

Abstract
Background:
Clinical studies have shown that celecoxib can significantly inhibit the development of tumors, and basic experiments and in vitro experiments also provide a certain basis, but it is not clear how celecoxib inhibits tumor development in detail.
Methods:
A literature search of all major academic databases was conducted (PubMed, China National Knowledge Internet (CNKI), Wan-fang, China Science and Technology Journal Database (VIP), including the main research on the mechanisms of celecoxib on tumors.
Results:
Celecoxib can intervene in tumor development and reduce the formation of drug resistance through multiple molecular mechanisms.
Conclusion:
Celecoxib mainly regulates the proliferation, migration, and invasion of tumor cells by inhibiting the cyclooxygenases-2/prostaglandin E2 signal axis and thereby inhibiting the phosphorylation of nuclear factor-κ-gene binding, Akt, signal transducer and activator of transcription and the expression of matrix metalloproteinase 2 and matrix metalloproteinase 9. Meanwhile, it was found that celecoxib could promote the apoptosis of tumor cells by enhancing mitochondrial oxidation, activating mitochondrial apoptosis process, promoting endoplasmic reticulum stress process, and autophagy. Celecoxib can also reduce the occurrence of drug resistance by increasing the sensitivity of cancer cells to chemotherapy drugs.
Keywords: autophagy, cancer stem cells, celecoxib, cyclooxygenase-2, endoplasmic reticulum stress, prostaglandin E2, reactive oxygen species, tumor
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) target the inhibition of cyclooxygenases (COXs) to achieve analgesic, antipyretic and anti-inflammatory effects. COX consists of 2 isoforms, COX-1 and COX-2, and COX-1 mainly to maintain homeostasis, COX-2 functions under the stress of inflammatory factors and pain.[1,2]
COX can catalyze the synthesis of thromboxane, prostacyclin, and prostaglandin (PG) from arachidonic acid. Prostaglandins include prostaglandin D2, prostaglandin I2 (PGI2), prostaglandin E2 (PGE2), and prostaglandin F2 receptor α, of which PGE2 and PGI2 are important inflammatory factors that regulate the body's pain stress, immune and inflammatory response.[3] NSAIDs achieve anti-inflammatory effects by inhibiting the activity of the inflammatory factor COX-2 and the synthesis of PGE2. Numerous studies have confirmed that NSAIDs also have chemopreventive effects on tumors.[4,5,6] For example, indomethacin can promote the apoptosis of colorectal cancer cells by inhibiting the activity of peroxisome proliferator-activated receptor δ.[7] At the same time, many case-control studies have shown that patients who take NSAIDs for a long time have a significantly lower incidence of colorectal cancer and breast cancer.[8,9]
The first generation of NSAIDs showed different degrees of side effects (including gastrointestinal reactions, nephrotoxicity, cardiovascular diseases, central nervous system diseases, and platelet-related diseases).[10] COX-2 produced drug resistance by upregulating the transport protein, reducing the concentration of intracellular drugs.[11] The second-generation selective COX-2 inhibitor celecoxib appears in such an environment. Celecoxib is one of the commonly used NSAIDs in the clinic, mainly for the treatment of inflammatory diseases such as osteoarthritis, rheumatoid arthritis, and inflammatory musculoskeletal.[12,13] Celecoxib is a 1,5-diaryl substituted pyrazole compound (Fig. 1), chemically named 4- [5- (4-methylphenyl) -3- (trifluoromethyl)-1H- Pyrazolyl-1-yl], which plays a role by selectively inhibiting COX-2.[14] Compared with other NSAIDs, celecoxib shows lower toxicity side effects (such as the most common gastrointestinal bleeding and gastric ulcer).[15] Early studies have shown that celecoxib can effectively reduce the incidence of colorectal cancer, especially inhibiting the development of familial adenomatous polyposis to colorectal cancer.[16] So far, celecoxib has been used in various cancer chemoprevention research, including non-small cell lung cancer (NSCLC), breast cancer, hepatic carcinoma, prostate cancer, colorectal cancer, bladder cancer, head, and neck cancer, esophageal cancer, cervical cancer, non-Melanoma, and other cancers (Table 1).
Figure 1.
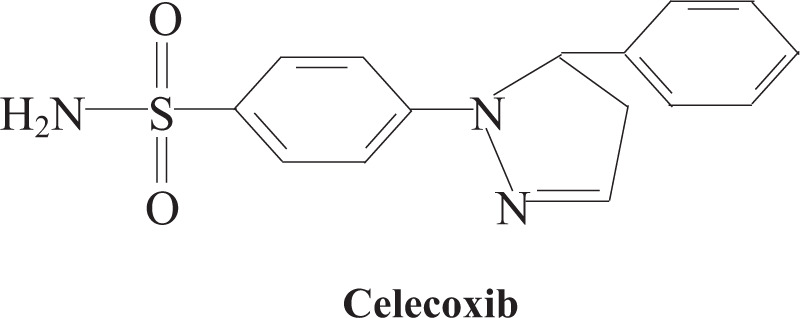
Chemical formula of celecoxib.
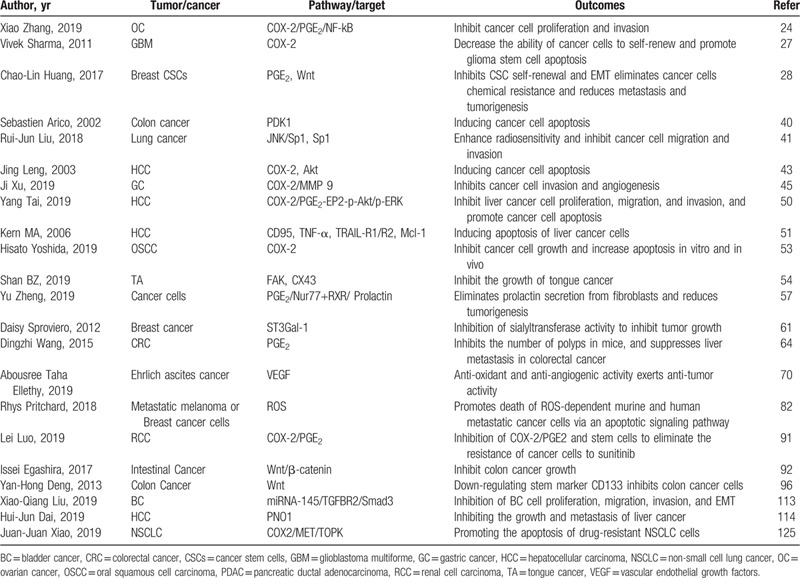
Table 1.
In vivo and in vitro studies of celecoxib intervention in tumors through different mechanisms.
