

Abstract
Introduction:
Polycythemia vera (PV), a Philadelphia chromosome-negative myeloproliferative neoplasm, is characterized by panmyelosis, pancytosis and a JAK2 mutation. Patients are at increased risk of thrombohemorrhagic events, and progression to myelofibrosis or acute leukemia. Current treatments include aspirin, phlebotomy and cytoreductive drugs (most commonly hydroxyurea). Givinostat is a potent, class I/II histone deacetylase (HDAC) inhibitor that is in phase I/II clinical trials in PV, was well-tolerated and yielded promising clinico-hematological responses. A phase III study versus hydroxyurea in high-risk PV patients is planned.
Areas Covered:
We present an overview of PV, current treatment guidelines, and the putative mechanism(s) of action of givinostat. We discuss the preclinical and clinical studies of givinostat in PV and briefly review approved and investigational competitor compounds.
Expert Opinion:
HDAC inhibitors have long been known to be active in PV, but chronic toxicities can be challenging. Givinostat, however, is active and well-tolerated, and is entering a pivotal Phase III randomized trial. It offers the possibility of replacing hydroxyurea as the standard first line cytoreductive choice for PV patients. This would transform the current therapeutic paradigm and guidelines for PV management. Although surrogate clinical study endpoints may suffice for regulatory purposes, thrombosis reduction and the prevention of disease progression are most important to patients and clinicians.
Keywords: clinical trial, epigenetic, givinostat, HDAC, histone deacetylase inhibitor, HSP90, JAK2V617F, myeloproliferative neoplasm (MPN), polycythemia vera (PV)
1. Introduction
Polycythemia vera (PV) [1-3], essential thrombocythemia (ET) and primary myelofibrosis (PMF) are the three classic Philadelphia-chromosome-negative chronic myeloproliferative neoplasms (MPNs) [4,5]. MPNs are caused by clonal proliferation arising at the hematopoietic stem cell level, and manifest distinct clinical phenotypes [4]. PMF and ET are the most aggressive and indolent classic MPNs, respectively [3]. PV, the most prevalent MPN [6], is primarily characterized by erythrocytosis, but leukocytosis and thrombocytosis are common. PV is associated with a substantial risk of thrombosis and hemorrhage. The survival of patients with PV is significantly inferior to that of an age- and sex-matched population (median 18.9 years) [7,8] and the cumulative incidence risk of disease progression to post-PV acute myeloid leukemia (AML), the most devastating complication, is 5.5-18.7% and 7.9-17% at 15 and 20 years, respectively [9], with or without an intervening post-PV MF phase. Progression to post-PV MF, transformation to AML [10,11] and major thrombotic events are the leading causes of PV mortality. In addition, patients with PV exhibit abnormal levels of inflammatory cytokines and a broad range of symptoms [12], including pruritus, fatigue, splenomegaly, and bone pain, which compromise quality of life.
The majority of MPN patients harbor the mutation V617F in the pseudokinase domain of the Janus kinase 2 (JAK2) gene [13,14]. JAK2V617F is considered a ‘driver’ mutation [15] that plays a pivotal role in PV pathogenesis by constitutively activating the JAK-signal transducer and activator of transcription 3/5 (STAT3/5) signaling pathway, leading to ligand-independent proliferation of hematopoietic stem and progenitor cells [13,16,17]. JAK2 mutations are detected in virtually all PV patients (≈ 95% harbor JAK2V617F in exon 14 and ~4% bear other JAK2 mutations in exon 12) and 50-60% of PMF/ET patients (all V617F) [14]. The pathogenetic role of some non-canonical mutations in JAK2 has been increasingly recognized in recent years [18]. Mutations (indels) in exon 13 of JAK2 have recently been shown to give rise to a novel MPN characterized by a PV-like phenotype, accompanied by eosinophilia [19]. Among the seven DNA-binding STAT proteins regulating transcription of genes that are important in cell proliferation and survival upon STAT activation (phosphorylation) [20], deregulated STAT5 activation induced by JAK2V617F is absolutely necessary for PV pathogenesis [21,22,23,24].
2. Overview of the market
Currently, no curative treatments are available for PV, and therapeutic options are focused on prevention of thrombotic/hemorrhagic events, and alleviating symptom burden [3,25]. Strict control of the hematocrit to values <45% is a key management goal in PV that was established by the seminal CYTO-PV study [26]. Prior history of thrombosis and age ≥60 years are considered high-risk features in PV. The guidelines from the European LeukemiaNet (ELN) [27] and the National Comprehensive Cancer Network (NCCN) [28] recommend cytoreductive therapy in high-risk patients, while phlebotomy is typically indicated for low-risk patients. In general, all patients with PV should have daily low dose aspirin, unless contraindicated (e.g., in patients with acquired von Willebrand’s disease) [29]. Cytoreduction may be required in low-risk patients with new thrombosis or disease-related major bleeding, progressive leukocytosis, symptomatic thrombocytosis, poor tolerance of phlebotomy, intractable symptoms or progressive splenomegaly [28,30].
Hydroxyurea (HU) [27,28] and, more recently, recombinant pegylated interferon-alpha [31] have been recommended as frontline cytoreductive treatments for high-risk PV patients. The ELN [27] and NCCN [28] guidelines suggest consideration of non-pegylated and pegylated interferons in younger patients given concerns for skin cancer and leukemic transformation with prolonged HU use. Interferons, which are non-teratogenic, are also preferred in pregnancy. However, about 24% of PV patients treated with HU develop resistance [32] or intolerance (development of mucocutaneous ulcers, fever or myelosuppression). Interferon alpha is an option for HU-resistant/intolerant PV patients [33]; however, the high incidence of adverse events has limited widespread or long-term use [34]. Pegylated formulations of interferon alpha have been used off-label for PV treatment because they are better tolerated and more stable, resulting in lower injection frequencies compared to conventional interferon; however, even pegylated interferon alpha-2a has been associated with considerable adverse events and relatively high rates of discontinuation [33,34]. Of interest, the longer acting ropeginterferon alpha-2b (monopegylated proline interferon), administered every 2 weeks, could ultimately replace currently used interferon formulations [35]. A recent study showed that ropeginterferon alpha-2b potently targets JAK2V617F+ cells in vivo and in vitro [36]. The phase III PROUD/CONTINUATION-PV randomized trial and its extension study clearly demonstrated that ropeginterferon alpha-2b was well-tolerated and superior to HU at 3 years of follow-up (and beyond, though not at 1 year) for the treatment of high-risk PV patients (treatment naïve or minimally treated with HU) [37]. Ropeginterferon alpha-2b was significantly superior to HU in terms of complete hematological (CHR) and molecular (decrease of JAK2V617F allele burden) responses; and regarding the composite outcome of complete hematological responses plus reduction in disease burden (splenomegaly and PV-associated symptoms) in high-risk PV patients [37]. In February 2019, the European Medicines Agency (EMA) approved ropeginterferon alpha-2b (Table 1) for the frontline treatment of PV patients without symptomatic splenomegaly.
Table 1.
Classes/agents in development for treatment of PV besides HDAC inhibitors.
| Agent | Mechanism of action | Phase | References |
|---|---|---|---|
| Ropeginterferon alpha-2b | Targets JAK2V617F+ BM progenitors in PV | III | [35,36,37] |
| KRT-232 | HDM2 inhibitor | II | [52] |
| PTG-300 | Hepcidin mimetic | II | [54,55] |
Abbreviations: BM: bone marrow; HDM2: human double minute 2; PV: polycythemia vera.
In December 2014, the U.S. Federal Drug Administration (FDA) approved ruxolitinib (oral JAK1/2 inhibitor) as a second line treatment for HU-intolerant/resistant PV patients, on the basis of the findings of the RESPONSE trial [38]. The 5-year follow-up of this phase 3 trial [39] further underscored the fact that ruxolitinib confers durable and effective hematocrit control, and significantly improves splenomegaly, symptoms and overall quality of life in PV patients not responding to or intolerant of HU [40,41,42]. Ruxolitinib also demonstrated efficacy as a first-line treatment in patients with high-risk PV (in terms of decreasing erythrocytosis, phlebotomy, splenomegaly and pruritus) in the ongoing Ruxo-BEAT clinical trial that is being conducted in Germany (NCT02577926) [43]. Despite the clinical benefits of ruxolitinib in PV, it does not appear to alter the natural history of the disease [44]. Furthermore, hard data are still lacking to support a statistically significant reduction by ruxolitinib in thromboembolic events in patients with PV [45] although, intriguingly, in the laboratory, ruxolitinib impairs neutrophil extracellular trap formation in MPN patient samples [46]. Ruxolitinib also controls leukocytosis, which has been found to independently predict thrombosis in PV [47], although not all studies have found this to be the case [48].
