective active site, which allows them to degrade virtually any protein irrespective of amino acid sequence (Stingele et al., 2016; Vaz et al., 2016). This, however, creates the need to prohibit unwanted cleavage of non-crosslinked cellular proteins. Accordingly, the human DPC protease SPRTN appears to be highly regulated. Mono-ubiquitinated SPRTN is excluded from chromatin, with the presence of DPCs triggering deubiquitylation and concurrent relocalization to chromatin (Stingele et al., 2016). Moreover, SPRTN’s protease activity depends entirely on the presence of DNA. SPRTN is inactive in vitro when incubated on its own but becomes strongly activated upon DNA binding (Stingele et al., 2016; Vaz et al., 2016). DNA is thought to act as a scaffold bringing substrate and enzyme together, triggering non-specific degradation of DNA-bound proteins (non-DNA-binding proteins are not targeted by SPRTN even in the presence of DNA). If true in vivo, then recruiting SPRTN to DNA would carry enormous risks because all nearby chromatin proteins would potentially be subjected to uncontrolled degradation. However, insights obtained using a model system of replication-coupled DPC repair (using frog egg extracts) indicate that proteolytic action is exquisitely controlled; SPRTN cleaves plasmid-borne DPCs only when the replisome has passed over the lesion and when the daughter strand has been extended on the DPC, whereas replisome and chromatin factors remain untouched (Duxin et al., 2014; Larsen et al., 2019; Sparks et al., 2019). How this specificity is achieved and whether it requires sophisticated regulation is unknown.
Here we identify an entirely unexpected DNA structure specificity of SPRTN by analyzing its activity for the first time using defined model DNA-protein conjugates. Moreover, NMR experiments suggest that SPRTN achieves such high precision using a unique bipartite strategy: two distinct DNA-binding interfaces reliably read out structural features and DNA context and couple it to activation of the enzyme. This regulatory mechanism results in tight spatial restriction of SPRTN’s activity, which allows degradation of crosslinked proteins in a controlled and safe manner.
Results
SPRTN Cleaves DPCs at dsDNA Ends
To understand how SPRTN’s activity is influenced by different types of DNA, we initially focused on an intriguing conundrum. SPRTN has been reported to be efficiently activated by DNA oligonucleotides, whether they were single- or double-stranded (Lopez-Mosqueda et al., 2016; Vaz et al., 2016). In contrast, others observed a striking difference using long circular DNA for activation. Single-stranded DNA (ssDNA) circles were found to activate SPRTN much more strongly than dsDNA circles (Stingele et al., 2016). Remarkably, these seemingly contradictory results hold true when conducted in the same experiment. ssDNA circles (ФX174 phage DNA, 5.4 kb) induce SPRTN activity much more efficiently than dsDNA circles, as judged by autocleavage and cleavage of histone H1 (Figures 1A and S1A–S1C). However, 60-mer single- and double-stranded oligonucleotides activate SPRTN very similarly, although generally less than ssDNA circles. The specific inability of long circular dsDNA to activate SPRTN becomes even more obvious under more stringent high-salt assay conditions (150 mM KCl). Denaturation of dsDNA circles (ФX174 phage DNA or pMAX-GFP plasmids) to ssDNA by heating and snap-cooling on ice restores their activation potential (Figures 1B and S1D–S1G). We conclude that it is indeed the double-strandedness that prohibits SPRTN activation by dsDNA circles. To test whether the reason for the differential activation of SPRTN by dsDNA circles and double-stranded oligonucleotides is simply the difference in length, we next tested PCR-generated dsDNA fragments of decreasing size for activation. Strikingly, the shorter the dsDNA fragment, the more strongly it activates SPRTN under high-salt conditions (Figures 1C, S1H, and S1I). Of note, histone H1 cleavage cannot be observed, which indicates that it requires stronger activation of SPRTN or reflects the binding preference of H1 itself. Importantly, when using shorter DNA fragments, the total amount of DNA was kept constant. Thus, the number of dsDNA ends increases when shorter fragments are used, which raises the possibility that SPRTN is activated by dsDNA ends (Figure 1D).
Figure 1.

SPRTN Cleaves DPCs at dsDNA Ends
(A) Recombinant SPRTN (500 nM) and histone H1 (500 nM) were incubated alone or in the presence of DNA (5.4 kb circles [ФX174] or 60-mer oligonucleotides, each single-stranded or double-stranded) for 2 h at 25°C. DNA concentrations were 1 μM for 60-mer oligonucleotides or the corresponding amount of circular DNA (11.4 nM). Reactions were analyzed by SDS-PAGE followed by western blotting and Coomassie staining. Cleaved fragments of SPRTN and H1 are indicated by asterisks. Quantification of western blots results of SPRTN and histone H1 cleavage: values represent the mean ± SD of three independent experiments. The p values were calculated using an unpaired t test.
(B) Reactions and quantification were conducted as in (A) but also included dsDNA (ФX174) denatured by heating and snap-cooling on ice.
(C) PCR-generated dsDNA fragments were tested for activation of SPRTN as in (A).
(D) Schematic representation of SPRTN’s activation by dsDNA and its correlation with DNA length and the number of dsDNA ends.
(E) Schematic of the model DPCs used in (F) and (H). Protein G was conjugated site-specifically to fluorescently labeled 30-mer oligonucleotides prior to annealing complementary reverse oligonucleotides.
(F) Free DNA or the indicated model DPCs (25 nM) were incubated alone or in the presence of recombinant SPRTN (5 nM, WT or the catalytically inactive E112Q [EQ] variant) for 2 h at 25°C prior to separation by native PAGE.
(G) Quantification of the DPC cleavage assay shown in (F). Values represent the mean ± SD of three independent experiments.
(H) EMSAs were used to assess binding of catalytically inactive SPRTN EQ (12.5 and 50 nM) to free dsDNA or the indicated DPCs (25 nM).
See also Figure S1.
To test whether SPRTN is indeed active at dsDNA ends, we generated defined model DPCs: protein G conjugated in a site-specific manner to Cy5-labeled 30-mer oligonucleotides followed by purification via ion-exchange chromatography (Figure 1E). Drastically reduced enzyme concentrations in the low nanomolar range (100-fold less compared with previous assays) can be used to assess cleavage of these substrates. Wild-type (WT) SPRTN, but not the catalytically inactive E112Q (EQ) variant, efficiently cleaves the protein adduct when crosslinked to the terminal base at the 3′ or 5′ end of a dsDNA oligonucleotide (Figures 1F and 1G). In stark contrast, the adduct is not processed at an internal position despite SPRTN binding to it very similarly, as determined by electrophoretic mobility shift assays (EMSAs) (Figure 1H). This apparent specificity of SPRTN is striking and potentially explains how dsDNA-bound chromatin proteins are protected from cleavage.
SPRTN Cleaves DPCs at Hairpins and ssDNA to dsDNA Junctions
It is unlikely that activation takes place exclusively at dsDNA ends because ssDNA circles activate SPRTN very efficiently. To gain insights into activation of SPRTN by ssDNA, we assessed cleavage of the same model DPCs in their single-stranded versions (Figure 2A). Remarkably, the cleavage preference shifts dramatically. The internal adduct is cleaved most efficiently, the 5′ adduct is still processed but to a lower degree, and the 3′ adduct is barely cleaved at all (Figures 2B and 2C). Again, SPRTN binds similarly to all substrates (Figure 2D). Next we wanted to find out whether cleavage preference is related to secondary structures forming within the ssDNA (the long ssDNA circles that efficiently activate SPRTN contain various hairpin structures). The sequence used for the model DPCs is predicted to form a stable hairpin at assay temperature (25°C), and the cleavage efficiency of the protein G adduct appeared to correlate with the proximity to the hairpin. Thus, we tested the isolated hairpin for activation of SPRTN and observed efficient induction of autocleavage and histone cleavage (Figures 2E–2G and S2A). A mutation predicted to result in collapse of the hairpin strongly reduces activation, whereas the double-stranded versions of both sequences activate indistinguishably. Notably, abolishment of hairpin formation does not only reduce activation but also binding by SPRTN (Figure S2B). Furthermore, strictly ssDNAs (poly(dA) or poly(dT)) do not induce SPRTN autocleavage but do so when annealed to each other (Figures 2H, 2I, and S2C). Finally, we tested cleavage of a model DPC substrate containing strictly ssDNA (C3A11XA12C3) (Figure 2J) and observed neither cleavage nor efficient binding by SPRTN (Figures 2K–2M). Taken together, these data indicate that formation of secondary structures is required for binding and activation of SPRTN by ssDNA. Next we annealed complementary 15-mer or 30-mer oligonucleotides to the single-stranded model DPC, which restored strong binding by SPRTN (Figures 2J and 2M). However, efficient cleavage of the DPC occurs only at the ss/dsDNA junction (Figures 2K and 2L). We conclude that a short section of paired DNA bases is needed for SPRTN to bind efficiently. Cleavage, however, appears to also require the presence of DPCs at specific DNA structures, either at dsDNA ends, in proximity to the stem loop of a hairpin, or at a ss/dsDNA junction.
