

Abstract
Optimal attention performance requires cholinergic modulation of corticothalamic neurons in the prefrontal cortex. These pyramidal cells express specialized nicotinic acetylcholine receptors containing the α5 subunit encoded by Chrna5. Disruption of this gene impairs attention, but the advantage α5 confers on endogenous cholinergic signaling is unknown. To ascertain this underlying mechanism, we used optogenetics to stimulate cholinergic afferents in prefrontal cortex brain slices from compound-transgenic wild-type and Chrna5 knock-out mice of both sexes. These electrophysiological experiments identify that Chrna5 is critical for the rapid onset of the postsynaptic cholinergic response. Loss of α5 slows cholinergic excitation and delays its peak, and these effects are observed in two different optogenetic mouse lines. Disruption of Chrna5 does not otherwise perturb the magnitude of the response, which remains strongly mediated by nicotinic receptors and tightly controlled by autoinhibition via muscarinic M2 receptors. However, when conditions are altered to promote sustained cholinergic receptor stimulation, it becomes evident that α5 also works to protect nicotinic responses against desensitization. Rescuing Chrna5 disruption thus presents the double challenge of improving the onset of nicotinic signaling without triggering desensitization. Here, we identify that an agonist for the unorthodox α-α nicotinic binding site can allosterically enhance the cholinergic pathway considered vital for attention. Treatment with NS9283 restores the rapid onset of the postsynaptic cholinergic response without triggering desensitization. Together, this work demonstrates the advantages of speed and resilience that Chrna5 confers on endogenous cholinergic signaling, defining a critical window of interest for cue detection and attentional processing.
SIGNIFICANCE STATEMENT The α5 nicotinic receptor subunit (Chrna5) is important for attention, but its advantage in detecting endogenous cholinergic signals is unknown. Here, we show that α5 subunits permit rapid cholinergic responses in prefrontal cortex and protect these responses from desensitization. Our findings clarify why Chrna5 is required for optimal attentional performance under demanding conditions. To treat the deficit arising from Chrna5 disruption without triggering desensitization, we enhanced nicotinic receptor affinity using NS9283 stimulation at the unorthodox α-α nicotinic binding site. This approach successfully restored the rapid-onset kinetics of endogenous cholinergic neurotransmission. In summary, we reveal a previously unknown role of Chrna5 as well as an effective approach to compensate for genetic disruption and permit fast cholinergic excitation of prefrontal attention circuits.
Keywords: acetylcholine, Chrna5, nicotinic receptors, NS9283, optogenetics, prefrontal cortex
Introduction
The medial prefrontal cortex (PFC) is essential for working memory and top-down attention (Goldman-Rakic, 1995; Miller and Cohen, 2001; Dalley et al., 2004). Cholinergic neuromodulation of the prefrontal cortex by projections from the basal forebrain is required for attention (Dalley et al., 2004). These projections release acetylcholine during successful cue detection and performance of sustained attention tasks (Himmelheber et al., 2000; Parikh et al., 2007; Gritton et al., 2016; Howe et al., 2017). Corticothalamic neurons in layer 6 are excited by such acetylcholine release (Kassam et al., 2008; Hedrick and Waters, 2015; Sparks et al., 2018). They express the α5 nicotinic receptor subunit encoded by Chrna5 (Wada et al., 1990; Winzer-Serhan and Leslie, 2005) in addition to the more commonly expressed α4 and β2 subunits of high-affinity nicotinic receptors. In contrast to other nicotinic receptor subunits, the α5 subunit cannot participate in the traditional cholinergic binding site, and instead it exhibits specialized functions such as increasing calcium permeability and sensitivity of α4β2* nicotinic receptors to acetylcholine in cell systems (Tapia et al., 2007; Kuryatov et al., 2008). It also boosts the response to exogenous stimulation ex vivo in mouse prefrontal cortex (Bailey et al., 2010; Tian et al., 2011). However, the impact of the α5 subunit on synaptic cholinergic neurotransmission in prefrontal cortex is unknown.
Behavioral and genetic evidence suggests that the α5 nicotinic subunit plays a role in attention and more generally in prefrontal executive function. Mice lacking Chrna5 display attention deficits in the five-choice serial reaction time test, exhibiting a failure to detect cues when the task difficulty is increased to make cue duration shorter (Bailey et al., 2010). Work in rats has also confirmed that Chrna5 is important for performing demanding attention tasks (Howe et al., 2018). Human polymorphisms in Chrna5 that affect receptor functionality are associated with a cognitive phenotype that increases early experimentation with smoking and risk for addiction (Bierut et al., 2008), as well as increased risks of schizophrenia, cognitive impairments, and attention deficit hyperactivity disorder (Hong et al., 2011; Schuch et al., 2016; Han et al., 2019).
Despite the link with attention and prefrontal cognitive processes, the role of Chrna5 in responding to endogenous acetylcholine release has yet to be examined. Most α5 characterization relies on heterologous expression systems and not the natural environment of nicotinic receptors in neurons (Baenziger and daCosta, 2013). While there is tight control over trafficking of cholinergic receptors (Matta et al., 2017), the lack of validated antibodies for immunoelectron microscopy means the relationship between α5 and cholinergic afferents is unknown. Functional examination lacks a specific pharmacological tool to discriminate α5 subunit-containing nicotinic receptors. Studies characterizing cholinergic functionality in α5 knock-out mice have used exogenous applications of acetylcholine that differ vastly from the rapid timescales of endogenous neurotransmission (Parikh et al., 2007). Thus, there is a gap in our understanding of the role of Chrna5 in cholinergic modulation of attention circuits. The development of optogenetic tools to specifically express channelrhodopsin in cholinergic neurons (Zhao et al., 2011; Hedrick et al., 2016) allows us to overcome this gap and measure the function of the α5 subunit in endogenous cholinergic modulation.
To probe the advantage that α5 confers on endogenous cholinergic signaling, we optogenetically stimulated cholinergic afferents in prefrontal brain slices of compound transgenic wild-type and α5 knock-out mice. Concurrent whole-cell electrophysiology shows that the α5 subunit is essential for achieving rapid kinetics of cholinergic neurotransmission. Under conditions of prolonged stimulation, the α5 subunit preserves the synaptic cholinergic response from desensitization. A pharmacological approach targeting the recently discovered α-α acetylcholine binding site on nicotinic receptors (Harpsøe et al., 2011; Wang et al., 2015; Mazzaferro et al., 2017) rescues onset kinetics of the cholinergic response after α5 disruption. Recent perspectives on cholinergic modulation have sought to shed light on the temporal scales of cholinergic signaling (Disney and Higley, 2020; Sarter and Lustig, 2020; Venkatesan et al., 2020). In this context, our work reveals a critical and specialized role for the α5 nicotinic receptor subunit in initiating rapid cholinergic signaling.
Materials and Methods
Animals.
To elicit endogenous acetylcholine release optogenetically and examine responses in α5 wild-type (α5WT) and α5 knock-out (α5KO) littermate mice, we created two compound transgenic mouse lines. Mouse crosses are illustrated in the respective figures using these mice. The first was achieved by crossing ChAT-ChR2 (catalog #014546, The Jackson Laboratory; Zhao et al., 2011) with α5KO mice (Salas et al., 2003) to achieve parents. These mice were crossed with α5HET mice to generate α5WT and α5KO ChAT-ChR2/+ mice. We independently verified the results of optogenetic cholinergic stimulation using a different line of mice to express channelrhodopsin in cholinergic neurons. ChAT-IRES-Cre (catalog #031661, The Jackson Laboratory) and Ai32 mice (catalog #012569, The Jackson Laboratory) were each crossed with α5HET mice, and their offspring were crossed with each other to generate littermate α5WT and α5KO ChAT-IRES-Cre/+ Ai32/+ mice. All animals were bred on a C57BL/6 background. Both male and female animals age greater than postnatal day 60 (P60) were used. Mice were weaned at P21, separated based on sex and group housed (2–4 mice/cage), and given ad libitum access to food and water on a 12 h light/dark cycle with lights on at 7:00 A.M. Guidelines of the Canadian Council on Animal Care were followed, and all experimental procedures were approved by the Faculty of Medicine Animal Care Committee at the University of Toronto. A total of 97 mice were used for this study, with similar numbers of males and females.
Electrophysiology.
Animals were anesthetized with an intraperitoneal injection of chloral hydrate (400 mg/kg) and then decapitated. The brain was rapidly extracted in ice-cold sucrose ACSF (254 mm sucrose, 10 mm d-glucose, 26 mm NaHCO3, 2 mm CaCl2, 2 mm MgSO4, 3 mm KCl, and 1.25 mm NaH2PO4). The 400-μm-thick coronal slices of prefrontal cortex (bregma 2.2-1.1) were obtained on a Dosaka linear slicer (SciMedia). Slices were allowed to recover for at least 2 h in oxygenated (95% O2, 5% CO2) ACSF (128 mm NaCl, 10 mm d-glucose, 26 mm NaHCO3, 2 mm CaCl2, 2 mm MgSO4, 3 mm KCl, and 1.25 mm NaH2PO4) at 30°C before being used for electrophysiology.
For whole-cell patch-clamp electrophysiology, brain slices were transferred to a chamber mounted on the stage of a BX51WI microscope (Olympus) and constantly perfused with oxygenated ACSF at 30°C. Layer 6 pyramidal neurons were patched based on their morphology and the proximity to white matter. The recording electrodes (2-4 MΩ) were filled with patch solution composed of 120 mm potassium gluconate, 5 mm KCl, 10 mm HEPES, 2 mm MgCl2, 4 mm K2-ATP, 0.4 mm Na2-GTP, and 10 mm sodium phosphocreatine, with pH adjusted to 7.3 using KOH. Data were acquired with a Multiclamp 700B amplifier at 20 kHz with a Digidata 1440A digitizer and pClamp 10.7 acquisition software (Molecular Devices). All recordings were compensated for the liquid junction potential (14 mV). Layer 6 pyramidal responses were examined in voltage clamp at −75 mV and in current clamp at rest or starting from −70 mV.
There are two distinct population of pyramidal neurons in layer 6, which differ in their spiking pattern to current injection—regular spiking corticothalamic neurons and doublet spiking corticocortical neurons (Kumar and Ohana, 2008; Ledergerber and Larkum, 2010; Thomson, 2010). As previously reported using endogenous (Hedrick and Waters, 2015) and exogenous acetylcholine (Yang et al., 2019), we found that the corticocortical neurons exhibit a purely muscarinic receptor-mediated hyperpolarization, while regular spiking corticothalamic neurons exhibit nicotinic receptor-mediated depolarization. Therefore, our experiments focused on the regular spiking corticothalamic pyramidal neurons to characterize the role of α5 subunit containing nicotinic receptors in the response to endogenous acetylcholine release. In some of these neurons, the responses to optogenetic stimulation were characterized only in voltage clamp, some only in current clamp, and some in both recording modalities.
Optogenetics.
To excite channelrhodopsin-containing axonal fibers, blue light (473 nm) was delivered in brief pulses (5 ms) with an LED (2 mW; Thorlabs) through the 60× objective lens. Eight pulses of light, 5 ms each, were delivered in a frequency-accommodating manner, starting at 50 Hz and ending at 10 Hz to stimulate the cholinergic axons (Fig. 1A, experimental schematic). This paradigm was intended to replicate the accommodating firing pattern of cholinergic neurons (Unal et al., 2012). As indicated, a subset of experiments alternatively used only a single pulse of light (5 ms).
Figure 1.