The quest for disease-modifying agents in PV, along with the significant unmet needs of the subgroup of HU-resistant/intolerant patients (at increased risk of disease transformation and death [49]) spurred the exploration of other novel PV treatments. In this respect, alternate mechanism-based agents, such as human double minute 2 (HDM2) inhibitors, hepcidin mimetics, and histone deacetylase (HDAC) inhibitors have been investigated.
HDM2, the physiologic negative regulator of p53, is upregulated in MPN hematopoietic progenitor cells that harbor JAK2V617F and wild-type TP53 [50]. HDM2 inhibitors prevent the proteasomal degradation of p53 and thus trigger p53-dependent apoptosis. In preclinical studies, activation of p53, achieved through blockade of the p53-HDM2 interaction in CD34+ progenitor cells from PV and MPN patients by nutlins, increased apoptosis [50, 51]. The oral HDM2 inhibitor KRT-232 is currently in clinical development for HU-resistant/intolerant patients with phlebotomy-dependent PV [52] (Table 1), following demonstration of activity and safety of another HDM2 inhibitor, idasanutlin, in a small, investigator-initiated study [53]; however, a subsequent global phase II trial of the latter drug (NCT03287245) was prematurely terminated. Injectable peptide mimetics of hepcidin, such as PTG-300, are also being studied for the treatment of phlebotomy-dependent patients with PV (Table 1). The peptide hormone hepcidin, primarily secreted by hepatocytes, is a master regulator of iron absorption and metabolism [54]; preclinical studies in PV mice demonstrated that exogenous minihepcidin normalized hematocrit levels and improved splenomegaly [55].
The interest in HDACs as novel targets for drug development stems from the pleiotropic roles they play in neoplastic cells [56], including their regulatory role in the expression of numerous genes (for example, HDACs modulate expression of pro-apoptotic or tumor suppressor genes), and aberrant activity in many cancer types [57], including MPNs. Besides universally activating the JAK/STAT pathway, JAK2V617F can promote myeloproliferation by phosphorylating histone H3 after translocating to the nucleus [13,58]. This action, in turn, impairs the binding of HP1α to chromatin, ultimately promoting the transcription of thousands of genes. Furthermore, significantly upregulated HDAC expression [57] has been reported in patients with chronic MPNs; two studies showed that HDAC4, HDAC5, HDAC6, HDAC9, and HDAC11 were elevated in patients with PV [59,60]. Splenomegaly was associated with increased levels of HDACs in MF patients [60], and progressive increase of HDAC6 levels was reported during disease evolution from PV to MF [59]. A recent study demonstrated that HDAC11 is required for proliferation and survival of oncogenic JAK-driven MPN cell lines and MPN patient specimens [61]. Collectively, the involvement of HDACs and epigenetic dysregulation in MPN pathogenesis [62,63,64,65] together with the critical role of JAK2 and its downregulation by HDAC6 inhibitors via interference with acetylation of the chaperone protein heat shock protein 90 (HSP90) [66], spurred interest in exploring HDAC inhibitors as a novel treatment for MPNs [57,67,68,69,70].
3. Givinostat
3.1. Introduction
Givinostat (Box 1) is a synthetic, orally bioavailable, potent HDAC inhibitor, bearing the hydroxamate group ─CO-NH(OH) [71]. Givinostat inhibits the function of the zinc-dependent Class I and II HDACs (there are ten members in classes I and II, namely HDAC 1 through 10) [69]. The high inhibitory potency of givinostat is attributed to chelation of the zinc ion at the HDAC hydrophobic catalytic binding domain by the hydroxamate moiety [72,73,74]. The inhibition constants (Ki) of givinostat for HDACs range between 0.004 and 0.39 μM (givinostat binds strongly to HDAC1, 2, and 6) [73,75]. HDAC inactivation results in high acetylation of lysine residues at N-termini of histones, inducing conformational changes of condensed chromatin to its open form, and activation of gene transcription [70]; therefore, HDACs are prominent drug targets. Deregulated histone deacetylation is associated with compact chromatin and transcriptional repression (silencing of tumor suppressor genes) [67,70]. HDACs also deacetylate a plethora of non-histone proteins (cytosolic and nuclear) that regulate major cellular functions, for example, HSP90, p53, GATA1, α-tubulin, and many others [76,77].
Box 1. Drug summary.
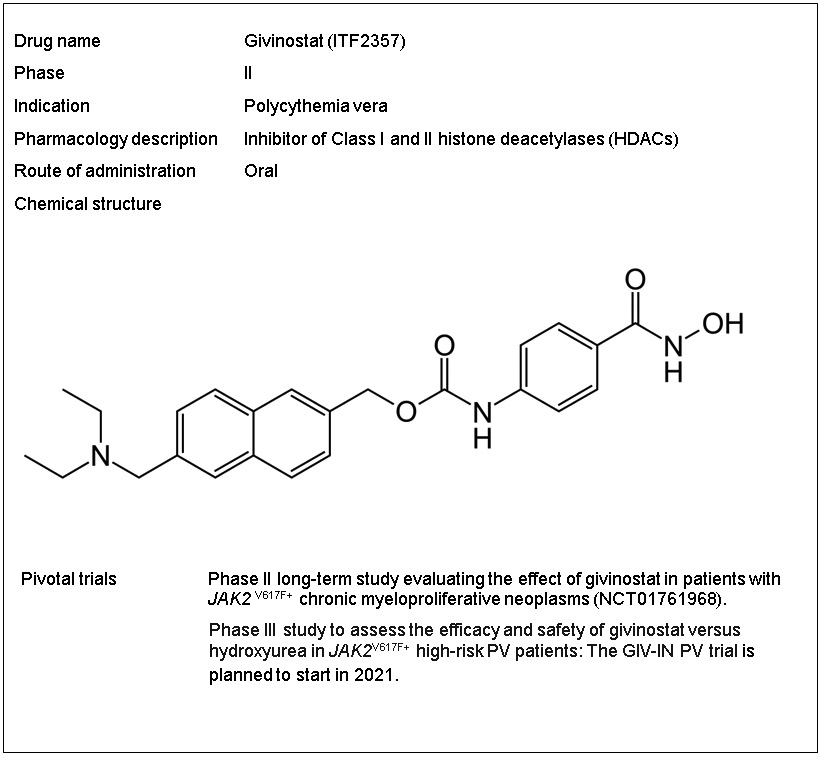
3.2. Mechanisms of action
Phosphorylation of JAK2 leads to recruitment of the transcription factors STAT3/STAT5, dimerization, and translocation to the nucleus [17]. Beyond pharmacologic inhibition of the JAK2 kinase, JAK2/STAT signaling can also be suppressed by interfering with JAK2 expression. Givinostat downregulates JAK2 by acetylation of the chaperone protein HSP90, resulting in disruption of its function (JAK2 is a client of the chaperone protein, HSP90) [56,66,69,78]. HSP90 acetylation is regulated by HDAC6 (Class II HDAC) [57], which is strongly inhibited by givinostat [73]. Inhibition of HSP90 in MPN cell lines and murine models disrupted JAK2 stability, induced proteasomal degradation and depletion of JAK2V617F, decreased p-JAK2 and p-STAT5, and inhibited JAK-STAT downstream signaling [78,79]. HDACs are also required for STAT5-induced activation of transcription [24,56,69,80,81]. Several studies unequivocally demonstrated that STAT5 is essential for induction of PV by JAK2V617F, and other hematologic malignancies [21,22,23,24]; interference with STAT5 function, thus, represents another mode of HDACi action beyond down-regulation of JAK2V617F via acetylation of HSP90.
The activity of givinostat in JAK2-positive MPN cells extends to non-canonical, nuclear (epigenetic) effects of JAK2 [17]. Upon translocation to the nucleus, wild-type JAK2 and JAK2V617F phosphorylate histone H3 at residue Tyr41; H3Y41 phosphorylation blocks binding of the transcription modulator heterochromatin protein HP1α to chromatin, which induces changes in gene transcription, and aberrant gene expression (oncogenesis) in hematopoietic progenitors [13,82,83]. In the nucleus, JAK2V617F also phosphorylates the arginine methyltransferase PRMT5 [84], preventing its interaction with MEP50 and decreasing global arginine methylation of histones H2A/H4, thus promoting myeloproliferation [82, 83,84].