Figure 2.

SPRTN Cleaves DPCs at Hairpins and ss/dsDNA Junctions
(A) Schematic of the model DPCs used in (B) and (D). Protein G was conjugated site-specifically to fluorescently labeled 30-mer oligonucleotides. Secondary structures and respective melting temperatures (TM) were predicted using the mfold webserver.
(B) Free DNA or the indicated model DPCs (25 nM) were incubated alone or in the presence of recombinant SPRTN (5 nM, WT or the catalytically inactive EQ variant) for 2 h at 25°C prior to separation by native PAGE.
(C) Quantification of the DPC cleavage assay shown in (B). Values represent the mean ± SD of three independent experiments.
(D) EMSA assays were used to assess binding of catalytically inactive SPRTN EQ (12.5 and 50 nM) to free ssDNA and the indicated DPCs (25 nM).
(E) Schematic of the 15-mer DNA hairpin and its mutant variant used for activation of SPRTN in (F).
(F and G) Recombinant SPRTN (500 nM) and histone H1 (500 nM) were incubated alone or in the presence of the indicated DNAs (4 μM) for 2 h at 25°C and 80 mM KCl. Reactions were analyzed by SDS-PAGE, followed by western blotting and Coomassie staining. Cleaved fragments of SPRTN and H1 are indicated by asterisks. Quantification of western blots results of SPRTN and histone H1 cleavage: values represent the mean ± SD of three independent experiments. The p values were calculated using an unpaired t test.
(H and I) 15-mer poly(dA) or poly(dT) oligonucleotides (4 μM) were tested for activation of SPRTN. Reactions and quantification were as in (F) and (G).
(J) Schematic of the model DPCs used in (K) and (M). Protein G was conjugated site-specifically to fluorescently labeled 30-mer oligonucleotides prior to annealing complementary reverse oligonucleotides.
(K) The indicated model DPCs (25 nM) were incubated alone or in the presence of recombinant SPRTN (12.5 nM, WT or the catalytically inactive EQ variant) for 2 h at 25°C prior to separation by native PAGE.
(L) Quantification of the DPC cleavage assay shown in (K). Values represent the mean ± SD of three independent experiments.
(M) EMSAs were used to assess binding of catalytically inactive SPRTN EQ (12.5 and 50 nM) to the indicated model DPCs (25 nM). An asterisk indicates non-resolvable high-molecular-weight aggregates.
See also Figure S2.
SPRTN’s Structure-Specific Activity Requires Two Distinct DNA-Binding Interfaces
Having established that SPRTN’s protease activity displays strict preferences for certain DNA contexts, we wanted to find out how specificity is achieved. SPRTN is a 55-kDa protein, with the N-terminal part of the enzyme bearing the catalytic metalloprotease domain (Figure 3A). The largely unstructured C-terminal tail contains several protein-protein interaction domains (a ubiquitin-binding zinc finger, a proliferating cell nuclear antigen (PCNA)-interacting protein motif, and a SHP box required for binding to the chaperone-like protein p97) (Centore et al., 2012; Davis et al., 2012; Mosbech et al., 2012; Stingele et al., 2015). Between the tail and protease domain, a basic DNA-binding region (BR) of low complexity was identified that bears several positively charged amino acids (Mórocz et al., 2017; Stingele et al., 2016; Toth et al., 2017). A recent crystal structure of an N-terminal SPRTN fragment revealed an unexpected zinc-binding domain (ZBD) immediately after the protease domain and preceding the BR (PDB: 6MDX; Li et al., 2019). The ZBD was speculated to constitute a ssDNA-binding domain, which is interesting given that we cannot detect efficient binding of SPRTN to substrates containing only ssDNA. Consistent with previous data, we observed reduced autocleavage in SPRTN variants with specific amino acid replacements in the ZBD domain (the ZBD∗2 [R185A] variant displays a more severe effect than ZBD∗1 [Y179A_W197A]) (Figure 3B; Li et al., 2019). Similarly, a SPRTN variant with amino acid replacements in the BR domain (BR∗; K220A_K221E_G222A_K223A) shows a comparable reduction in activity. Consistent with their crucial role in vitro, the more severe ZBD∗2 variant and the BR∗ variant display decreased autocleavage when expressed in cells, although recruitment to chromatin after DPC induction by formaldehyde is not affected (Figures 3C, S3A, and S3B). To test whether ZBD and BR contribute to SPRTN’s essential function in cells, we expressed cDNAs of the respective SPRTN variants with a retroviral vector in human haploid HAP1 cells (Figure 3D). Next we transfected these cells with recombinant nuclear localization signal (NLS)-Cas9/guide RNA (gRNA) complexes targeting the 5′ and 3′ UTR of the endogenous allele (Figure S3C). The persistence of the resulting SPRTN KO allele was then monitored over time using qPCR. HAP1 cells complemented with WT SPRTN or ZBD∗1 can tolerate loss of the endogenous SPRTN allele whereas cells transduced with an empty vector (EV) or catalytically inactive SPRTN-EQ cannot (Figure 3E). SPRTN-BR∗ and ZBD∗2 display only partial complementation, highlighting the importance of both modules.
Figure 3.

SPRTN’s Structure-Specific Activity Requires Two Distinct DNA-Binding Domains
(A) Schematic of SPRTN’s domain structure, highlighting the zinc-binding domain (ZBD), the basic DNA-binding region (BR), the SHP box (p97 binding), the PCNA-interacting motif (PIP), and the ubiquitin-binding zinc finger (UBZ). Asterisks indicate the zinc-coordinating residues within the ZBD, and plus signs indicate positively charged amino acids within the BR. The function of the ZBD and BR were tested in this study using the indicated amino acid replacements (ZBD∗1, Y179A/W197A; ZBD∗2, R185A; BR∗, K220A/K221E/G222A/K223A).
(B) Recombinant SPRTN (500 nM, WT or the indicated variants) and histone H1 (500 nM) were incubated alone or in the presence of ssDNA circles (ФX174 virion) for 2 h at 25°C in the presence of 80 or 150 mM KCl. Reactions were analyzed by SDS-PAGE followed by Coomassie staining.
(C) SPRTN autocleavage assessed in cells. The indicated YFP-SPRTN-Strep variants were transiently transfected in HeLa Flp-In TRex cells. SPRTN autocleavage fragments were enriched on GFP trap resins, followed by western blotting against the N-terminal YFP tag. Western blotting against GAPDH of cell lysates served as loading control.
(D) HAP1 cell lines complemented by retroviral transduction with cDNAs encoding the indicated C-terminally Strep-tagged SPRTN variants. SPRTN-Strep was enriched on Strep-tactin beads prior to western blotting because of low expression levels. Western blotting against GAPDH of cell lysates served as loading control.
(E) The indicated cell lines were transfected with NLS-Cas9/gRNA complexes targeting the UTRs of the endogenous SPRTN allele. The ratio between the resulting knockout (KO) allele compared with the WT allele was monitored over time using qPCR. A schematic of the genotyping strategy is depicted in Figure S3C. Values represent the mean ± SD of three technical replicates normalized to day 2.
(F–H) The indicated fluorescently labeled model DPCs (25 nM) were incubated alone or in the presence of recombinant SPRTN (WT or the indicated variants) for 2 h at 25°C prior to separation by native PAGE. SPRTN concentrations were 5 nM in (F) and (H) and 12.5 nM in (G). Quantification: values represent the mean ± SD of three independent experiments.
(I–K) EMSAs were used to assess binding of catalytically inactive SPRTN EQ (alone or in combination with the indicated amino acid replacements in the ZBD/BR) to the indicated DPCs (25 nM). SPRTN concentrations were 3.125, 6.25, 12.5, 25, and 50 nM. Asterisks indicate non-resolvable high-molecular-weight aggregates.
See also Figure S3.
To understand how ZBD and BR contribute to SPRTN’s activity, we tested the respective SPRTN variants for DPC cleavage and binding. Cleavage of a protein adduct at a dsDNA end, a ss/dsDNA junction, or a hairpin structure is severely reduced in the BR∗ and ZBD∗2 variants (Figures 3F–3H). The less stringent ZBD∗1 mutation mostly affects cleavage of the hairpin DPC. Remarkably, despite being crucial for proteolytic activity, the SPRTN-ZBD∗ and BR∗ variants do not show observable defects in substrate binding (Figures 3I–3K and S3D). A severe effect on binding is only observed upon introduction of simultaneous alterations in both DNA binding regions (ZBD∗2/BR∗). Taken together, these results demonstrate that both DNA binding regions are required for activity and also suggest that recognition of substrates by SPRTN depends on two distinct features recognized by the ZBD and BR, respectively.
