

Abstract
Inhibitors against the NS3-4A protease of hepatitis C virus (HCV) have proven to be useful drugs in the treatment of HCV infection. Although variants have been identified with mutations that confer resistance to these inhibitors, the mutations do not restore replicative fitness and no secondary mutations that rescue fitness have been found. To gain insight into the molecular mechanisms underlying the lack of fitness compensation, we screened known resistance mutations in infectious HCV cell culture with different genomic backgrounds. We observed that the Q41R mutation of NS3-4A efficiently rescues the replicative fitness in cell culture for virus variants containing mutations at NS3-Asp168. To understand how the Q41R mutation rescues activity, we performed protease activity assays complemented by molecular dynamics simulations, which showed that protease-peptide interactions far outside the targeted peptide cleavage sites mediate substrate recognition by NS3-4A and support protease cleavage kinetics. These interactions shed new light on the mechanisms by which NS3-4A cleaves its substrates, viral polyproteins and a prime cellular antiviral adaptor protein, the mitochondrial antiviral signaling protein MAVS. Peptide binding is mediated by an extended hydrogen-bond network in NS3-4A that was effectively optimized for protease-MAVS binding in Asp168 variants with rescued replicative fitness from NS3-Q41R. In the protease harboring NS3-Q41R, the N-terminal cleavage products of MAVS retained high affinity to the active site, rendering the protease susceptible for potential product inhibition. Our findings reveal delicately balanced protease-peptide interactions in viral replication and immune escape that likely restrict the protease adaptive capability and narrow the virus evolutionary space.
Keywords: hepatitis C virus (HCV), serine protease (NS3-4A), structure constraints, evolution, adaptation, mitochondrial antiviral signaling protein (MAVS), protease inhibitor, resistance mutation, replicative fitness, drug resistance, molecular biology, molecular dynamics, molecular adaptation
Viruses under treatment with specific antivirals undergo rapid adaptive selection to maximize their fit for escape from drug pressure. Thereby, increased replicative fitness can lead to decreased drug sensitivity of viral variants (1) and determine the probability with which resistant variants will persist (2). Based on a deterministic model (3–6), hepatitis C virus (HCV) variants under selective pressure with direct-acting antiviral agents are selected by their degree of resistance and capability to replicate and assemble infectious virus particles (7). Protease inhibitors (PI) against the serine protease NS3-4A of HCV are burdened with a particularly high risk for drug resistance development that inflicts considerable fitness costs (7, 8). Although compensatory second-site mutations that rescue fitness deficits from drug resistance mutations are pivotal drivers of viral quasispecies evolution (3, 9, 10), they are virtually not observed upon PI treatment failure in the NS3-4A protease of HCV. Although the impact of mutations on PI resistance has been well-characterized, their role in replicative fitness of viral variants is less understood. The NS3-4A protease plays an essential role in the HCV replication cycle (11–13). In particular, NS3-4A is involved in RNA synthesis by proteolytically processing nonstructural proteins from the viral polyprotein. In addition, the capacity to cleave and inactivate mitochondrial antiviral signaling protein (MAVS) (13) (also called Cardif, VISA, and IPS-1 (14–16)) in the RIG-I-signaling pathway is a cardinal feature of NS3-4A, by which HCV blocks induction of IFN-β. MAVS cleavage is a key mechanism in the selection of drug-resistance mutations against macrocyclic PIs and is important for promoting viral persistence. Novel PI-based treatment regimens still appear to fail in some patients, specifically those with advanced liver disease or liver cirrhosis (17). Importantly, most of these difficult-to-treat patients show dominant resistant variants with D168A or D168V amino acid substitutions in NS3-4A at the time of virologic failure (17). We previously showed that the Asp168 mutant proteases are characterized by increased capacity to cleave MAVS and suppress IFN-β induction (18). The reconstitution of a tight network of electrostatic interactions between protease and the peptide substrate in those variants allows much stronger binding of MAVS than in other protease variants or WT HCV (18).
In the present study, we unraveled a molecular mechanism in PI-resistant Asp168 variants that affect the cutting efficiency of peptide substrates by a second-site mutation in the viral protease, NS3-Q41R. By interference with critically balanced protease-peptide interactions, the second-site mutation rescues replicative fitness deficits of Asp168 variants in infectious cell culture. The finding that NS3-Q41R, which itself is reported to confer resistance to macrocyclic PIs (19), has a fitness-compensatory effect in Asp168 variants is surprising, given the very low prevalence of variants harboring Q41R in PI-experienced patients (<0.1%) (20). Our findings highlight a previously unknown molecular mechanism for MAVS that potentially restricts virus evolution under PI pressure, providing a new paradigm for variant selection from HCV-infected cells.
Results
Rational approach to characterize replicative fitness of PI-resistant variants
We aimed to identify PI resistance–associated amino acid substitutions in NS3-4A that lead to high replicative fitness and potentially confer a general drug resistance phenotype to the virus (1). Resistance mutations were identified by a review of published data from both cell culture–based and clinical studies of PIs (7).
Replicative fitness of Asp168 variants is enhanced by the second-site mutation NS3-Q41R
To characterize the impact of PI-resistance mutations on RNA replication, we constructed two comprehensive panels, each with 23 resistance mutations in derivatives of pH77S (21) (Fig. 1, A and B). The two panels only differed by the cell culture adaptive mutation Q41R in their NS3-4A protease backbone (7) (Fig. 1B). We measured Gaussia luciferase (GLuc) activity from NS3-Gln41/GLuc2A RNA and NS3-Q41R/GLuc2A RNA in media collected at 24-h intervals after transfection. NS3-Q41R showed no loss of replication capacity compared with NS3-Gln41, although the replication capacity of mutants in both backbones, in general, was more compromised in NS3-Q41R (Fig. 1C). Interestingly, NS3-Q41R served to promote the replication of the NS3–Asp168 variants Ala/Glu/His/Val despite acting synergistically to impair the replication of other key PI-resistant variants, such as R155K. Compared with WT, the GLuc activity of D168A was compensated to ∼91% by NS3-Q41R (161% increase compared with NS3-Gln41; fold change (FC) 2.6) and that in D168E was boosted to ∼141% by NS3-Q41R (44% increase compared with NS3-Gln41; FC 1.4). For the clinically relevant D168A/E mutants with increased replicative fitness by NS3-Q41R, we further measured the ability to produce infectious viruses in NS3-Gln41 and NS3-Q41R protease backbones using a sensitive GLuc assay (Fig. 2). Both mutants produced more infectious viruses in the NS3-Q41R backbone than in NS3-Gln41, which corresponded with the respective results on their RNA replication capacity (Fig. 2). Asp168 mutants are signature mutations selected from pressure with macrocyclic PIs such as simeprevir, asunaprevir, paritaprevir and glecaprevir (17, 22). To assess the ability of a macrocyclic PI to inhibit the RNA replication of PI-resistant variants in NS3-Gln41 and NS3-Q41R protease backbones, antiviral EC50 values were determined from the concentration of simeprevir required to cause a 50% reduction in the secretion of GLuc by RNA-transfected cells. The NS3-Q41R backbone caused an increase in EC50 for most PI-resistance mutations tested with on-average-higher–level PI resistance than the respective mutants in an NS3-Gln41 genomic background. All Asp168 mutants tested in the NS3-Q41R backbone showed high-level PI resistance against simeprevir (FC 32–156 compared with the WT protease NS3-Gln41); see Table 1 and Fig. S1 for details on the replicative fitness and drug resistance.
Figure 1.