Givinostat exhibits anti-inflammatory activity similar to other hydroxamic acid-containing HDACs [85]. The inhibitory activity of givinostat on pro-inflammatory cytokines and systemic inflammation in vitro and in vivo (mice), respectively, was first reported in 2005 [86]. Pro-inflammatory cytokines have been implicated in MPNs (including PV) pathophysiology [12], and their activity is mediated through the JAK/STAT pathway. Givinostat inhibits the production/release of the pro-inflammatory cytokines tumor necrosis factor alpha (TNFα), interleukin (IL)-1α, IL-1β, IL-6, IFN-γ, without affecting production of anti-inflammatory cytokines [75,86, 87, 88,89], and inhibits the production of vascular endothelial growth factor (VEGF), a cytokine with a critical role in tumor angiogenesis [68,88]. TNFα enhances clonal expansion of JAK2 V617F+ cells from PV, ET, and MF patients (JAK2 V617F allele burden correlates with TNFα expression) [90]. The lowest values of TNFα, IL-1β, IL-6, and IFN-γ were noted 4h after the administration of 50/100 mg doses of givinostat in healthy humans [87]. Additionally, givinostat upregulated the endogenous cyclin-dependent kinase (CDK) inhibitor p21 in multiple myeloma and AML cells [68,88], and induced apoptosis in human Ph+ B-cell ALL cells [91].
4. Clinical development
4.1. Inhibitory activity of givinostat on JAK2 V617F+ cell lines and preclinical studies in PV
Rambaldi et al. pioneered the preclinical and clinical development of givinostat in PV [71]. In the first preclinical study, givinostat inhibited the clonogenic activity of JAK2V617F+ cells from PV/ET patients at 100- to 250-fold lower concentrations (IC50 in the range of 10−3-10−2 μM) as compared to JAK2 wild-type cells (K562) [92]. Inhibition occurred through specific and significant down-modulation of JAK2V617F, phosphorylated JAK2V617F, as well as STAT5 and STAT3 in human erythroleukemia (HEL) cells, which are homozygous for the V617F mutation; however, high levels of phosphorylated wild-type JAK2 were not affected in cells of PV patients and K562 cells (not harboring JAK2V617F), and expression of JAK2V617F mRNA was not modulated in granulocytes from PV patients (i.e., inhibition occurred at the post-translational level) [92]. In another report, givinostat inhibited proliferation and induced apoptosis of JAK2V617F MPN cells at approximately threefold lower concentrations compared to JAK2 wild-type myeloid leukemia cells [93]. In the same report, givinostat downregulated the hematopoietic transcription factors NFE2 (overexpressed in MPN cells and patients) and C-MYB (myeloblastosis gene product), at the mRNA and protein levels, in JAK2V617F+ MPN cell lines (global gene expression analysis showed that givinostat down-modulated 33 genes implicated in hematopoiesis in HEL and UKE1 cells); and proliferation and erythroid differentiation were inhibited in freshly isolated CD34+ cells from MPN patients [93]. Similar findings were noted in a small cohort of MPN patients who exhibited downregulated NFE2 mRNA and normalized platelet counts after 84 days of treatment with givinostat [94]. NFE2 has an important role in the control of erythroid progenitor cell proliferation and differentiation, and is overexpressed in MPN patients [93]. In NFE2 transgenic mice (exhibiting MPN features), the level of acetylation was restored in hypo-acetylated H3 histones upon treatment with the HDAC inhibitor [94]. Furthermore, givinostat enriched acetylated H3 histones at lysine 9 on the NFE2 promoters in UKE1 and SET2 cells, and inhibited JAK2/STAT5-extrcellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation in JAK2V617F+ MPN cells [93]. Inhibition of JAK2 and STAT5 phosphorylation in JAK2V617F+ HEL and UKE1 cell lines by givinostat alone as well as in combination with HU was shown in another study [95]. Givinostat in combination with HU demonstrated strong synergistic activity in inducing apoptosis of JAK2V617F+ HEL and UKE1 cell lines, which occurred through inhibition of the JAK2/STAT5 pathway, caspase 3 activation, and p21 (CDKN1A) downregulation (HU prevented p21 upregulation by givinostat) [95]. Low concentrations of givinostat inhibited STAT5 phosphorylation and JAK/STAT pathway gene expression in B-cell precursor acute lymphoblastic leukemia (pre-B-ALL) CRLF2-rearranged cell lines (harboring JAK2R683G and JAK2I682F) [96]; and induced apoptosis in SUP-B15 (Ph+ B-cell ALL) [91], T-ALL [97], AML [88], and glioblastoma [98] cell lines. Finally, givinostat strongly hyperacetylated histones H3/H4 and α-tubulin (mediated via HDAC6 inhibition) in leukemic cell lines [88,89].
4.2. Clinical trials
4.2.1. Pharmacokinetics and metabolism.
In a phase I clinical trial conducted in healthy volunteers, givinostat demonstrated dose-dependent (linear) pharmacokinetics. The study tested a single dose of 600 mg, and repeat doses of 50, 100, or 200 mg for 7 consecutive days. Givinostat was quickly absorbed, and the maximum plasma concentration (Cmax) was recorded within 2.1-2.6 h (time to maximum plasma concentration, Tmax) after oral administration (50-200 mg/day); the drug did not accumulate in the body [87]. In this trial, the half-life of givinostat was approximately 6-7 h, indicating that twice daily dosing is appropriate for clinical use. A dose-dependent decrease in platelet counts was noted (on day 9, platelets dropped by 17, 25, and 35% below baseline in the cohorts receiving 50, 100, and 200 mg/day, respectively); the effect was transient, and platelet counts partially and fully recovered in 2 and 4 weeks, respectively [87].
In a recent clinical study, Rambaldi et al. characterized the pharmacokinetics of givinostat in PV patients [99]. The plasma parameters recorded in 34 patients after a single dose at the maximum tolerated dose (MTD, 100 mg twice daily) during part B of the study were the following: Tmax = 2 h, Cmax = 71.5 ± 34.4 ng/ml, and time to last detectable concentration (Tlast) = 7.42 ± 1.61 h; repeat-dose plasma pharmacokinetic parameters on day 28 of cycle 2 (8 weeks) for 17 patients were: Tmax = 2 h, Cmax = 90.8 ± 33.5 ng/ml, and Tlast = 8.00 ± 0.0340 h [99]. The median Tmax for givinostat was 1.5 – 4 h, and steady state was reached by day 28 of cycle 1 [99]. Givinostat has two main metabolites in the plasma (ITF2374 and ITF2375) [87,99].
4.2.2. Clinical trials of givinostat in patients with MPNs.
The aforementioned promising preclinical studies showing anti-proliferative activity of givinostat in JAK2V617F+ MPN cells [92,93,95] led to three clinical trials in patients with MPNs, primarily PV, that have been completed so far [99-103] and one long-term clinical trial, which was expanded and is ongoing [104] (Table 2).
Table 2.
Clinical trials of givinostat in MPNs.