NMR Analysis Reveals Bipartite Recognition of DNA Structures by SPRTN
To probe the structural contributions of ZBD and BR for DNA binding, we analyzed two constructs comprising the entire ZBD-BR module or just the ZBD using NMR. NMR backbone chemical shift assignments enabled analysis of the DNA interactions (Figures 4 and S4). First, when comparing ZBD-BR and ZBD in the absence of DNA, we observed significant chemical shift differences in the β sheet of the ZBD (Figure 4A, top; 4B; and S4A). This suggests transient contacts between the BR and the β sheet of the ZBD. This is further supported by the NMR relaxation experiments, which show that the BR is less flexible on a sub-nanosecond timescale, especially in comparison with the C-terminal end (Figure 4A, bottom). Together, these data suggest a dynamic interaction of the intrinsically disordered BR with the ZBD. Next we monitored chemical shift perturbations (CSPs) for ZBD-BR and ZBD in 1H,15N correlation experiments upon adding 15-mer ssDNA (poly(dA)) or dsDNA (the same sequence as used in Figures 2F and S2B for binding and activation assays). Binding to ssDNA and dsDNA by ZBD-BR and ZBD is readily observed, as evidenced by significant chemical shift changes and line broadening (intensity changes; Figures 4C, S4B, and S4C). Notably, however, large CSPs for the BR region are only observed upon binding dsDNA but not ssDNA, whereas CSPs of the ZBD are observed with ssDNA and dsDNA (Figures 4C, 4D, S4B, and S4C). This is in line with electrostatic interactions of positively charged side chains within the BR with the negatively charged phosphate backbone of the double-stranded region of the dsDNA ligand. Consistent with this interpretation, the interaction between BR and dsDNA is strongly reduced when titration is performed at higher salt concentrations (500 mM) (Figures 4E–4G and S4B). In contrast, the interaction between ZBD and DNA is much less affected, which is in line with the previous observation that the ZBD binds to DNA bases through stacking of its aromatic residues (Li et al., 2019).
Figure 4.

NMR Analysis Reveals Bipartite Recognition of DNA Structures by SPRTN
(A) Comparison of NMR data for two SPRTN constructs comprising the ZBD only or ZBD and the BR (ZBD-BR). Top: chemical shift differences of the backbone amide resonances between ZBD and ZBD-BR. Bottom: backbone flexibility of ZBD-BR from {1H}-15N-heteronuclear NOE data. Errors for heteronuclear NOE values were estimated from error propagation of peak height uncertainties based on average noise levels (six randomly chosen positions in each NMR spectra). The dotted area indicates the BR region.
(B) Mapping of chemical shift differences of ZBD in the presence of BR from (A) onto the ZBD structure (PDB: 6MDW). Red color highlights residues with CSPs of more than 0.025 ppm in (A).
(C) Chemical shift perturbations (CSPs) and intensity differences (line broadening) of backbone amides in ZBD-BR upon addition of an equimolar ratio of ssDNA (gray) and dsDNA (red). Errors for intensity ratios upon DNA-binding were estimated from error propagation of peak height uncertainties based on average noise levels (six randomly chosen positions in each NMR spectra). The dotted area indicates the BR region. No boxes are shown for prolines, unassigned, or ambiguous (overlapped) residues. Spectral overlays are show in Figure S4B.
(D) Spectral changes upon DNA binding are mapped onto the ZBD structure (PDB: 6MDW). Changes observed for binding of ZBD-BR to ssDNA (top) or dsDNA (bottom) are shown in red for residues with an intensity ratio of less than 0.15 (85% intensity loss) or CSPs of more than 0.05 ppm.
(E) CSP and intensity changes of ZBD-BR upon addition of an equimolar dsDNA at 100 mM (low salt, red) and 500 mM (high salt, green) salt concentrations. Errors as in (C). Spectral overlays are shown in Figure S4B.
(F) Spectral changes upon dsDNA binding at high salt concentration are mapped onto the ZBD structure (PDB: 6MDW), where the 10 residues with the highest intensity or CSP changes are shown in green. Red spheres indicate changes with an intensity ratio of less than 0.15 (85% intensity loss) or CSPs of more than 0.05 ppm (as in D).
(G) NMR signals (black, free; red, bound) in Figure S4A, highlighting BR residues upon addition of an equimolar ssDNA, dsDNA at 100 mM salt concentration, and dsDNA at 500 mM salt concentration. See Figure S4B for the experimental conditions.
(H) Top: 1H-NMR spectrum of the 15-mer dsDNA. Assignments of the imino resonances of T and G in base pairs in the dsDNA ligand are shown in bold in the sequence. Only 13 signals are observed because of fraying of the terminal base pairs (underlined in the sequence). Center and bottom: 1H-NMR imino spectra of the dsDNA in the presence of an equimolar amount of ZBD or ZBD-BR, respectively. The gray box indicates strongly affected signals (line-broadening) upon addition of the ZBD. NMR spectra were recorded with 100-μM sample concentration in 100 mM potassium chloride, 50 mM HEPES (pH 7.5), 2 mM TCEP at 298 K on a 600-MHz spectrometer.
See also Figure S4.
Next we asked which features in the DNA are recognized by SPRTN’s ZBD. To this end, we monitored spectral changes of the imino NMR signals in the base pairs of the 15-mer dsDNA upon binding to ZBD-BR or ZBD (Figure 4H). Intriguingly, binding of the isolated ZBD mainly affects NMR signals of base pairs at one end of the dsDNA (i.e., T13 and T14). In contrast, when the low-complexity and highly charged BR is present, most imino signals are affected and experience line broadening. This further indicates that the BR contributes binding to the double-stranded part of the oligonucleotide. Thus, we hypothesize that the ZBD interacts specifically with unpaired DNA bases available for interaction at the dsDNA end. This idea is in agreement with the fact that the ZBD interacts with the presumably less stable end of the oligonucleotide (GAT versus CCT). Accordingly, we argue that the common feature recognized by the ZBD is the presence of ssDNA at “frayed” dsDNA ends, ss/dsDNA junctions, or at the ends of a DNA hairpin, whereas the BR enhances binding through non-specific interactions with the double-stranded parts of these structures. If correct, then DPC processing by SPRTN should be enabled by introduction of DNA disruptions that allow local unwinding and, thus, result in the presence of unpaired DNA bases in the vicinity of the DPC.
SPRTN Cleaves DPCs in Close Proximity to Disruptions within dsDNA
To test this hypothesis, we generated model DPCs containing specific disruptions expected to result in local opening of duplex DNA in close proximity to the DPC. First, we disrupted the duplex by a nick, a nick combined with a mismatch (1 bp), or a gap (1 bp) opposite the protein adduct (Figure 5A). Strikingly, this enables cleavage of the DPC depending on SPRTN’s ZBD and BR domain (Figures 5A–5C and S5A). Second, we inserted a bubble of increasing size opposite the protein adduct (Figure 5D). Disrupting the 30-mer duplex by a bubble larger than 2 bp enables efficient DPC cleavage by SPRTN (Figures 5D, 5E and S5B). Cleavage of the protein adduct within the bubble again depends on both DNA binding domains (Figure 5F). Having established the requirement for discontinuities within duplex DNA for DPC cleavage by SPRTN, we investigated the spatial interdependency between the activating structure and the position of the protein adduct. To this end, we recessed the DNA strand opposite a 3′ DPC in small steps, moving the putatively activating ss/dsDNA junction farther and farther away from the protein adduct (Figure 5G). Remarkably, an initial increase in cleavage (with a peak around 5 bp between the junction and the adduct) is followed by a sharp decrease when the junction is moved farther away from the adduct (Figures 5G and 5H), whereas binding to the substrates is only mildly affected (Figure 5I). Next we assessed the inverted scenario, in which we brought the activating junction closer to an internal adduct (Figure 5J). In this scenario, cleavage of the protein adduct again depends on close proximity between the junction and the adduct; DPC proteolysis increases sharply at distance smaller than 5 bp (Figures 5J–5L). Again, this effect did not correlate with binding to the substrate (Figure 5L). We conclude that activation of SPRTN happens in a spatially confined manner that restricts substrate cleavage to a very narrow window around specific DNA structures.
Figure 5.