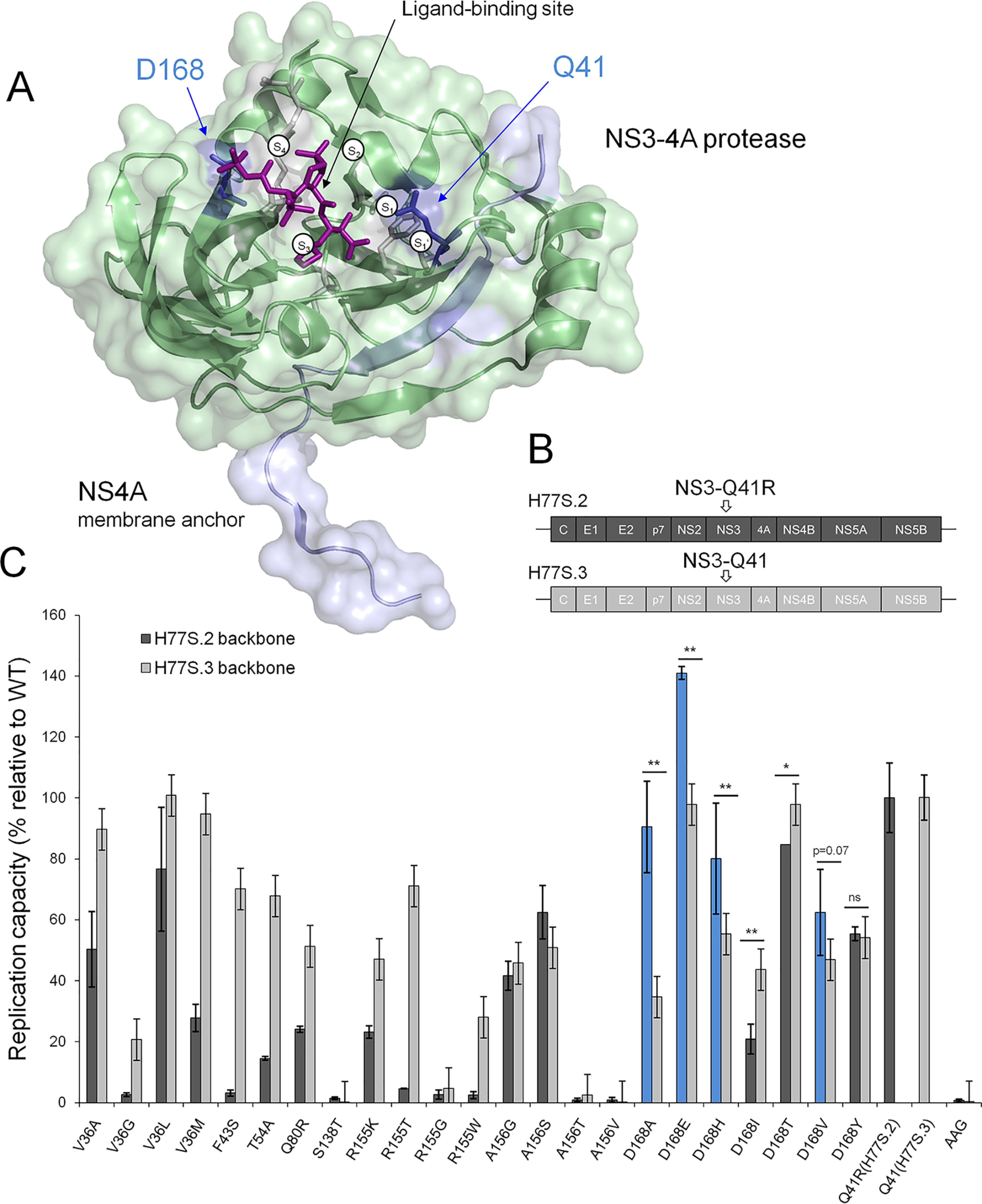
Replicative fitness of PI-resistant variants in NS3 ± Q41R protease genomic backgrounds. A, structural context of PI-resistance mutations depicted on the NS3-4A protease structure from PDB 2OC8 (50) with PI bound to the ligand-binding site (purple stick model); ligand-binding pockets S4 to S1′ (51). The protein surface is partially transparent; protease domain: green, NS4A: light blue. Gln41 and Asp168 are shown as blue stick models located at opposite ends of the protease substrate–binding pocket. B, the H77S.2 molecular clone contains six cell culture adaptive mutations (not shown) throughout the viral genome; the only cell culture adaptive mutation located in the NS3-4A protease domain, NS3-Q41R (depicted by arrow), is reverted back to NS3-Gln41 WT in H77S.3 (6). C, GLuc activity secreted by RNA-transfected cells normalized to pH77S.3 and pH77S.2 WT. Blue shading indicates PI-resistant variants with enhanced replicative fitness by NS3-Q41R. Data shown represent the mean ± S.D. from at least three independent experiments; *p < 0.001; **p < 0.0001; by two-sided t test. AAG, negative control.
Figure 2.

Impact of NS3 ± Q41R on the infectious virus production of Asp168 variants. Comparison of infectious virus yield from D168A/E-protease mutants within NS3-Gln41 and NS3-Q41R backbones. Infectious virus production determined from supernatant fluids at 72 and 96 h infected to naïve Huh-7.5 cells as determined by GLuc activity assay. Data shown represent the mean ± S.D. from at least three independent experiments.
Table 1.
HCV RNA replication and EC50 of simeprevir for PI-resistant variants
HCV RNA replication and EC50 of simeprevir for PI-resistant variants in an NS3-Gln41 (H77S.3) or NS3-Q41R (H77S.2) genomic background.
| Residue | Variant | Replication capacity (%*) |
EC50 (nm§) |
||
|---|---|---|---|---|---|
| NS3-Q41R | NS3-Q41 | NS3-Q41R | NS3-Q41 | ||
| WT | 100 | 100 | 6.4, 0.3 | 6.4, 0.3 | |
| V36 | A | 50.4 | 89.7 | 56, 3.5 | 14, 6.2 |
| G | 2.6 | 20.8 | n/a | 42, 5.2 | |
| L | 76.7 | 100.8 | 38, 2.6 | 12, 2.0 | |
| M | 27.8 | 94.7 | 37, 2.6 | 11, 1.3 | |
| Q41 | R# | n/a | 99.9 | 29, 0.4 | 29, 0.4 |
| F43 | S | 3.2 | 70.2 | 197, 24 | 67, 3.2 |
| T54 | A | 14.5 | 68.0 | 7.3, 0.2 | 3.2, 0.6 |
| Q80 | R | 24.1 | 51.4 | 239, 16 | 48, 5.3 |
| S138 | T | 1.4 | 0.2 | 185, 35 | n/a |
| R155 | K | 23.2 | 47.1 | >1000 | 259, 8.8 |
| T | 4.6 | 71.1 | 162, 21 | 85, 3.7 | |
| G | 2.7 | 45.8 | n/a | 74, 5.7 | |
| W | 2.5 | 28.1 | n/a | 544, 20 | |
| A156 | G | 41.7 | 45.8 | 329, 26 | 84, 7.7 |
| S | 62.5 | 50.9 | 5.8, 1.0 | 2.1, 1.0 | |
| T | 0.88 | 2.5 | n/a | 270, 21 | |
| V | 1.0 | 0.3 | n/a | n/a | |
| D168 | A | 90.5 | 34.7 | >1000 | 2047, 270 |
| E | 141.0 | 97.9 | 504, 65 | 120, 4.8 | |
| H | 80.0 | 55.4 | >1000 | 2228, 103 | |
| I | 20.1 | 43.7 | >1000 | 6935, 469 | |
| T | 84.7 | 97.9 | >1000 | 2025, 77 | |
| V | 62.5 | 47.0 | >1000 | 8070, 624 | |
| Y | 55.4 | 54.2 | n/d | 2640, 584 | |
*Replication capacity relative to WT (%).
§EC50 (nm; mean, S.D.)