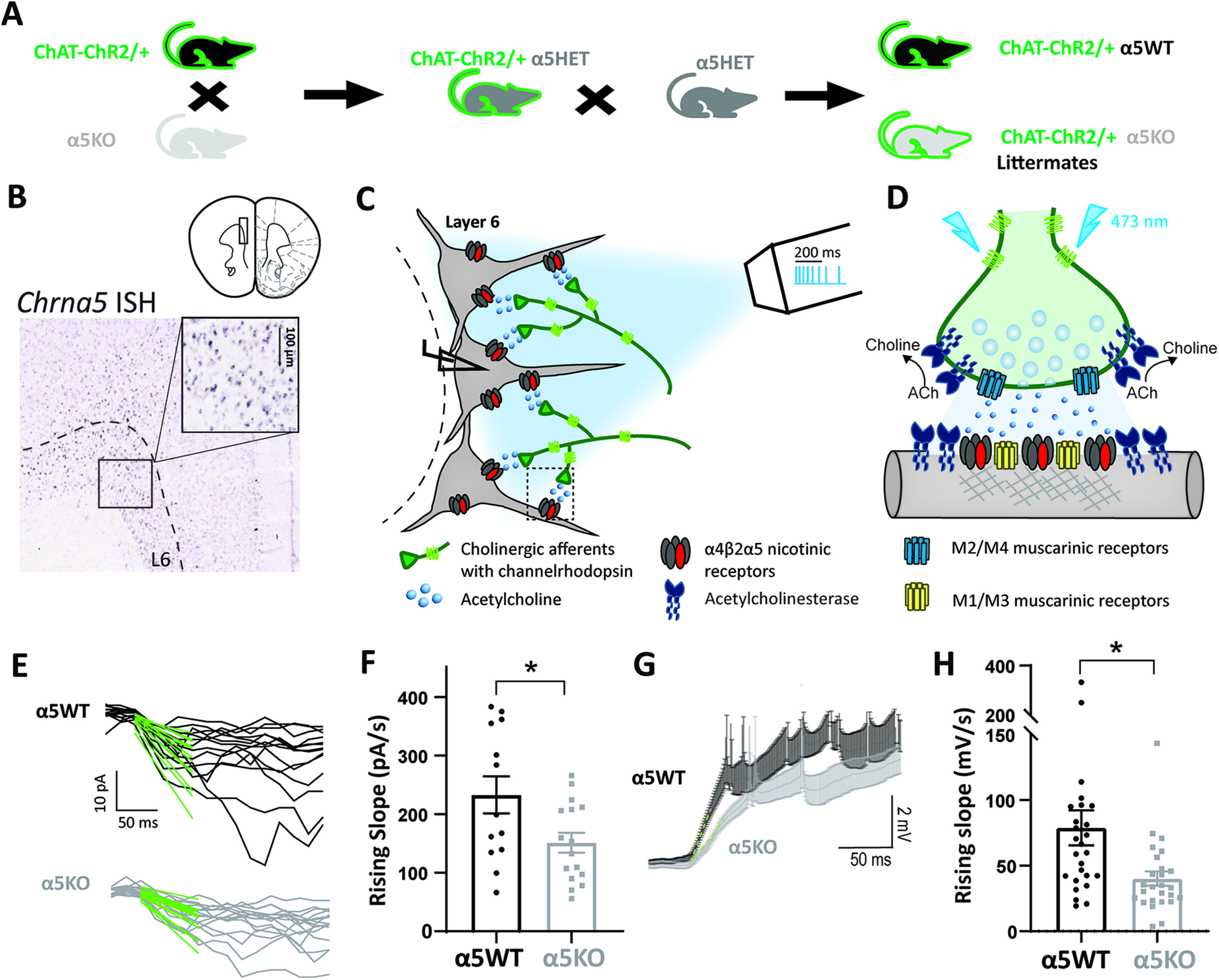
Chrna5 is critical for maintaining rapid onset of the response to optogenetic acetylcholine release. A, Schematic showing mouse crosses to obtain littermate wild-type (α5WT) and α5KO ChAT-ChR2 mice. B, In situ hybridization for Chrna5 mRNA in mouse prefrontal cortex shows expression in layer 6 neurons (image from the Allen Institute for Brain Science). Schematic showing coronal slice of mouse brain is adapted from the study by Paxinos and Franklin (2004). C, Schematic depicting experimental approach of whole-cell patch clamping of layer 6 pyramidal neurons in brain slices to measure responses to optogenetic release of acetylcholine from cholinergic afferents expressing channelrhodopsin. Optogenetic stimulation pattern of eight pulses (5 ms) in a frequency-accommodating manner (50 Hz decreasing to 10 Hz) is shown with scale bar (legend and detailed illustration of cholinergic synapses in D). D, Schematic showing optogenetic stimulation of a cholinergic synapse causing acetylcholine release onto layer 6 pyramidal neurons in the prefrontal cortex. The different effectors shown are postsynaptic nicotinic receptors with the Chrna5 subunit [(α4)2(β2)2α5 receptors], M1/M3 muscarinic receptors, presynaptic M2/M4 autoinhibitory muscarinic receptors, and acetylcholinesterase. E, Fast-rising phase of cholinergic responses in voltage clamp (−75 mV) in α5WT (top) and α5KO (bottom), and linear fit (green) to the first 50 ms of the response from light onset. F, Bar graph showing the rising slope (in pA/s) of the current determined from the linear fit in α5WT and α5KO. α5KO mice show significantly slower onset of cholinergic responses. G, Average current-clamp response of α5WT and α5KO (n = 26 cells each) H, Bar graph showing the rising slope (mV/s) of the depolarization determined from the linear fit in α5WT and α5KO (**p < 0.01, *p < 0.05, unpaired t test).
Pharmacology.
Atropine (200 nm; Sigma-Aldrich) was applied to block muscarinic receptors. AFDX-116 (300 nm; Tocris Bioscience) was used to block M2 muscarinic receptors. Dihydro-β-erythroidine (DhβE; 3 μm; Tocris Bioscience) was used to block β2-containing nicotinic receptors. CNQX (20 μm; Alomone Labs), APV (50 μm; Alomone Labs), and picrotoxin (50 μm, Alomone Labs) were used to block glutamate and GABAA receptors. Diisopropylfluorophosphate (DFP; 20 μm; Toronto Research Chemicals) was used to block acetylcholinesterase and induce spillover of acetylcholine. Nicotine (100 nm; Sigma-Aldrich) was used for desensitization experiments. All experiments with nicotine and DFP were done in the presence of atropine to isolate the nicotinic response. The selective agonist of the α4-α4 binding site, NS9283 (100 nm; Tocris Bioscience) was used to restore the rapid rise time of the nicotinic response in layer 6 neurons of the α5KO mice.
Experimental design, analysis, and statistics.
Age- and sex-matched mice were used for the main investigations of the effect of Chrna5 genotype. These experiments drew data from at least three mice per genotype, and recordings were made from one to two cells per slice and two to four cells per mouse. Results observed were consistent across the mice recorded within each genotype. Analysis of postsynaptic cholinergic responses was performed in Clampfit 10.2 (Molecular Devices) and Axograph. Raw traces were used for calculating the rising slope of the voltage-clamp response within 50 ms of light onset to get accurate measurement of the fast-onset kinetics. Downsampled traces were used to fit double exponentials to the cholinergic responses and for representation. Exponential and linear fits to the responses were performed on Axograph. Magnitude of the cholinergic responses in voltage clamp were determined by the peak current (in picoamperes) as well as the charge transferred (in picocoulombs) into the cell, which is measured as the area under the current response for 1 s starting from the light onset. GraphPad Prism 8 was used for statistical analysis and plotting graphs. Genotype differences in parameters of the cholinergic responses between α5WT and α5KO mice were compared with two-tailed unpaired t tests, where applicable. The effects of pharmacological manipulations in α5WT and α5KO mice were compared with Sidak's post hoc test, and repeated-measures/paired t tests were used if the recordings were obtained from the same cell prepharmacology and postpharmacology. When comparing the effect of a drug (e.g., nicotine) across time between α5WT and α5KO mice, data were analyzed using two-way repeated-measures ANOVA (or mixed-effects analysis in the case of missing time points for some cells) and Sidak's multiple-comparisons test used to compare α5WT and α5KO mice at each time point or between the baseline condition and different time points of drug application within each genotype. All ANOVAs were performed with the Geisser–Greenhouse correction for sphericity. To measure the time course of desensitization, fits of one-phase exponential decay were compared using the extra sum of squares F test (Prism 8). p values <0.05 were considered statistically significant, and effect sizes (Cohen's d) were included for the major results. Data are reported as the mean ± SEM.
Results
Chrna5 is critical for maintaining a rapid-onset response to endogenous acetylcholine
To assess the contribution of the α5 nicotinic subunit to endogenous cholinergic neurotransmission, we bred compound transgenic mice to achieve channelrhodopsin-labeled cholinergic fibers in littermate α5WT and α5KO mice, as illustrated in Figure 1A. We recorded regular-spiking pyramidal neurons in layer 6 to obtain a population of neurons known to have nicotinic acetylcholine receptors enriched for α5 (Bailey et al., 2010), as illustrated by Chrna5 expression in Figure 1B. Figure 1D shows a schematic of the hypothesized components of the cholinergic synapse onto these layer 6 pyramidal neurons based on available data (Levey et al., 1991; Zhang et al., 2002; Hedrick and Waters, 2015; Sparks et al., 2018). Since basal forebrain cholinergic neurons innervating the prefrontal cortex are thought to fire in brief bursts with spike frequency accommodation (Lee et al., 2005; Unal et al., 2012), we chose a pattern of optogenetic stimulation to mimic burst firing with eight pulses of blue light (473 nm) delivered in a frequency-accommodating manner as illustrated in the schematic in Figure 1C.
Our examination of layer 6 neuron cholinergic responses to optogenetic stimulation revealed that there were distinct differences in the onset kinetics between the α5WT and α5KO neurons. We fit a line to the fast-rising phase of the cholinergic responses (initial 50 ms) to calculate the rising slope of the response (Fig. 1E,G). The rising slope of the cholinergic current is significantly smaller in α5KO neurons (151 ± 17 pA/s) compared with the α5WT neurons (233 ± 32 pA/s; Cohen's d: 0.9; unpaired t test: t(27) = 2.34, p = 0.02; Fig. 1F). We expanded this examination and found that the genotype difference in rising kinetics is also reflected in the current-clamp responses, with the α5KO neurons having a significantly slower rising slope (40 ± 5 mV/s), ∼50% when compared with the α5WT neurons (79 ± 14 mV/s; Cohen's d: 0.7; unpaired t test: t(50)=2.68, p = 0.001; Fig. 1H). Of all the neurons recorded for optogenetic cholinergic responses in current clamp (n = 26 cells, 7 mice/genotype), similar proportions in α5WT (31%) and α5KO (27%) neurons show direct cholinergic-induced spiking. In this subset, the slower onset of cholinergic responses in the α5KO neurons translates to a significant delay in cholinergic activation-induced spiking (delay in onset of first spike in α5KO neurons: 87 ± 39 ms; unpaired t test comparing onset of first spike in α5WT and α5KO: t(14) = 2.25, p = 0.04). For both voltage-clamp and current-clamp examination, the peak response amplitudes themselves are not significantly different between the two genotypes (peak current, unpaired t test: t(26) = 0.38, p = 0.70; peak depolarization, unpaired t test: t(50) = 0.94, p = 0.35). There are no sex differences or sex-by-genotype interactions on any measure of the endogenous cholinergic response (data not shown). Furthermore, genotype differences in response-onset kinetics are observed in the absence of genotype differences in passive electrophysiological properties (Table 1). These results indicate that the α5 subunit is critical for the rapid onset of cholinergic activation in layer 6 of the prefrontal cortex.
Table 1.
Electrophysiological properties of α5WT and α5KO layer 6 pyramidal neurons
| α5WT | α5KO | Unpaired t test | Summary | |
|---|---|---|---|---|
| Resting membrane potential (mV) | −88 ± 1 | −88 ± 1 | t(50) = 0.1, p = 0.9 | NS |
| Spike amplitude (mV) | 76 ± 2 | 76 ± 2 | t(50) = 0.1, p = 0.9 | NS |
| Input resistance (MΩ) | 134 ± 8 | 142 ± 11 | t(50) = 0.6, p = 0.6 | NS |
| Capacitance (pF) | 64 ± 2 | 61 ± 2 | t(50) = 1.1, p = 0.3 | NS |
| Membrane time constant (ms) | 9 ± 1 | 9 ± 1 | t(50) = 0.1, p = 0.9 | NS |
The intrinsic properties of layer 6 pyramidal neurons do not differ statistically between α5WT and α5KO neurons. The endogenous cholinergic responses for these neurons are illustrated in Figure 1 The α5WT and α5KO groups each contain 26 neurons from 7 mice.