| Study Title | ClinicalTrials.gov Identifier | Phase | Study Design |
Population studied | Efficacy† | Toxicity | Ref. |
|---|---|---|---|---|---|---|---|
| Phase IIA study of the HDAC inhibitor ITF2357 in patients with JAK2V617F+ chronic myeloproliferative diseases | NCT00606307 | IIA | Pilot study multicenter, open-label, non-randomized study | 29 patients in total: 16 with MF; 12 with JAK2V617F+ PV and 1 ET, needing cytoreductive therapy, intolerant/refractory to HU, platelet count > 100 x 109/L | At 24 weeks of treatment at a starting dose of 50 mg twice daily: ITT analysis for 13 PV/ET patients: ORR: 54% (1 CR + 6 PR) For evaluable PV patients: Hct < 45% without phlebotomy: 50% (5/10) WBC count < 10 x109/L: 50% (5/10) Platelet count < 400 x109/L: 20% (2/10) no splenomegaly: 70% (7/10) no pruritus: 90% (9/10) |
No grade 4 AE Mostly grade 2 gastrointestinal disorders and one grade 3; ≤ grade 2 anemia (21%) and 1 grade 3, thrombocytopenia (10%), QTc elongation (17%) | [100] |
| Phase II study of the HDAC inhibitor givinostat (ITF2357) in combination with HU in patients with JAK2V617F+ polycythemia vera not responding to HU | NCT00928707 | II | Multicenter, open-label, randomized study between two groups that received 50 or 100 mg givinostat/day in combination with the MTD of HU (A Simon phase-II dose selection design was applied) | 44 patients with JAK2V617F+ PV not responding to the MTD of HU monotherapy§ | Arm A: 50 mg givinostat/day + MTD of HU at 12 weeks (ITT analysis): ORR: 55% (12 PR) Hct < 45% without phlebotomy: 23% (5/22) WBC < 10 x109/L: 21% (3/14) Platelet count < 400 x 109/L: 42% (5/12) No splenomegaly: 5% (1/19) Pruritus resolution: 64% (7/11) Arm B: 100 mg givinostat/day + MTD of HU at 12 weeks (ITT analysis): ORR: 50% (2 CR + 9 PR) Hct < 45% without phlebotomy: 36% (8/22) WBC count < 10 x109/L: 23% (3/13) Platelet count < 400 x 109/L:37% (3/8) No splenomegaly: 11% (2/18) Pruritus resolution: 67% (8/12) |
No grade 4 AE; 5% of the patients had AE, grade 2 thrombocytopenia and gastrointestinal disorders, 1 grade 3 (nausea, anemia) | [101] |
| A two-part (Phase Ib/II) study to assess the safety and preliminary efficacy of givinostat in patients with JAK2V617F+ polycythemia vera | NCT01901432 | Ib/II | Multinational, open-label, non-randomized Part A: MTD¥ Dose was escalated according to a 3+3 design, adopting a modified Fibonacci escalation scheme Part B: efficacy and safety of MTD (Simon’s two-stage design was employed)‡ | Part A: 12 patients (9 completed 6 cycles) Part B: 35 patients (27 completed 6 cycles) Active/uncontrolled JAK2V617F+ PV: hematocrit ≥ 45% or < 45% with phlebotomy, platelet count > 400 x 109/L, and WBC > 10 x 109/L |
Part A (ITT population): ORR: 72.7%; 1 patient achieved CR after 3 and 1 after 6 cycles. Parameters after 3 and 6 cycles (x 4 weeks) respectively: Hct < 45% without phlebotomy: 54.5 and 27.3%; WBC count ≤ 10 x109/L: 54.5 and 18.2%; platelet count ≤ 400 x109/L: 45.5% for both cycles; normal spleen: 54.5% for both cycles; no pruritus, headache, microvascular disturbances: 63.6 and 72.7% Part B (ITT population). ORR: 80.6%; 3 patients achieved CR after 3 cycles and 1 after 6. Parameters after 3 and 6 cycles‡ (x 4 weeks) respectively: Hct < 45% without phlebotomy: 77.4 and 48.4%; WBC count ≤ 10 x109/L: 90.3 and 67.7%; platelet count ≤ 400 x109/L: 74.2 and 71.0%; normal spleen: 16.1% for both cycles; no pruritus, headache, microvascular disturbances: 74.2% and 61.3% |
Part A: 1 case of grade 4 thrombocytopenia and two grade 3 dyspepsia, grade 1 or 2 thrombocytopenia (33.3%) Part B: No grade 4 AE; one grade 3 neutropenia; thrombocytopenia (45.7%), diarrhea (51.4%), and high serum creatinine (37.1%) | [99,102,103] |
| Long-term Phase II study evaluating the effect of givinostat in patients with JAK2V617F+ chronic myeloproliferative neoplasms* | NCT01761968 EudraCT# 2012-003499-37 | II | Multicenter, international, open label core trial that was expanded to long-term Expanded access* | A total of 45 patients with PV were treated for a median of 4 years, # including 32% of patients treated for ≥ 7 years 33 out of 45 PV patients were still on treatment at the cut-off date (July 2017), and all MF patients (3) had dropped out of the study | Interim data on treatment for a median of 4 years: CR: 11%, PR: 89% Hct < 45% without phlebotomy: 56% WBC count ≤ 10 x109/L: 56% Platelet count ≤ 400 x109/L: 78% normal spleen: 56% no pruritus: 89% 22% JAK2V617F+ allele burden decrease overall incidence of thrombosis: 2.3% patients/year |
No grade 4 AE One grade 3 anemia, and two other grade 3 AEs (asthenia, asymptomatic QTc prolongation) | [104] |
Patients who tolerated givinostat and had achieved clinical benefit at the end of the core protocols (and/or while participating in a compassionate use program) were given the opportunity to continue treatment in a long-term, multicenter, international study. The most common dose of givinostat was 100 mg twice daily [104].
The patients received givinostat 50 or 100 mg/day combined with the MTD of HU for each patient (the median dose of HU was 1 g) [101].
According to ELN response criteria [105].
MTD, maximum tolerated dose: 100 mg twice daily.
The median dose of givinostat in Part B of the study was 150 mg/day [99].
Interim data at a median of 4 years (range 6 months to 9 years) were reported [104].
Abbreviations: AE: adverse event; CR: complete response; ET: essential thrombocythemia; Hct: hematocrit; HDAC: histone deacetylase; HU: hydroxyurea; ITT: intention-to-treat; MF: myelofibrosis; MTD: maximum tolerated dose; ORR: overall response rate; PR: partial response; PV: polycythemia vera; QTc: QT interval in electrocardiogram; WBC: white blood cell.
4.2.2.a. Phase IIA, pilot study of givinostat in JAK2V617F+ MPN patients [100].
In a phase IIA, multicenter, open-label, pilot non-randomized study (NCT00606307), the efficacy and safety of givinostat was assessed in 12 PV, 1 ET, and 16 MF patients refractory/intolerant to HU; all patients were JAK2V617F+. The starting dose was 50 mg twice daily; escalation to 50 mg three times daily was permitted if required [100]. The enrolled patients had platelet counts >100×109/L, required cytoreductive treatment, and were intolerant/refractory to HU per ELN criteria [32]. The cohort comprised heavily pretreated patients except for 5 who joined the trial to avoid front-line treatment with HU.
Among 13 patients with PV/ET, the complete response rate was 54% (intention-to-treat analysis) according to ELN criteria [105]: one PV patient had a complete response (CR), 6 had partial responses, and 2 discontinued treatment. Splenomegaly and pruritus were resolved in 70% (7/10) and 90% (9/10), respectively, of evaluable PV patients at 24 weeks (Table 2). The rate of complete resolution of pruritus that givinostat induced is striking, given that this symptom is often debilitating and responds poorly to conventional treatments (the effect of givinostat on pruritus is likely attributed to the reduction of inflammatory cytokines) [71, 87]. Seven out of ten PV patients (70%) became phlebotomy-independent on treatment. A major response was documented in 3 (19%) MF patients (2 post-PV MF and 1 post-ET MF), and spleen size decreased in 5/14 (31%). A progressive decrease of the JAK2V617F allele burden was recorded in the PV/ET patients with givinostat treatment: 55% (mean) at study entry versus 47% and 41% after 12 and 24 weeks, respectively; in the PV patient who achieved CR, the JAK2V617F allele burden mean decreased from 39% to 18% in 24 weeks [100]. In the MF cohort, the mean JAK2V617F allele burden remained unchanged (56%) at 24 weeks compared to baseline.
4.2.2.b. Phase II study of givinostat combined with HU in JAK2V617F+ PV patients [101]
In a subsequent phase II, multicenter clinical trial, the efficacy of givinostat (50 or 100 mg/day) in combination with HU was evaluated in 44 JAK2V617F+ PV patients in need of cytoreductive therapy and not responding to the MTD of HU monotherapy, administered for at least 3 months (NCT00928707), according to ELN criteria [32]. Forty-four patients were randomized into two arms and received 50 or 100 mg givinostat/day combined with their individual MTD of HU. At baseline, all patients had hematocrit ≤ 45% (achieved with phlebotomies if necessary). At the time of the primary response assessment (12 weeks) according to ELN criteria [105], the overall response rates (ORR) were 55% and 50% (2 CR) in the subgroups receiving 50 and 100 mg/day, respectively (Table 2) [101]. Similar to the pilot trial of givinostat monotherapy, pruritus control was notable: 7/11 (64%) and 8/12 (67%) achieved complete resolution in the subgroups treated with 50 and 100 mg/day, respectively. Thrombocytopenia and gastrointestinal disorders were the most common (grade 2) adverse events (Table 2) [101].