There are 2 isozymes in COX, COX-1 is generally expressed in many parts, COX-2 is rapidly stimulated and expressed by mitogens, growth factors, and cytokines.[17] After being stimulated, COX-2 expresses and promotes the synthesis of prostaglandins, thromboxane, prostacyclin, and leukotriene, which can lead to inflammation, even malignant proliferation, and tumor formation.[18] COX-2 not only regulates inflammation and pain stress response but also regulates mitosis, cell differentiation, and angiogenesis, as well as inhibits apoptosis and changes cell adhesion. Under steady-state, its expression and activity are low. It is stimulated by growth factors, cytokines, tumor promoters, and hormones to increase the activity and expression of COX-2 and promote the synthesis of PGE2.[19] PGE2 plays a key role in the process of tumorigenesis by maintaining inflammatory state and promoting cell proliferation, angiogenesis, and metastasis.[20] Studies have confirmed that the expression of COX-2 in cancer cells is significantly increased, which promotes the increase of prostacyclin and prostaglandin synthesis and promotes the proliferation of tumor cells.[21] PGE2 synthesis requires 2 key enzymes, microsomal prostaglandin E synthetase-1 and COX-2.[22] All 3 substances can promote the growth of tumor cells. It was found that microsomal prostaglandin E synthetase-1 can directly promote the proliferation of human hepatoma cells, increase the expression of β-catenin and early growth response 1, and promote the proliferation, migration, and invasion of hepatoma cells through the synthesis of PGE2 and the activation of β - catenin and early growth response 1 signaling pathway.[23] Studies have found that in ovarian cancer, COX-2 and its downstream gene PGE2 can promote the expression of matrix metalloproteinase 2 and 9 (MMP 2 and MMP 9) and regulate the metastasis of ovarian cancer cells.[24] The overexpression of MMP will destroy the structure of the extracellular matrix (ECM) and basement membrane, promote the invasion and metastasis of cancer cells.[25] Besides, COX-2 activates the PGE2 signal, which is involved in the regulation of inflammation and cancer. The imbalance of the COX-2/PGE2 signal axis induces the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K)/Akt pathway, promotes the development of tumor and induces the stimulation of COX-2 expression to form a loop feedback effect.[17] At present, studies have confirmed that celecoxib, a COX-2 inhibitor, can effectively reduce the risk of several cancers (including breast cancer, colon cancer, prostate cancer), and also has a therapeutic effect on neurodegenerative diseases (Parkinson disease and Alzheimer disease).[14]
Cancer cells can proliferate and self-renew indefinitely. A few cancer cells display the characteristics of stem cells and have the stem cell-like ability to maintain tumor growth and metastasis, similar to stem cells, called cancer stem cells (CSCs) or tumor-initiating cells.[26] CSCs not only have the ability to self-renew and differentiate, which enhance the heterogeneity of tumor cells but also play an anti-radiation effect by potentially activating the stress response of the DNA damage site and enhancing the DNA repair ability, as well as being preserved during chemotherapy treatment and continue to proliferate into drug-resistant stem cell populations.[27] With more and more studies finding that celecoxib can effectively inhibit the dry expression of tumors, including breast cancer and liver cancer.[28,29] Also, celecoxib also showed a combination of chemotherapeutic drugs cisplatin and gefitinib, which enhanced the lethality of chemotherapeutic drugs to cancer cells, while significantly reducing the multidrug resistance response (Table 2). For example, celecoxib can significantly enhance the therapeutic effect of cisplatin on drug-resistant gastric cancer cells by inhibiting the expression of COX-2 and the anti-apoptotic gene BCL-2.[30]
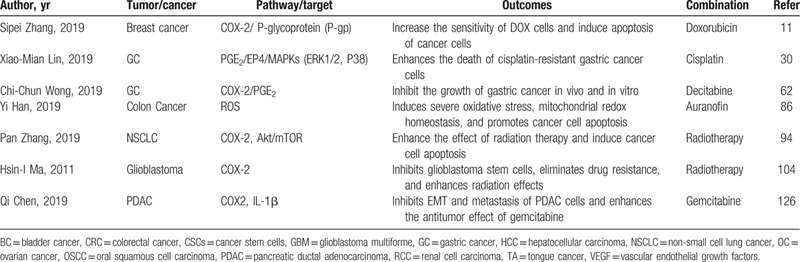
Table 2.
Studies of celecoxib combined with chemotherapy and radiation to suppress tumors.
In eukaryotic cells, autophagy degrades organelles, proteins, and macromolecular substances in the cytoplasm through lysosomes, and recycles the decomposition products to form a dynamic equilibrium.[31] There are 3 types of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy. Microautophagy is the use of lysosomal membrane invasion or protrusion to capture targets, and chaperone-mediated autophagy is the use of partners to identify target protein of a specific sequence after the target protein stretches, it directly transmembrane into the lysosome. Macroautophagy first forms a double-membrane autophagosome, which contains the target protein and then fuses with the lysosome to form autophagy Lysosomes, while the target protein in the autophagosome is transported to the lysosome is degraded and recovered.[32,33,34] Autophagy clears misfolded or aggregated proteins in the endoplasmic reticulum (ER), damaged organelles (including ER, mitochondria, and peroxisomes), and pathogens that enter the cell.[35] Autophagy has 2 sides to cancer. On the one hand, it suppresses tumors through the elimination mechanism to prevent oxidative stress and maintain genome stability, On the other hand, it provides cancer cells with materials that can resist nutritional deficiency and hypoxia to promote the development of cancer cells.[36] LC3 and P62 are classic autophagy-related markers. LC3, also known as autophagy-related protein 8, includes LC3-I and LC3-II (LC3-I is cleaved into lipidated LC3-II during the autophagosome maturation process), and when autophagy induction occurs, the ratio of LC3-II/LC3-I protein level increases.[37,38] P62, also known as SQSTM1, can bind to LC3 and ubiquitinated proteins and degrade in autolysosomes.[39]
In the past, it was believed that celecoxib could intervene tumor mainly by inhibiting the COX-2 pathway. As the study found, the anticancer effect of celecoxib can also be caused by the COX-2 independent mechanism. For example, studies have shown that celecoxib can promote cancer cell apoptosis by inhibiting the signal pathway of 3-phosphoinositide-dependent kinase-1 and downstream protein kinase B (Akt) in human colon cancer cells.[40] In addition, the study on NSCLC shows that celecoxib enhances the sensitivity of cancer cells to radiation therapy and inhibits cancer cell migration and invasion by inhibiting the activity of C-Jun amino-terminal kinase and downregulating the expression of specific protein 1.[41] Therefore, this review describes the mechanism research of celecoxib on cancer intervention through the COX-2 pathway, and also summarizes the in vitro and in vivo research of COX-2 independent mechanism, to explore how celecoxib can play a better role in clinical anticancer treatment.
2. Celecoxib inhibits tumors through COX-2/PGE2 signaling axis
As a common inducible enzyme in inflammatory tissues, COX-2 is involved in the carcinogenic pathway of many tissues and organs. The expression of COX-2 is high in various cancer tissues (liver cancer, breast cancer, gastric cancer, esophageal cancer, colon cancer, lung cancer, head, and neck squamous cell carcinoma), the release of prostaglandins in tumor area is increased, which can directly induce cell mitosis and cause cancer.[42] On the one hand, high expression of COX-2 and PGE2 can promote the proliferation of cancer cells, and COX-2/PGE2 can induce Akt phosphorylation and inhibit cancer cell apoptosis. On the other hand, PGE2 can promote the proliferation and metastasis of cancer cells by activating β-catenin signal pathway.[23,43,44] COX-2 is highly expressed in ovarian cancer, which promotes the invasion ability of cancer cells by promoting the phosphorylation of nuclear factor-κ-gene binding (NF-κb), up-regulating the expression of C-myc and phosphorylated STAT, and increasing the expression of MMP 2 and MMP 9.[24] COX-2/MMP 9 can mediate tumor-associated macrophages in the tumor microenvironment of gastric cancer cells and promote cancer cell invasion and metastasis.[45] Similarly, PGE2 can activate NF-κb, MMP 2, and MMP 9, and promote the phosphorylation of signal transducer and activator of transcription to promote cancer cells proliferation, migration, invasion, and distant angiogenesis.[46,47,48] In turn, NF-κb can regulate COX-2/PGE2 to mediate the migration and invasion of malignant breast cancer cells.[49] Precisely, celecoxib, a selective COX-2 inhibitor, can effectively inhibit the proliferation and metastasis of cancer cells by inhibiting the COX-2/PGE2 signal axis and influencing the downstream target genes or pathways.