Autoinhibitory control of layer 6 cholinergic release is strong and Chrna5 independent
Cholinergic synaptic transmission is likely to include activation of presynaptic and postsynaptic muscarinic receptors, as shown in Figure 2A. Previous studies have shown M2 muscarinic autoinhibitory modulation of cholinergic transmission indirectly from brain tissue superfusates (Iannazzo and Majewski, 2000; Zhang et al., 2002; Brown, 2010). However, the extent of autoinhibitory control on layer 6 cholinergic transmission in the PFC is unknown and has not been examined directly using endogenous release combined with postsynaptic measurements (Obermayer et al., 2017; Colangelo et al., 2019) and is a question of particular interest because of the firing pattern of cholinergic neurons (Unal et al., 2012). Therefore, we examined the contribution of muscarinic receptors in the response to endogenous cholinergic neurotransmission to determine whether differences in muscarinic autoinhibition could underlie the altered kinetics in the α5KO. Of note, the pan-muscarinic receptor antagonist atropine increases the magnitude of the cholinergic responses in both genotypes (two-way repeated-measures ANOVA, atropine × genotype interaction: F(1,14) = 0.73, p = 0.41; effect of atropine: F(1,14) = 30.87, p < 0.0001, N = 5, 4 mice for α5WT, α5KO; Fig. 2B,C). We hypothesized that this overall response potentiation is because of the block of presynaptic autoinhibitory M2/M4 muscarinic receptors on cholinergic terminals. We tested this hypothesis by specifically blocking the M2 muscarinic receptor that is the main autoinhibitory receptor in the cortex (Levey et al., 1991; Zhang et al., 2002) using AFDX-116. Blocking M2 muscarinic receptors significantly potentiates the cholinergic response in both α5WT and α5KO (two-way repeated-measures ANOVA: effect of AFDX-116: F(1,6) = 16.32, p = 0.007) with no significant interaction between the effect of AFDX-116 and genotype (AFDX-116 × genotype interaction: F(1,6) = 3.50, p = 0.11; effect of genotype: F(1,6) = 0.18, p = 0.69). We compared the effects of atropine and AFDX-116, and found that the increase in cholinergic response magnitude caused by AFDX-116 (6.9 ± 2.0 pC) does not differ significantly from atropine (5.4 ± 1.0 pC; unpaired t test: t(26) = 0.04, p = 0.96; data are combined across both genotypes as there are no genotype differences). We conclude that both AFDX and atropine are equally effective in enhancing layer 6 cholinergic responses in both α5WT and α5KO. Our findings show for the first time that endogenous cholinergic synaptic transmission in layer 6 is ∼40% suppressed because of M2 muscarinic-mediated autoinhibition.
Figure 2.
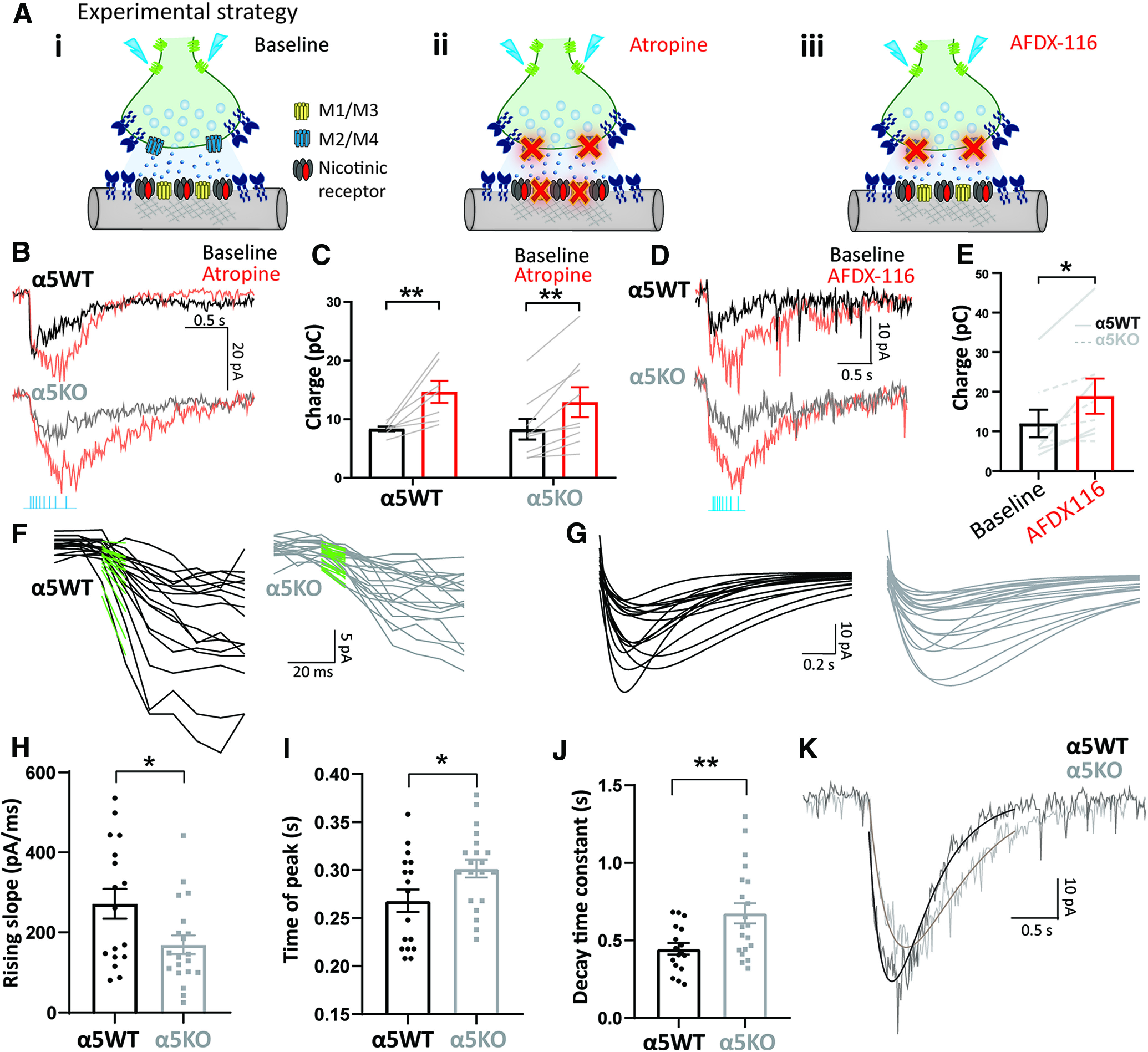
Autoinhibition of optogenetically released acetylcholine by M2 muscarinic receptors is strong and Chrna5 independent. A, i–iii, Experimental schematic illustrating cholinergic synaptic transmission at baseline (i), when all muscarinic receptors both postsynaptic M1/M3 and presynaptic M2/M4 are blocked by atropine (ii), and when presynaptic autoinhibitory M2/M4 muscarinic receptors are selectively blocked by AFDX-116 (iii). B, Cholinergic response of an α5WT (top) and α5KO neuron (bottom) in voltage clamp before and after the application of atropine (200 nm). C, Bar graph showing the cholinergic current charge at baseline and after atropine in α5WT and α5KO. Atropine significantly increases cholinergic current charge in both α5WT and α5KO (**p < 0.01, Sidak's post hoc test) to the same extent. D, Cholinergic response of an α5WT neuron (top) and α5KO neuron (bottom) in voltage clamp before and after the application of M2 antagonist AFDX-116 (300 nm). E, Bar graph showing cholinergic current charge before and after the application of AFDX-116 in α5WT and α5KO. Blocking M2 muscarinic receptors is sufficient to significantly increase cholinergic response magnitude in both α5WT and α5KO (paired t test: t(7) = 3.47, *p = 0.01). F, Fast-rising phase of cholinergic responses in α5WT (top) and α5KO (bottom), and linear fit (green) to the first 50 ms of the response from light onset after the application of atropine. G, Double-exponential fits to cholinergic responses in α5WT (left) and α5KO (right) neurons. H–J, Bar graph showing the rising slope (pA/s) of the current determined from the linear fit (H), time of peak current (I), and decay time constant (J) determined from double-exponential fits of the cholinergic responses in α5WT and α5KO (*p < 0.05, **p < 0.01, unpaired t test). K, Example cholinergic response with exponential fit of an α5WT and α5KO neuron illustrates slower onset, delayed peak, and slower decay in α5KO.
The genotype differences in the kinetics of the cholinergic responses are still evident following block of muscarinic receptors, with the α5KO neurons showing significantly slower rising slope (170 ± 23 pA/s) compared with the α5WT neurons (272 ± 37 pA/s; Cohen's d: 0.8; unpaired t test: t(35) = 2.40, p = 0.02; Fig. 2F,H). The time of peak current, as measured from the exponential fits to the responses (Fig. 2G,I) is significantly delayed by 33.5 ± 14.7 ms in the α5KO compared with the α5WT (unpaired t test: t(35) = 2.27, p = 0.03; Fig. 2I). In addition to this slower onset observed both at baseline and in the presence of atropine, we also find that the α5KO neurons show a significantly greater decay time constant (675 ± 65 ms) compared with α5WT neurons (446 ± 37 ms; unpaired t test: t(35) = 2.93, p = 0.006; Fig. 2J). Although the response kinetics are different between genotypes, the charge transfer does not differ by genotype either before or with atropine (Fig. 2C); nor are there differences in the response pharmacology. Nicotinic receptor-mediated responses to acetylcholine release are completely eliminated in both α5WT and α5KO (98% reduction) by the β2 nicotinic receptor antagonist DhβE (two-way repeated-measures ANOVA; effect of DhβE: F(1,4) = 38.96, p = 0.003), with no significant interaction or main effect of genotype, indicating that nicotinic receptors in the α5KO remain β2-containing receptors (effect of genotype: F(1,4) = 0.27, p = 0.63; genotype × DhβE interaction: F(1,4) = 0.11, p = 0.76). These findings show that even with muscarinic autoinhibition blocked, α5KO mice show aberrant kinetics of endogenous cholinergic responses, necessitating the need for alternative approaches targeting nicotinic receptors directly to correct cholinergic transmission.
Replication in a different optogenetic model: Chrna5 is required for rapid cholinergic kinetics
To test the robustness of the finding that α5KO mice show a selective deficit in kinetics of cholinergic activation, we repeated our experiments in a different line of mice targeting channelrhodopsin to cholinergic neurons. To create α5WT and α5KO offspring for these experiments, we bred compound crosses of α5het/ChAT-IRES-Cre and α5het/Ai32 mice, as illustrated in Figure 3. It has been reported recently that this fate-mapping approach will include a subset of neurons that are cholinergic only transiently during development and release glutamate in the adult (Nasirova et al., 2020). Therefore, we examined all recordings obtained in response to optogenetic stimulation for evidence of fast glutamatergic EPSCs time locked to the stimulus onset and included only recordings without such light-evoked EPSCs. We additionally performed a subset of experiments in the presence of glutamate receptor blockers CNQX and APV and found no significant differences from the data acquired without glutamate blockers (two-way repeated-measures ANOVA; effect of CNQX+APV: F(1,7) = 2.49, p = 0.16).
Figure 3.