4.2.2.c. Phase Ib/II study evaluating safety and efficacy of the maximum tolerated dose of givinostat in PV patients [99,102,103]
Rambaldi et al. recently reported the results of the phase Ib/II, international, open-label, non-randomized trial evaluating givinostat in JAK2V617F+ adult PV patients with active/uncontrolled disease (NCT01901432) [99]. In part A (Phase Ib) of the trial, the MTD was determined at 200 mg/day (100 mg twice daily) [99,102,103], whereas in part B (Phase II) of the trial, the safety, efficacy and tolerability of givinostat at this dose were evaluated after 3 and 6 cycles [99]. To be eligible, patients had to have hematocrit ≥ 45% or < 45% with phlebotomy, platelet count > 400 ×109/L, and white blood cells (WBC) > 10 ×109/L; exclusion criteria included absolute neutrophil count (ANC) < 1.2 ×109/L and previous treatment with JAK2 or HDAC inhibitors. After 12 and 24 weeks of treatment, in parts A and B of the trial, 72.7 and 80.6% of the intention-to-treat populations, respectively, had complete or partial responses (Table 2) per ELN response criteria [105], and givinostat was well-tolerated [99,102,103]. The majority of the patients achieved normal hematological parameters (Table 2); in part B of the study, nearly 20% of the patients experienced ≥35% spleen volume reduction (SVR); a good proportion (~40%) exhibited reduction of severe pruritus. Microvascular symptoms and pruritus were eliminated in 13% and 19% of the intention-to-treat population, respectively, compared to baseline, after Cycle 6 (Part B) [99]. After 3 and 6 cycles of treatment, the JAK2V617F allele burden exhibited a moderate reduction compared to baseline, and differential gene expression was observed (HDAC3, GLRX, and STAT4 were upregulated, and MYC was downregulated) in part B of the study [99].
4.2.2.d. Long-term phase II study of givinostat in JAK2V617F+ PV patients [104].
Eligible patients with JAK2V617F+ chronic MPNs who tolerated givinostat treatment and had achieved clinical benefit at the end of the core protocols (and/or while participating in a compassionate use program) were given the opportunity to continue treatment in a long-term, multicenter, international study (NCT01761968). Patients continued givinostat treatment at their last tolerable dose and schedule [104]. The highest dose that was administered for long-term treatment of JAK2V617F+ PV patients was 100 mg twice daily, and the median time on treatment was 4 years (range 6 months to 9 years); evaluations were performed quarterly, and 73% of the patients were still receiving treatment at the time of the last assessment (July 2017). Interim data from this long-term phase II study were presented at the 59th Meeting of the American Society of Hematology (ASH) [104]. The interim analysis showed that 11% of PV patients had a complete response and 89% of them had a partial response, according to the ELN response criteria [105], after treatment for a median of 4 years (Table 2) [104]. The hematocrit was normal without phlebotomy in 56% of PV patients, whereas platelet and WBC counts were normalized in 78% and 56% [104]. Furthermore, after a median of 4 years, 56% of the patients exhibited a normal spleen (evaluated by palpation and/or imaging), pruritus was eliminated in 89% of the patients, and no patients reported headaches or microvascular symptoms [104]. The JAK2V617F allele burden was reduced by 22% compared to baseline after a median of 4 years of treatment. During the treatment period, 4 patients developed 5 thrombotic events (3 deep vein thromboses of the legs, 1 myocardial infarction, and 1 transient ischemic attack); the overall incidence of thrombosis with givinostat was 2.3% [104], which compares favorably to the 5.8% and 3% patients/year reported with phlebotomy and HU, respectively [30]. The promising clinical efficacy and good tolerability of long-term treatment with givinostat in patients with PV observed in this study serves as the basis for the pivotal phase III clinical trial that will evaluate the efficacy/safety of givinostat as compared to HU in JAK2V617F+ PV patients; this study will be launched in the near future.
4.3. Safety and tolerability
Over the course of nearly 10 years, more than 500 patients have been treated with givinostat on more than 20 clinical trials [71]. As noted above, a phase I trial was conducted in healthy volunteers (doses ranged between 50 and 600 mg administered once or twice daily) to determine the safety of givinostat in 2011 [87]. That trial showed that 100 mg twice daily was a safe and well-tolerated dose in healthy individuals [87]. In part A of the phase Ib/II study (NCT01901432), Rambaldi et al. also determined the MTD of givinostat in PV patients to be 100 mg twice daily [99]. The safety of this dose (100 mg twice daily) was additionally confirmed in the long-term clinical trial (NCT01761968) [104].
Overall, givinostat was well tolerated and did not present any major safety concerns in any of the clinical studies that evaluated the drug in adult PV patients (Table 2) [99,100,102, 103,104], as well as in juvenile patients with idiopathic arthritis [106]. No hemorrhagic effects, organ toxicities, or deaths occurred [71,87,99,100,102, 103,104]. The most common drug-related adverse events in healthy volunteers [87] and PV patients [71,99,100,104] were thrombocytopenia and gastrointestinal disorders (Table 2), which were generally mild to moderate, and reversible with discontinuation of the drug. In the study that Furlan et al. conducted in healthy individuals, the mean platelet counts reached their nadir 9 days after treatment (17, 25, and 35% below baseline corresponding to daily doses of 50, 100, and 200 mg, respectively) but recovered back to baseline within 2-3 weeks after drug discontinuation [87].
In the pilot phase IIA study of givinostat in PV patients (NCT00606307), mild gastrointestinal side effects were the most common; grade ≤ 2 anemia and thrombocytopenia were noted in 21% and 10% of the patients, respectively, but there were no grade 4 drug-related adverse events (AEs) [100]. In the more recent phase II clinical trial (NCT01901432), the majority of drug-related AEs were grade 1 or 2, and they were noted during the first three treatment cycles; the most common ones were thrombocytopenia (45.7%), diarrhea (51.4%), and increased serum creatinine (37.1%), except for one case of grade 4 thrombocytopenia and two cases of grade 3 dyspepsia that were noted during phase A of the trial [99]. In the long term phase II study in PV patients (NCT01761968), givinostat was well tolerated, and no drug-related grade 4 AE occurred; one drug-related grade 3 AE (anemia) that resolved in 8 days and two other drug-related grade 3 AEs (QTc prolongation and asthenia) were recorded [104]. A few episodes of QTc prolongation (a known “class” effect with HDACis) [107] have been noted in PV patients but not in healthy individuals [71].
4.4. Regulatory affairs
The EMA granted orphan drug designation to givinostat for treatment of PV in September 2017.
5. Conclusions
Givinostat is an orally bioavailable, potent inhibitor of Class I and II HDACs that exhibits antineoplastic activity against several hematologic malignancies, and is under clinical development for PV. Preclinical studies clearly demonstrated that givinostat decreases JAK2/STAT5 phosphorylation, attenuates JAK2/STAT5 signaling, and induces apoptosis of JAK2V617F+ MPN cell lines at very low concentrations. The clinical experience shows that givinostat is well-tolerated and efficacious in PV patients. The phase Ib/II trial established the MTD of givinostat in PV patients (100 mg twice daily); the majority of the patients in this trial had normalization of hematological parameters and symptom improvement (particularly of pruritus and microvascular symptoms) compared to baseline [99]. The long-term phase II clinical trial established the durability of response to givinostat and its long-term tolerability in JAK2V617F+ PV patients [104]. The great majority of the patients maintained at least a partial response for more than 4 years on treatment (and 11% was in CR) [104]. Givinostat reduced symptoms, particularly pruritus, and the overall incidence of thrombotic events compared to historical controls treated with HU or phlebotomy (2.3% patients per year with givinostat versus 5.8% with phlebotomy and 3% patients per year with HU [30]). A global phase III trial that will evaluate the efficacy of givinostat versus HU in high-risk (≥ 60 years old and/or with history of thrombosis) JAK2V617F+ PV patients has been planned for the near future based on these encouraging efficacy and safety data.
6. Expert Opinion
Drug development for indolent malignancies such as PV is difficult. In the absence of disease-modifying agents, the main goals of therapy are reduction of thrombohemorrhagic complications and alleviation of symptoms. HU represents the mainstay of frontline therapy for high risk PV, although ropeginterferon alpha-2b was recently approved in the first-line setting in Europe for patients without symptomatic splenomegaly and may receive FDA approval in the US. The FDA recently accepted a Biologics License Application for ropeginterferon alpha-2b as a treatment for PV patients without symptomatic splenomegaly, based on the findings of the phase III PROUD-PV/CONTINUATION-PV studies [37]. Data support the use of pegylated interferon alpha in the frontline setting as well, although discontinuation rates can be high due to toxicity. Ruxolitinib is the only licensed agent after failure of HU, although pegylated interferon alfa is also efficacious in this setting. In a disease with a long natural history such as PV, a good safety and tolerability profile for an investigational agent is imperative: a global, multi-center, phase 2 trial of the HDM2 inhibitor idasanutlin (NCT03287245) was recently terminated owing to concerns over gastrointestinal toxicities, despite this being an active drug.