Celecoxib, as a selective COX-2 inhibitor, should not only be used for anti-inflammatory treatment, many potential pharmacological properties need to be taken seriously. Recent studies have used celecoxib to act on liver cancer cells and found that Celecoxib not only inhibits the growth of cancer cells by inhibiting the proliferation of liver cancer cells, promoting apoptosis and inducing the blockage of the G0/G1 cell cycle but also inhibits the effects of COX-2/PGE2/PGE2 (EP2)/p-Akt/p-ERK signaling pathway to up-regulate E-cadherin, which can significantly inhibit the migration and invasion of liver cancer cells.[50] Celecoxib promotes apoptosis in hepatocellular carcinoma by inhibiting the expression of COX-2 to induce the expression of CD95, tumor necrosis factor receptor, tumor necrosis factor-related apoptosis-inducing ligand (TRAILR1) and TRAIL-R2 death receptor.[51] Moreover, celecoxib inhibited COX-2 to significantly down-regulate the expression of myeloid leukemia-1, an anti-apoptotic member of the Bcl-2 family, release Bax translocation to mitochondria and cytochrome C to induce apoptosis of tumor cells.[51,52] In the study of squamous cell carcinoma of head and neck, it was found that lipopolysaccharide in Porphyromonas gingivalis (the main pathogen of periodontitis) promotes the expression of COX-2 in oral squamous cell carcinoma, while celecoxib can significantly inhibit the development of tumor, and inhibit the transformation of G0/G1-S phase and reduce the proliferation of tumor cells by increasing the expression of p21.[53,54] Furthermore, celecoxib can promote T cells to play a role in the tumor microenvironment through the COX-2 pathway. Since 2,3-dioxygenase (IDO) can decompose L-tryptophan into L-kynurenine, a large amount of IDO decomposition of tryptophan in the tumor microenvironment will cause immune T lymphocytes to not effectively recognize tumor cells, and even accelerate T lymphocyte apoptosis.[55] In breast cancer experiments, it was found that the inhibition of COX-2 with celecoxib could significantly reduce the level of IDO in breast cancer, and the regulation of downstream PGE2 of COX-2 can also inhibit the process of tryptophan consumption by IDO.[56] In addition, the association of tumors with ECM plays an important role in the early stages of metastasis. Some studies have found that COX-2-expressing cancer cells induce the expression of orphan nuclear receptor NR4A (Nur77) in ECM fibroblasts through PGE2, NR4A combines with retinoic acid X receptor to form a dimer and binds to the prolactin promoter, and prolactin stimulates the metastasis signal in tumor cells, and prolactin receptor promotes the proliferation of metastatic cancer cells, while celecoxib can directly inhibit COX-2/PGE2 and then inhibit the metastasis and proliferation of cancer cells.[57]
Most proteins expressed or secreted on the plasma membrane carry polysaccharides, and the structure and function of glycoproteins are changed by glycosylation, where abnormal glycosylation is one of the early events of cancer.[58] Glycosylation is involved in the regulation of cell adhesion, cell-matrix interaction and cell metastasis mediated by transmembrane protein receptor integrin.[59] Cancer cells use the integrin family to bind molecules in ECM to help with metastasis.[60] COX-2/PGE2 can regulate glycosylation and promote the development of cancer. Studies on breast cancer cells found that PGE2 produced by COX-2 pathway can induce the increase of mRNA expression of α-2,3 sialyltransferase-3, and α-2,3 sialyltransferase-3 is highly expressed in tumors, especially in bladder cancer, colon cancer, and breast cancer, which leads to the increase of truncated sialidase expression and then promotes the development of cancer.[61] At the same time, studies have also shown that celecoxib can effectively reverse the effect of the COX-2 pathway on sialylation.[61] Aberrant methylation of promoter DNA is not only one of the early events of cancer, but also occurs throughout cancer. Studies have found that COX-2/PGE2 signaling axis can induce 5-methylcytosine and enhance the methylation of O-6-methylguanine-DNA and methyltransferase promoters in gastric cancer, while celecoxib can significantly inhibit the process of DNA methylation.[62]
At present, clinical cancer treatment mainly depends on the surgery and postoperative radiotherapy and chemotherapy. On the one hand, chemotherapy drugs (such as cisplatin and doxorubicin) can cause cancer cell death through different drug mechanisms, on the other hand, they can up-regulate the COX-2/PGE2 signal axis to activate CSCs in the tumor microenvironment, and the synthesized PGE2 mediates multidrug resistance of cancer cells by enhancing the expression of P-glycoprotein.[63,64] Therefore, the drug rationality of celecoxib can fully exert the inhibitory effect of the COX-2/PGE2 signal axis, and the combination of celecoxib and chemotherapy drugs can not only effectively promote the sensitivity of chemotherapy drugs, but also prevent the emergence of chemotherapy resistance. Recently, it has been reported that celecoxib combined with chemotherapy has obvious advantages in targeting anticancer therapy.[65]
3. Celecoxib promotes cancer cell death through mitochondrial apoptosis mechanism
The energy metabolism of mitochondria is mainly composed of the Tricarboxylic acid cycle and electron transport chain, and Aconitic acid hydratase (ACO2), which contains iron-sulfur cluster, is sensitive to the change of reactive oxygen species (ROS).[66] ROS is an active oxygenate, including superoxide, hydroxyl radical, and non-radical molecules.[67] ROS can regulate hypoxia-inducible factor-1 α and effectively regulate the mRNA of related proteins such as glycolysis, erythropoiesis, cell proliferation, and angiogenesis.[68,69] In the normal state, oxidants and antioxidants can maintain a dynamic balance, which can maintain the redox balance in cells by removing the active substances that cause oxidative stress and cell damage, and high concentration of ROS leads to the destruction of balance and cell structure.[70] Normal cells use reduced glutathione, catalase, superoxide dismutase, acetaldehyde, and other substances to form a strong antioxidant defense and resist high concentration ROS.[71,72] In cancer cells, the content of ROS is significantly higher than that in normal cells, because the proliferation of cancer cells requires more ATP. The side effect of this continuous energy production is the accumulation of ROS, and the antioxidant system in the cancer cells is maintained at a high level to ensure that the content of ROS does not exceed the threshold that the cancer cells can tolerate, and because of the high expression of ROS scavenging system, it shows a low level of ROS in cancer stem cells.[73,74] Under the stimulation of hypoxia and hypoglycemia, the synthesis of mitochondrial ATP was significantly blocked, especially the oxidative phosphorylation process (Oxphos), and inhibition of the respiratory chain leads to a significant increase in ROS release, which can cause mutations and promote cancer cell invasion and migration.[75,76] However, the reduction of oxidative phosphorylation can induce autophagy, and the cells can eliminate part of ER and reduce the quality of mitochondria when cells receive electrons in the respiratory chain, which can reduce the production of ROS in mitochondrial.[35] The supply of blood vessels and nutrients around cancer cells is far less than the demand of rapidly proliferating cancer cells, and the lack of local oxygen leads to the instability of tumor blood vessels and accelerates angiogenesis and distant metastasis.[77] The main way for cancer cells to obtain energy is glycolysis, while the hypoxia environment will seriously damage the process of oxidative phosphorylation, resulting in a large amount of ROS released, which will destroy mtDNA, a subunit encoding OXPHOS enzyme, and its mutation may enhance tumor cell metastasis potential to promote tumor development, forming a circular negative feedback effect.[78] Moreover, hypoxia-inducible factor-2 α and its target genes Oct-4, c-myc, and Nanog can increase the number of CSC in the hypoxia environment, and they have stronger differentiation and proliferation ability to adapt to various poor environments.[79,80] Therefore, the high level of ROS in cancer cells may be the result of the inhibition of oxidative phosphorylation, insufficient energy supply, and increased ROS release stimulated by hypoxia and starvation. However, the ROS expression level is higher than that of normal cells, which will not affect the proliferation and metastasis of cancer cells. It will still get more energy by transferring to the vicinity of blood vessels or generating new blood vessels. After using exogenous intervention, continuing to release the ROS level will lead to cellular oxidative stress and cell death.