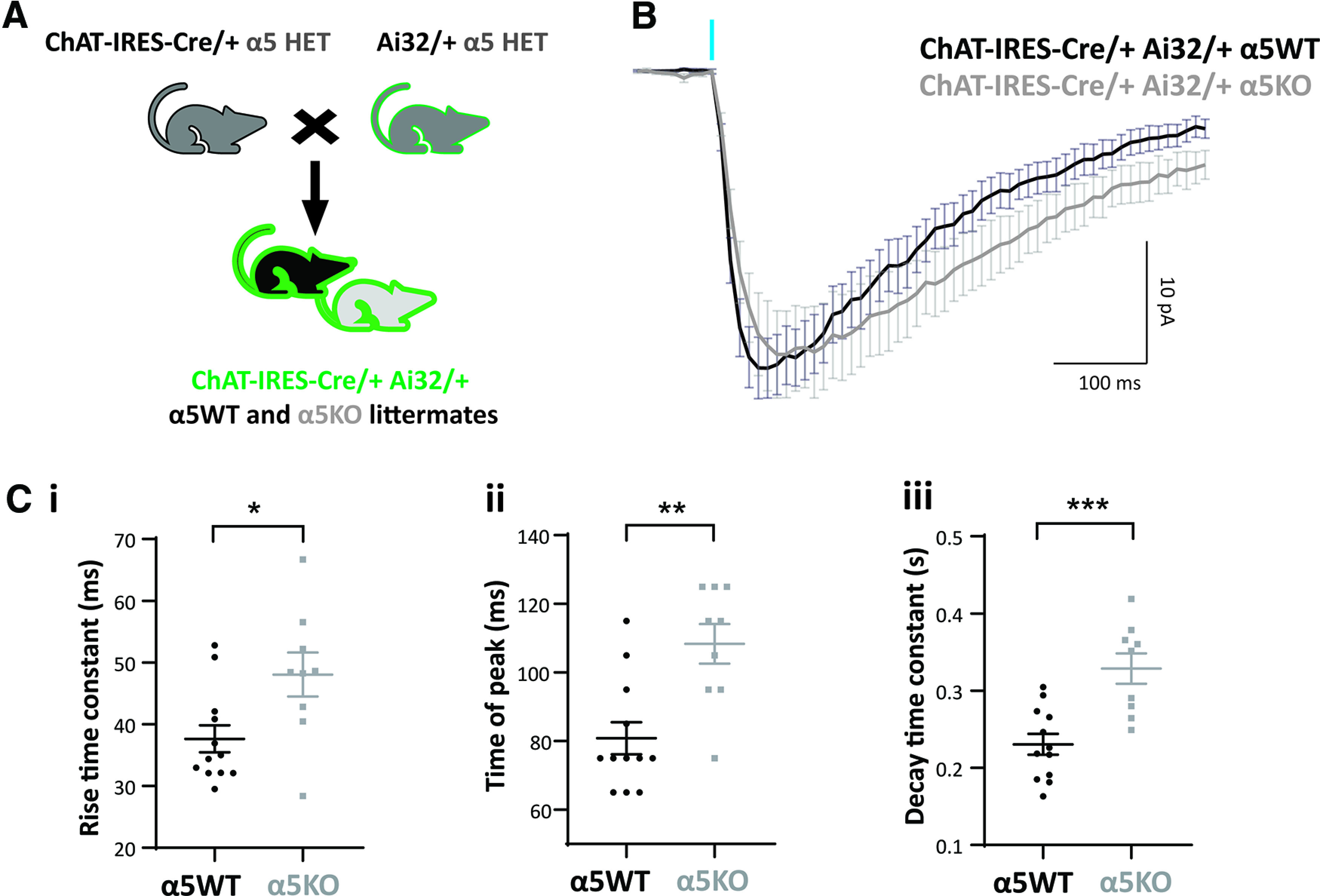
Chrna5 maintains rapid cholinergic kinetics in a different optogenetic model. A, Schematic showing mouse crosses to obtain littermate wild-type (α5WT) and α5 knock-out (α5KO) ChAT-IRES-Cre/+ Ai32/+ mice. B, Average cholinergic current obtained in response to one 5 ms flash of optogenetic stimulation in α5WT (n = 12) and α5KO (n = 9) neurons in the presence of atropine. C, i–iii, Bar graph showing rise time constant (i), time of peak (ii), and decay time constant (iii) in α5WT and α5KO (*p < 0.05, **p < 0.01, ***p < 0.001, unpaired t test). Cholinergic responses in the α5KO have a slower rise, a delayed peak, and a slower decay compared with α5WT.
The ROSA26 promoter in ChAT-IRES-Cre/+ Ai32/+ α5WT and α5KO expressed channelrhodopsin more strongly in the cholinergic afferents than the ChAT promoter, and a single pulse of light (5 ms) was sufficient to generate a cholinergic response of comparable magnitude to that examined in Figure 2. The average cholinergic current elicited in layer 6 pyramidal neurons by a single 5 ms pulse of light stimulation in α5WT (n = 12 cells) and α5KO (n = 9 cells) is shown in Figure 3B (N = 4 mice/genotype). Consistent with results from the α5KO ChAT-ChR2 mice, the cholinergic response in the α5KO is delayed compared with α5WT, with the rise time constant significantly greater in α5KO (48 ± 11 ms) compared with α5WT (38 ± 8 ms; unpaired t test: t(19) = 2.6, p = 0.02; Fig. 3C, i). Although the α5KO neurons show slower rise, they attain a peak magnitude (28 ± 4 pA) similar to that of α5WT neurons (29 ± 3 pA; unpaired t test: t(19) = 0.22, p = 0.8), but the α5KO peak occurs at a significantly delayed time point (delay in time of peak in α5KO: 27.5 ± 7 ms; unpaired t test t(19) = 3.74, p = 0.001; Fig. 3C, ii). The α5KO neurons also show a significantly slower decay time constant compared with α5WT (α5WT: 231 ± 13 ms; vs α5KO: 329 ± 20 ms; unpaired t test: t(19) = 4.27, p = 0.0004; Fig. 3C, iii). We are thus able to replicate the key deficits in cholinergic response kinetics observed in α5KO ChAT-ChR2 mice in α5KO ChAT-IRES-Cre/+ Ai32/+ mice. We conclude that Chrna5 is essential to maintain the rapid onset of response to acetylcholine release in layer 6 pyramidal neurons.
Chrna5 protects endogenous nicotinic signaling against desensitization
Acetylcholine levels in the prefrontal cortex increase during attention, arousal, exploration, and other cognitive tasks, as well as during stress (Pepeu and Giovannini, 2004; Gritton et al., 2016; Teles-Grilo Ruivo et al., 2017). To understand the consequences of prolonged acetylcholine presence occurring under situations of high cognitive effort, we blocked acetylcholine breakdown by inhibiting acetylcholinesterase irreversibly with DFP. The increase in cortical acetylcholine levels caused by DFP (Testylier and Dykes, 1996) has been shown to be similar in magnitude to that observed during successful cue detection in animals performing sustained attention tasks (Parikh et al., 2007). We measured layer 6 neuron responses to a train of optogenetic stimulation in α5WT and α5KO ChAT-ChR2 mice before and after acetylcholinesterase inhibition with DFP, in the continuous presence of atropine (Fig. 4A–C). Blocking acetylcholinesterase causes the optogenetic cholinergic responses to nearly double in the α5WT (cholinergic current charge at baseline: 16 ± 2 pC; after DFP: 32 ± 5 pC; Sidak's post hoc test: t(58) = 2.92, p = 0.01; N = 5 mice), but the increase is not significant in the α5KO (cholinergic current charge at baseline: 13 ± 2 pC; after DFP: 18 ± 3 pC; t(58) = 0.85, p = 0.64; N = 3 mice; Fig. 4C). Overall, there are significant main effects of DFP and genotype on the charge transfer from the optogenetic cholinergic response (two-way ANOVA; effect of DFP: F(1,58) = 7.29, p = 0.009; effect of genotype: F(1,58) = 5.90, p = 0.02; DFP × genotype interaction: F(1,58) = 2.34, p = 0.13). Post hoc comparison shows that after acetylcholinesterase inhibition, the cholinergic charge transfer is significantly lower in α5KO (18 ± 3 pC) compared with α5WT (32 ± 5 pC; Cohen's d: 0.8; Sidak's post hoc test: t(58) = 3, p = 0.008). These responses to endogenous acetylcholine release in the presence of cholinesterase inhibitors are reminiscent of genotype differences observed for direct responses to exogenous acetylcholine application (Bailey et al., 2010), where there is a prolonged high concentration of acetylcholine at the synapse because of saturation of acetylcholinesterase. We inferred that α5KO neurons show smaller cholinergic responses compared with α5WT neurons following cholinesterase block because they are susceptible to greater desensitization in the prolonged presence of high acetylcholine levels at the synapse.
Figure 4.
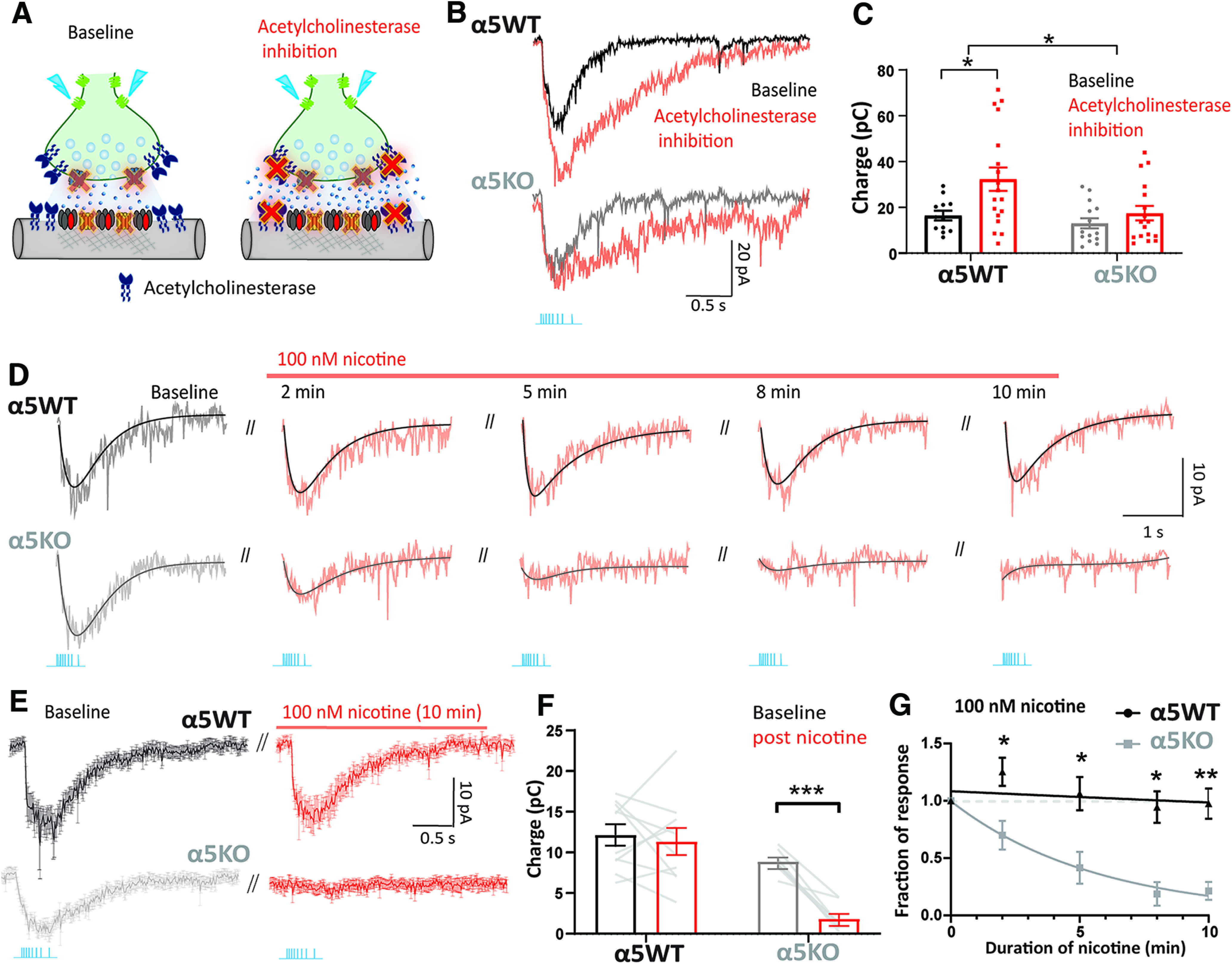
Chrna5 protects endogenous cholinergic signaling against desensitization. A, Experimental schematic illustrating cholinergic synaptic transmission at baseline when muscarinic receptors are blocked by atropine and following the application of an acetylcholinesterase inhibitor (DFP) to prevent the breakdown of acetylcholine. B, Nicotinic response of an α5WT neuron (top) and α5KO neuron (bottom) in voltage clamp before and after the application of the acetylcholinesterase inhibitor DFP (20 μm). C, Bar graph showing nicotinic current charge in α5WT and α5KO at baseline and in the presence of DFP. Cholinesterase inhibition causes a significant increase in the magnitude of cholinergic responses in α5WT (*p < 0.05, Sidak's post hoc test), but does not enhance cholinergic responses in the α5KO. D, Optogenetically evoked nicotinic responses with their exponential fits in α5WT and α5KO at different time points during the application of 100 nm nicotine for 10 min. E, Average nicotinic current in response to optogenetic acetylcholine release in α5WT (n = 5) and α5KO (n = 6) neurons before and after 10 min of nicotine administration. F, Bar graph showing nicotinic current charge before and after 10 min of nicotine administration in α5WT and α5KO (***p < 0.001, Sidak's post hoc test). G, Time course of change in endogenous cholinergic response during nicotine application is fit to an exponential decay. Best fit τdecay for α5KO: 5.7 min; for α5WT: 105.0 min (*p < 0.05 **p < 0.01, Sidak's post hoc test comparing α5WT and α5KO).