SPRTN Cleaves DPCs in Close Proximity to Disruptions within dsDNA
(A, D, G, and J) Cleavage of model DPCs. Protein G was conjugated site-specifically to fluorescently labeled 30-mer oligonucleotides prior to annealing complementary reverse oligonucleotides to generate the indicated substrates. Model DPCs (25 nM) were incubated alone or in the presence of recombinant SPRTN (WT, 5 nM) for 2 h at 25°C prior to separation by native PAGE.
(B, E, H, and K) Quantifications of DPC cleavage assays shown in (A), (D), (G), and (J). Values represent the mean ± SD of three independent experiments.
(C and F) Both DNA binding domains of SPRTN are required for DPC processing. The indicated fluorescently labeled model DPCs (25 nM) were incubated alone or in the presence of recombinant SPRTN (WT or the indicated variants, 5 nM) for 2 h at 25°C prior to separation by native PAGE. Quantification: values represent the mean ± SD of three independent experiments.
(I and L) SPRTN binds similarly to the model DPCs shown in (G) and (J). EMSA assays were used to assess binding of catalytically inactive SPRTN EQ (25 nM) to the indicated model DPCs (25 nM).
See also Figure S5.
Discussion
Many DNA repair mechanisms (e.g., nucleotide excision repair or the Fanconi anemia pathway) are dispensable for viability unless cells are exposed to high levels of damage (Langevin et al., 2011; Setlow et al., 1969). In contrast, loss of the DPC protease SPRTN is lethal in mammalian cells, indicating constant life-threatening levels of DPCs (Hart et al., 2015; Maskey et al., 2014). Detection and repair of those crosslinks is complicated by several challenges. The diversity of these lesions (type of protein adduct/DNA structure) makes it difficult to evolve sensor proteins with high affinity for DPCs. The exception is enzymes specifically involved in repairing only certain protein adducts, such as TDP1 and TDP2, which target TOP1 and TOP2 adducts, respectively (Cortes Ledesma et al., 2009; Pouliot et al., 1999). Moreover, the DPC repair machinery must reliably distinguish covalent adducts from mere DNA-bound proteins (which are present in very large excess). Here we discovered that such specificity is achieved by recognition of DNA context, which is directly coupled to DPC cleavage. Importantly, several types of frequent DPCs form specifically at those structures, which trigger SPRTN activation. First, SPRTN protects cells against the toxicity of drugs (e.g., etoposide) inducing entrapment of TOP2 and appears to also be important for processing covalent SPO11 adducts during meiosis (Dokshin et al., 2020; Lopez-Mosqueda et al., 2016; Vaz et al., 2016). In both scenarios, TOP2 and SPO11 form covalent adducts with the 5′ ends of a dsDNA end. Second, SPRTN repairs covalent TOP1 adducts (induced by compounds such as camptothecin), which occur at DNA nicks (Maskey et al., 2017; Pommier, 2006). Third, polymerase ß can become covalently trapped at DNA gaps during base excision repair (SPRTN’s role in repairing those adducts has not yet been assessed) (Quiñones et al., 2015). In all of these cases, the DPC already encompasses a DNA structure, which allows activation of SPRTN. The situation is different for non-specific DPCs induced by reactive metabolites, such as formaldehyde or acetaldehyde, which are expected to form within intact dsDNA. These lesions require pre-processing to make them amenable to cleavage by SPRTN. Recent data obtained using frog egg extracts indicate that this happens in a replication-dependent manner (Larsen et al., 2019; Sparks et al., 2019). A leading-strand DPC initially stalls progression of the replicative helicase, but the crosslink is eventually bypassed (presumably depending on a second helicase, RTEL1, unwinding the stalled fork) (Figure 6A). This transfers the protein adduct into ssDNA. However, proteolysis of the DPC only occurs when the DNA polymerase extends the newly synthesized strand to the lesion, creating a ss/dsDNA junction at the DPC, a DNA structure allowing activation of SPRTN. Thus, the structure-specific activity of SPRTN enables controlled repair of various DPCs and allows its coupling to processes such as replication.
Figure 6.

Model of SPRTN’s DNA Structure-Specific Protease Activity
(A) Model of replication-coupled transfer of DPCs from dsDNA into a ss/dsDNA junction. The ss/dsDNA junction bears both features required for SPRTN activation: dsDNA, which is recognized by the BR, and unpaired DNA bases, which engage the ZBD.
(B) Schematic overview of the DNA structures activating SPRTN. DNA nicks, gaps, ends, bubbles, and junctions contain both features required for SPRTN activation: dsDNA and unpaired DNA bases.
SPRTN achieves precision through a flexible, bipartite strategy based on two distinct DNA binding interfaces. SPRTN binds efficiently to DPCs within dsDNA (Figure 1H). However, binding alone is not sufficient to induce substrate cleavage. This may explain why chromatin proteins are not subjected to random cleavage by SPRTN in vivo. Induction of activity requires simultaneous engagement of ZBD and BR with DNA, which is only possible when the DNA has single- and double-stranded character. Our NMR analysis shows that the BR mediates sequence-independent electrostatically driven interactions with the negatively charged phosphate backbone of the dsDNA. In contrast, the ZBD binds to ssDNA—either to unpaired DNA bases at ss/dsDNA junctions and bubbles or unpaired bases formed by unwinding/breathing of the terminal base pairs at DNA nicks or dsDNA ends (Figure 6B). The exact molecular nature of the resulting activation remains to be determined, but previous results suggest that it involves conformational changes within SPRTN (Stingele et al., 2016). In agreement, the ZBD appears to constrain access to SPRTN’s active site and would likely need to move aside for efficient substrate processing (Li et al., 2019). Taken together, the principles discovered here shift the current paradigm that DPC proteases are non-specific enzymes. On the contrary, our data demonstrate that SPRTN is a precise tool whose activation is spatially restricted, only allowing DPC cleavage in a very narrow window around the activating DNA structure. Furthermore, our results raise interesting questions regarding recruitment of SPRTN to sites of DPC formation in cells. SPRTN appears to have no specific affinity for its target structures. For example, it is activated similarly by a short DNA hairpin and 15-mer duplex DNA despite binding more strongly to dsDNA (Figures 2F and S2B). Thus, we favor a model in which SPRTN is initially recruited via protein-protein interactions and not through DNA binding. In agreement, it has been proposed that recruitment of SPRTN to chromatin upon formaldehyde exposure requires a ubiquitylation signal (Borgermann et al., 2019). Moreover, SPRTN recruitment to TOP1 DPCs depends on direct interaction between the protease and the adaptor protein TEX264 (Fielden et al., 2020). Hence, initial recruitment appears to be highly context-dependent. When recruited, SPRTN can utilize its non-specific DNA binding ability to scan the DNA in the vicinity for the presence of activating structures, which then trigger local activation of the protease and concurrent cleavage of protein adducts.