Celecoxib is more likely to act as an uncoupling agent of OXPHOS and respiratory chain inhibitor in the study of mitochondria.[71,81,82] Celecoxib targets mitochondria and promotes the release of ROS by significantly increased oxidative stress. Some studies have confirmed that celecoxib can significantly improve the synthesis of mitochondrial superoxide in metastatic melanoma and breast cancer cells within a few minutes, and lead to the decrease of cell consumption and mitochondrial transmembrane potential (△ ψ m), increasing mitochondrial membrane permeability to promote the release of ROS, and a certain level of ROS can help the growth of cancer cells, but the continuous increase beyond the threshold will activation of the mitochondrial pathway for apoptosis or programmed cell death.[82,83] Celecoxib can reduce the antioxidant mechanism and promote the oxidation of mitochondria, affect mitochondrial function, promote Ca2+ influx, produce a higher pro-oxidation state, increase the accumulation of ROS in cancer cell mitochondria, and the cancer cells are overloaded, and by activating mitochondria excessive pore permeability, promoting the formation of mitochondria excessive pore permeability complex, activating the apoptotic pathway of cancer cells, and leading to cancer cell death.[84,85] Also in colon cancer, celecoxib causes an increase in cellular ROS release, leading to oxidation of mitochondrial antioxidant enzymes (Trxs), hexokinase (HK), cytochrome c oxidase II (Mtco2), and inhibits the glycolysis process, ATP synthesis is significantly reduced, leading to cancer cell death.[86]
In addition to cancer cells, celecoxib can also inhibit CSCs. Low ROS level can protect CSCs from oxidative damage, preserve the characteristics of cancer stem cells and proliferate in a favorable environment, while celecoxib can effectively inhibit CSCs by increasing the release of mitochondrial ROS and inhibiting glycolysis.[87]
4. Celecoxib promotes tumor cell apoptosis by enhancing endoplasmic reticulum stress
The proteins secreted by ER are folded and assembled through various mechanisms. When the program is damaged, misfolded and unfolded proteins appear. Some unfolded proteins will be transferred to the cytoplasm, absorbed by autophagy lysosomes and degraded, while the remaining unfolded and misfolded proteins will accumulate in the ER and generate ER stress response.[88] ER stress response is a process that loses homeostasis, hinders normal cell function, and is a mechanism by which cells respond to excessive unfolded proteins in the ER.[89] Stimulation such as starvation, hypoxia, altered glycosylation, and oxidative stress all interfere with the protein folding process, in which unfolded proteins (exogenous membrane proteins or secreted proteins are overexpressed) are deposited in the ER, causing ER stress response.[90] ER stress activates the unfolded protein response (UPR), causing the activation of 3 UPR sensors, including the Activating transcription factor 6, the Inositol dependent enzyme 1α, and the PKR-like ER kinase, through limits the accumulation of misfolded proteins, enhances the function of eliminating unfolded proteins and increases the ability of the ER to reduce the misfolded of proteins, and for cancer cells, long-term UPR activation can induce cell death mechanisms.[91,92] UPR can also induce macro-autophagy in tumor microenvironment macrophages, which is not a single outcome. According to current research, mega-autophagy can enhance cell survival in some experiments and promote apoptosis in others.[93]
The early induction of apoptosis by celecoxib is through the activation of the endoplasmic reticulum stress response.[94] During ER stress, ER calcium is released, ROS increase, the nuclear shift of NF-κb occurs, p38-mitogen-activated protein kinase phosphorylation, activated NF-κb can regulate the expression of COX-2, and promote proliferation in cancer cells.[95] However, the release of calcium in the ER, the uptake of Ca2+ by mitochondria, and the apoptosis of cells are regulated by Bcl-2, which is located in mitochondria and endoplasmic reticulum. There are 2 outcomes of Bcl-2. One is the loss of anti-apoptosis after phosphorylation by JNK, and the inability to bind pro-apoptotic molecules after phosphorylation, increase the release of Ca2+ in ER, increase the uptake of Ca2+ by mitochondria, and lead to cells apoptosis, the other is the PI3K complex that aggregates into the autophagy pathway to help cells survive.[33] It has been shown that COX-2 can regulate ER stress and produce drug resistance in hepatocellular carcinoma cells through p38/PI3K/Akt pathway, and celecoxib can effectively block this pathway.[92] In addition, celecoxib can inhibit the activity of ER calcium-ATP binding enzyme and inactivate the protein kinase Akt, increase the binding of IL-12 to calcium reticulum, and promote the apoptosis of cancer cells.[96]
5. Celecoxib inhibits tumor through Wnt pathway
COX-2 and prostaglandins induce tumor microenvironment inflammation and activate various signaling pathways, including Wnt/β-catenin signaling.[97,98] Early studies of celecoxib's prevention and impact on tumors focused on the COX-2 pathway.[99] However, recent studies using celecoxib and a celecoxib analog (2,5-dimethyl celecoxib) that does not inhibit COX-2 have shown that colon cancer can affect celecoxib inhibition does not depend on the COX-2 pathway, the target points to the Wnt signaling pathway.[100] Therefore, celecoxib can inhibit the transduction of Wnt/β-catenin signaling pathway through COX-2 dependent and non-dependent mechanisms and has anti-cancer effects.[101]
As a stem cell regulator, PGE2 can promote stem cell function and has no specific regulation on the steady-state maintenance of normal stem cells and the proliferation of tumor stem cells. PGE2 can enhance the expansion of stem cells in the hematopoietic system and tumor stem cells in colorectal tumors by up-regulating the activity of the Wnt pathway.[64,102] Celecoxib can effectively inhibit the characteristics of breast CSCs and tumorigenesis in vivo by significantly inhibiting PGE2 to downregulate the Wnt pathway.[28] Studies have confirmed that autocrine and paracrine synthesis of PGE2 induces CSC formation by activating the Wnt pathway and that celecoxib interferes with colorectal cancer by down-regulating the expression of the CSC surface marker CD133 through a COX-2 independent mechanism.[103] Moreover, celecoxib can enhance the sensitivity of glioblastoma to radiotherapy by regulating the expression of CD133.[104]
6. Celecoxib inhibits cancer in other ways
In osteosarcoma, celecoxib induced the transformation of LC3-I to LC3-II, which can induce autophagy of osteosarcoma in a dose-dependent manner.[105] It is found that celecoxib can inhibit autophagy flux by inhibiting lysosomal function, increase the level of autophagy-related proteins LC3-II and p62, and act as autophagy inhibitor to induce apoptosis and necrosis of tumor cells.[106,107] The accumulation of p62 was observed when autophagy was inhibited, and the expression of p62 decreased when autophagy occurred.[107] Meanwhile, some experimental studies have confirmed that celecoxib can promote cancer cell apoptosis by inhibiting the autophagy process of urothelial cancer and colorectal cancer.[108,109]