We hypothesized that this Chrna5 genotype difference in the ability of the optogenetic response to withstand prolonged exposure to acetylcholine reflects a difference in nicotinic receptor desensitization (Bailey et al., 2010; Grady et al., 2012). To test this hypothesis, we treated the brain slice with the drug nicotine, which is well known to desensitize nicotinic receptors in the cortex (Quick and Lester, 2002; Paradiso and Steinbach, 2003; Picciotto et al., 2008), for 10 min at a concentration known to predominantly exert desensitizing effects in this neuronal population (100 nm; Bailey et al., 2010). The α5WT optogenetic cholinergic response is unchanged by the application of nicotine, whereas the α5KO optogenetic response is rapidly attenuated (Fig. 4D–G). The cholinergic current charge measured in the α5WT and α5KO before and after the application of nicotine shows a significant interaction between the effect of nicotine and the genotype (two-way repeated-measures ANOVA; nicotine exposure × genotype interaction: F(1,16) = 9.8, p = 0.006). Post hoc comparisons show that the α5WT response is not significantly different before and after nicotine (12 ± 1 vs 11 ± 2 pC; Sidak's post hoc test: t(16) = 0.61, p = 0.8; N = 7 mice), whereas the α5KO response is greatly reduced post-nicotine administration (9 ± 0.5 vs 2 ± 0.6 pC; Cohen's d: 2.1; t(16) = 5.39, p < 0.001; N = 5 mice; Fig. 4F). An exponential decay fit to the graph of normalized response magnitude against nicotine exposure time reveals significant differences in the time course of desensitization between α5WT and α5KO (Fig. 4G). While α5KO neurons exhibit 80% desensitization at the end of a 10 min exposure and a τdecay of 5.7 min, α5WT neurons show only 2.5% desensitization (comparison of fits between α5WT and α5KO; extra sum of F squares test: F(1,70) = 32.6, p < 0.0001). We conclude that the elimination of endogenous cholinergic responses in the α5KO following acute exposure to 100 nm nicotine is because of the desensitization of nicotinic receptors lacking the α5 subunit. We further examined the effect of nicotine concentration on the ability of the α5 subunit to protect against desensitization, testing 500 nm nicotine in α5WT and α5KO ChAT-ChR2 mice. We observe that this higher concentration of nicotine can evoke desensitization in the α5WT mice (77% desensitization at the end of a 10 min exposure; τdecay of 6.7 min; n = 4 cells); however, the desensitization is markedly greater and faster in the α5KO (98% desensitization at the end of 10 min exposure; τdecay of 1.7 min; n = 3 cells; comparison of fits between α5WT and α5KO, Extra sum of F squares test: F(1,31) = 23.8, p < 0.0001). Furthermore, there is 50% recovery from 500 nm nicotine-induced desensitization in the α5WT mice after a 10 min washout period versus only 10% recovery in α5KO mice after the same washout period. We conclude that Chrna5 is protective against desensitization by low agonist concentrations, and more resilient than non-alpha5-containing nicotinic receptors found in the α5KO at higher agonist concentrations. Thus, Chrna5 is essential to protect prefrontal endogenous cholinergic signaling from desensitization induced either by high acetylcholine levels or acute exposure to nicotine.
Replication in a different optogenetic model: Chrna5 limits desensitization
We tested the robustness of our observation that α5KO endogenous cholinergic responses are more vulnerable to desensitization by examining whether it was independent of transgenic strategy for optogenetic release. Accordingly, we repeated experiments to measure nicotinic receptor desensitization in ChAT-IRES-Cre/+ Ai32/+ α5WT and α5KO mice, isolating the nicotinic response with atropine, and using the same accommodating-frequency optogenetic stimulus train used above. In these ChAT-IRES-Cre/+ Ai32/+ mice, the train response is stronger than in the ChATChR2 mice but is replicable and shows a significant genotype difference in the timing of its peak (t(29) = 2.20, p = 0.03), with α5KO peak occurring 37.2 ± 16.9 ms after the α5WT (N = 4 mice/genotype). The delay is observed in the absence of a difference in the peak cholinergic current (α5WT: 54 ± 6 pA; vs α5KO: 51 ± 4 pA; unpaired t test: t(29) = 0.32, p = 0.7) or in the cholinergic charge transfer (α5WT: 21 ± 3 pC; vs α5KO: 24 ± 2 pC; unpaired t test: t(29) = 0.90, p = 0.4). In short, the properties of the train responses in the ChAT-IRES-Cre/+ Ai32/+ α5WT and α5KO mice are suitable for testing whether loss of α5 increases vulnerability to nicotine desensitization.
For this experiment, the cholinergic response magnitude in voltage clamp to a train of optogenetic stimulation was measured at different time points during the application of 100 nm nicotine (Fig. 5). In this optogenetic line, there is again a significant interaction between the effect of nicotine exposure and genotype (two-way repeated-measures ANOVA: nicotine × genotype interaction: F(4,40) = 10.19, p < 0.0001). Similar to results obtained in ChAT-ChR2 α5KO mice, we see that ChAT-IRES-Cre/+ Ai32/+ α5KO mice show almost complete desensitization at the end of a 10 min exposure to 100 nm nicotine (fraction of response at 5 min: 0.36 ± 0.06; Sidak's post hoc test: t(5) = 10.80, p = 0.0005; fraction of response at 10 min: 0.16 ± 0.04; Sidak's post hoc test: t(5) = 20.06, p < 0.0001). While considerably milder, there is also significant nicotine-elicited desensitization in ChAT-IRES-Cre/+ Ai32/+ α5WT mice (fraction of response at 5 min: 0.61 ± 0.09; Sidak's post hoc test: t(5) = 4.20, p = 0.03; fraction of response at 10 min: 0.57 ± 0.08; Sidak's post hoc test: t(5) = 5.77, p = 0.009). The stronger optogenetic release of acetylcholine in this mouse line helps to illustrate the degree to which Chrna5 enables the α5WT to resist desensitization. The time course of desensitization is significantly different between α5WT and α5KO mice: while α5KO mice exhibit 83% desensitization at the end of 10 min nicotine exposure and a τdecay of 4.8 min, α5WT mice show only 43% desensitization and a τdecay of 16.3 min (comparison of fits between α5WT and α5KO mice; extra sum of F squares test: F(1,70) = 32.6, p < 0.0001; Fig. 5B).
Figure 5.
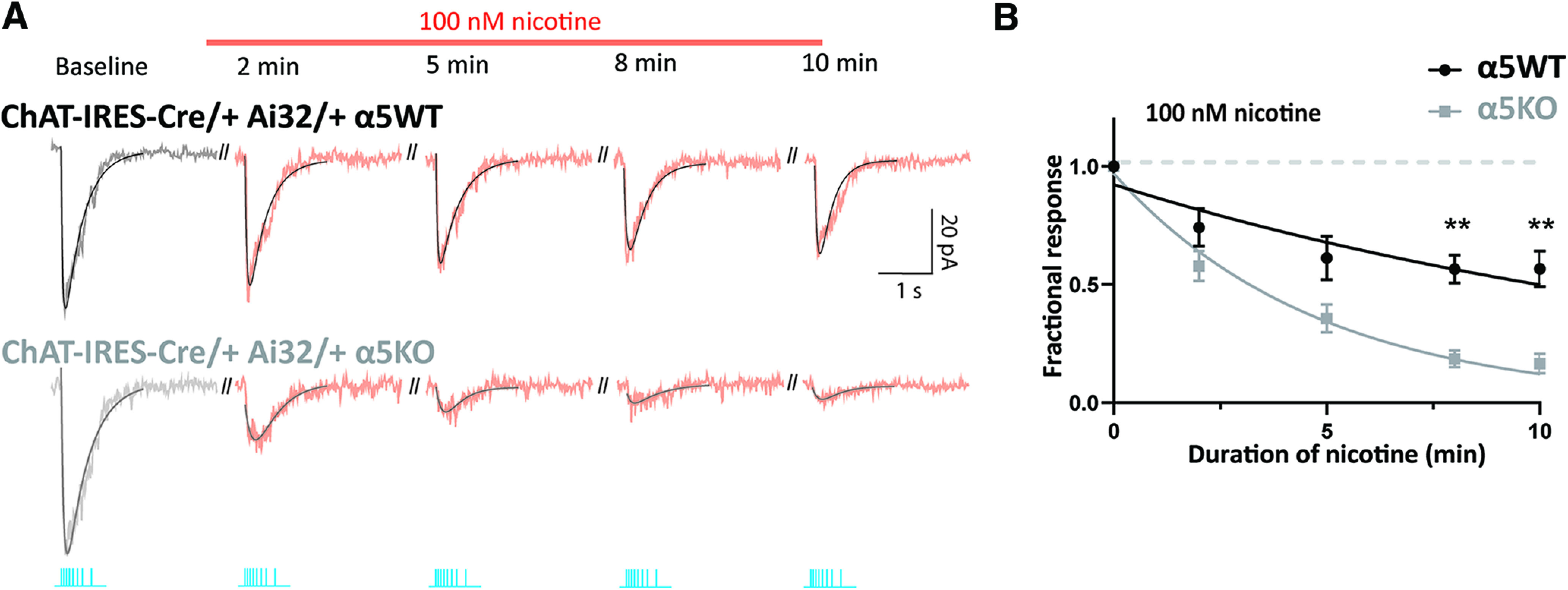
Chrna5 attenuates desensitization of endogenous cholinergic signals in a different optogenetic model. A, Optogenetically evoked cholinergic responses in an α5WT and α5KO ChAT-IRES-Cre/+ Ai32/+ neuron when muscarinic receptors are blocked by atropine, recorded at different time points during the application of 100 nm nicotine. B, Time course of change in endogenous nicotinic response as nicotine is applied is fit to an exponential decay. Best fit τdecay for α5KO: 4.8 min, for α5WT: 16.3 min (**p < 0.01, Sidak's post hoc test comparing α5WT and α5KO).
These results confirm the critical role of Chrna5 in protecting endogenous cholinergic signaling from desensitization. Together with the results in Figures 1–5, we are able to show using two different transgenic strategies for optogenetic acetylcholine release that Chrna5 in layer 6 of the prefrontal cortex has the following two roles: (1) Chrna5 is essential for a rapid onset of cholinergic activation; and (2) Chrna5 protects prefrontal endogenous cholinergic signaling from desensitization induced either by high acetylcholine levels or by acute exposure to nicotine.
Rescuing rapid cholinergic response onset by targeting an unorthodox binding site
The α5KO mice show a deficit in the timing of cholinergic excitation that would delay input integration and the activation of postsynaptic partners, a finding consistent with the attention deficits observed in α5KO mice (Bailey et al., 2010). The manipulations that we have tested, including blocking presynaptic muscarinic autoinhibition (Fig. 2) and inhibiting acetylcholinesterase (Fig. 4), fail to rescue this slow onset of cholinergic excitation. Our optogenetic results suggest that attempts to rescue endogenous nicotinic signaling in α5KO mice must navigate a careful path between hastening response onset and avoiding desensitization. Therefore, we chose to examine a strategy of positive allosteric modulation, aiming to potentiate the nicotinic response without altering the duration of nicotinic receptor stimulation. In the absence of the α5 subunit, α4β2 nicotinic receptors in α5KO mice can adopt the following two possible stoichiometries: (α4)2(β2)3 receptors with greater affinity to acetylcholine (EC50, ∼1 μm); or (α4)3(β2)2 receptors with lower affinity to acetylcholine (EC50, ∼100 μm; Tapia et al., 2007). Accordingly, we took advantage of NS9283, a selective agonist for the unorthodox α4-α4 nicotinic receptor binding site found in the lower-affinity (α4)3(β2)2 nicotinic receptors (Grupe et al., 2013; Olsen et al., 2013, 2014; Jain et al., 2016). Stimulation of this nontraditional nicotinic receptor binding site does not produce a current in itself but instead enhances the affinity for acetylcholine at the orthodox α4-β2 binding sites (Wang et al., 2015; Indurthi et al., 2016; Wang and Lindstrom, 2018). Such enhancement in wild-type mice improves cognitive performance, bringing significant improvement in social recognition memory and a decrease in omission errors on attention tasks (Timmermann et al., 2012).