Our data raise the intriguing additional possibility that DPCs can be made “degradable” by DNA nicking or by creating a DNA bubble, which would be sufficient to allow activation of SPRTN and cleavage of the protein adduct. In this context, it is tempting to speculate that bubble-generating processes, such as transcription, might enable activation of SPRTN. In line with this idea, genetic evidence obtained in flies and worms suggest that SPRTN does not act exclusively in a replication-dependent manner (Delabaere et al., 2014; Stingele et al., 2016). Finally, recent revelations of additional cellular proteases acting on DPCs raise the exciting possibility that specific proteases target DPCs in specific DNA contexts, analogous to cleavage of diverse DNA structures by various structure-specific endonucleases (Borgermann et al., 2019; Dehé and Gaillard, 2017; Kojima et al., 2020; Serbyn et al., 2020; Svoboda et al., 2019). To understand the increasing complexity of DPC repair, it will be paramount to understand the in vitro specificity of these enzymes, which appear to have distinct but also partially overlapping functions in vivo. Given that these enzymes protect cells against various chemotherapeutic agents, they constitute promising novel drug targets to serve as adjuvants for anti-cancer therapies.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Strep-tag II antibody | Abcam | Cat#ab76949; RRID:AB_1524455 |
| Anti-Histone H3 antibody | Abcam | Cat#ab10799; RRID:AB_470239 |
| Anti-GFP from mouse IgG1κ (used for YFP detection) | Sigma | Cat#11814460001; RRID:AB_390913 |
| GFP antibody rabbit polyclonal (used for YFP detection) | Chromotek | Cat#PABG1; RRID:AB_2749857 |
| GAPDH (14C10) Rabbit mAb | Cell Signaling | Cat#2118; RRID:AB_561053 |
| Anit-H1.10 antibody | Abcam | Cat#ab11079; RRID:AB_2295032 |
| Anti-SPRTN mAB (6F2) | Stingele lab | Clone6F2 |
| Goat Anti-Rat Immunoglobulins/HRP | Sigma | Cat#A9037; RRID:AB_258429 |
| Goat Anti-Mouse Immunoglobulins/HRP | Dako | Cat#P0447; RRID:AB_2617137 |
| Swine Anti-Rabbit Immunoglobulins/HRP | Dako | Cat#P0399; RRID:AB_2617141 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Scientific | Cat#A-11001; RRID:AB_2534069 |
| Bacterial and Virus Strains | ||
| BL21(DE3) | Thermo Scientific | Cat#C600003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 16% Formaldehyde (w/v), Methanol-free | Thermo Scientific | Cat#28906 |
| InstantBlue | Sigma | Cat#ISB1L |
| Doxycycline Hyclate | Sigma | Cat#D9891 |
| ProLong Gold Antifade Reagent | Thermo Fisher | Cat#P10144 |
| DAPI Solution | Thermo Fisher | Cat#62248 |
| 4x NuPAGE LDS sample buffer | Thermo Scientific | Cat#NP0007 |
| Phusion HF enzyme | NEB | Cat#M0530 |
| UltraPure BSA | Thermo Scientific | Cat#AM2616 |
| Histone H1° Human | NEB | Cat#M2501S |
| Protein G | BioVision | Cat#6510 |
| Lipofectamine 2000 | Thermo Scientific | Cat#11668030 |
| IGEPAL | Sigma | Cat#I8896 |
| Biotin | IBA Lifesciences | Cat#2-1016-005 |
| Pefabloc SC | Merck | Cat#11585916001 |
| TCEP | ROTH | Cat#HN95.2 |
| cOmplete EDTA-free protease inhibitor cocktail | Merck | Cat#4693132001 |
| Critical Commercial Assays | ||
| proFIRE Amine Coupling Kit | Dynamic Biosensors | Cat#PF-NH2-1 |
| NucleoSpin® Gel and PCR Clean-up | MACHEREY-NAGEL | Cat#740609 |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Scientific | Cat#P11496 |
| GeneJET Genomic DNA purification kit | Thermo Scientific | Cat#K0722 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat#1725271 |
| Experimental Models: Cell Lines | ||
| Human HeLa Flp-In-T-REx | The Francis Crick Institute Cell Services | N/A |
| Human HAP1 | Thijn Brummelkamp, NKI Amsterdam | N/A |
| Oligonucleotides | ||
| Oligonucleotide sequences used in this study are provided in Table S1 | N/A | N/A |
| Recombinant DNA | ||
| Ds ФX174 phage DNA RFI | NEB | Cat#N3021S |
| Ss ФX174 phage DNA virion | NEB | Cat#N3023S |
| pMAX-GFP | LONZA | Cat#VDC-1040 |
| pNIC-STREP-ZB-SPRTN-WT | This study | N/A |
| pNIC-STREP-ZB-SPRTN-Equation (E112Q) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-ZBD1∗ (Y179A_W197A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-ZBD2∗ (R185A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-BR∗ (K220A_K221E_G222A_K223A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-EQ-ZBD1∗ (E112Q_Y179A_W197A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-EQ-ZBD2∗ (E112Q_R185A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-EQ-BR∗ (E112Q_ K220A_K221E_G222A_K223A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-EQ-ZBD2∗-BR∗ (E112Q_R185A_K220A_K221E_G222A_K223A) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-ZBD-BR (aa151-245) | This study | N/A |
| pNIC-STREP-ZB-SPRTN-ZBD (aa151-215) | This study | N/A |
| pcDNA5-FRT/TO-YFP-SPRTN-WT-Strep | Stingele et al., 2016 | N/A |
| pcDNA5-FRT/TO-YFP-SPRTN-Equation (E112Q)-Strep | Stingele et al., 2016 | N/A |
| pcDNA5-FRT/TO-YFP-SPRTN-ZBD1∗ (Y179A_W197A)-Strep | This study | N/A |
| pcDNA5-FRT/TO-YFP-SPRTN-ZBD2∗ (R185A)-Strep | This study | N/A |
| pcDNA5-FRT/TO-YFP-SPRTN-BR∗ (K220A_K221E_G222A_K223A)-Strep | This study | N/A |
| pOG44 | Thermo Scientific | Cat#V600520 |
| pBABE-puro | Addgene | Cat#1764 |
| pBABE-puro-SPRTN-WT-Strep | This study | N/A |
| pBABE- puro-SPRTN-Equation (E112Q)-Strep | This study | N/A |
| pBABE- puro-SPRTN-ZBD1∗ (Y179A_W197A)-Strep | This study | N/A |
| pBABE- puro-SPRTN-ZBD2∗ (R185A)-Strep | This study | N/A |
| Software and Algorithms | ||
| Prism 7 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | NIH | https://imagej.net/Fiji/Downloads |
| Adobe Photoshop CC2018 | Adobe | https://www.adobe.com/es/products/photoshop.html |
| Other | ||
| HiTrap Heparin HP affinity columns | GE Healthcare | Cat#17040701 |
| PD-10 Desalting columns | GE Healthcare | Cat#17085101 |
| Strep-Tactin®XT Superflow® high capacity cartridges | IBA Lifesciences | Cat#2-4026-001 |
| HiLoad 16/600 Superdex 200 pg column | GE Healthcare | Cat#GE28-9893-35 |
| 10 kDa cutoff Amicon Ultra centrifugal filters | Merck | Cat#UFC801096 |
| GFP-Trap Magnetic Agarose | Chromotek | Cat#gtma-10 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Julian Stingele (stingele@genzentrum.lmu.de).
Materials Availability
All plasmids are available on request.
Data and Code Availability
This study did not generate code or reposited datasets.
Experimental Model and Subject Details
Cell Lines
Human HeLa Flp-In-T-Rex (female) cells were obtained from and authenticated by Francis Crick Institute Cell Services. HeLa Flp-In-T-Rex cells expressing YFP-SPRTN-Twin-Strep-tag variants were generated using the Flp-In-T-REx system (Thermo Fisher) using pOG44 and the respective pcDNA5-FRT/TO plasmids according to manufacturer’s instructions and grown in Dulbecco’s modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). Protein expression was induced by overnight incubation with doxycycline (final concentration 1 μg/mL). Human HAP1 (male) cells (generated and kindly provided by Thijn Brummelkamp, NKI Amsterdam) stably expressing SPRTN variants were generated by transduction as described preciously (Jae et al., 2014). In brief, HEK293T cells were transfected with pBabe-puro (Addgene #1764) empty vector or containing the coding sequence for SPRTN variants together with pAdvantage (Clontech) and the standard retroviral packaging plasmids VSV-g and Gag-pol. 48h after transfection, viral supernatant was collected and HAP1 cells were transduced with the 0.45 μm filtrate in the presence of 8 μg/mL protamine sulfate (Sigma). After 24 h transduced HAP1 cells were selected with 1 μg/mL puromycin (Invivogen).
Method Details
Purification of Recombinant SPRTN
The sequence of full-length human SPRTN in the pNIC-ZB-SPRTN plasmid (Vaz et al., 2016) was replaced with a version codon-optimized for bacterial expression and the His-tag was replaced by a Twin-Strep-tag. For protein expression plasmids were transformed into BL21(DE3) Escherichia coli cells and grown at 37°C in Terrific broth (TB) medium until they reached OD 0.7. Protein expression was induced by addition of 0.5 mM IPTG over night at 18°C. Next, cells were harvested, resuspended in buffer A (50 mM HEPES/KOH pH 7.2, 500 mM KCl, 1 mM MgCl2, 10% Glycerol, 0.1% IGEPAL, 0.04 mg/mL Pefabloc SC, cOmplete EDTA-free protease inhibitor cocktail tablets, 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), pH 7.2) and lysed by sonication. All steps were carried out at 4°C. Cell lysate was incubated with benzonase (45 U/ mL lysate) for 30 min on ice prior to the removal of cell debris by centrifugation at 18000 g for 30 min. Cleared supernatant was applied to Strep-Tactin®XT Superflow® high capacity cartridges, washed with 3 column volumes (CV) of buffer A and 4 CV of buffer B (50 mM HEPES/KOH pH 7.2, 500 mM KCl, 10% Glycerol, 1 mM TCEP, pH 7.2). Proteins were eluted in 6 CV buffer C (50 mM HEPES/KOH pH 7.2, 500 mM KCl, 10% Glycerol, 1 mM TCEP and 50 mM Biotin, pH 7.2). Eluted proteins were further applied to HiTrap Heparin HP affinity columns and washed with 3 CV buffer B before eluting in buffer D (50 mM HEPES/KOH pH 7.2, 1 M KCl, 10% Glycerol, 1 mM TCEP, pH 7.2). Eluted fractions containing recombinant SPRTN protein were desalted against buffer B using PD-10 desalting columns. The affinity tag was cleaved off over night at 4°C by the addition of His-tagged TEV protease with 1:10 mass ratio. Cleaved recombinant SPRTN protein was further purified by size exclusion chromatography using a HiLoad 16/600 Superdex 200 pg column equilibrated in buffer E (50 mM HEPES/KOH pH 7.2, 500 mM KCl, 10% Glycerol, 0.5 mM TCEP, pH 7.2). Eluted proteins were concentrated with 10 kDa cutoff Amicon Ultra centrifugal filters before snap-freezing in liquid nitrogen and storing at −80°C. Proteins used for NMR analysis were expressed in 15N or 13C-/15N-containing media and purified as described above including minor changes. After cleavage of the affinity tag the samples were applied again on Strep-Tactin®XT Superflow® high capacity cartridges. The flow through was collected and further purified by size exclusion chromatography.