Epithelial-mesenchymal transition (EMT) is a process of adaptive changes in various tissues during embryonic development, and epithelial cells and mesenchymal cells are transformed into each other, and it is worth noting that EMT can enhance the mobility and invasion of cancer cells and promote metastasis, and EMT is also closely related to the production of CSCs.[110,111,112] Recently, it has been found that celecoxib can inhibit the process of EMT and hinders the metastasis of cancer cells in vivo by up-regulating microRNA-145 and down-regulating the expression of transforming growth factor-β receptor 2 and Smad family member 3, and inhibits cancer cell metastasis in vivo.[113]
In addition, studies have confirmed that NOB1's RNA-binding gene chaperone –-PNO1 (mainly expressed in liver, lung, spleen, and kidney) has increased expression in liver cancer and exerts carcinogenesis through the AKT/mTOR pathway. In vivo and in vitro experiments have found that celecoxib can effectively inhibit PNO1 to exert an antitumor effect, reduce tumor growth, and reduce lung metastasis.[114] It is also found in colorectal cancer research that PNO1, as a ribosome assembly factor, can activate the ribosomal protein- MDM2-p53 pathway to significantly promote the proliferation of cancer cells, while deletion or low expression can inhibit cell proliferation and induce apoptosis to inhibit tumor growth in vivo.[115]
7. Side effects
Although celecoxib shows anti-cancer potential, it has been found in current clinical studies that long-term use of celecoxib can cause damage to vital organs.[116,117] Long-term use of celecoxib significantly reduces the risk of recurrence and transition to colorectal cancer in high-risk colorectal adenomas, but long-term use increases the risk of hypertension among participants who already have cardiovascular risk factors.[118] This is due to celecoxib can reduce PGI2 synthesis, which affects vasodilation and platelet activation, causing side effects of the cardiovascular system.[119] Similarly, celecoxib can reduce the synthesis of PGE2 and PGI2, while PGE2 is mainly involved in the regulation of water and ion transport, PGI2 is involved in the dynamic balance of renal vascular tension, glomerular filtration rate, and renin release, and the synthesis of the 2 substances is affected, leading to side effects of the renal system.[120] However, it is worth mentioning that celecoxib shows fewer gastrointestinal adverse reactions than other NSAIDs.[121,122]
8. Conclusions
Generally speaking, celecoxib is a routine clinical drug, which is easy to obtain and low in price. It has good pharmacological properties whether it inhibits the development and metastasis of tumors through various mechanisms or it is combined with other treatment methods to hinder the growth of the tumor. Celecoxib inhibits the classic COX-2/PGE2 signal axis and affects some downstream pathways or mechanisms (such as Akt, NF-κb, STAT3, MMP), and inhibits tumor proliferation and metastasis. Furthermore, there is a close link between celecoxib's enhancement of mitochondrial oxidative stress and induction of the ER stress program, while ROS has become the medium of 2 kinds of organelles to activate the apoptosis program, Bcl-2 is also involved in regulating the association between mitochondria and the endoplasmic reticulum. The balance of oxidation and antioxidation maintains the activity of tumor cells and CSCs, while celecoxib promotes tumor cells apoptosis by breaking the balance of oxidation and antioxidation. It also includes the regulation of CSCs through the Wnt pathway to reduce the production of drug-resistant cells, and inhibit the proliferation of tumor cells. Although it has been observed in clinical trials that long-term use can cause cardiovascular disease, the rational arrangement of medication can avoid adverse reactions. The most important thing is to find a clear mechanism and target, and the final purpose is to synthesize more accurate drugs for clinical use.
9. Discussion
For a long time, celecoxib, as a commonly used clinical drug, mainly relieves the symptoms and signs of osteoarthritis, rheumatoid arthritis, and other inflammatory diseases. With the deepening and expansion of research, its hidden drug potential has been gradually explored. For example, celecoxib can change the cell membrane potential and permeability to enhance the efficacy of antibiotics against Staphylococcus aureus.[123] Celecoxib combined with gentamicin can reduce NF-κb and COX-2 immune response and anti-apoptotic effects to reduce the renal toxicity of gentamicin.[124] Celecoxib can enhance the chemotherapy effect of gefitinib on NSCLC by inhibiting COX-2/MET (hepatocyte growth factor receptor, HGFR)/T-lymphokine-activated killer cell-originated protein kinase (TOPK) signaling pathway.[125] Celecoxib can also be combined with gemcitabine to increase the clinical effect of pancreatic cancer treatment.[126] Not only that, but Celecoxib can also treat refractory lymphoid malformations by inhibiting the mechanism of COX-2/PGE2, and no adverse reactions were observed during this period.[127] Celecoxib has shown the effect of interventional treatment of tumors, whether it is used alone or in combination with other interventional treatments. Of course, we have also paid attention to some clinical experiments and found that long-term use of celecoxib has shown good tumor intervention effects, but also has adverse reactions.[118] Celecoxib is an FDA approved drug with good advantages. Moreover, our focus is not entirely on the celecoxib itself, but on the role of its pharmacological mechanism in the process of tumor intervention. Through in-depth exploration and study of more detailed mechanisms and targets, it is our goal to find and even develop more reasonable drugs. Ye et al. first used the NSAIDs sulindac to interfere with colon cancer, and found its corresponding target tRXRα (retinoic acid X receptor α was hydrolyzed by protein to form a truncated RXR α in cancer cells), and identified a sulindac analog k-80003, which enhanced the binding to tRXRα and weakened the COX inhibitory activity.[128] The toxic and side effects of celecoxib are closely related to the intake dose, duration, frequency of medication, and the high-risk factors for the patient's disease. Avoiding these factors can help us better use celecoxib. Moreover, there have been many recent studies on the development of celecoxib drug-related derivatives or combinations to help better target therapy and reduce drug side effects.[129]
Acknowledgment
The authors thank Professor Biguang Tuo (Department of Gastroenterology, Affiliated Hospital to Zunyi Medical College) for highly professional services.
Author contributions
Bin Wen contributed to search and write the manuscript, Ying-ting Wei contributed to sort out the form, Lan-lan Mu contributed to sort and check the information of the form, Guo-rong Wen contributed to check the manuscript, and Kui Zhao contributed to revise the manuscript.
Footnotes
Abbreviations: COXs = cyclooxygenases, CSCs = cancer stem cells, ECM = extracellular matrix, ER = endoplasmic reticulum, IDO = 2,3-dioxygenase, NSAIDs = non-steroidal anti-inflammatory drugs, Oxphos = oxidative phosphorylation process, PGE2 = prostaglandin E2, PGI2 = prostaglandin I2, ROS = reactive oxygen species, UPR = unfolded protein response.
How to cite this article: Wen B, Wei YT, Mu LL, Wen GR, Zhao K. The molecular mechanisms of celecoxib in tumor development. Medicine. 2020;99:40(e22544).
This study was supported by research grants from the National Natural Science Foundation of China (No. 81560467 to KZ).
Ethics committee or institutional is not applicable. Because this article does not involve the operation of patients or animals directly, but focuses on the molecular mechanism of drug intervention and drug resistance.
The authors have no conflicts of interest to disclose.