Here, we asked whether NS9283 can rescue the onset kinetics of the endogenous cholinergic response in α5KO mice without triggering desensitization. One requirement for this approach to work is that at least a proportion of nicotinic receptors in α5KO mice must have adopted the (α4)3(β2)2 stoichiometry (Fig. 6A). We measured cholinergic responses to a train of optogenetic stimulation in ChAT-ChR2 α5KO mice in the presence of atropine before and after the application of 100 nm NS9283 for 5 min (Fig. 6B–E). We found that this low concentration of NS9283 is effective at speeding the onset of nicotinic activation (increase in rising slope: 80 ± 30 pA/s; N = 3 mice; paired t test: t(8) = 2.66, p = 0.03; Fig. 6C). NS9283 also increases the peak amplitude of the response (change: 12 ± 2 pA; paired t test: t(9) = 4.99, p = 0.0008). Further assessment of the restorative capacity of NS9283 in current clamp again shows that it significantly increases the onset speed of cholinergic responses (increase in rising slope: 12 ± 5 mV/s; paired t test: t(9) = 2.41, p = 0.04; Fig. 6E) and the response amplitude (change: 4 ± 1 mV; paired t test: t(9) = 5.18, p = 0.0006). Figure 6F shows example exponential fits to cholinergic responses in an α5WT, α5KO, and the same α5KO neuron after application of 100 nm NS9283. We see that the application of 100 nm NS9283 is able to rescue the slow-onset kinetics of α5KO cholinergic responses to match the α5WT response. We then examined the effect of 100 nm NS9283 in the absence of any muscarinic block in a subset of neurons and found a similar enhancement of the rising slope as observed with atropine (increase in rising slope without atropine: 110 ± 50 pA/s; unpaired t test comparing increase with and without atropine: t(13) = 0.53, p = 0.60). The increase in peak amplitude was also similar (increase in peak current without atropine: 13 ± 3 pA; unpaired t test comparing increase with and without atropine: t(14) = 0.16, p = 0.88). We conclude that the ability of NS9283 to correct aberrant cholinergic kinetics in the α5KO is effective even under baseline conditions and thus represents a generalizable result with implications for the treatment of attention disorders.
Figure 6.

Pharmacological restoration of rapid kinetics of endogenous cholinergic responses after disruption of Chrna5. A, Schematic illustrating the two possible stoichiometries of nicotinic receptors in the α5KO. NS9283 is a selective agonist for the α4-α4 site found in (α4)3(β2)2 receptors. B, Average endogenous cholinergic current traces before and after administration of 100 nm NS9283 in the α5KO (n = 10 cells), with inset showing the fast-rising phase of the response with linear fit (green) to the first 50 ms of the response from light onset. C, Bar graph showing the rising slopes for the optogenetically evoked currents before and after NS9283 in the α5KO. D, Average endogenous cholinergic depolarizations (inset, fast-rising phase of the response with linear fit in green, n = 10 cells). E, Bar graph showing the rising slopes for the optogenetically evoked depolarizations before and after NS9283 administration in the α5KO. Atropine was present throughout to isolate the nicotinic response; but similar potentiation by NS9283 of the onset speed and peak current of the endogenous cholinergic response was also observed without muscarinic blockade (see Results). F, Example exponential fits to nicotinic responses in α5WT and α5KO neurons, as well as in the same α5KO neuron following application of 100 nm NS9283. The application of NS9283 rescues onset kinetics of α5KO cholinergic responses to match the α5WT. G, Optogenetically evoked nicotinic responses along with linear fits of the same α5KO neuron shown in F at 5 and 10 min of NS9283 application and following a 15 min washout period. Potentiation caused by NS9283 is long lasting, and optogenetic release of endogenous acetylcholine can be repeated without triggering the desensitization of nicotinic receptors (*p < 0.05, paired t test).
Furthermore, we observe that the rescue of onset kinetics and potentiation caused by NS9283 is long lasting and does not trigger significant desensitization of nicotinic receptors on subsequent endogenous acetylcholine release (Fig. 6G). There is no significant effect of continuous NS9283 application on the amplitude of endogenous cholinergic responses obtained in α5KO (one-way repeated-measures ANOVA: F(1.9,3.9) = 2.74, p = 0.18). We finally tested higher concentrations (300 nm, 1 μm) of NS9283 in the presence of atropine to examine the dose dependency of its potentiation of endogenous nicotinic stimulation (Fig. 7). We observe that NS9283 potentiates the rising slope of the endogenous cholinergic responses in the α5KO in a dose-dependent manner (increase in rising slope: 100 nm: 80 ± 30 pA/s; 300 nm: 404 ± 72 pA/s; 1 μm: 837 ± 138 pA/s; one-way ANOVA: F(2,18) = 23.75, p < 0.0001; Sidak's post hoc test: 100 nm vs 300 nm: t(18) = 3.60, p = 0.006; 300 nm vs 1 μm: t(18) = 3.83, p = 0.004; 100 nm vs 1 μm: t(18) = 6.81, p < 0.0001; Fig. 7A,B). The peak amplitude also increases concurrently, without desensitization on serial application of increasing NS9283 concentrations (increase in peak current: 100 nm: 12 ± 2 pA; 300 nm: 54 ± 12 pA; 1 μm: 130 ± 26; one-way ANOVA: F(2,19) = 23.8, p < 0.0001). A similar pattern was observed in a sample investigated in current clamp. In this sample, increasing rising slope was also evident (increase in rising slope: 100 nm: 12 ± 5 mV/s; 300 nm: 30 ± 10 mV/s; 1 μm: 64 ± 24 mV/s; one-way ANOVA: F(2,19) = 4.88, p = 0.02; Sidak's post hoc test: 100 nm vs 1 μm: t(19) = 3.12, p = 0.02; Fig. 7C,D) and greater depolarization leading to spiking (increase in peak depolarization: 100 nm: 4 ± 1 mV; 300 nm: 56 ± 12 mV; 1 μm: 76 ± 10 mV; one-way ANOVA: F(2,19) = 20.7, p < 0.0001). The persistent improvement in the endogenous cholinergic excitation of layer 6 neurons with prolonged application and increasing concentrations of NS9283 highlights its capacity to restore disrupted nicotinic signaling. Using NS9283, we are able to rescue the onset of cholinergic responses in α5KO to achieve the rapid timing observed in α5WT without engaging desensitization mechanisms.
Figure 7.
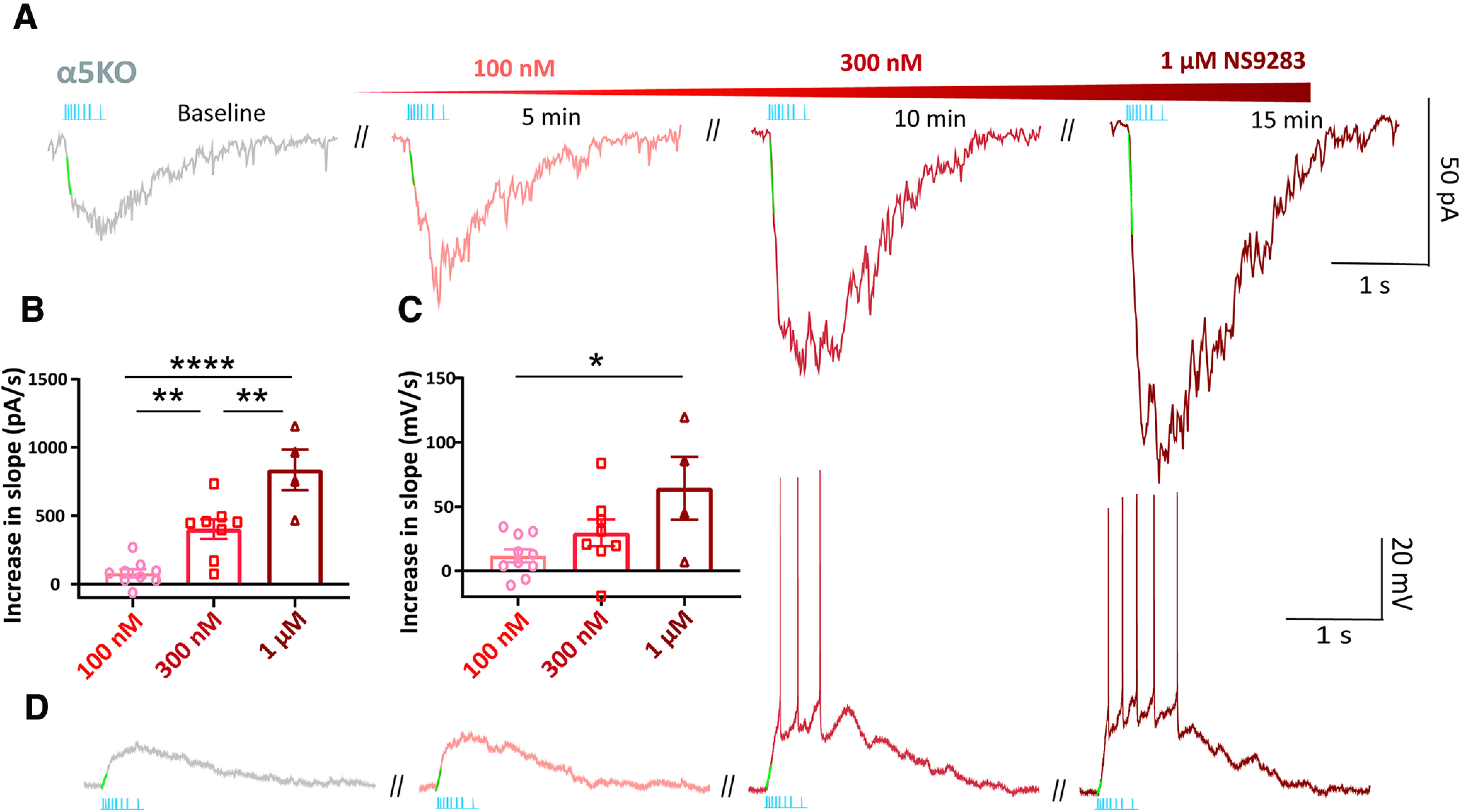
NS9283 enhances endogenous cholinergic signaling in prefrontal cortex of α5KO mice. Dose-dependent increase in the effects of NS9283 (100 nm, 300 nm, and 1 μm) to potentiate the endogenous cholinergic response, in terms of rising slope and amplitude. A, Example voltage-clamp responses from one neuron exposed serially to increasing NS9283 concentrations. B, C, Summary bar graph showing the effects of increasing doses of NS9283 on the rising slope of the currents (B) and depolarization (C) evoked by optogenetic stimulation of endogenous cholinergic release. D, Example current-clamp nicotinic responses from one neuron exposed serially to increasing NS9283. Atropine was present throughout to isolate the nicotinic component of the response. Clearly, NS9283 retains the ability to enhance the endogenous nicotinic response over time without triggering desensitization of this response (*p < 0.05, **p < 0.01, ****p < 0.0001, Sidak's post hoc test).
Discussion
Our results reveal that the α5 subunit encoded by Chrna5 is necessary to generate rapid onset of responses to endogenous acetylcholine released optogenetically. In this way, it regulates the timing of the peak cholinergic modulation of layer 6 pyramidal neurons in prefrontal cortex, but not its magnitude. In addition, the α5 subunit protects the endogenous cholinergic signaling from desensitization induced by prolonged exposure to acetylcholine or acute nicotine. Finally, we show that the slow onset of cholinergic responses in mice lacking the α5 subunit can be rescued using NS9283, a selective agonist for the unorthodox α-α binding site on (α4)3(β2)2 nicotinic receptors.
Chrna5 permits a rapid response to endogenous cholinergic signaling
Rapid cholinergic modulation of the prefrontal cortex is critical for attention. Layer 6 pyramidal neurons are key players in this phenomenon, since a large proportion are corticothalamic and can exert a direct top-down influence on incoming sensory inputs (Kassam et al., 2008; Thomson, 2010). Layer 6 corticothalamic neurons express the α5 nicotinic receptor subunit encoded by Chrna5, which is critical for the performance of demanding attention tasks (Bailey et al., 2010). We observe that neurons lacking α5 show significantly impaired kinetics in responding to endogenous acetylcholine release, exhibiting a much slower rise and delayed time of peak. The slow onset of cholinergic activation in α5KO results in a delay of up to ∼100 ms in initiation of acetylcholine-induced spiking in layer 6 neurons. We posit that the delay in cholinergic activation in the α5KO could result in the failure to integrate inputs or activate postsynaptic targets within a window critical for the detection of sensory cues, leading to attention deficits observed in these mice (Bailey et al., 2010).