HDAC inhibitors, which have pleiotropic anti-neoplastic effects, have long been of interest for development as anti-cancer drugs. Despite extensive study over many years across diverse tumor types, agents of this class are currently approved only for the treatment of peripheral and cutaneous T-cell lymphomas and, in the case of panobinostat, multiple myeloma. Interest in developing these agents for myeloid malignancies diminished after negative results in large phase III trials in acute myeloid leukemia and myelodysplastic syndromes [108,109]. Within MPNs, where the ability of HDAC inhibitors to acetylate HSP90 and disrupt its chaperone function, thereby down-regulating JAK2, is particularly appealing, the experience in myelofibrosis has been disappointing, with chronic, low-grade long-term toxicities often precluding long-term delivery, necessary for disease-modifying effects to appear [110,111]. However, givinostat (formerly ITF2357) has stood out as an HDAC inhibitor with good tolerability and clear evidence of efficacy, particularly in PV, an almost universally JAK2-driven disease.
As discussed in this review, givinostat has produced high response rates (albeit partial in the majority of cases) in early phase trials in PV. The drug appears to be particularly effective for pruritus, a “cytokine cluster” symptom also responsive to ruxolitinib. The key question as to what extent givinostat may reduce thromboembolic events remains to be answered, and requires a randomized trial. Of note, clear data supporting a reduced risk of thrombosis are not available thus far either for ruxolitinib in the second-line setting or for ropeginterferon alpha-2b in the upfront/early setting [37]. Hopefully, the planned phase 3 head-to-head randomized clinical trial of givinostat versus HU will be adequately powered to assess this very important endpoint. Other (as yet elusive) goals of therapy in PV include prevention or delay of progression to MF and transformation to AML. Owing to the long-term nature of these complications, extended follow-up of the planned phase 3 randomized clinical trial will be required to establish benefits in these respects.
Other approaches/mechanisms of action continue to be pursued in this space. The oral HDM2 inhibitor KRT-232 is being studied in the HU-resistant/intolerant setting and, once a safe and efficacious dose/schedule is identified, KRT-232 may be compared head-to-head against ruxolitinib, using a study design and endpoints very similar to those employed in RESPONSE (NCT03669965). PTG-300 is a hepcidin-mimetic that is being studied in phlebotomy-requiring patients who continue their baseline cytoreductive therapy on the trial (NCT04057040); while this approach could generate preliminary evidence of activity of this novel drug class in PV, a registration-directed strategy is not apparent yet. In comparison, givinostat has been in clinical trials in patients with PV for a much longer period, and given the efficacy and safety data available so far, a pivotal trial in the frontline setting versus HU appears reasonable and justified. Most importantly, after decades of no new drug approvals in PV apart from that of ruxolitinib in December 2014, it is gratifying to see multiple potentially disease-modifying agents of different classes enter the clinic. Hopefully, givinostat will expand our frontline options for treating high risk PV beyond HU and various interferon formulations.
Article Highlights.
Polycythemia vera (PV) is a chronic myeloproliferative neoplasm, characterized by trilineage myeloproliferation and JAK2 mutations in approximately 99% of patients. While considered an indolent malignancy, survival in PV is inferior to that of an age- and sex-matched population. The main clinical concerns in patients with PV are thrombohemorrhagic complications, troublesome symptoms, progression to myelofibrosis and transformation to acute myeloid leukemia (AML).
There is a major unmet need for drugs that can prevent PV progression to myelofibrosis and AML.
Givinostat is a potent histone deacetylase inhibitor that is under clinical development for treatment of patients with PV. Givinostat has been evaluated in several phase I/II clinical trials in patients with myeloproliferative neoplasms (MPNs), primarily PV, where it showed the highest efficacy.
Givinostat down-regulated JAK2 and STAT5 phosphorylation, reduced JAK2/STAT5 signaling, and selectively induced apoptosis of JAK2V617F+ MPN cell lines at very low concentrations.
The maximum tolerated dose of givinostat was determined to be 100 mg twice daily. The overall response rate to givinostat in a phase II study evaluating its efficacy in JAK2V617F+ patients with PV was 80.6% and all hematological parameters normalized in the majority of the patients, at the end of 3 and 6 treatment cycles. Reduction of severe pruritus was noted in 40% of the intention-to-treat population, and the symptom was completely resolved in 19%, compared to baseline, after 6 treatment cycles. Givinostat was well-tolerated; the most common adverse effects being grade 1/2 thrombocytopenia and gastrointestinal disorders.
Long-term results of the phase II clinical trial demonstrated 11% complete and 89% partial response rates with significant clinical (phlebotomy independence) and molecular responses (reduction in JAK2V617F+ allele burden compared to baseline), considerable reduction in pruritus and a lower incidence of thrombotic events compared to historical controls treated with hydroxyurea; and good tolerability in JAK2V617F+ PV patients.
A global, phase III clinical trial, evaluating the efficacy and safety of givinostat compared to hydroxyurea in JAK2V617F+ high-risk PV patients, has been planned to start in 2021.
Acknowledgments
Funding
This article was supported, in part, by the MD Anderson Cancer Center Support Grant P30 CA016672 from the National Cancer Institute (National Institutes of Health).
Footnotes
Declaration of Interest
P Bose has received research support from Incyte Corporation, Celgene, CTI Biopharma, Kartos Therapeutics, Blueprint Medicines, Constellation Pharmaceuticals, NS Pharma, Promedior, Astellas, and Pfizer. P. Bose has received honoraria from Incyte Corporation, Celgene, CTI Biopharma, Kartos Therapeutics, and Blueprint Medicines. S. Verstovsek has received research support from Incyte Corporation, Roche, Celgene, Gilead, Promedior, CTI Biopharma, Genetech, Blueprint Medicines, NS Pharma, Novartis, Sierra Oncology, Pharma Essentia, Astra Zeneca, Italfarmaco, and Protagonist Therapeutics. S. Verstovsek has received consultancy fees from Constellation Pharmaceuticals, Pragmatist, Sierra Oncology, Incyte, Novartis, and Celgene. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**) to readers
- 1.Falchi L, Newberry KJ, Verstovsek S. New therapeutic approaches in polycythemia vera. Clin Lymph Myeloma Leukemia. 2015;15(S1):S27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verstovsek S, Komrokji RS. Novel and emerging therapies for the treatment of polycythemia vera. Expert Rev Hematol. 2015;8(1):101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bose P, Verstovsek S. Updates in the management of polycythemia vera and essential thrombocythemia. Ther Adv Hematol. 2019;10:1–13.*Recent review of the state of the art in the management of PV and ET
- 4.Verstovsek S, Newberry KJ, Amin HM. Philadelphia chromosome-negative myeloproliferative neoplasms (Chapter 6) In: The MD Anderson Manual of Medical Oncology, Kantarjian HM, Wolff RA. eds., 3rd edition, McGraw-Hill Education, 2016, pp. 103–130. [Google Scholar]
- 5.Spivak JL. Myeloproliferative neoplasms. New Engl J Med. 2017;376(22):2168–2181. [DOI] [PubMed] [Google Scholar]
- 6.Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of Myeloproliferative Neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595–600. [DOI] [PubMed] [Google Scholar]
- 7.Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cerquozzi S and Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5:e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105(7):2664–2670. [DOI] [PubMed] [Google Scholar]
- 11.Kiladjian JJ, Chevret S, Dosquet C, et al. Treatment of polycythemia vera with hydroxyurea and pipobroman: Final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29(29):3907–3913. [DOI] [PubMed] [Google Scholar]
- 12.Geyer H, Scherber RM, Kosiorek H, et al. Symptomatic profiles of patients with polycythemia vera: Implications of inadequately controlled disease. J Clin Oncol. 2016;34(2):151–159. [DOI] [PubMed] [Google Scholar]
- 13.Quintás-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10(2):127–140. [DOI] [PubMed] [Google Scholar]
- 14.Gäbler K, Behrmann I, Haan C. JAK2 mutants (e.g., JAK2V617F) and their importance as drug targets in myeloproliferative neoplasms. JAK-STAT. 2013;2(3):e25025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullally A Lane SW, Ball B, et al. Physiological JAK2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17(6):584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleppe M, Koppikar P, Riester M, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Disc. 2015;5:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170.**Important comprehensive review of the JAK and STAT protein families and their role in hematological malignancies and immunodeficiency disorders.