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
- [1].Lipsky PE, Brooks P, Crofford LJ, et al. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch Intern Med 2000;160:913–20. [DOI] [PubMed] [Google Scholar]
- [2].Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. FASEB J 1998;12:1063–73. [PubMed] [Google Scholar]
- [3].Lee HS, Yun SJ, Ha JM, et al. Prostaglandin D2 stimulates phenotypic changes in vascular smooth muscle cells. Exp Mol Med 2019;51:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 2012;13:518–27. [DOI] [PubMed] [Google Scholar]
- [5].Hull MA, Sprange K, Hepburn T, et al. Eicosapentaenoic acid and aspirin, alone and in combination, for the prevention of colorectal adenomas (seAFOod Polyp Prevention trial): a multicentre, randomised, double-blind, placebo-controlled, 2 × 2 factorial trial. Lancet 2018;392:2583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Roy HK, Turzhitsky V, Wali R, et al. Spectral biomarkers for chemoprevention of colonic neoplasia: a placebo-controlled double-blinded trial with aspirin. Gut 2017;66:285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gurpinar E, Grizzle WE, Piazza GA. COX-independent mechanisms of cancer chemoprevention by anti-inflammatory drugs. Front Oncol 2013;3:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tomić T, Domínguez-López S, Barrios-Rodríguez R. Non-aspirin non-steroidal anti-inflammatory drugs in prevention of colorectal cancer in people aged 40 or older: a systematic review and meta-analysis. Cancer Epidemiol 2019;58:52–62. [DOI] [PubMed] [Google Scholar]
- [9].Fraser DM, Sullivan FM, Thompson AM, et al. Aspirin use and survival after the diagnosis of breast cancer: a population-based cohort study. Br J Cancer 2014;111:623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tsioulias GJ, Go MF, Rigas B. NSAIDs and colorectal cancer control: promise and challenges. Curr Pharmacol Rep 2015;1:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang S, Guo N, Wan G, et al. pH and redox dual-responsive nanoparticles based on disulfide-containing poly(beta-amino ester) for combining chemotherapy and COX-2 inhibitor to overcome drug resistance in breast cancer. J Nanobiotechnology 2019;17:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kaushik K, Das A. Cycloxygenase-2 inhibition potentiates trans-differentiation of Wharton's jelly-mesenchymal stromal cells into endothelial cells: transplantation enhances neovascularization-mediated wound repair. Cytotherapy 2019;21:260–73. [DOI] [PubMed] [Google Scholar]
- [13].Baselyous Y, De Cocinis M, Ibrahim M, et al. Potentially inappropriate concomitant medicine use with the selective COX-2 inhibitor celecoxib: Analysis and comparison of spontaneous adverse event reports from Australia, Canada and the USA. Expert Opin Drug Saf 2019;18:153–61. [DOI] [PubMed] [Google Scholar]
- [14].Arefi H, Naderi N, Shemirani ABI, et al. Design, synthesis, and biological evaluation of new 1,4-diarylazetidin-2-one derivatives (β-lactams) as selective cyclooxygenase-2 inhibitors. Arch Pharm (Weinheim) 2020;35:e1900293. [DOI] [PubMed] [Google Scholar]
- [15].Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. celecoxib long-term arthritis safety study. JAMA 2000;284:1247–55. [DOI] [PubMed] [Google Scholar]
- [16].Veettil SK, Lim KG, Ching SM, et al. Effects of aspirin and non-aspirin nonsteroidal anti-inflammatory drugs on the incidence of recurrent colorectal adenomas: a systematic review with meta-analysis and trial sequential analysis of randomized clinical trials. BMC Cancer 2017;17:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yamahana H, Takino T, Endo Y, et al. A novel celecoxib analog UTX-121 inhibits HT1080 cell invasion by modulating membrane-type 1 matrix metalloproteinase. Biochem Biophys Res Commun 2020;521:137–44. [DOI] [PubMed] [Google Scholar]
- [18].Sethi R, Gomez-Coronado N, Walker AJ, et al. Neurobiology and therapeutic potential of cyclooxygenase-2 (COX-2) inhibitors for inflammation in neuropsychiatric disorders. Front Psychiatry 2019;10:605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mrena J, Wiksten JP, Kokkola A, et al. COX-2 is associated with proliferation and apoptosis markers and serves as an independent prognostic factor in gastric cancer. Tumour Biol 2010;31:1–7. [DOI] [PubMed] [Google Scholar]
- [21].Tong D, Liu Q, Liu G, et al. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett 2017;389:23–32. [DOI] [PubMed] [Google Scholar]
- [22].Kamei D, Murakami M, Sasaki Y, et al. Microsomal prostaglandin E synthase-1 in both cancer cells and hosts contributes to tumour growth, invasion and metastasis. Biochem J 2009;425:361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lu D, Han C, Wu T. Microsomal prostaglandin E synthase-1 promotes hepatocarcinogenesis through activation of a novel EGR1/beta-catenin signaling axis. Oncogene 2012;31:842–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang X, Yan K, Deng L, et al. Cyclooxygenase 2 promotes proliferation and invasion in ovarian cancer cells via the PGE2/NF-kappaB pathway. Cell Transplant 2019;28: 1 Suppl: 1S–3S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou X, Shi X, Ren K, et al. Celecoxib inhibits cell growth and modulates the expression of matrix metalloproteinases in human osteosarcoma MG-63 cell line. Eur Rev Med Pharmacol Sci 2015;19:4087–97. [PubMed] [Google Scholar]
- [26].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- [27].Sharma V, Dixit D, Ghosh S, et al. COX-2 regulates the proliferation of glioma stem like cells. Neurochem Int 2011;59:567–71. [DOI] [PubMed] [Google Scholar]
- [28].Huang C, Chen Y, Liu H, et al. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget 2017;8:115254–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chu TH, Chan HH, Kuo HM, et al. Celecoxib suppresses hepatoma stemness and progression by up-regulating PTEN. Oncotarget 2014;5:1475–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin XM, Li S, Zhou C, et al. Cisplatin induces chemoresistance through the PTGS2-mediated anti-apoptosis in gastric cancer. Int J Biochem Cell Biol 2019;116:105610. [DOI] [PubMed] [Google Scholar]
- [31].Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014;20:460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013;368:651–62. [DOI] [PubMed] [Google Scholar]
- [33].Ravanan P, Srikumar IF, Talwar P. Autophagy: The spotlight for cellular stress responses. Life Sci 2017;188:53–67. [DOI] [PubMed] [Google Scholar]
- [34].Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer 2017;17:528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014;10:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang ZJ, Chee CE, Huang S, et al. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011;10:1533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19:5720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131–45. [DOI] [PubMed] [Google Scholar]
- [40].Arico S, Pattingre S, Bauvy C, et al. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J Biol Chem 2002;277:27613–21. [DOI] [PubMed] [Google Scholar]
- [41].Liu R, Tan Q, Luo Q. Decreased expression level and DNA-binding activity of specificity protein 1 via cyclooxygenase-2 inhibition antagonizes radiation resistance, cell migration and invasion in radiation-resistant lung cancer cells. Oncol Lett 2018;16:3029–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Saxena P, Sharma PK, Purohit P. A journey of celecoxib from pain to cancer. Prostaglandins Other Lipid Mediat 2019;147:106379. [DOI] [PubMed] [Google Scholar]
- [43].Leng J, Han C, Demetris AJ, et al. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 2003;38:756–68. [DOI] [PubMed] [Google Scholar]
- [44].Kern MA, Schubert D, Sahi D, et al. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology 2002;36:885–94. [DOI] [PubMed] [Google Scholar]