Temporal constraints on endogenous cholinergic signaling
We demonstrate for the first time that the cholinergic inputs to the layer 6 pyramidal neurons are under strong M2 muscarinic receptor-mediated autoinhibition of release leading to ∼40% suppression of the full response by active presynaptic M2 receptors. Releasing the autoinhibitory brake on acetylcholine release does not improve the aberrant cholinergic kinetics in the α5KO. However, the strong muscarinic autoinhibition of endogenous cholinergic release onto layer 6 pyramidal neurons would be predicted to restrict the emphasis onto the fast-rising phase of a response to a train of cholinergic stimuli. The acetylcholine released to the first stimulus will activate the presynaptic M2 receptors and suppress further release because of the subsequent stimuli. The potential for such autoinhibition highlights the importance of the α5 nicotinic subunit in generating an initial rapid response to the acetylcholine release. Together with the high expression of the metabolic enzyme acetylcholinesterase in deep layers of the prefrontal cortex (Sendemir et al., 1996; Anderson et al., 2009), our results suggest that prefrontal layer 6 cholinergic modulation is hardwired for rapid transient effects with Chrna5 ensuring rapid postsynaptic activation.
Cognitive ramifications of rapid cholinergic signaling
Acetylcholine release in the cortex has been shown to vary on rapid timescales with behavioral state–cholinergic axon activation in the barrel cortex correlates rapidly with whisking behavior (Eggermann et al., 2014). Similarly, the activity of cholinergic axons in the auditory cortex rapidly shifts and is predictive of behavioral context (Kuchibhotla et al., 2017). Fast cholinergic transients are observed in the prefrontal cortex in association with rewards and cue detection on sustained attention tasks (Parikh et al., 2007; Gritton et al., 2016; Teles-Grilo Ruivo et al., 2017). Prefrontal nicotinic receptors are required for the initial transition from low-gamma to high-gamma states coinciding with cue presentation in an attention task (Howe et al., 2017). The attention deficits observed in mice lacking Chrna5 performing a five-choice serial reaction time test were also critically dependent on timing (Bailey et al., 2010): α5KO mice were impaired only at the briefest and most demanding stimulus durations. The slower cholinergic activation of layer 6 corticothalamic neurons in α5KO would be consistent with a failure to detect brief cues within a critical window for integration.
Chrna5 to protect the synaptic cholinergic response
While rapid cholinergic signaling in the PFC is critical for the detection of sensory cues, cholinergic tone in the PFC is important under challenging conditions of sustained attention (Sarter and Lustig, 2019, 2020). High cholinergic tone in the PFC is associated with sustained attention and top-down attentional control in the presence of distractor challenges, and can last well beyond the task duration (Himmelheber et al., 2000; St. Peters et al., 2011; Paolone et al., 2012). Prefrontal acetylcholine levels also greatly increase during conditions requiring high cognitive effort and stress (Mark et al., 1996; Pepeu and Giovannini, 2004; Teles-Grilo Ruivo et al., 2017). To replicate this scenario ex vivo, we prolonged acetylcholine presence by blocking acetylcholinesterase irreversibly and examined the role of Chrna5. This experiment revealed a sharp dichotomy between the genotypes, where cholinergic responses after acetylcholinesterase block were much smaller in the α5KO mice compared with the α5WT mice. We interpret this as a greater vulnerability of non-α5 nicotinic receptors in the α5KO mice to desensitization in the prolonged presence of high acetylcholine concentrations when optogenetically stimulated after cholinesterase block. Furthermore, acute exposure to low levels of nicotine thought to mimic the concentrations seen in smokers (Rose et al., 2010) sharply attenuated synaptic cholinergic transmission in the α5KO mice, while α5WT cholinergic transmission was resilient, revealing that the α5 nicotinic subunit has a critical role in protecting against desensitization. These experiments demonstrate using endogenous acetylcholine release to physiological stimulation patterns, a critical role for the α5 subunit in conferring a protective role against desensitization during elevated cholinergic tone or acute nicotine exposure.
A novel treatment approach and clinical relevance
The loss of Chrna5 causes profound attention deficits (Bailey et al., 2010; Howe et al., 2018), and it is of great interest to identify pharmacological interventions to correct this dysfunction. However, the vulnerability of α5KO animals to complete desensitization of their endogenous cholinergic signaling is of utmost importance when considering approaches to treat the attention deficits with cholinergic modulators. Treatment with cholinesterase inhibitors in animals lacking the α5 subunit is problematic as it could engage powerful desensitization of endogenous prefrontal cholinergic signaling. We instead show that aberrant cholinergic kinetics, which may underlie attention deficits in α5KO animals, can be rescued partly by NS9283, an agonist for the unorthodox α-α binding site that allosterically enhances nicotinic receptor affinity without causing desensitization. A low concentration of NS9283 (100 nm) was able to restore the slow onset of synaptic cholinergic responses in α5KO to α5WT levels. Unlike nicotine, which is an agonist, or DFP, which increases overall cholinergic tone thereby causing greater activation of nicotinic receptors, NS9283 does not activate the nicotinic receptors by itself. Instead, the action of NS9283 at the unorthodox α4-α4 binding site facilitates efficient activation by acetylcholine at the canonical α4β2 sites (Wang et al., 2015; Indurthi et al., 2016). We find that NS9283 by itself does not induce desensitization in the α5KO but exerts dose-dependent increases in cholinergic response speed and amplitude. NS9283 has been previously shown to improve cognitive performance in wild-type animals (Timmermann et al., 2012), pointing to an underestimated potential of this drug to improve attention in compromised states. Our work provides a rationale to consider NS9283 to physiologically manipulate endogenous cholinergic signaling to improve attention in pathologic states. This may be particularly relevant for the treatment of attention disorders in humans carrying prevalent nonfunctional polymorphisms in the Chrna5 gene (Bierut et al., 2008).
Recent examinations of the cholinergic system have shown great interest in mechanisms underlying diverse spatiotemporal scales of cholinergic signaling in the cortex (Disney and Higley, 2020; Sarter and Lustig, 2020; Venkatesan et al., 2020). Although much remains unknown about patterns of cortical acetylcholine release and subsequent network-level effects (Laszlovszky et al., 2020), we performed a direct characterization of Chrna5 in ex vivo brain slices using optogenetic acetylcholine release. Our study reveals a specialized role of the α5 nicotinic receptor subunit in generating the rapid cholinergic modulation of the prefrontal cortex known to be critical for cognition. Such kinetic properties may define critical windows for cognitive processing. We also show that the α5 nicotinic subunit protects rapid cholinergic signaling from desensitization induced by elevated acetylcholine levels or nicotine exposure. Finally, we demonstrate that rapid cholinergic signaling can be rescued in the absence of α5 without triggering desensitization by allosterically enhancing nicotinic receptors with NS9283, an agonist for the unorthodox binding site. Future work investigating the role of Chrna5 in vivo, where modulators and local excitatory and inhibitory circuitry converge onto pyramidal cells, is of interest to further elucidate its contributions to physiological response parameters. The current approach improves our understanding of cholinergic modulation of attention circuits and identifies a pharmacological target to restore the rapid kinetics of cholinergic signaling in pathologic conditions.
Footnotes
The authors declare no competing financial interests.
This research was funded by the Canadian Institutes of Health Research (Grant CIHR MOP 89825, to E.K.L.; Grant CIHR PJT-153101, to E.K.L.), the Canada Research Chair in Developmental Cortical Physiology (E.K.L.), an Ontario Graduate Scholarship (S.V.), and the Mary Beatty Fellowship from the University of Toronto (S.V.). We thank Janice McNabb and Ha-Seul Jeoung for expert technical assistance. Earlier presentations of this work received valuable feedback from Dr. Junchul Kim, Dr. Beverly Orser, and Dr. Steve Prescott of the University of Toronto.
References
- Anderson LA, Christianson GB, Linden JF (2009) Mouse auditory cortex differs from visual and somatosensory cortices in the laminar distribution of cytochrome oxidase and acetylcholinesterase. Brain Res 1252:130–142. 10.1016/j.brainres.2008.11.037 [DOI] [PubMed] [Google Scholar]
- Baenziger JE, daCosta CJB (2013) Molecular mechanisms of acetylcholine receptor-lipid interactions: from model membranes to human biology. Biophys Rev 5:1–9. 10.1007/s12551-012-0078-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CDC, De Biasi M, Fletcher PJ, Lambe EK (2010) The nicotinic acetylcholine receptor alpha5 subunit plays a key role in attention circuitry and accuracy. J Neurosci 30:9241–9252. 10.1523/JNEUROSCI.2258-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, et al. (2008) Variants in the nicotinic receptors alter the risk for nicotine dependence. Am J Psychiatry 165:1163–1171. 10.1176/appi.ajp.2008.07111711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA. (2010) Muscarinic acetylcholine receptors (mAChRs) in the nervous system: some functions and mechanisms. J Mol Neurosci 41:340–346. 10.1007/s12031-010-9377-2 [DOI] [PubMed] [Google Scholar]
- Colangelo C, Shichkova P, Keller D, Markram H, Ramaswamy S (2019) Cellular, synaptic and network effects of acetylcholine in the neocortex. Front Neural Circuits 13:24. 10.3389/fncir.2019.00024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Bouger P, Chudasama Y, Cardinal RN, Robbins TW (2004) Cortical cholinergic function and deficits in visual attentional performance in rats following 192 IgG-saporin-induced lesions of the medial prefrontal cortex. Cereb Cortex 14:922–932. 10.1093/cercor/bhh052 [DOI] [PubMed] [Google Scholar]
- Disney AA, Higley MJ (2020) Diverse spatiotemporal scales of cholinergic signaling in the neocortex. J Neurosci 40:720–725. 10.1523/JNEUROSCI.1306-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Kremer Y, Crochet S, Petersen CCH (2014) Cholinergic signals in mouse barrel cortex during active whisker sensing. Cell Rep 9:1654–1660. 10.1016/j.celrep.2014.11.005 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. (1995) Cellular basis of working memory. Neuron 14:477–485. 10.1016/0896-6273(95)90304-6 [DOI] [PubMed] [Google Scholar]
- Grady SR, Wageman CR, Patzlaff NE, Marks MJ (2012) Low concentrations of nicotine differentially desensitize nicotinic acetylcholine receptors that include α5 or α6 subunits and that mediate synaptosomal neurotransmitter release. Neuropharmacology 62:1935–1943. 10.1016/j.neuropharm.2011.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritton HJ, Howe WM, Mallory CS, Hetrick VL, Berke JD, Sarter M (2016) Cortical cholinergic signaling controls the detection of cues. Proc Natl Acad Sci U S A 113:E1089–E1097. 10.1073/pnas.1516134113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupe M, Jensen AA, Ahring PK, Christensen JK, Grunnet M (2013) Unravelling the mechanism of action of NS9283, a positive allosteric modulator of (α4)3(β2)2 nicotinic ACh receptors. Br J Pharmacol 168:2000–2010. 10.1111/bph.12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Zhang T, Ni T, Zhu L, Liu D, Chen G, Lin H, Chen T, Guan F (2019) Relationship of common variants in CHRNA5 with early-onset schizophrenia and executive function. Schizophr Res 206:407–412. 10.1016/j.schres.2018.10.011 [DOI] [PubMed] [Google Scholar]