DNAs for Activation Assays
Oligonucleotides were used as follows: 60-mer ssDNA = oJS_63, 60-mer ds DNA = oJS_63 + oJS_64, 15-mer hairpin = oJS_106, 15-mer hairpin mutant = oJS_119, 15-mer hairpin dsDNA = oJS_106 + oJS_107, 15-mer hairpin mutant dsDNA = oJS_119 + oJS_120 (sequences are provided in Table S1). Single-stranded DNAs were incubated for 10 min at 95°C before snap-cooling on ice. Double-stranded DNAs were annealed in a PCR machine (5 min incubation at 95°C followed by a decrease in temperature of 2°C/min until 10°C was reached). A standard PCR protocol using Phusion HF enzyme was used to generate PCR fragments with double-stranded ФX174 (RF I) DNA as template and the following primer combinations: oJS_31 + oJS_30, oJS_122 + oJS_30, oJS_35 + oJS_34, oJS_123 + oJS_34. PCR fragments were gel purified (NucleoSpin Gel and PCR Clean-up) before used in activation assays. Denaturation of double-stranded DNA circles (ФX174 (RF I) or pMAX-GFP) was induced by incubation at 95°C for 10 min followed by immediate snap-cooling on ice. Successful denaturation was confirmed using PicoGreen a fluorescent dye specific for double-stranded DNA.
Protein-Oligonucleotide Conjugation
Protein G was crosslinked to oligonucleotides X1, X15, X30 and C3A11XA12C3, which contained a 5′-Cy5 label and a 3′ phosphate group. An Amino-C6-dT was incorporated at the intended crosslinking position and its terminal primary amine group was further processed to yield a reduced thiol (SH-C9-dT) (Ella Biotech GmbH). Conjugation was carried out with 3 nmol oligonucleotide and 50 μL of 5 mg/mL Protein G using the proFIRE Amine Coupling Kit. During the coupling reaction, the terminal thiol group of SH-C9-dT was further functionalized to an NHS-ester, which can react with a primary amine group of proteins. Crosslinked oligonucleotides (conjugates) were purified by ion exchange chromatography using a proFIRE device (Dynamic Biosensors) according to the manufacturer’s instructions. Next, the conjugates were desalted against storage buffer (50 mM HEPES/KOH pH 7.2, 100 mM KCl and 10% Glycerol, pH 7.2) and then snap-frozen in liquid nitrogen and stored at −80°C. Conjugate concentration was determined by measuring Cy5 absorbance with a SpectraMax Paradigm Multi-Mode Detection platform (Molecular Devices). The conjugates were used to generate model DPCs by annealing complementary reverse oligonucleotides (see scheme in Table S2 for details). Annealing was carried out directly prior to cleavage reactions or EMSAs. Conjugates were annealed with complementary reverse oligonucleotides by mixing them at a ratio of 1:1.2 (conjugate:oligonucleotide) in a reaction buffer of 25 mM HEPES/KOH pH 7.2, 50 mM KCl, 5% Glycerol, and 0.2 mg/mL BSA. Annealing was accomplished by incubating the reaction for 20 minutes at 37°C for X1, X15 or X30 conjugates. C3A11XA12C3 conjugates were annealed by incubating the reaction for 2 min at 37°C followed by a decrease in temperature of 1°C/min to 25°C.
SPRTN Autocleavage/Histone H1 Cleavage Assays
Reactions were performed at 25°C in 20 μl containing 500 nM SPRTN, 500 nM histone H1 and DNA (amount was kept constant in all assays and corresponded to 1 μM of a 60-mer oligonucleotide). The reaction buffer comprised 50 mM HEPES/KOH pH 7.2, 2.9% glycerol and either 80 or 150 mM KCl. Reactions were stopped by addition of 4 x LDS sample buffer supplemented with β-mercaptoethanol and boiling at 95°C for 10 min, resolved on 4%–12% Bis-Tris gradient gels using MOPS buffer and stained with InstantBlue or analyzed by western blotting using anti-SPRTN and anti-H1 antibodies and HRP-conjugated anti-rat IgG or anti-mouse IgG, respectively, as secondary antibodies. The intensity of western blots and scanned gels was adjusted globally using Adobe Photoshop. Cleavage reactions were quantified by dividing the amount of cleaved protein by the total amount of protein (cleaved and uncleaved) as determined by analysis of western blot results using ImageJ.
Model DNA-Protein Crosslink Cleavage Assays
Cleavage of model DPCs by SPRTN was performed in a reaction volume of 10 μl containing 5 nM SPRTN (or as indicated in the figure legend) and 25 nM DPC in a final reaction buffer of 17.5 mM HEPES/KOH pH 7.2, 80 mM KCl, 3.5% Glycerol, 5 mM TCEP and 0.1 mg/mL BSA. Reactions were incubated for 2 h at 25°C. 2 μL of 6x Orange G loading dye was added and cleaved DPC fragments were resolved on 20% TBE gels using 1X TBE as running buffer at 4°C. Gels were photographed using a BioRad Chemidoc MP system using filter settings for Cy5 fluorescence. The intensity of scanned gels was adjusted globally using ImageJ, which was also used to quantify cleavage by dividing the amount of cleaved conjugate by the total amount of conjugate (cleaved and uncleaved) and subtraction of background signal (determined from lanes without SPRTN).
DNA Binding Assays
Electrophoretic mobility shift assays (EMSAs) were used to analyze DNA binding of recombinant proteins. Assay composition was exactly as in SPRTN autocleavage assays with varying amounts of catalytically inactive SPRTN-E112Q. Binding reactions were incubated for 20 min on ice prior to separation on 6% native PAGE gels with 0.5x TBE as running buffer at 4°C. Gels were photographed using a BioRad Chemidoc MP system using filter settings for Cy5 fluorescence. The intensity of the scanned images was adjusted globally using ImageJ.
DNA-Protein Crosslink Binding Assays
Electrophoretic mobility shift assays (EMSAs) were used to analyze binding of catalytically inactive SPRTN-E112Q variants to diverse model DPCs. Therefore 25 nM model DPC was incubated with varying concentrations of recombinant SPRTN proteins for 15 minutes on ice. The total reaction volume was kept to 10 μL with a final reaction buffer of 17.5 mM HEPES/KOH pH 7.2, 80 mM KCl, 3.5% Glycerol, 5 mM TCEP and 0.1 mg/mL BSA. SPRTN-bound DPCs were separated on 6% native PAGE gels in 0.5x TBE running buffer at 4°C. Gels were photographed using a BioRad Chemidoc MP system using filter settings for Cy5 fluorescence. The intensity of the scanned images was adjusted globally using ImageJ.
Cellular Autocleavage Assay
pcDNA5-FRT/TO plasmids encoding YFP-SPRTN-Strep variants (3 μg) were transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Protein expression was induced by overnight (16h) incubation with doxycycline (final concentration 1 μg/mL). SPRTN autocleavage was induced by treating with 200 μM Formaldehyde for 2 hours. Cells lysed on ice in 500 μl lysis buffer (50 mM HEPES pH 7.5, 1 M NaCl, 10% glycerol, 1% IGEPAL CA-630, 20 mM iodoacetamide, 0.04 mg/ml PefaBloc SC and cOmplete EDTA- free protease inhibitor cocktail tablets (1 tablet/50 ml)). After addition of benzonase (4U/ml), lysates were incubated for 30 min on ice. Lysates were cleared by centrifugation at 4°C and applied to 15 μL of GFP-Trap Magnetic Agarose (Chromotek) according to manufacturer’s instructions. Finally, samples were resuspended in 40 μl 1X LDS-sample buffer, subjected analysis by SDS-PAGE and western blotting with anti-GFP antibody (PABG1, Chromotek) and peroxidase-conjugated anti-rabbit IgG as secondary antibody. Input samples were analyzed by SDS-PAGE and western blotting with anti-GAPDH antibody (Cell Signaling) and peroxidase-conjugated anti-rabbit IgG as secondary antibody.