- [45].Xu J, Yu Y, He X, et al. Tumor-associated macrophages induce invasion and poor prognosis in human gastric cancer in a cyclooxygenase-2/MMP9-dependent manner. Am J Transl Res 2019;11:6040–54. [PMC free article] [PubMed] [Google Scholar]
- [46].Poligone B, Baldwin AS. Positive and negative regulation of NF-kappaB by COX-2: roles of different prostaglandins. J Biol Chem 2001;276:38658–64. [DOI] [PubMed] [Google Scholar]
- [47].Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol 2004;31: 2 Suppl 7: 2–11. [DOI] [PubMed] [Google Scholar]
- [48].Li T, Zhong J, Dong X, et al. Meloxicam suppresses hepatocellular carcinoma cell proliferation and migration by targeting COX-2/PGE2-regulated activation of the beta-catenin signaling pathway. Oncol Rep 2016;35:3614–22. [DOI] [PubMed] [Google Scholar]
- [49].Kim MJ, Kim HS, Lee SH, et al. NDRG2 controls COX-2/PGE(2)-mediated breast cancer cell migration and invasion. Mol Cells 2014;37:759–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Tai Y, Zhang LH, Gao JH, et al. Suppressing growth and invasion of human hepatocellular carcinoma cells by celecoxib through inhibition of cyclooxygenase-2. Cancer Manag Res 2019;11:2831–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kern MA, Haugg AM, Koch AF, et al. Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res 2006;66:7059–66. [DOI] [PubMed] [Google Scholar]
- [52].Ahmed S, Khan H, Fratantonio D, et al. Apoptosis induced by luteolin in breast cancer: mechanistic and therapeutic perspectives. Phytomedicine 2019;59:152883. [DOI] [PubMed] [Google Scholar]
- [53].Yoshida H, Yoshimura H, Matsuda S, et al. Celecoxib suppresses lipopolysaccharide-stimulated oral squamous cell carcinoma proliferation in vitro and in vivo. Oncol Lett 2019;18:5793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shan BZ, Guo B, Li YS, et al. Effect of celecoxib on protein expression of FAK and Cx43 in DMBA induced rat tongue carcinoma cells. Eur Rev Med Pharmacol Sci 2019;23:9454–63. [DOI] [PubMed] [Google Scholar]
- [55].Muller AJ, Prendergast GC. Indoleamine 2,3-dioxygenase in immune suppression and cancer. Curr Cancer Drug Targets 2007;7:31–40. [DOI] [PubMed] [Google Scholar]
- [56].Curry JM, Besmer DM, Erick TK, et al. Indomethacin enhances anti-tumor efficacy of a MUC1 peptide vaccine against breast cancer in MUC1 transgenic mice. PLoS One 2019;14:e0224309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zheng Y, Comaills V, Burr R, et al. COX-2 mediates tumor-stromal prolactin signaling to initiate tumorigenesis. Proc Natl Acad Sci U S A 2019;116:5223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Oliveira-Ferrer L, Legler K, Milde-Langosch K. Role of protein glycosylation in cancer metastasis. Semin Cancer Biol 2017;44:141–52. [DOI] [PubMed] [Google Scholar]
- [59].Isaji T, Sato Y, Fukuda T, et al. N-glycosylation of the I-like domain of beta1 integrin is essential for beta1 integrin expression and biological function: identification of the minimal N-glycosylation requirement for alpha5beta1. J Biol Chem 2009;284:12207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer 2002;2:91–100. [DOI] [PubMed] [Google Scholar]
- [61].Sproviero D, Julien S, Burford B, et al. Cyclooxygenase-2 enzyme induces the expression of the alpha-2,3-sialyltransferase-3 (ST3Gal-I) in breast cancer. J Biol Chem 2012;287:44490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wong CC, Kang W, Xu J, et al. Prostaglandin E2 induces DNA hypermethylation in gastric cancer in vitro and in vivo. Theranostics 2019;9:6256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007;446:749–57. [DOI] [PubMed] [Google Scholar]
- [64].Wang D, Fu L, Sun H, et al. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 2015;149:1884–95.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Liu J, Chang B, Li Q, et al. Redox-responsive dual drug delivery nanosystem suppresses cancer repopulation by abrogating doxorubicin-promoted cancer stemness, metastasis, and drug resistance. Adv Sci (Weinh) 2019;6:1801987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang P, Mai C, Wei YL, et al. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med Oncol 2013;30:552. [DOI] [PubMed] [Google Scholar]
- [67].Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem 2012;287:4434–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jung S-N, Yang WK, Kim J, et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008;29:713–21. [DOI] [PubMed] [Google Scholar]
- [69].Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, et al. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 2009;9:1084–101. [DOI] [PubMed] [Google Scholar]
- [70].Ellethy AT. Potential antitumor activity of nonsteroidal anti-inflammatory drugs against Ehrlich ascites carcinoma in experimental animals. Int J Health Sci (Qassim) 2019;13:11–7. [PMC free article] [PubMed] [Google Scholar]
- [71].Ralph SJ, Pritchard R, Rodriguez-Enriquez S, et al. Hitting the bull's-eye in metastatic cancers-NSAIDs elevate ROS in mitochondria, inducing malignant cell death. Pharmaceuticals (Basel) 2015;8:62–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Porporato PE, Sonveaux P. Paving the way for therapeutic prevention of tumor metastasis with agents targeting mitochondrial superoxide. Mol Cell Oncol 2015;2:e968043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013;12:931–47. [DOI] [PubMed] [Google Scholar]
- [74].Reczek CR, Chandel NS. The two faces of reactive oxygen species in cancer. Ann Rev Cancer Biol 2017;1:79–98. [Google Scholar]
- [75].Ishikawa K, Takenaga K, Akimoto M, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008;320:661–4. [DOI] [PubMed] [Google Scholar]
- [76].Nunes JB, Peixoto J, Soares P, et al. OXPHOS dysfunction regulates integrin-beta1 modifications and enhances cell motility and migration. Hum Mol Genet 2015;24:1977–90. [DOI] [PubMed] [Google Scholar]
- [77].Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010;40:294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rodriguez-Enriquez S, Carreno-Fuentes L, Gallardo-Perez JC, et al. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int J Biochem Cell Biol 2010;42:1744–51. [DOI] [PubMed] [Google Scholar]
- [79].Heddleston JM, Li Z, Lathia JD, et al. Hypoxia inducible factors in cancer stem cells. Br J Cancer 2010;102:789–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liang D, Ma Y, Liu J, et al. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012;12:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Moreno-Sanchez R, Bravo C, Vasquez C, et al. Inhibition and uncoupling of oxidative phosphorylation by nonsteroidal anti-inflammatory drugs: study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem Pharmacol 1999;57:743–52. [DOI] [PubMed] [Google Scholar]
- [82].Pritchard R, Rodriguez-Enriquez S, Pacheco-Velazquez SC, et al. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem Pharmacol 2018;154:318–34. [DOI] [PubMed] [Google Scholar]
- [83].Mullarky E, Cantley LC. Diverting glycolysis to combat oxidative stress. In: Nakao K, et al. Innovative Medicine: Basic Research and Development. Springer, 2015. pp. 3–23. [PubMed] [Google Scholar]
- [84].Ralph SJ, Nozuhur S, Ra AL, et al. Repurposing drugs as pro-oxidant redox modifiers to eliminate cancer stem cells and improve the treatment of advanced stage cancers. Med Res Rev 2019;39:2397–426. [DOI] [PubMed] [Google Scholar]
- [85].Barsukova A, Komarov A, Hajnoczky G, et al. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur J Neurosci 2011;33:831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Han Y, Chen P, Zhang Y, et al. Synergy between auranofin and celecoxib against colon cancer in vitro and in vivo through a novel redox-mediated mechanism. Cancers 2019;11:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hou GX, Liu PP, Zhang S, et al. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell Death Dis 2018;9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012;13:89–102. [DOI] [PubMed] [Google Scholar]