#H77S.2 is Arg41 and H77S.3 is Gln41
n/a, replication too low to measure EC50; n/d, mutant not prepared.
For further in vitro characterization, we selected the clinically most relevant PI-resistance–associated mutations D168A and D168E that showed rescued replicative fitness due to NS3-Q41R, but omitted D168H (clinically less important) and D168V (only weak fitness compensation). D168T was selected from the Asp168 mutants as a negative control without impact on replicative fitness in protease backbones ± Q41R.
NS3-Q41R had no effect on the protease fold
To characterize the fitness-compensatory mechanism of NS3-Q41R in Asp168 variants, we expressed and purified the NS3-4A WT fusion and different mutant forms (see “Experimental procedures” and Fig. S2). We first tested if D168A/E/T ± NS3-Q41R had an effect on the protease stability (Fig. 3 and Table 2). The impact of mutants on protein-folding was determined by a thermal shift assay, where protein denaturation at increasing temperatures was revealed by increased fluorescence (see “Experimental procedures”). The NS3-Gln41 protease WT was stable to temperature with a melting temperature, Tm, at 54.9 °C (Fig. 3). NS3-Q41R had no effect on the protein stability. A significant difference in protein stability was observed between D168E and D168A/T mutant proteases irrespective of the NS3-4A protease background ± Q41R (p < 0.0001) (Fig. 3). As we showed previously (18), the impact on protein fold in Asp168 mutants is likely related to weakening or stabilization of a local salt bridge and H-bond network in the outer ligand-binding site of the protease. In contrast to D168A, the D168E mutant can compensate the missing interactions of the Asp168 residue with Arg155 and Arg123 and stabilize the protein fold (18). Our present study confirms the modeling results published earlier (18). The melting temperatures reflect a stable protease fold in D168E versus destabilization in D168A (Fig. 3). Importantly, NS3-Q41R showed no difference to the NS3-Gln41 WT background either as a single mutant or as a second-site mutation with D168A/E/T (Fig. 3 and Table 2). Thus, we considered it unlikely that the increased replicative fitness in the NS3-Q41R background in D168A/E could be due to a modulation of protein stability in the NS3-4A protease.
Figure 3.

Impact of Asp168 mutants on the protein fold of the NS3-4A protease ± NS3-Q41R. Data from real-time thermal stability assay using Sypro Orange, a temperature-stable fluorophore that exhibits enhanced fluorescence upon interacting with unfolded proteins. The thermal stability of WT protease and mutants was assessed under increasing incubation temperatures. The impact of mutations on protein unfolding patterns is characterized by fluorescence emission curves (left). Melting temperatures (Tm) from purified NS3-4A protease mutants are determined by fitting the sigmoidal melt curve to the Boltzmann equation (right). Error bars represent the mean ± S.D. from at least three independent experiments; *p ≤ 0.05; **p ≤ 0.01; by two-sided t test.
Table 2.
Enzyme kinetic constants and protein melting temperatures for NS3-4A protease variants
Kinetic constants (left panel) and melting temperatures (right panel) were obtained by a FRET-based protease and thermal shift assay, respectively. Data shown represent the mean ± S.D. from at least three independent experiments.
| Substrate | Variant | Enzyme kinetics |
Variant | Melting temperature Tm (°C) | ||
|---|---|---|---|---|---|---|
| Km (μm) | kcat (/s) | kcat/Km (1/(s*µm)) | ||||
| NS4A/4B | WT (Q41) | 3.11 ± 0.29 | 0.19 ± 0.011 | 0.062 ± 0.008 | WT (Q41) | 54.9 ± 0.3 |
| Q41R | 2.11 ± 0.14 | 0.18 ± 0.006 | 0.085 ± 0.008 | Q41R | 55.1 ± 0.3 | |
| D168A | 2.45 ± 0.23 | 0.09 ± 0.004 | 0.035 ± 0.004 | D168A | 48.7 ± 0.1 | |
| Q41R-D168A | 1.71 ± 0.23 | 0.15 ± 0.009 | 0.087 ± 0.015 | Q41R-D168A | 48.3 ± 0.2 | |
| D168E | 1.79 ± 0.11 | 0.12 ± 0.003 | 0.067 ± 0.005 | D168E | 55.9 ± 0.2 | |
| Q41R-D168E | 1.83 ± 0.10 | 0.14 ± 0.001 | 0.074 ± 0.005 | Q41R-D168E | 55.8 ± 0.3 | |
| D168T | 2.03 ± 0.15 | 0.13 ± 0.005 | 0.064 ± 0.006 | D168T | 47.1 ± 0.1 | |
| Q41R-D168T | 1.69 ± 0.14 | 0.12 ± 0.004 | 0.071 ± 0.007 | Q41R-D168T | 48.5 ± 0.2 | |
| MAVS | WT (Q41) | 7.82 ± 0.70 | 0.058 ± 0.002 | 0.008 ± 0.001 | ||
| Q41R | 37.32 ± 4.42 | 0.275 ± 0.021 | 0.007 ± 0.001 | |||
| D168A | 14.41 ± 2.91 | 0.029 ± 0.003 | 0.002 ± 0.000 | |||
| Q41R-D168A | 1.77 ± 0.14 | 0.507 ± 0.011 | 0.287 ± 0.024 | |||
NS3-Q41R can modulate the kinetic properties of the protease for viral polyprotein substrates and MAVS
Next, we analyzed the protease enzymatic activity using an in vitro FRET-based assay that measures the cleavage of a viral polyprotein substrate. We tested the kinetic properties of NS3-Gln41 WT and D168A/E/T ± NS3-Q41R mutants using a peptide substrate derived from the viral NS4A/4B polyprotein cleavage site (Fig. 4A and Table 2) comprising an excited donor molecule and a quencher (see “Experimental procedures”). In this assay, fluorescence is released when the protease recognizes and cleaves the FRET substrate. We determined the amount of substrate needed to obtain half of the protease maximum rate of reaction (Km) and substrate turnover rates (kcat). The protease with the NS3-Q41R backbone exhibited a lower Km than the NS3-Gln41 WT (p = 0.08; FC 0.5). No difference was found in the substrate turnover kcat between the two genomic backgrounds. The D168A mutant in NS3-Gln41 showed a significantly lower kcat (p = 0.0008; FC 1.1) and a slight decrease in Km (p = 0.24; FC 0.3) compared with Asp168-WT. A further decrease in Km (p = 0.13; FC 0.8) paralleled by a significant increase in kcat (p = 0.003; FC 0.7) for D168A was found in the NS3-Q41R backbone. Whereas differences in Km were not significant for D168E ± NS3-Q41R, kcat was significantly higher for D168E in the NS3-Q41R versus NS3-Gln41 backbone (p = 0.01; FC 0.2) (Fig. 4A). No such effect was observed for D168T ± NS3-Q41R (Fig. 4A), which was tested as a Asp168 variant not compensated for replicative fitness deficits by NS3-Q41R (Fig. 1C). Although Km and kcat varied in rather narrow ranges between 1.7 and 3.1 µm and 0.09 and 0.19, respectively, the kinetic constants reflect the fitness-compensatory effect of NS3-Q41R for D168A/E variants that we observed in infectious cell culture (Fig. 1C).
Figure 4.

Impact of Asp168 mutants and NS3 ± Q41R on protease enzymatic function. A, reaction velocity and Michealis-Menten kinetics as assessed from purified protein of protease WT and Asp168 mutants ± NS3-Q41R and the natural polyprotein substrate NS4A/4B and (B) assessed for the MAVS peptide (each at different peptide concentrations). Reaction constants Km and kcat were calculated after nonlinear regression curve fitting. Error bars represent the mean ± S.D. from at least three independent experiments; *p < 0.05; **p ≤ 0.01; ***p ≤ 0.001; by two-sided t test.