Strep-Tactin Pull-down
Cells were lysed on ice in lysis buffer (10 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 2 mM MgCl2, 20 mM iodoacetamide, 0.04 mg/ml PefaBloc SC and cOmplete EDTA-free protease inhibitor cocktail tablets (1 tablet/50 ml)). After addition of benzonase (4U/ml), lysates were incubated for 30 min on ice. Lysates were cleared by centrifugation at 4°C and incubated with Strep-Tactin®XT Superflow® beads for 4h. Beads were washed three times with lysis buffer before resuspension in 30 μl 2x LDS-sample buffer. Finally, samples were subjected to analysis by SDS-PAGE and western blotting with Anti-Strep-tag II antibody (Abcam).
Cas9/gRNA RNP Transfection and qPCR Analysis
Human HAP1 cells expressing cDNA encoding C-terminally Strep-tagged SPRTN variants cells were electroporated with NLS-Cas9/gRNA RNPs using a 4D-Nucleofector (Lonza). In brief, crRNA1 and crRNA2 are incubated with tracRNA (95°C, 5 minutes), respectively, to generate gRNAs. gRNAs were mixed with NLS-Cas9 and incubated for 10 minutes at RT to generate RNPs. 1x106 cells were resuspended in 20 μl Nucleofection Solution (Lonza, SE. Cell line 4D-Nucleofector X Kit). Suspended cells were then mixed with RNPs and electroporated (program EN-138). Cells were plated and samples collected every 48 hours after electroporation for genomic DNA extraction (GeneJET Genomic DNA purification kit, Thermo Scientific). The relative amount of KO and WT allele was monitored for each cell line at each time point by qPCR analysis. Each 10 μl reaction contained 20 ng genomic DNA, 0.4 μl forward and reverse primer (10 μM) and 5 μl SYBR Green Supermix (Bio-Rad). PCR reaction was performed in technical triplicates using primers amplifying either WT or KO allele. For analysis, CqWT was subtracted from CqKO to obtain ΔCq. 2-(ΔCq) was calculated for each time point and normalized to the day 2 value (2-(ΔΔCq)).
Chromatin Fractionation
Chromatin fractionation experiments were performed as described before (Bellelli et al., 2014). In brief, cells in the mid-exponential phase of growth were collected by scraping in ice-cold 1x phosphate-buffered saline (PBS). Cells were then equally split and either directly resuspended in 1x LDS buffer or incubated for 10 min in ice-cold CSK buffer (10 mM PIPES, 100 mM NaCl, 1.5 mM MgCl2, 5 mM EDTA, 300 mM sucrose and 0.5% Triton X-100, protease inhibitors and phosphatase inhibitors). Chromatin-bound proteins were isolated by low speed centrifugation (3,000 rpm, 3 min at 4°C). Finally, samples were subjected to analysis by SDS-PAGE and western blotting with Anti-Strep-tag II (Abcam) and anti-histone H3 (Sigma) antibodies.
Immunofluorescence Staining
For indirect immunofluorescence, cells were pre-extracted in CSK buffer containing 0.5% Triton X-100 (10 min on ice) and/or fixed in 4% paraformaldehyde, permeabilized with PGBT buffer (PBS, 0.2% fish skin gelatin, 0.5% BSA, 0.5% Triton X-100) (45 min at room temperature) and then incubated with anti-GFP antibody (Sigma) overnight at 4°C. Coverslips were then washed 3 times for 5 min in PGBT buffer and incubated with Alexa Fluor 488 goat anti-mouse antibody (Thermo Scientifc) and DAPI counterstaining (0.5 μg/ml) for 1h at room temperature. Coverslips were mounted in Prolong Gold Antifade Mountant (Thermo Fisher). Pictures were acquired with a ZEISS LSM710 confocal microscope.
NMR Spectroscopy
NMR samples (non-labeled or uniformly 15N13C-/15N-labeled for SPRTN-ZBD/ZBD, non-labeled for dsDNA) were prepared at protein concentrations of 100 – 350 μM in three buffer conditions (100 mM KCl, 50 mM HEPES pH 7.5, 2 mM TCEP; 20 mM NaCl, 50 mM sodium phosphate pH 6.5, 2 mM TCEP; 500 mM KCl, 50 mM HEPES pH 7.5, 2 mM TCEP) with 10% D2O added as lock signal. NMR experiments were recorded at 278 K and 298 K on 900-, 800-, 600-MHz Bruker Avance NMR spectrometers, equipped with cryogenic or room-temperature triple resonance gradient probes. NMR spectra were processed by TOPSPIN3.5 (Bruker), then analyzed using NMRFAM-SPARKY (Lee et al., 2015). Backbone resonance assignments of both SPRTN-ZBD and SPRTN-ZBD-BR were obtained from a uniformly 15N,13C-labeled protein employing standard triple resonance experiments HNCA, HNCACO, HNCACB and CBCA(CO)NH (Sattler et al., 1999). 1H-15N Heteronuclear NOE experiments were recorded on a 600-MHz spectrometer at 298 K with an interleaved manner with and without proton saturation. Imino resonances were obtained through 2D 1H-1H NOESY with mixing time of 150 - 200 msec at 278 K and 298 K on 600- and 900-MHz spectrometers. CSP values were calculated based on the following,, where 6.5 was applied to the chemical shift change of 15N as Rscale factor, as suggested previously (Mulder et al., 1999).
Quantification and Statistical Analysis
Statistical analyses (unpaired t test) were performed using Prism 7 (GraphPad Software). Statistical details of each experiment (including the exact value of n, what n represents and precision measures) can be found in the figure legends.
Acknowledgments
We thank K. Ramadan for providing the pNIC-ZB SPRTN plasmid; D. Yaneva for help with protein purification; and S. Panier, G. Hewitt, R. Bellelli, and P. Wolf for discussions and comments on the manuscript. H.-Y.L. is supported by the Peter and Traudl Engelhorn Foundation, S.Z. by the LMU – China Scholarship Council Program, and A.C.A. and P.W. by the International Max-Planck Research School for Molecular Life Sciences. J.S. is supported by the European Research Council (ERC Starting Grant 801750 DNAProteinCrosslinks), by the Alfried Krupp Prize for Young University Teachers awarded by the Alfried-Krupp von Bohlen und Halbach-Stiftung, and the Deutsche Forschungsgemeinschaft (CRC1064). L.T.J. is supported by the European Research Council (ERC Starting Grant SOLID). J.S., L.T.J., and M.S. acknowledge support from the Center for Integrated Protein Science Munich (CIPSM).
Author Contributions
Conceptualization, H.K.R. and J.S.; Investigation, H.K.R., H.-S.K., M.J.G., A.K., H.-Y.L., S.Z., A.C.A., P.W., E.F., L.T.J., and J.S.; Writing – Original Draft, J.S.; Writing – Review & Editing, H.K.R., H.-S.K., M.S., and J.S.; Funding Acquisition, M.S. and J.S.; Supervision, M.S. and J.S.
Declaration of Interests
The authors declare no competing interests.
Published: August 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.08.003.