- 18.Benton CB, Boddu PC, DiNardo CD, et al. Janus kinase 2 variants associated with the transformation of myeloproliferative neoplasms into acute myeloid leukemia. Cancer. 2019. 125(11):1855–1866. [DOI] [PubMed] [Google Scholar]
- 19.Patel AB, Franzini A, Leroy E, et al. JAK (ex13InDel) drives oncogenic transformation and is associated with chronic eosinophilic leukemia and polycythemia vera. Blood. 2019;134(26):2388–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perner F, Perner C, Ernst T, et al. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells. 2019;8:854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan D, Hutchison RE, Mohi G. Critical requirement for STAT5 in a mouse model of polycythemia vera. Blood. 2012;119(15):3539–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walz C, Ahmed W, Lazarides K, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2V617F in mice. Blood. 2012;119(15):3550–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Recio C, Guerra B, Guerra-Rodriguez M, et al. Signal transducer and activator of transcription (STAT)-5: an opportunity for drug development in oncohematology. Oncogene. 2019;38:4657–4668. [DOI] [PubMed] [Google Scholar]
- 24.Wingelhofer B, Neubauer HA, Valent P, et al. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia.2018;32:1713–1728.**Articles 21–24 demonstrate the essential role of STAT5 in PV pathogenesis.
- 25.Vannucchi AM. How I treat polycythemia vera. Blood. 2014;124(22):3212–3220. [DOI] [PubMed] [Google Scholar]
- 26.Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33.**Seminal CYTO-PV study that established the association of hematocrit <45% in PV with lower thrombotic and death risks.
- 27.Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. 2018;32(5):1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerds AT, Gotlib J, Bose P, et al. NCCN Clinical Practice Guidelines in Oncology: Myeloproliferative Neoplasms: NCCN clinical guidelines in oncology. Version 32019. https://www.nccn.org/professionals/physician_gls/default.aspx (accessed: Feb 2020). [Google Scholar]
- 29.Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. [DOI] [PubMed] [Google Scholar]
- 30.Barbui T, Vannucchi AM, Finazzi G, et al. A reappraisal of the benefit-risk profile of hydroxyurea in polycythemia vera: A propensity-matched study. Am J. Hematol. 2017;92:1131–1136. [DOI] [PubMed] [Google Scholar]
- 31.Mascarenhas J, Kosiorek HE, Prchal JT, et al. Results of the myeloproliferative neoplasms - Research Consortium (MPN-RC) 112 randomized trial of pegylated interferon alfa-2a (PEG) versus hydroxyurea (HU) therapy for the treatment of high risk polycythemia vera (PV) and high risk essential thrombocythemia (ET). Blood. 2018. 132;(Suppl.1), abstract #577. [Google Scholar]
- 32.Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. British J Haematol. 2010;148(6):961–963. [DOI] [PubMed] [Google Scholar]
- 33.Yacoub A, Mascarenhas J, Kosiorek H, et al. Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood. 2019;134(18):1498–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masarova L, Patel KP, Newberry KJ, et al. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017;4(4):e165–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, et al. Ropeginterferon alfa-2b, a novel IFNα−2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015;126(15):1762–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verger E, Soret-Dulphy J, Maslah N, et al. Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018;8:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–e208.**Pivotal phase 3 clinical trial that showed the superiority of ropeginterferon alfa-2b versus hydroxyurea in PV patients after 3 years of treatment.
- 38.Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435.**Pivotal clinical trial that led to approval of ruxolitinib for treatment of hydroxyurea-resistant/ intolerant PV patients.
- 39.Kiladjian JJ, Zachee P, Hino M, et al. Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. 2020;7(3):e226–e237.**Important follow-up of the phase 3 clinical trial that evidenced the durable efficacy of ruxolitinib in PV patients who are resistant or intolerant to hydroxyurea.
- 40.Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120(4):513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80 week follow-up from the RESPONSE trial. Haematologica. 2016;101(7):821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mesa R, Verstovsek S, Kiladjian JJ, et al. Changes in quality of life and disease-related symptoms in patients with polycythemia vera receiving ruxolitinib or standard therapy. Eur J Haematol. 2016;97(2):192–200. [DOI] [PubMed] [Google Scholar]
- 43.Koschmieder S, Isfort S, Wolfjavascript D, et al. Ruxolitinib shows efficacy in patients with newly-diagnosed polycythemia vera: futility analysis of the randomized Ruxo-BEAT clinical trial of the German study group for myeloproliferative neoplasms. Blood. 2019;134 (Suppl.1):2944. [Google Scholar]
- 44.Bose P, Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next? Blood. 2017;130(2):115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masciulli A, Ferrari A, Carobbio A, et al. Ruxolitinib for the prevention of thrombosis in polycythemia vera: a systematic review and meta-analysis. Blood Advances. 2020;4(2):380–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Science Transl. Res. 2018;10(436):eaan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbui T, Masciulli A, Rosa M, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126(4):560–561. [DOI] [PubMed] [Google Scholar]
- 48.Ronner L, Podoltsev NA, Gotlib J, et al. Persistent leukocytosis in polycythemia vera is associated with disease evolution but not thrombosis: An analysis from a 520-patient retrospective multi-center database. Blood.2019;134(Suppl.1):2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alvarez-Larrán A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119(6):1363–1369. [DOI] [PubMed] [Google Scholar]
- 50.Nakatake M, Monte-Mor B, Debili M, et al. JAK2(V617F) negatively regulates p53 stabilization by enhancing MDM2 via La expression in myeloproliferative neoplasms. Oncogene. 2012;31(10):1323–1333. [DOI] [PubMed] [Google Scholar]
- 51.Lu M, Li Y, Wang X, Hoffman R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon α 2a target JAK2V617F-positive progenitor and stem cells. Blood. 2014;124(5):771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gotlib J, Gabrail N, O’Connell CL, et al. A randomized, open-label, multicenter, phase 2 study to evaluate the efficacy, safety, and pharmacokinetics of KRT-232 compared with ruxolitinib in patients with phlebotomy-dependent polycythemia vera. Blood. 2019;134(Suppl.1):4168. [Google Scholar]
- 53.Mascarenhas J, Lu M, Kosiorek H, et al. Oral idasanutlin in patients with polycythemia vera. Blood. 2019;134(6):525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ginzburg YZ, Feola M, Zimran E, et al. Dysregulated iron metabolism in polycythemia vera: etiology and consequences. Leukemia. 2018;32:2105–2116.*Useful review article that explains the etiology and consequences of dysregulated iron metabolism in PV.
- 55.Cascu C, Oikonomidou PR, Chen H, et al. Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood. 2016;128(2):265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol Therap. 2014;143:323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.West AC and Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39.*Important article reviewing HDAC inhibitors (Classes I, II, and IV) in cancer.
- 58.Dawson MA, Bannister AJ, Göttgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature. 2009;461(7265):819–822.**Seminal article showing nuclear action of JAK2 and regulation of gene expression via histone phosphorylation.
- 59.Skov V, Stauffer Larsen T, Thomassen M., et al. Increased gene expression of HDACs in patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Lymphoma. 2012;53(1):123–129. [DOI] [PubMed] [Google Scholar]
- 60.Wang JC, Chen C, Dumlao T, et al. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk Lymphoma. 2008;49(12):2321–2327. [DOI] [PubMed] [Google Scholar]
- 61.Yue L, Sharma V, Horvat NP, et al. HDAC11 deficiency disrupts oncogene-induced hematopoiesis in myeloproliferative neoplasms. Blood. 2020;135(3):191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Greenfield G, McPherson S, Mills K, et al. The ruxolitinib effect: understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J Transl Med. 2018;16:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mascarenhas J, Roper N, Chaurasia P, Hoffman R. Epigenetic abnormalities in myeloproliferative neoplasms: a target for novel therapeutic strategies. Clin Epigenet. 2011;2:197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27:1861–1869. [DOI] [PubMed] [Google Scholar]
- 65.Milosevic JD and Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97:183–197. [DOI] [PubMed] [Google Scholar]
- 66.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005;280(29):26729–34.**Study that established disruption of HSP90 chaperone function via HDAC6 inhibition.