Notably, NS3-Q41R is not observed as a fitness-compensatory mutation in PI-failure patients with Asp168 variants (23). As we showed previously, efficient MAVS cleavage is a characteristic feature of Asp168 variants and a key escape mechanism under selective pressure with macrocyclic PIs (18). Hence, we reasoned that NS3-Q41R could have varied the kinetic properties of D168A variants toward MAVS. To characterize the effect of D168A and NS3-Q41R on the protease-MAVS cleavage kinetics, we designed a novel FRET peptide incorporating the MAVS cleavage site. As done for NS4A/4B, Km and kcat of D168A ± NS3-Q41R were determined for the MAVS peptide (Fig. 4B). In contrast to NS4A/4B, we observed vast variations in the kinetic constants for MAVS. Compared with the NS3-Gln41 protease WT, NS3-Q41R showed an ∼5-fold increase in kcat (p = 0.008; FC 4.7), although the Km was significantly higher than in the WT (p = 0.02; FC 4.8). A highly significant and fundamental change with inverse kinetic constants was observed when NS3-Q41R was present as a second-site mutation in the D168A protease (Fig. 4B). Here, D168A showed a more than 16-fold increase in kcat compared with D168A without NS3-Q41R (p = 0.0003; FC 16.5). In parallel, the Km was significantly decreased (p < 0.05; FC 7.1) (Fig. 4B). Thus, there was large reversal in Km and kcat for D168A upon the addition of NS3-Q41R. Next, we calculated the ratio kcat/Km, the specificity constant, to compare the relative rates of NS3-4A protease cleavage on the competing substrates NS4A/4B and MAVS (Table 2). We found that the specificity constant of the D168A-protease for MAVS increased more than 100-fold when harboring NS3-Q41R and 3.3-fold higher than for NS4A/4B.
NS3-Q41R increases the ability of the protease and Asp168 variants to cleave MAVS
The above results using peptide substrates in vitro suggested a fitness advantage due to NS3-Q41R in Asp168 variants via increased MAVS cleavage capacity. To test this hypothesis in cells, we prepared expressed constructs for Asp168-WT and D168A/E/T mutants in protease genomic backgrounds ± NS3-Q41R. The expressed constructs were transfected into human bone osteosarcoma epithelial cell line U-2 OS cells that express high levels of endogenous MAVS (18). Western blotting was employed after transfection with NS3-4A expression constructs to detect full-length and cleaved MAVS (Fig. 5A). Our data revealed that NS3-Gln41 constructs harboring D168E/T had a comparable capacity to cleave endogenous MAVS compared with the NS3-Gln41 protease WT. A notable exception is D168A in the NS3-Gln41 protease that displayed a significantly higher potency to cleave MAVS than the NS3-Gln41 protease WT, which is in line with our previous findings (18). Interestingly, the addition of NS3-Q41R increased the capacity of the protease to cleave MAVS for all Asp168 variants tested (Fig. 5A). Most notably, full-length MAVS was not visible already after 12 h of transfection in D168A + NS3-Q41R. We observed complete cleavage of MAVS after 16 h of transfection for D168A/E/T in the NS3-Q41R background. Remarkably, we observed no degradation of the cleavage products for D168A/E/T in NS3-Q41R after 12 h of transfection (Fig. 5A).
Figure 5.

Impact of Asp168 mutants and NS3 ± Q41R on MAVS cleavage capacity and IFN-β activation. A, U-2 OS cells were left untransfected (mock), transfected with a GFP expression plasmid (control of transfection efficiency), and transfected with expression constructs harboring NS3-Gln41 or NS3-Q41R ± D168A/E/T. MAVS cleavage as assessed by Western blotting; one of five representative experiments is shown. B, U-2 OS cells co-transfected with IFN-β reporter plasmids, a full-length MAVS expression construct, and D168A/E/T mutant protease constructs ± NS3-Q41R, subsequently analyzed for IFN-β-dependent luciferase activity. Renilla luciferase (RLuc) activity determined to normalized IFN-β activity. Error bars represent the mean ± S.D. from at least three independent experiments; *p ≤ 0.05; **p ≤ 0.01; by two-sided t test; f.l., full-length; cl., cleaved form; FI, fold induction; ns, not significant.
Next, we tested the capacity of MAVS in U-2 OS cells to induce IFN-β for Asp168-WT and D168A/E/T mutants in protease genomic backgrounds ± NS3-Q41R after transfection with an IFN-β–reporter plasmid (Fig. 5B). As expected, over-expression of MAVS resulted in strong activation of the IFN-β reporter. As previously shown (18), we found IFN-β activity significantly inhibited by D168A in the NS3-Gln41 protease backbone. Moreover, we found that proteases harboring NS3-Q41R or NS3-Q41R + D168E significantly attenuated the IFN-β activity. The competence of the Q41R-D168A protease to interfere with MAVS-induced IFN-β–reporter activation did not differ significantly from the Gln41 background. A potential explanation is that peptides from MAVS cleavage could play an inhibitory role at the protease active site. Such a model could explain that the Q41R-D168A protease with high kcat but low Km for MAVS was not superior in inhibiting IFN-β activity compared with the D168A mutant protease (Fig. 4B and Fig. 5B).
An extended hydrogen-bond network around the protease active site plays a critical role in substrate recognition
We previously showed that the mutant D168A reconstitutes electrostatic interactions between the protease and MAVS, facilitating stronger binding of the MAVS peptide to the D168A mutant than to the Asp168-WT protease (18). To characterize the molecular mechanisms of fitness compensation by NS3-Q41R as a second-site mutation in the D168A protease, we performed molecular dynamics (MD) simulations of the NS3-Gln41 protease WT and the Q41R-D168A mutant protease, bound to the viral polyprotein junction NS4A/4B or to MAVS (Fig. 6, left panel). For each simulation, we analyzed the H-bond network surrounding the protease active center (Fig. 6, right panel) and calculated peptide-protease interaction energies (Table 3).
Figure 6.