- Harpsøe K, Ahring PK, Christensen JK, Jensen ML, Peters D, Balle T (2011) Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci 31:10759–10766. 10.1523/JNEUROSCI.1509-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick T, Waters J (2015) Acetylcholine excites neocortical pyramidal neurons via nicotinic receptors. J Neurophysiol 113:2195–2209. 10.1152/jn.00716.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick T, Danskin B, Larsen RS, Ollerenshaw D, Groblewski P, Valley M, Olsen S, Waters J (2016) Characterization of channelrhodopsin and archaerhodopsin in cholinergic neurons of Cre-Lox transgenic mice. PLoS One 11:e0156596. 10.1371/journal.pone.0156596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmelheber AM, Sarter M, Bruno JP (2000) Increases in cortical acetylcholine release during sustained attention performance in rats. Brain Res Cogn Brain Res 9:313–325. 10.1016/s0926-6410(00)00012-4 [DOI] [PubMed] [Google Scholar]
- Hong LE, Yang X, Wonodi I, Hodgkinson CA, Goldman D, Stine OC, Stein ES, Thaker GK (2011) A CHRNA5 allele related to nicotine addiction and schizophrenia. Genes Brain Behav 10:530–535. 10.1111/j.1601-183X.2011.00689.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe WM, Gritton HJ, Lusk NA, Roberts EA, Hetrick VL, Berke JD, Sarter M (2017) Acetylcholine release in prefrontal cortex promotes gamma oscillations and theta–gamma coupling during cue detection. J Neurosci 37:3215–3230. 10.1523/JNEUROSCI.2737-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe WM, Brooks JL, Tierney PL, Pang J, Rossi A, Young D, Dlugolenski K, Guillmette E, Roy M, Hales K, Kozak R (2018) α5 nAChR modulation of the prefrontal cortex makes attention resilient. Brain Struct Funct 223:1035–1047. 10.1007/s00429-017-1601-1 [DOI] [PubMed] [Google Scholar]
- Iannazzo L, Majewski H (2000) M2/M4-muscarinic receptors mediate automodulation of acetylcholine outflow from mouse cortex. Neurosci Lett 287:129–132. 10.1016/S0304-3940(00)01163-0 [DOI] [PubMed] [Google Scholar]
- Indurthi DC, Lewis TM, Ahring PK, Balle T, Chebib M, Absalom NL (2016) Ligand binding at the α4-α4 agonist-binding site of the α4β2 nAChR triggers receptor activation through a pre-activated conformational state. PLoS One 11:e0161154 10.1371/journal.pone.0161154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Kuryatov A, Wang J, Kamenecka TM, Lindstrom J (2016) Unorthodox acetylcholine binding sites formed by α5 and β3 accessory subunits in α4α2* nicotinic acetylcholine receptors. J Biol Chem 291:23452–23463. 10.1074/jbc.M116.749150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK (2008) Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci 28:8756–8764. 10.1523/JNEUROSCI.2645-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Gill JV, Lindsay GW, Papadoyannis ES, Field RE, Sten TAH, Miller KD, Froemke RC (2017) Parallel processing by cortical inhibition enables context-dependent behavior. Nat Neurosci 20:62–71. 10.1038/nn.4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Ohana O (2008) Inter- and intralaminar subcircuits of excitatory and inhibitory neurons in layer 6a of the rat barrel cortex. J Neurophysiol 100:1909–1922. 10.1152/jn.90684.2008 [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Onksen J, Lindstrom J (2008) Roles of accessory subunits in α4β2* nicotinic receptors. Mol Pharmacol 74:132–143. 10.1124/mol.108.046789 [DOI] [PubMed] [Google Scholar]
- Laszlovszky T, Schlingloff D, Hegedüs P, Freund TF, Gulyás A, Kepecs A, Hangya B (2020) Distinct synchronization, cortical coupling and behavioral function of two basal forebrain cholinergic neuron types. Nat Neurosci 23:992–1003. 10.1038/s41593-020-0648-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledergerber D, Larkum ME (2010) Properties of layer 6 pyramidal neuron apical dendrites. J Neurosci 30:13031–13044. 10.1523/JNEUROSCI.2254-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Alonso A, Jones BE (2005) Cholinergic basal forebrain neurons burst with theta during waking and paradoxical sleep. J Neurosci 25:4365–4369. 10.1523/JNEUROSCI.0178-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991) Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11:3218–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark G, Rada P, Shors T (1996) Inescapable stress enhances extracellular acetylcholine in the rat hippocampus and prefrontal cortex but not the nucleus accumbens or amygdala. Neuroscience 74:767–774. 10.1016/0306-4522(96)00211-4 [DOI] [PubMed] [Google Scholar]
- Matta JA, Gu S, Davini WB, Lord B, Siuda ER, Harrington AW, Bredt DS (2017) NACHO mediates nicotinic acetylcholine receptor function throughout the brain. Cell Rep 19:688–696. 10.1016/j.celrep.2017.04.008 [DOI] [PubMed] [Google Scholar]
- Mazzaferro S, Bermudez I, Sine SM (2017) α4β2 nicotinic acetylcholine receptors: relationships between subunit stoichiometry and function at the single channel level. J Biol Chem 292:2729–2740. 10.1074/jbc.M116.764183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, Cohen JD (2001) An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24:167–202. 10.1146/annurev.neuro.24.1.167 [DOI] [PubMed] [Google Scholar]
- Nasirova N, Quina LA, Agosto‐Marlin IM, Ramirez J, Lambe EK, Turner EE (2020) Dual recombinase fate mapping reveals a transient cholinergic phenotype in multiple populations of developing glutamatergic neurons. J Comp Neurol 528:283–307. 10.1002/cne.24753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermayer J, Verhoog MB, Luchicchi A, Mansvelder HD (2017) Cholinergic modulation of cortical microcircuits is layer-specific: evidence from rodent, monkey and human brain. Front Neural Circuits 11:100. 10.3389/fncir.2017.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JA, Kastrup JS, Peters D, Gajhede M, Balle T, Ahring PK (2013) Two distinct allosteric binding sites at α4β2 nicotinic acetylcholine receptors revealed by NS206 and NS9283 give unique insights to binding activity-associated linkage at cys-loop receptors. J Biol Chem 288:35997–36006. 10.1074/jbc.M113.498618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JA, Ahring PK, Kastrup JS, Gajhede M, Balle T (2014) Structural and functional studies of the modulator NS9283 reveal agonist-like mechanism of action at α4β2 nicotinic acetylcholine receptors. J Biol Chem 289:24911–24921. 10.1074/jbc.M114.568097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolone G, Lee TM, Sarter M (2012) Time to pay attention: attentional performance time-stamped prefrontal cholinergic activation, diurnality, and performance. J Neurosci 32:12115–12128. 10.1523/JNEUROSCI.2271-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso KG, Steinbach JH (2003) Nicotine is highly effective at producing desensitization of rat alpha4beta2 neuronal nicotinic receptors. J Physiol 553:857–871. 10.1113/jphysiol.2003.053447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M (2007) Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron 56:141–154. 10.1016/j.neuron.2007.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K (2004) The mouse brain in stereotaxic coordinates. Amsterdam: Academic. [Google Scholar]
- Pepeu G, Giovannini MG (2004) Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem 11:21–27. 10.1101/lm.68104 [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH (2008) It is not “either/or”: activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol 84:329–342. 10.1016/j.pneurobio.2007.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RAJ (2002) Desensitization of neuronal nicotinic receptors. J Neurobiol 53:457–478. 10.1002/neu.10109 [DOI] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK (2010) Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci U S A 107:5190–5195. 10.1073/pnas.0909184107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas R, Orr-Urtreger A, Broide RS, Beaudet A, Paylor R, De Biasi M (2003) The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol 63:1059–1066. [DOI] [PubMed] [Google Scholar]
- Sarter M, Lustig C (2019) Cholinergic double duty: cue detection and attentional control. Curr Opin Psychol 29:102–107. 10.1016/j.copsyc.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Lustig C (2020) Forebrain cholinergic signaling: wired and phasic, not tonic, and causing behavior. J Neurosci 40:712–719. 10.1523/JNEUROSCI.1305-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuch JB, Polina ER, Rovaris DL, Kappel DB, Mota NR, Cupertino RB, Silva KL, Guimarães-da-Silva PO, Karam RG, Salgado CAI, White MJ, Rohde LA, Grevet EH, Bau CHD (2016) Pleiotropic effects of Chr15q25 nicotinic gene cluster and the relationship between smoking, cognition and ADHD. J Psychiatr Res 80:73–78. 10.1016/j.jpsychires.2016.06.002 [DOI] [PubMed] [Google Scholar]
- Sendemir E, Erzurumlu RS, Jhaveri S (1996) Differential expression of acetylcholinesterase in the developing barrel cortex of three rodent species. Cereb Cortex 6:377–387. 10.1093/cercor/6.3.377 [DOI] [PubMed] [Google Scholar]
- Sparks DW, Tian MK, Sargin D, Venkatesan S, Intson K, Lambe EK (2018) Opposing cholinergic and serotonergic modulation of layer 6 in prefrontal cortex. Front Neural Circuits 11:107. 10.3389/fncir.2017.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Peters M, Demeter E, Lustig C, Bruno JP, Sarter M (2011) Enhanced control of attention by stimulating mesolimbic–corticopetal cholinergic circuitry. J Neurosci 31:9760–9771. 10.1523/JNEUROSCI.1902-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J (2007) Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol 71:769–776. 10.1124/mol.106.030445 [DOI] [PubMed] [Google Scholar]
- Teles-Grilo Ruivo LM, Baker KL, Conway MW, Kinsley PJ, Gilmour G, Phillips KG, Isaac JTR, Lowry JP, Mellor JR (2017) Coordinated acetylcholine release in prefrontal cortex and hippocampus is associated with arousal and reward on distinct timescales. Cell Rep 18:905–917. 10.1016/j.celrep.2016.12.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testylier G, Dykes RW (1996) Acetylcholine release from frontal cortex in the waking rat measured by microdialysis without acetylcholinesterase inhibitors: effects of diisopropylfluorophosphate. Brain Res 740:307–315. 10.1016/s0006-8993(96)00893-1 [DOI] [PubMed] [Google Scholar]
- Thomson AM. (2010) Neocortical layer 6, a review. Front Neuroanat 4:13. 10.3389/fnana.2010.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian MK, Bailey CDC, De Biasi M, Picciotto MR, Lambe EK (2011) Plasticity of prefrontal attention circuitry: upregulated muscarinic excitability in response to decreased nicotinic signaling following deletion of α5 or β2 subunits. J Neurosci 31:16458–16463. 10.1523/JNEUROSCI.3600-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann D, Sandager-Nielsen K, Dyhring T, Smith M, Jacobsen A-M, Nielsen E, Grunnet M, Christensen J, Peters D, Kohlhaas K, Olsen G, Ahring P (2012) Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of α2- and α4-containing nicotinic acetylcholine receptors. Br J Pharmacol 167:164–182. 10.1111/j.1476-5381.2012.01989.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal CT, Golowasch JP, Zaborszky L (2012) Adult mouse basal forebrain harbors two distinct cholinergic populations defined by their electrophysiology. Front Behav Neurosci 6:21. 10.3389/fnbeh.2012.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S, Jeoung H-S, Chen T, Power SK, Liu Y, Lambe EK (2020) Endogenous acetylcholine and its modulation of cortical microcircuits to enhance cognition. In: Current topics in behavioral neuroscience, pp 1–23. Berlin, Heidelberg: Springer. [DOI] [PubMed] [Google Scholar]
- Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW (1990) The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (α5) in the rat central nervous system. Brain Res 526:45–53. 10.1016/0006-8993(90)90248-A [DOI] [PubMed] [Google Scholar]
- Wang J, Lindstrom J (2018) Orthosteric and allosteric potentiation of heteromeric neuronal nicotinic acetylcholine receptors. Br J Pharmacol 175:1805–1821. 10.1111/bph.13745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kuryatov A, Sriram A, Jin Z, Kamenecka TM, Kenny PJ, Lindstrom J (2015) An accessory agonist binding site promotes activation of α4β2* nicotinic acetylcholine receptors. J Biol Chem 290:13907–13918. 10.1074/jbc.M115.646786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM (2005) Expression of alpha5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol 481:19–30. 10.1002/cne.20357 [DOI] [PubMed] [Google Scholar]
- Yang D, Günter R, Qi G, Radnikow G, Feldmeyer D (2019) Cell type-specific modulation of layer 6A excitatory microcircuits by acetylcholine in rat barrel cortex. bioRxiv. Advance online publication. Retrieved August 17, 2020 DOI: 10.1101/701318 10.1101/701318 [DOI] [Google Scholar]
- Zhang W, Basile AS, Gomeza J, Volpicelli LA, Levey AI, Wess J (2002) Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice. J Neurosci 22:1709–1717. 10.1523/JNEUROSCI.22-05-01709.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G (2011) Cell type–specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods 8:745–752. 10.1038/nmeth.1668 [DOI] [PMC free article] [PubMed] [Google Scholar]