- 67.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J. Clin Oncol 2005;23(17):3971–3993. [DOI] [PubMed] [Google Scholar]
- 68.Vannucchi AM, Guglielmelli P, Rambaldi A, et al. Epigenetic therapy in myeloproliferative neoplasms: evidence and perspectives. J Cell Mol Med. 2009;13(8A):1437–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bose P and Verstovsek S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opinion Invest Drugs. 2016;25(12):1393–1403.*Comprehensive review article on the mechanisms of action of HDAC inhibitors in MPN, along with a summary of the preclinical and clinical studies in MPN.
- 70.San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC inhibitors in acute myeloid leukemia. Cancers. 2019;11(11):pii: E1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pieri L, Guglielmelli P, Finazzi G, Vannucchi AM. Givinostat for the treatment of polycythemia vera. Expert Opin Orphan Drugs. 2014;2(8):841–850.*First comprehensive review article on givinostat in treatment of PV.
- 72.Vannini A, Volpari C, Filocamo G, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Nat Acad Sci. USA, 2004;101(42):15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wagner FF, Weiwer M, Lewis MC, Holson EB. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics. 2013;10(4):589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li S, Fossati G, Marchetti C, et al. Specific inhibition of histone deacetylase 8 reduces gene expression and production of pro-inflammatory cytokines in vitro and in vivo. J Biol Chem. 2015;290(4):2368–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. [DOI] [PubMed] [Google Scholar]
- 77.Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nature Rev Mol Cell Biology. 2019;20:156–174.**Comprehensive review article on non-histone protein acetylation.
- 78.Marubayashi S, Koppikar P, Taldone T, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin Invest. 2010;120(10):3578–3593.**Study that established chaperone protein HSP90 as a therapeutic target in MPN.
- 79.Fiskus W, Verstovsek S, Manshouri T, et al. Heat shock protein 90 is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17(23):7347–7358.**Important article showing synergism of HSP90 and JAK2 inhibitors in human MPN cells.
- 80.Rascle A, Johnston JA, Amati B. Deacteylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol Cell Biol. 2003;23(12):4162–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinz S, Unser S, Buob D, et al. Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extraterminal (BET) protein function. Nucleic Acids Res. 2015;43:3524–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McPherson S, McMullin MF, Mills K. Epigenetics in myeloproliferative neoplasms. J Cell Mol Med. 2017;21(9):1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quintás-Cardama A, Verstovsek S. Molecular pathways: JAK/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19(8):1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu F, Zhao X, Perna F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19(2):283–294.**Important article describing a nuclear action of JAK2 and regulation of gene expression via PRMT5 phosphorylation.
- 85.Leoni F, Zaliani A, Bertolini G, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits anti-inflammatory properties via suppression of cytokines. Proc Nat Acad Sci. USA 2002;99(5):2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leoni F, Fossati G, Lewis EC, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systematic inflammation in vivo. Mol Med. 2005;11(1–12):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Furlan A, Monazani V, Reznikov LL, et al. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (Givinostat). Mol Med. 2011;17(5–6):353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Golay J, Cuppini L, Leoni F, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21:1892–1900. [DOI] [PubMed] [Google Scholar]
- 89.Carta S, Semino C, Fossati G, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1β-containing secretory lysosomes: role of microtubules. Blood. 2006;108(5):1618–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fleischman AG, Aichberger KI, Luty SB, et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011;118:6392–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Zhao K, Yao C, et al. Givinostat, a type II histone deacetylase inhibitor, induces potent caspase-dependent apoptosis in human lymphoblastic leukemia. Genes Cancer. 2016;7(9–10):292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guerini V, Barbui V, Spinelli O, et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2 V617F. Leukemia. 2008;22:740–747.*First preclinical study demonstrating the antitumor activity of givinostat in JAK2 V617F-mutated MPN cells.
- 93.Amaru Calzada A, Todoerti K, Donadoni L, et al. The HDAC inhibitor givinostat modulates the hematopoietic transcription factors NFE2 and C-MYB in JAK2V617F myeloproliferative neoplasm cells. Exper Hematol. 2012;40:634–645. [DOI] [PubMed] [Google Scholar]
- 94.Kaufmann KB, Grunder A, Hadlich T, et al. A novel murine model of myeloproliferative disorders generated by overexpression of the transcription factor NFE2. J Exper Med. 2012;209:35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amaru Calzada A, Pedrini O, Finazzi G, et al. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2V617F myeloproliferative neoplasm patients. Exper Hematol. 2013;41:253–260.e2. [DOI] [PubMed] [Google Scholar]
- 96.Savino AM, Trentin L, Vieri M, et al. The histone deacetylase inhibitor givinostat (ITF2357) exhibits potent anti-tumor activity against CRLF2-rearranged BCP-ALL. Leukemia. 2017;31:2365–2375. [DOI] [PubMed] [Google Scholar]
- 97.Pinazza M, Borga C, Agnusdei V, et al. An immediate transcriptional signature associated with response to the histone deacetylase inhibitor givinostat in T acute lymphoblastic leukemia xenografts. Cell Death & Disease. 2016;7:e2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marampon F, Leoni F, Mancini A, et al. Histone deacetylase inhibitor ITF2357 (givinostat) reverts transformed phenotype and counteracts stemness in in vitro and in vivo models of human glioblastoma. J Cancer Res Clin Oncol. 2019;145:393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rambaldi A, Iurlo A, Vannucchi AM, et al. Safety and efficacy of the maximum tolerated dose of givinostat in polycythemia vera: a two-part phase Ib/II study. Leukemia. 2020;34:2234–2237.*Important study that determined the MTD of givinostat and demonstrated its considerable efficacy in PV patients.
- 100.Rambaldi A, Dellacasa CM, Finazzi G, et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. British J Haematol. 2010;150(4):446–455. [DOI] [PubMed] [Google Scholar]
- 101.Finazzi G, Vannucchi AM, Martinelli V, et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hyrdoxycarbamide monotherapy. British J Haematol. 2013;161(5):688–694. [DOI] [PubMed] [Google Scholar]
- 102.Finazzi G, Martino B, Pezzuto A, et al. A two-part study of givinostat in patients with polycythemia vera: maximum tolerated dose definition and preliminary efficacy results. Blood. 2016;128(22):4261. [Google Scholar]
- 103.Rambaldi A, Iurlo A, Vannucchi AM, et al. A two-part study of givinostat in patients with polycythemia vera: the maximum tolerated dose selection and the proof-of-concept final results. Blood. 2017;130(S1):253 (#100916). [Google Scholar]
- 104.Finazzi G, Iurlo A, Martino B, et al. A long-term safety and efficacy study of givinostat in patients with polycythemia vera: the first 4 years of treatment. Blood. 2017;130(S1):1648 (#101752). [Google Scholar]
- 105.Barosi G, Birgegard G, Finazzi G, et al. Response criteria for essential thrombocythemia and polycythemia vera: Result of a European LeukemiaNet consensus conference. Blood. 2009;113(20):4829–4833. [DOI] [PubMed] [Google Scholar]
- 106.Vojinovic J, Damjanov N, D’Urzo C, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthr. Rheumatol. 2011;63(5):1452–1458. [DOI] [PubMed] [Google Scholar]
- 107.Strevel EL, Ing DJ, Siu LL. Molecularly targeted oncology therapeutics and prolongation of the QT interval. J. Clin. Oncol. 2007;25(22):3362–3371. [DOI] [PubMed] [Google Scholar]
- 108.Sekeres MA, Othus M, List AF, et al. Randomized phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: North American Intergroup Study SWOG S1117. J Clin Oncol. 2017;35(24):2745–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garcia-Manero G, Othus M, Pagel JM, et al. SWOG S1203: A randomized phase III study of standard cytarabine plus daunorubicin (7+3) therapy versus idarubicin with high dose cytarabine (IA) with or without vorinostat (IA+V) in younger patients with previously untreated acute myeloid leukemia (AML). Blood. 2016;128(22):901. [Google Scholar]
- 110.Bose P, Swaminathan M, Pemmaraju N, et al. A phase 2 study of pracinostat combined with ruxolitinib in patients with myelofibrosis. Leuk Lymphoma. 2019;60(7):1767–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mascarenhas J, Marcellino BK, Lu M, et al. A phase I study of panobinostat and ruxolitinib in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk Res. 2020;88:106272. [DOI] [PubMed] [Google Scholar]