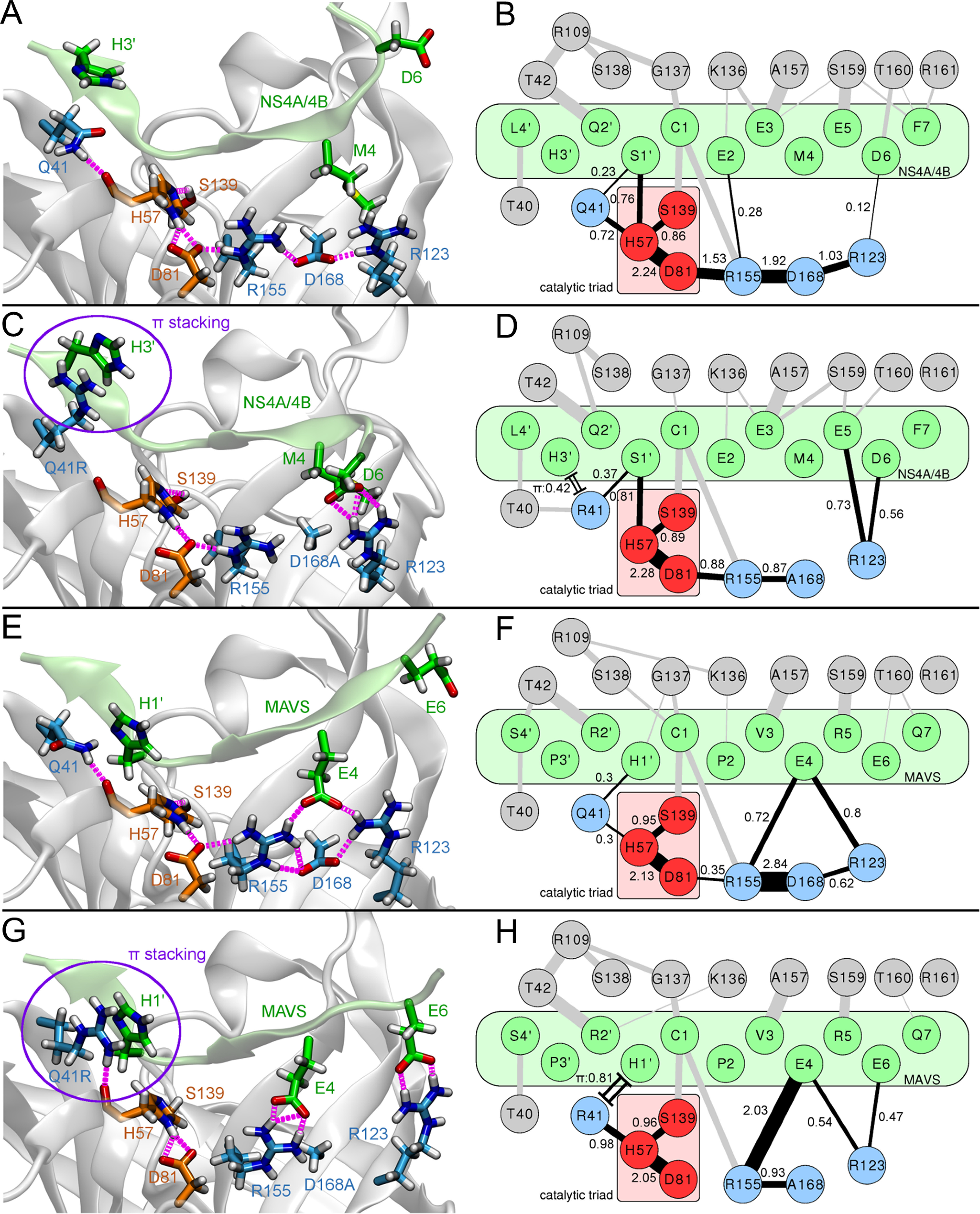
Molecular characterization of protease-substrate interactions. Illustrations of representative protease binding-site conformations (left panel: A, C, E and G) and H-bond networks of protease-substrate complex simulations (right panel: B, D, F and H). (A) 3D structure and (B) residue-interaction network of the NS4A/4B substrate bound to the NS3-Gln41 protease WT or (C) 3D structure and (D) residue-interaction network for NS4A/4B bound to the Q41R-D168A protease; (E) 3D structure and (F) residue-interaction network for MAVS bound to the NS3-Gln41 protease WT or (G) 3D structure and (H) residue-interaction network for MAVS bound to the Q41R-D168A protease. Hydrogen bonds present in each time frame were extracted from MD trajectories. Edge widths (single lines) in the residue networks shown represent the average number of H-bonds during simulation time and correspond to the individual bond strength; a quantitative measure for the bond strength was provided by corresponding numbers. Only edges above a minimum occurrence limit of 0.1 are shown. Cation-π stacking interactions between arginine and histidine residues (time fraction within π stacking distance cutoff) are shown as double lines with caps. Atom colors: Substrate (green); protease catalytic triad (red); extended H-bond network of the catalytic triad (blue). H-bonds are shown in magenta (A, C, E and G) and black (B, D, F and H).
Table 3.
Average interaction energies of the NS3-4A protease-substrate complexes
Average interaction energies in kcal/mol from the last 100 ns of MD simulations of the NS3-4A protease-substrate complexes.
| Variant | Substrate | ΔEb | ΔΔEc | σe(ΔE)d |
|---|---|---|---|---|
| WT (Q41) | NS4A/4B | −558.85 | −3% | 8.34 |
| N-term NS4A/4Ba | −541.94 | 3.79 | ||
| Q41R-D168A | NS4A/4B | −719.46 | +9% | 11.33 |
| N-term NS4A/4Ba | −783.06 | 7.80 | ||
| WT (Q41) | MAVS | −184.57 | +49% | 6.16 |
| N-term MAVSa | −274.70 | 6.22 | ||
| Q41R-D168A | MAVS | −237.56 | +77% | 4.22 |
| N-term MAVSa | −420.34 | 3.29 |
aN-terminal cleavage product.
bAverage interaction energy without solvent.
cRelative interaction energy difference of the Q41R-D168A mutant compared with WT.
dError of uncorrelated samples.
A comparison of the simulated systems showed that the Asp168-WT protease with NS3-Gln41 bound to NS4A/4B formed a well-balanced and stable extended H-bond network (Fig. 6, A and B). The core of this network consisted of H-bonds within the protease catalytic triad Asp81-His57-Ser139. This core network was extended by H-bond contacts to Gln41 and Arg155-Asp168-Arg123. The two mutants D168A and NS3-Q41R substantially disturbed this extended network, weakening its coupling to the protease active site (Fig. 6, C and D). Instead, Arg41 and Arg123 participated in more H-bonds (∼1.3 more) and π–π contacts (+42% of simulation time) to the NS4A/4B peptide substrate than in the WT protease. For the Q41R-D168A mutant protease, the interaction energy between protease and the NS4A/4B noncleaved substrate increased by ∼29% compared with the WT protease (Table 3).
A similar analysis of the H-bond network for the WT protease bound to MAVS identified a substantially weaker stabilization of the catalytic site (Fig. 6, E and F) than in the NS4A/4B-protease complex (Fig. 6, A and B). The destabilization was a direct consequence of polar MAVS residues competing with intraprotease H-bonds within the extended network. This changed considerably because of the mutants D168A and NS3-Q41R (Fig. 6, G and H). Q41R participated in strong H-bonds (+0.98) with the protease active site while establishing π–π interactions (+81%) to MAVS at the same time, rigidifying the complex around the active site (Fig. 6H). In addition, D168A gained strong H-bonds between MAVS and Arg155 (+1.31)/Arg123 (+0.21). These changes in the bonding network leveraged the affinity of the N-terminal MAVS and NS4A/4B cleavage products to the Q41R-D168A protease (77% versus 9% gain in protease-peptide interaction energy, respectively) (Table 3). Summing up effects from protease mutations and substrate binding, NS3-Q41R and D168A showed substantial impact on the H-bond network at the protease active site. Both mutations split the H-bond network and freed protease residues for additional interactions with the substrates, explaining the observed decreases in Km values. Binding of the natural substrate NS4A/4B yielded a stable extended H-bond network in the WT protease, which was weakened by NS3-Q41R and D168A. In contrast, MAVS binding to the protease naturally disturbed this network in the WT protease. Here, the PI-resistance mutation D168A and its second-site mutation NS3-Q41R showed unique compensatory interactions in the protease bound to MAVS and stabilization of the protease active site surroundings. This aligns well with the kcat trends from our enzyme activity assays and Michaelis-Menten kinetics (Fig. 4).
Discussion
Fitness compensation by a second-site mutation in the HCV protease NS3-4A, as we show here, has not been noted previously (20, 23). We find NS3-Q41R to permit a fitness recovery for viral variants with resistance-associated amino acid substitutions at Asp168 in infectious cell culture at high-level resistance against a macrocyclic PI (Fig. 1C and Table 1). Our findings emphasize the capability of NS3-Q41R to yield “superfit” PI-resistant Asp168 variants. However, NS3-Q41R is not observed in patients, though the selection of Asp168 variants in advanced-stage liver disease is responsible for the vast majority of PI treatment failures (17). We considered the underlying mechanisms to be important to understand structural constraints and adaptive capability in HCV NS3-4A.