Supplemental Information
References
- Barker S., Weinfeld M., Murray D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 2005;589:111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Bellelli R., Castellone M.D., Guida T., Limongello R., Dathan N.A., Merolla F., Cirafici A.M., Affuso A., Masai H., Costanzo V. NCOA4 transcriptional coactivator inhibits activation of DNA replication origins. Mol. Cell. 2014;55:123–137. doi: 10.1016/j.molcel.2014.04.031. [DOI] [PubMed] [Google Scholar]
- Borgermann N., Ackermann L., Schwertman P., Hendriks I.A., Thijssen K., Liu J.C., Lans H., Nielsen M.L., Mailand N. SUMOylation promotes protective responses to DNA-protein crosslinks. EMBO J. 2019;38:e101496. doi: 10.15252/embj.2019101496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centore R.C., Yazinski S.A., Tse A., Zou L. Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Mol. Cell. 2012;46:625–635. doi: 10.1016/j.molcel.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.H., Chan N.-L., Hsieh T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013;82:139–170. doi: 10.1146/annurev-biochem-061809-100002. [DOI] [PubMed] [Google Scholar]
- Cortes Ledesma F., El Khamisy S.F., Zuma M.C., Osborn K., Caldecott K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature. 2009;461:674–678. doi: 10.1038/nature08444. [DOI] [PubMed] [Google Scholar]
- Davis E.J., Lachaud C., Appleton P., Macartney T.J., Näthke I., Rouse J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat. Struct. Mol. Biol. 2012;19:1093–1100. doi: 10.1038/nsmb.2394. [DOI] [PubMed] [Google Scholar]
- Dehé P.-M., Gaillard P.H.L. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017;18:315–330. doi: 10.1038/nrm.2016.177. [DOI] [PubMed] [Google Scholar]
- Delabaere L., Orsi G.A., Sapey-Triomphe L., Horard B., Couble P., Loppin B. The Spartan ortholog maternal haploid is required for paternal chromosome integrity in the Drosophila zygote. Curr. Biol. 2014;24:2281–2287. doi: 10.1016/j.cub.2014.08.010. [DOI] [PubMed] [Google Scholar]
- Dokshin G.A., Davis G.M., Sawle A.D., Eldridge M.D., Nicholls P.K., Gourley T.E., Romer K.A., Molesworth L.W., Tatnell H.R., Ozturk A.R. GCNA Interacts with Spartan and Topoisomerase II to Regulate Genome Stability. Dev. Cell. 2020;52:53–68.e6. doi: 10.1016/j.devcel.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin J.P., Dewar J.M., Yardimci H., Walter J.C. Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell. 2014;159:346–357. doi: 10.1016/j.cell.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielden J., Wiseman K., Torrecilla I., Li S., Hume S., Chiang S.-C., Ruggiano A., Narayan Singh A., Freire R., Hassanieh S. TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts. Nat. Commun. 2020;11:1274. doi: 10.1038/s41467-020-15000-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.V., Yardimci H., Long D.T., Ho T.V., Guainazzi A., Bermudez V.P., Hurwitz J., van Oijen A., Schärer O.D., Walter J.C. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T., Chandrashekhar M., Aregger M., Steinhart Z., Brown K.R., MacLeod G., Mis M., Zimmermann M., Fradet-Turcotte A., Sun S. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jae L.T., Raaben M., Herbert A.S., Kuehne A.I., Wirchnianski A.S., Soh T.K., Stubbs S.H., Janssen H., Damme M., Saftig P. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science. 2014;344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y., Machida Y., Palani S., Caulfield T.R., Radisky E.S., Kaufmann S.H., Machida Y.J. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 2020;11:1318. doi: 10.1038/s41467-020-15170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F., Crossan G.P., Rosado I.V., Arends M.J., Patel K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- Larsen N.B., Gao A.O., Sparks J.L., Gallina I., Wu R.A., Mann M., Räschle M., Walter J.C., Duxin J.P. Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell. 2019;73:574–588.e7. doi: 10.1016/j.molcel.2018.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W., Tonelli M., Markley J.L. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015;31:1325–1327. doi: 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D., Vaz B., Halder S., Lockhart P.J., Marinovic-Terzic I., Lopez-Mosqueda J., Philipp M., Sim J.C., Smith K.R., Oehler J. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 2014;46:1239–1244. doi: 10.1038/ng.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Raczynska J.E., Chen Z., Yu H. Structural Insight into DNA-Dependent Activation of Human Metalloprotease Spartan. Cell Rep. 2019;26:3336–3346.e4. doi: 10.1016/j.celrep.2019.02.082. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Lopez-Mosqueda J., Maddi K., Prgomet S., Kalayil S., Marinovic-Terzic I., Terzic J., Dikic I. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife. 2016;5:e21491. doi: 10.7554/eLife.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Ye W., Zhou L., Collins L.B., Chen X., Gold A., Ball L.M., Swenberg J.A. Structural characterization of formaldehyde-induced cross-links between amino acids and deoxynucleosides and their oligomers. J. Am. Chem. Soc. 2010;132:3388–3399. doi: 10.1021/ja908282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey R.S., Kim M.S., Baker D.J., Childs B., Malureanu L.A., Jeganathan K.B., Machida Y., van Deursen J.M., Machida Y.J. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 2014;5:5744. doi: 10.1038/ncomms6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey R.S., Flatten K.S., Sieben C.J., Peterson K.L., Baker D.J., Nam H.-J., Kim M.S., Smyrk T.C., Kojima Y., Machida Y. Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 2017;45:4564–4576. doi: 10.1093/nar/gkx107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mórocz M., Zsigmond E., Tóth R., Enyedi M.Z., Pintér L., Haracska L. DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res. 2017;45:3172–3188. doi: 10.1093/nar/gkw1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbech A., Gibbs-Seymour I., Kagias K., Thorslund T., Beli P., Povlsen L., Nielsen S.V., Smedegaard S., Sedgwick G., Lukas C. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012;19:1084–1092. doi: 10.1038/nsmb.2395. [DOI] [PubMed] [Google Scholar]
- Mulder F.A., Schipper D., Bott R., Boelens R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 1999;292:111–123. doi: 10.1006/jmbi.1999.3034. [DOI] [PubMed] [Google Scholar]
- Nakano T., Ouchi R., Kawazoe J., Pack S.P., Makino K., Ide H. T7 RNA polymerases backed up by covalently trapped proteins catalyze highly error prone transcription. J. Biol. Chem. 2012;287:6562–6572. doi: 10.1074/jbc.M111.318410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T., Miyamoto-Matsubara M., Shoulkamy M.I., Salem A.M., Pack S.P., Ishimi Y., Ide H. Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J. Biol. Chem. 2013;288:4649–4658. doi: 10.1074/jbc.M112.419358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T., Xu X., Salem A.M.H., Shoulkamy M.I., Ide H. Radiation-induced DNA-protein cross-links: Mechanisms and biological significance. Free Radic. Biol. Med. 2017;107:136–145. doi: 10.1016/j.freeradbiomed.2016.11.041. [DOI] [PubMed] [Google Scholar]
- Neale M.J., Pan J., Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Pouliot J.J., Yao K.C., Robertson C.A., Nash H.A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- Quiñones J.L., Thapar U., Yu K., Fang Q., Sobol R.W., Demple B. Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase β in vivo. Proc. Natl. Acad. Sci. USA. 2015;112:8602–8607. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinking H.K., Hofmann K., Stingele J. Function and evolution of the DNA-protein crosslink proteases Wss1 and SPRTN. DNA Repair (Amst.) 2020;88:102822. doi: 10.1016/j.dnarep.2020.102822. [DOI] [PubMed] [Google Scholar]
- Sattler M., Schleucher J., Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magn. Reson. Spectrosc. 1999;34:93–158. [Google Scholar]
- Serbyn N., Noireterre A., Bagdiul I., Plank M., Michel A.H., Loewith R., Kornmann B., Stutz F. The Aspartic Protease Ddi1 Contributes to DNA-Protein Crosslink Repair in Yeast. Mol. Cell. 2020;77:1066–1079.e9. doi: 10.1016/j.molcel.2019.12.007. [DOI] [PubMed] [Google Scholar]
- Setlow R.B., Regan J.D., German J., Carrier W.L. Evidence that xeroderma pigmentosum cells do not perform the first step in the repair of ultraviolet damage to their DNA. Proc. Natl. Acad. Sci. USA. 1969;64:1035–1041. doi: 10.1073/pnas.64.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks J.L., Chistol G., Gao A.O., Räschle M., Larsen N.B., Mann M., Duxin J.P., Walter J.C. The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell. 2019;176:167–181.e21. doi: 10.1016/j.cell.2018.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J., Schwarz M.S., Bloemeke N., Wolf P.G., Jentsch S. A DNA-dependent protease involved in DNA-protein crosslink repair. Cell. 2014;158:327–338. doi: 10.1016/j.cell.2014.04.053. [DOI] [PubMed] [Google Scholar]
- Stingele J., Habermann B., Jentsch S. DNA-protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem. Sci. 2015;40:67–71. doi: 10.1016/j.tibs.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Stingele J., Bellelli R., Alte F., Hewitt G., Sarek G., Maslen S.L., Tsutakawa S.E., Borg A., Kjær S., Tainer J.A. Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell. 2016;64:688–703. doi: 10.1016/j.molcel.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J., Bellelli R., Boulton S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017;18:563–573. doi: 10.1038/nrm.2017.56. [DOI] [PubMed] [Google Scholar]
- Svoboda M., Konvalinka J., Trempe J.F., Grantz Saskova K. The yeast proteases Ddi1 and Wss1 are both involved in the DNA replication stress response. DNA Repair (Amst.) 2019;80:45–51. doi: 10.1016/j.dnarep.2019.06.008. [DOI] [PubMed] [Google Scholar]
- Toth A., Hegedus L., Juhasz S., Haracska L., Burkovics P. The DNA-binding box of human SPARTAN contributes to the targeting of Polη to DNA damage sites. DNA Repair (Amst.) 2017;49:33–42. doi: 10.1016/j.dnarep.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Vaz B., Popovic M., Newman J.A., Fielden J., Aitkenhead H., Halder S., Singh A.N., Vendrell I., Fischer R., Torrecilla I. Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol. Cell. 2016;64:704–719. doi: 10.1016/j.molcel.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code or reposited datasets.