- [89].Avril T, Vauleon E, Chevet E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017;6:e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 2009;14:996–1007. [DOI] [PubMed] [Google Scholar]
- [91].Mann MJ, Hendershot LM. UPR activation alters chemosensitivity of tumor cells. Cancer Biol Ther 2006;5:736–40. [DOI] [PubMed] [Google Scholar]
- [92].Yang MY, Wu CH, Hung TW, et al. Endoplasmic reticulum stress-induced resistance to doxorubicin is reversed by mulberry leaf polyphenol extract in hepatocellular carcinoma through inhibition of COX-2. Antioxidants (Basel) 2019;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ding WX, Ni HM, Gao W, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007;282:4702–10. [DOI] [PubMed] [Google Scholar]
- [94].Zhang P, He D, Song E, et al. Celecoxib enhances the sensitivity of non-small-cell lung cancer cells to radiation-induced apoptosis through downregulation of the Akt/mTOR signaling pathway and COX-2 expression. PLoS One 2019;14:e0223760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hung JH, Su IJ, Lei HY, et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J Biol Chem 2004;279:46384–92. [DOI] [PubMed] [Google Scholar]
- [96].Alloza I, Baxter A, Chen Q, et al. Celecoxib inhibits interleukin-12 alphabeta and beta2 folding and secretion by a novel COX2-independent mechanism involving chaperones of the endoplasmic reticulum. Mol Pharmacol 2006;69:1579–87. [DOI] [PubMed] [Google Scholar]
- [97].Greenhough A, Smartt HJ, Moore AE, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009;30:377–86. [DOI] [PubMed] [Google Scholar]
- [98].Shao J, Jung C, Liu C, et al. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem 2005;280:26565–72. [DOI] [PubMed] [Google Scholar]
- [99].Luo L, Liang Y, Ding X, et al. Significance of cyclooxygenase-2, prostaglandin E2 and CD133 levels in sunitinib-resistant renal cell carcinoma. Oncol Lett 2019;18:1442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Egashira I, Takahashi-Yanaga F, Nishida R, et al. Celecoxib and 2,5-dimethylcelecoxib inhibit intestinal cancer growth by suppressing the Wnt/β-catenin signaling pathway. Cancer Sci 2017;108:108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Uram L, Misiorek M, Pichla M, et al. The effect of biotinylated PAMAM G3 dendrimers conjugated with COX-2 inhibitor (celecoxib) and PPARγ agonist (Fmoc-L-Leucine) on human normal fibroblasts, immortalized keratinocytes and glioma cells in vitro. Molecules 2019;24:3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].North TE, Goessling W, Walkley CR, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007;447:1007–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Deng Y, Su Q, Mo J, et al. Celecoxib downregulates CD133 expression through inhibition of the Wnt signaling pathway in colon cancer cells. Cancer Invest 2013;31:97–102. [DOI] [PubMed] [Google Scholar]
- [104].Ma HI, Chiou SH, Hueng DY, et al. Celecoxib and radioresistant glioblastoma-derived CD133+ cells: improvement in radiotherapeutic effects. Laboratory investigation. J Neurosurg 2011;114:651–62. [DOI] [PubMed] [Google Scholar]
- [105].Zhou P, Li Y, Li B, et al. Autophagy inhibition enhances celecoxib-induced apoptosis in osteosarcoma. Cell Cycle 2018;17:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lu Y, Liu XF, Liu TR, et al. Celecoxib exerts antitumor effects in HL-60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed Pharmacother 2016;84:1551–7. [DOI] [PubMed] [Google Scholar]
- [107].Lu Y, Liu LL, Liu SS, et al. Celecoxib suppresses autophagy and enhances cytotoxicity of imatinib in imatinib-resistant chronic myeloid leukemia cells. J Transl Med 2016;14:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Huang S, Sinicrope FA. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 2010;6:256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Huang KH, Kuo KL, Ho IL, et al. Celecoxib-induced cytotoxic effect is potentiated by inhibition of autophagy in human urothelial carcinoma cells. PLoS One 2013;8:e82034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol 2018;13:395–412. [DOI] [PubMed] [Google Scholar]
- [112].Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Liu X, Wu Y, Zhou Z, et al. Celecoxib inhibits the epithelial-to-mesenchymal transition in bladder cancer via the miRNA-145/TGFBR2/Smad3 axis. Int J Mol Med 2019;44:683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dai H, Zhang S, Ma R, et al. Celecoxib inhibits hepatocellular carcinoma cell growth and migration by targeting PNO1. Med Sci Monit 2019;25:7351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Shen A, Chen Y, Liu L, et al. EBF1-mediated upregulation of ribosome assembly factor PNO1 contributes to cancer progression by negatively regulating the p53 signaling pathway. Cancer Res 2019;79:2257–70. [DOI] [PubMed] [Google Scholar]
- [116].Walker C, Biasucci LM. Cardiovascular safety of non-steroidal anti-inflammatory drugs revisited. Postgrad Med 2018;130:55–71. [DOI] [PubMed] [Google Scholar]
- [117].Mccarberg BH, Cryer B. Evolving therapeutic strategies to improve nonsteroidal anti-inflammatory drug safety. Am J Ther 2015;22:e167–78. [DOI] [PubMed] [Google Scholar]
- [118].Thompson PA, Ashbeck EL, Roe DJ, et al. Celecoxib for the prevention of colorectal adenomas: results of a suspended randomized controlled trial. J Natl Cancer Inst 2016;108:djw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Varga Z, Sabzwari SRA, Vargova V. Cardiovascular risk of nonsteroidal anti-inflammatory drugs: an under-recognized public health issue. Cureus 2017;9:e1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Horl WH. Nonsteroidal anti-inflammatory drugs and the kidney. Pharmaceuticals (Basel) 2010;3:2291–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Reed GW, Nissen SE. NSAID choice: lessons from PRECISION. Aging (Albany NY) 2019;11:2181–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Onuora S. Therapy: celecoxib reduces risk of ulcer bleeding. Nat Rev Rheumatol 2017;13:324. [DOI] [PubMed] [Google Scholar]
- [123].Varma GYN, Kummari G, Paik P, et al. Celecoxib potentiates antibiotic uptake by altering membrane potential and permeability in Staphylococcus aureus. J Antimicrob Chemother 2019;74:3462–72. [DOI] [PubMed] [Google Scholar]
- [124].Abdelrahman RS, Abdelmageed ME. Renoprotective effect of celecoxib against gentamicin-induced nephrotoxicity through suppressing NFkappaB and caspase-3 signaling pathways in rats. Chem Biol Interact 2020;315:108863. [DOI] [PubMed] [Google Scholar]
- [125].Xiao J, Wang F, Lu H, et al. Targeting the COX2/MET/TOPK signaling axis induces apoptosis in gefitinib-resistant NSCLC cells. Cell Death Dis 2019;10:777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Chen Q, Wang J, Zhang Q, et al. Tumour cell-derived debris and IgG synergistically promote metastasis of pancreatic cancer by inducing inflammation via tumour-associated macrophages. Br J Cancer 2019;121:786–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Imamura M, Okamoto Y, Nishikawa T, et al. Celecoxib as a potential treatment for intractable lymphatic malformation. Pediatrics 2019;144:e20190319. [DOI] [PubMed] [Google Scholar]
- [128].Ye X, Wu H, Sheng L, et al. Oncogenic potential of truncated RXRalpha during colitis-associated colorectal tumorigenesis by promoting IL-6-STAT3 signaling. Nat Commun 2019;10:1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Skaga E, Skaga IO, Grieg Z, et al. The efficacy of a coordinated pharmacological blockade in glioblastoma stem cells with nine repurposed drugs using the CUSP9 strategy. J Cancer Res Clin Oncol 2019;145:1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]