To characterize the molecular mechanism of fitness compensation by NS3-Q41R, we used enzyme kinetics from purified protein and complementary molecular dynamics simulations. As previously shown, a characteristic feature of Asp168-protease mutants is the molecular adaptation of the protease active site for cleavage and inactivation of MAVS (18), a key cellular antiviral adaptor protein whose expression level directly determines antiviral immunity (14–16, 24). Strikingly, we found that the D168A mutant protease boosted to 16-fold higher MAVS cleavage efficiency by NS3-Q41R, whereas the efficiency for the viral polyprotein substrate was only 2-fold increased (Fig. 4). This is important, given that blocking RIG-I signaling by targeting MAVS is a key strategy in several highly diverse classes of viruses to evade type I–interferon immune responses (25–27). Conversely, MAVS in primates evolved under strong positive selection, driven by antagonism from ancient “paleovirus” infections (28). As a consequence of protease-peptide coevolution, viral proteases can be highly adapted for MAVS cleavage already in the WT protein (28). In the HCV protease NS3-4A, we found that NS3-Q41R had a strong impact on the catalytic activity and turnover in Asp168 mutant proteases, thereby differentially tuning the protease for viral polyprotein substrates and MAVS. Of note, the changes in the protease kinetic constants for the viral polyprotein substrate NS4A/4B reflect the fitness-compensatory effect of NS3-Q41R that we observed in infectious cell culture. The hepatocyte-derived cellular carcinoma cell line Huh-7.5 cells that we were using here are defective for signaling via RIG-I and MAVS (29). Hence, the MAVS cleavage capacity of the protease mutants should not have made an impact on the replicative fitness in our cell culture model of HCV infection.
Mechanistically, we find a delicately balanced extended hydrogen-bond network around the protease active site that was altered by Asp168 mutants and the second-site mutation NS3-Q41R. Governed by changes on the opposite ends of this network (Fig. 6), the protease active site environment is effectively optimized for MAVS binding by D168A and NS3-Q41R (Fig. 6H and Table 3). In line with these findings, we show that NS3-Q41R in cell culture shifted the balance in protease-substrate recognition in the Asp168 variants toward MAVS (Fig. 5). The presence of hydrophilic and charged residues in the peptide substrate between the P4 and P3′ sites is likely a key factor determining how Asp168 and Gln41 mutations influence binding and catalysis. Therefore, based on our observations for NS4A/4B and MAVS, the binding of other cellular substrates of the NS3-4A protease might also be affected by mutation of these sites: Primarily hydrophobic substrates like the membrane-associated glutathione peroxidase 8, the E3 ubiquitin ligase component UV-damaged DNA-binding protein 1, and the cleavage site b within T-cell protein tyrosine phosphatase, a phosphatase involved in growth factor signaling, could behave similarly to NS4A/4B, whereas hydrophilic residues of the Toll/interleukin-1 receptor domain–containing adaptor inducing interferon-β or the T-cell protein tyrosine phosphatase cleavage site a might interact with the extended network along the lines of MAVS (Fig. S3).
Notably, we found that the MAVS-cleaved products did not get degraded quickly and retained high affinity to Asp168 protease variants when harboring NS3-Q41R (Fig. 5 and Table 3). Hence, we speculated that the cleaved products might compete with protease substrates at the active site, a mechanism that potentially could down-regulate the protease catalytic efficiency. Adding to this notion, we calculated protease-peptide interaction energy differences from the molecular dynamics simulations that in the Q41R-D168A mutant context showed a remarkable gain of 77% for the N-terminal MAVS cleavage product compared with only 9% for the viral polyprotein peptide (Table 3). In support of a direct impact of MAVS on the protease catalytic activity, preincubation of the purified protein with MAVS peptides showed a more potent blockade of enzyme activity in a protease harboring NS3-Q41R than in the WT protease (Fig. 7).
Figure 7.
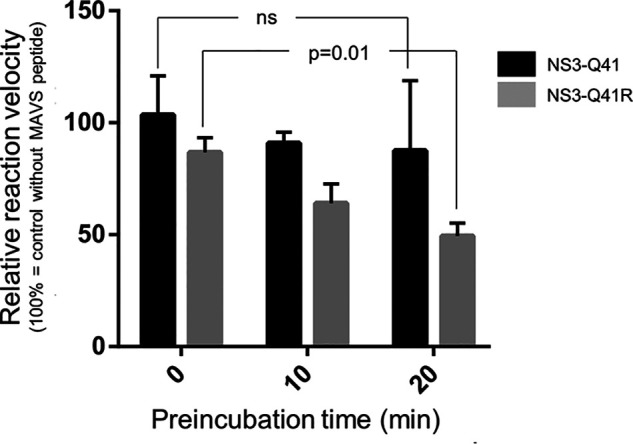
Inhibition of the NS3-4A protease ± Q41R by MAVS cleavage products. Relative reaction velocity for NS4A/4B cleavage as assessed for the purified NS3-Gln41 protease WT and NS3-Q41R mutant protease (measured by FRET protease assay) and different preincubation time with MAVS peptide (0, 10, and 20 min). Reference velocity (100%) is the cleavage reaction for the NS4A/4B FRET peptide without MAVS. Error bars represent the mean ± S.D. from at least three independent experiments; ns, not significant, p = 0.01; by two-sided t test.
It is difficult to compare directly the in vitro biochemistry with the cellular degradation of MAVS. There are several strong limitations for this comparison, such as ambiguous concentrations of viral protease, polyprotein substrates, and MAVS expression levels in cells. However, from the biochemistry data, we clearly conclude that mutations in the NS3-4A protease can have vast differences in the cutting efficiency of peptide substrates. Our data suggest that protease variants already optimized for MAVS cleavage can be easily overfitted for this peptide substrate by second-site mutations in NS3-4A. Such a mechanism could interfere with RNA synthesis and immune escape and hence prevent selection of fitness-compensatory mutations such as NS3-Q41R in patients. The mechanism could potentially explain previous observations from a chimpanzee infected with a cell culture–derived virus containing NS3-Q41R that initially was poorly fit but evolved to establish long-term persistent infection (30). Yi and coworkers observed an unusual 4–6-week delay before detectable viremia in this animal, during which the level of intrahepatic innate immune responses increased substantially. They found that the cell culture–adapted H77S.2 strain had reverted Arg41 back to Gln41 WT before viremia (30), whereas other chimpanzees infected by NS3-Gln41 WT strains were viremic within a week (31, 32).
Collectively, our data support a model in which balancing between MAVS cleavage capability and efficiency of polyprotein processing likely restricts the selection of drug-resistant HCV protease variants in vivo. Our data suggest that the negative selection of fitness-compensatory mutations in NS3-4A to some extent is a consequence of protease-MAVS overfitting, which is important because it can prevent fitness compensation and hence ultimately contribute to extinction of PI-resistant HCV variants upon treatment failure (2–5).
Experimental procedures
Cell culture studies
Central to our studies has been the use of an infectious molecular clone of genotype 1a H77 virus (21). The clone pH77S.2 was derived from pH77S and differs from pH77S.3 by the cell culture adaptive mutation NS3-Q41R in the NS3-4A protease domain (Fig. 1B). When transfected into permissive Huh-7.5 cells (21), genome-length RNA transcribed from pH77S.2 and pH77S.3 replicates efficiently and produces a virus that is infectious in naïve cells (21). To monitor replication, the GLuc sequence, fused at its C terminus to the foot-and-mouth disease virus 2A autoprotease, was inserted between p7 and NS2 (7).
Site-directed mutagenesis and transfection
Primers for NS3-4A amplification were used with restriction sites for NheI and EcoRI and designed as follows: genotype (GT)1a-NS3-NheI-For (ATGATGGCTAGCGCCACCATGGCGCCCATCACGGCGTACGCCCAGCAG) and GT1a-NS3-EcoRI-Rev (ATGATGGAATTCTCACTAGCACTCTTCCATCTCATCGAACTC). Amplified PCR products were subcloned into the expression vector pcDNA3.1(+) from Invitrogen. NS3-4A mutant constructs in NS3-Gln41 and NS3-Q41R backbones were generated by site-directed mutagenesis. Primers for the mutagenesis of Asp168 mutants were designed as follows: GT1a-NS3-D168A-For (GCTAAAGCGGTGGCCTTTATCCCTGTG) and GT1a-NS3-D168A-Rev (CACAGGGATAAAGGCCACCGCTTTAGC), GT1a-NS3-D168E-For (GCTAAAGCGGTGGAGTTTATCCCTGTG) and GT1a-NS3-D168E-Rev (CACAGGGATAAACTCCACCGCTTTAGC), and GT1a-NS3-D168T-For (GCTAAAGCGGTGACCTTTATCCCTGTG) and GT1a-NS3-D168T-Rev (CACAGGGATAAAGGTCACCGCTTTAGC). All generated constructs were controlled by sequencing with Eurofins. Transfection was performed using the X-tremeGene HP DNA transfection reagent (Roche) according to the manufacturer's instructions.
Viral fitness and antiviral resistance
Genome-length RNA was transcribed from the mutated pH77S.2, pH77S.3, pH77S.2/Gluc2A, and pH77S.3/GLuc2A plasmids in vitro, and studies to assess antiviral resistance and viral fitness were carried out as described previously (7).
MAVS cleavage and IFN-β promoter activation
To assess MAVS cleavage in cells, we used U-2 OS human osteosarcoma cells (obtained from the American Type Culture Collection) and Western blotting. U-2 OS cells express high levels of endogenous MAVS and are highly permissive for transfection with NS3-4A expression constructs as previously described (18). IFN-β promoter activation assays were based on luciferase reporter assays as described previously (33).
Antibodies
Western blot analyses were performed using the following antibodies: mouse mAb to Cardif/MAVS (Adri-1) (catalog no. A3002702, AdipoGen), anti-β-actin mouse mAb (AC-15) (Sigma-Aldrich), and goat antibody to HCV NS3 region (catalog no. 8C07014, Meridian Life Science).
Protein biochemistry
For transformation and recombinant NS3-4A protein expression, the pET-15b vector incorporating the single-chain proteolytic domain (NS4A21-32-GSGS-NS33-181) (34) of WT and mutant NS3-4A was transformed into Escherichia coli Rosetta (DE3)pLysS competent cells (Novagen) according to the manufacturer's protocol. Recombinant protein production and purification was carried out as described previously (Fig. S2) (34).
Thermal shift assay
Stability of the protein fold of expressed WT and mutant NS3-4A was measured by fluorescence-based thermal shift (35) using Sypro Orange, a temperature-stable fluorophore that exhibits enhanced fluorescence upon interacting with unfolded proteins. The impact of mutations on protein unfolding patterns is characterized by fluorescence emission curves. Melting temperatures from purified NS3-4A protease mutants are determined by fitting the sigmoidal melt curve to the Boltzmann equation.
Protease activity assay
The enzymatic activity and Michaelis-Menten kinetics of WT and mutant NS3-4A proteases were measured by a fluorescence-based protease assay (36, 37). To measure peptide cleavage kinetics, a viral polyprotein substrate derived from the NS4A/4B cleavage site was purchased from Anaspec (SensoLyte 520 HCV). To determine enzyme kinetics for MAVS cleavage, we designed a novel FRET peptide incorporating the MAVS cleavage site, produced by Anaspec (Ac–EREVPCHRPS–NH2).
MAVS product inhibition assay
We used an untagged MAVS peptide incorporating the MAVS cleavage site (Ac–EREVPCHRPS–NH2) to determine the influence of MAVS cleavage products on the cleavage kinetics of viral polyprotein substrates. Small amounts of purified NS3-4A protease (NS3-Gln41 WT or NS3-Q41R, 50 nm) were preincubated with MAVS peptide (50 µm) for 0, 10, or 20 min at room temperature to allow different time spans for cleavage product formation. After preincubation, 2 µm of the NS4A/4B FRET peptide (SensoLyte 520 HCV, Anaspec) was added and reaction velocity was measured as described above. Of note, MAVS and NS3-4A are shown to colocalize in infected cells (38). Hence, the effective concentration of MAVS can theoretically reach levels higher than 50 µm.
Statistical analyses
Comparisons of measurements were analyzed by two-sided t tests with a level of significance set at a minimum of p = 0.05.
Computational studies
Molecular systems were prepared from a crystal structure of the NS3-4A protease WT bound to the N-terminal cleavage product of MAVS (PDB 3RC5) (39). We modeled both full-length substrates, MAVS and NS4A/4B, prior to the peptide bond cleavage. The P′1–4 residues of MAVS, missing in the experimental structure, were rebuilt based on the crystal structure of bovine chymotrypsin bound to eglin C (PDB 1ACB) (40), as previously performed (41). Topologies and starting coordinates for molecular dynamics simulations of protease WT and mutants bound to MAVS and NS4A/4B substrates were prepared using the tleap program of AmberTools15 (42), minimized in gas phase while restraining the protein backbone. The structures were then solvated and neutralized in a box of SPC/E water molecules (43) using a minimum distance of 14.0 Å between the boundary and the protein atoms. All calculations were performed with the Amber14/AmberTools15 suite of programs (41) using the ff03.r1 Amber force field (44, 45). After minimization and adjustment of the box size to reach a target density of 1,000 kg/m3, the systems were gradually heated up to 300 K in the NVT ensemble (45). Finally, production runs were performed for 250 ns in the NPT ensemble at 300 K and 1 bar. Both heat-up and production runs used a time step of 1 fs, and SHAKE bond-length constraints were applied to all bonds involving hydrogen atoms. Temperature and pressure were controlled with Langevin dynamics (collision frequency of 4.0 ps−1) and isotropic position scaling, respectively.
Analysis of the trajectories was performed on frames extracted from the last 100 ns of the NPT production simulations. Analysis and visualization of residue-interaction networks was carried out using our in-house interaction network plugin CONAN (manuscript in preparation) for Cytoscape 3 (46). Hydrogen bonds present in each time frame were extracted from MD trajectories using cpptraj (46) with a maximum donor-acceptor distance of 3.5 Å and a minimum donor-hydrogen–acceptor angle of 135°. Edge widths in the networks shown represent the average number of H-bonds during simulation time. Only edges above a minimum simulation presence of 10% are shown. Cation-π stacking interactions between arginine and histidine residues were analyzed by considering side-chain distances and relative orientations. The side-chain plane was defined for the Arg and His residues based on the heavy atoms of the side-chain guanidinium group and imidazole ring, respectively. In this scheme, an interaction was counted as present if the distance between the center of mass of the two planes did not exceed 5.0 Å, and if the angle between the plane normal vectors was found in the ranges of 0–30° or 150–180°. Protein-substrate interaction energies were obtained from the “Delta G gas” term as calculated by MMPBSA.py (AmberTools17) (47–49). The error of uncorrelated samples σe was estimated by an autocorrelation time analysis (48), using a weighted single exponential fit (weight factor 1/(x + 1)), where x is the integer distance between frames. To reduce noise in the fit, only autocorrelation values larger than 0.1 were used.
Data availability
All data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank Hirai Feng and Stanley M. Lemon (The University of North Carolina at Chapel Hill) for helpful discussions and for critically reviewing the manuscript, Yolanda Martinez and Marie Fiedler for expert technical assistance, and Charles M. Rice (The Rockefeller University) for supplying the Huh-7.5 cells.
This article contains supporting information.
Author contributions—G. D., T. S., M. S., K. M., D. Y., A. M., T. M. Z., C. S., C. G., C. M. L., and C. W. formal analysis; G. D., T. S., M. S., A. M., T. M. Z., S. Z., R. T., I. A., C. M. L., and C. W. funding acquisition; G. D., T. S., M. S., K. M., D. Y., A. M., T. M. Z., C. S., C. G., R. M. R., K. B., M. Y., R. M. B., R. T., I. A., C. M. L., and C. W. investigation; G. D., T. S., M. S., D. Y., A. M., T. M. Z., R. T., I. A., C. M. L., and C. W. visualization; G. D., R. M. B., R. T., I. A., C. M. L., and C. W. methodology; G. D., T. S., M. S., K.M., D. Y., A. M., T. M. Z., C. S., C. G., R. M. R., K. B., M. Y., R. M. B., S. Z., R. T., I. A., C. M. L., and C. W. writing-review and editing; T. S., M. S., D. Y., A. M., T. M. Z., C. S., C. G., R. M. R., K. B., R. T., I. A., C. M. L., and C. W. validation; M. S., A. M., T. M. Z., and I. A. software; R. M. B., R. T., and C. W. conceptualization; S. Z., R. T., I. A., and C. W. resources; C. M. L. and C. W. supervision; C. W. writing-original draft; C. W. project administration.
Funding and additional information—This work was supported by the German Research Foundation and the Cluster of Excellence “Macromolecular Complexes”, CRC 807, SPP 1623, and RTG 1986 (to R. T.); the Center for Integrated Protein Science Munich (to I. A.), SFB 749/C8 (to A. M.), SFB 1035 (to M. S.), and WE 4388/6-1 (to C. W.); the IGSSE Graduate School (M. S.); a Goethe University-Frankfurt Tandem Research Scholarship (G. D.); and the DAAD-JSPS Joint Research Program (T. S. and C. W.). This work was also supported by the Hessian Initiative for the Development of Scientific-Economic Excellence (LOEWE), Research Center for Translational Medicine and Pharmacology; Novel Drug Targets against Poverty-Related and Neglected Tropical Infectious Diseases; and the Else Kröner-Fresenius Foundation, Graduate School for Translational Research Innovation-Pharma (C. M. L., C. W. and S. Z.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- FC
- fold change
- GLuc
- Gaussia luciferase
- GT
- genotype
- HCV
- hepatitis C virus
- kcat
- substrate turnover rate
- MAVS
- mitochondrial antiviral signaling protein
- MD
- molecular dynamics
- NS
- nonstructural
- PDB
- Protein Data Bank
- PI
- protease inhibitor
- RIG-I
- retinoic acid inducible gene I.
References
- 1. Sheldon J., Beach N. M., Moreno E., Gallego I., Piñeiro D., Martínez-Salas E., Gregori J., Quer J., Esteban J. I., Rice C. M., Domingo E., and Perales C. (2014) Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J. Virol. 88, 12098–12111 10.1128/JVI.01860-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doncheva N. T., Domingues F. S., McGivern D. R., Shimakami T., Zeuzem S., Lengauer T., Lange C. M., Albrecht M., and Welsch C. (2019) Near-neighbor interactions in the NS3-4A protease of HCV impact replicative fitness of drug-resistant viral variants. J. Mol. Biol. 431, 2354–2368 10.1016/j.jmb.2019.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Domingo E., Sheldon J., and Perales C. (2012) Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76, 159–216 10.1128/MMBR.05023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rong L., Dahari H., Ribeiro R. M., and Perelson A. S. (2010) Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2, 30ra32 10.1126/scitranslmed.3000544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pawlotsky J. M. (2016) Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 151, 70–86 10.1053/j.gastro.2016.04.003 [DOI] [PubMed] [Google Scholar]
- 6. Guedj J., and Neumann A. U. (2010) Understanding hepatitis C viral dynamics with direct-acting antiviral agents due to the interplay between intracellular replication and cellular infection dynamics. J. Theor. Biol. 267, 330–340 10.1016/j.jtbi.2010.08.036 [DOI] [PubMed] [Google Scholar]
- 7. Shimakami T., Welsch C., Yamane D., McGivern D. R., Yi M., Zeuzem S., and Lemon S. M. (2011) Protease inhibitor-resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 140, 667–675 10.1053/j.gastro.2010.10.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Götte M., Berghuis A. A., Matlashewski G., Sheppard D., and Wainberg M. A. (2017) Handbook of Antimicrobial Resistance, 1st Ed., Springer-Verlag, New York [Google Scholar]
- 9. Cervera H., Lalić J., and Elena S. F. (2016) Efficient escape from local optima in a highly rugged fitness landscape by evolving RNA virus populations. Proc. Biol. Sci. 283, 20160984 10.1098/rspb.2016.0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Acevedo A., Brodsky L., and Andino R. (2014) Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505, 686–690 10.1038/nature12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bartenschlager R., Lohmann V., and Penin F. (2013) The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 11, 482–496 10.1038/nrmicro3046 [DOI] [PubMed] [Google Scholar]
- 12. Ma Y., Yates J., Liang Y., Lemon S. M., and Yi M. (2008) NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 82, 7624–7639 10.1128/JVI.00724-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McGivern D. R., Masaki T., Lovell W., Hamlett C., Saalau-Bethell S., and Graham B. (2015) Protease inhibitors block multiple functions of the NS3/4A protease-helicase during the hepatitis C virus life cycle. J. Virol. 89, 5362–5370 10.1128/JVI.03188-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., and Tschopp J. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 10.1038/nature04193 [DOI] [PubMed] [Google Scholar]
- 15. Xu L. G., Wang Y. Y., Han K. J., Li L. Y., Zhai Z., and Shu H. B. (2005) VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell. 19, 727–740 10.1016/j.molcel.2005.08.014 [DOI] [PubMed] [Google Scholar]
- 16. Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., and Akira S. (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 10.1038/ni1243 [DOI] [PubMed] [Google Scholar]
- 17. Poordad F., Hezode C., Trinh R., Kowdley K. V., Zeuzem S., Agarwal K., Shiffman M. L., Wedemeyer H., Berg T., Yoshida E. M., Forns X., Lovell S. S., Da Silva-Tillmann B., Collins C. A., Campbell A. L., et al. (2014) ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N. Engl. J. Med. 370, 1973–1982 10.1056/NEJMoa1402869 [DOI] [PubMed] [Google Scholar]
- 18. Welsch C., Haselow K., Gouttenoire J., Schneider M., Morikawa K., Martinez Y., Susser S., Sarrazin C., Zeuzem S., Antes I., Moradpour D., and Lange C. M. (2015) Hepatitis C virus variants resistant to macrocyclic NS3-4A inhibitors subvert IFN-β induction by efficient MAVS cleavage. J. Hepatol. 62, 779–784 10.1016/j.jhep.2014.11.009 [DOI] [PubMed] [Google Scholar]
- 19. Jiang M., Mani N., Lin C., Ardzinski A., Nelson M., Reagan D., Bartels D., Zhou Y., Nicolas O., Rao B. G., Müh U., Hanzelka B., Tigges A., Rijnbrand R., and Kieffer T. L. (2013) In vitro phenotypic characterization of hepatitis C virus NS3 protease variants observed in clinical studies of telaprevir. Antimicrob. Agents Chemother. 57, 6236–6245 10.1128/AAC.01578-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu R., Geng D., Chi X., Wang X., Gao X., Xu H., Shi Y., Guan Y., Wang Y., Jin J., Ding Y., and Niu J. (2019) Computational analysis of naturally occurring resistance-associated substitutions in genes NS3, NS5A, and NS5B among 86 subtypes of hepatitis C virus worldwide. Infect. Drug Resist. 12, 2987–3015 10.2147/IDR.S218584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yi M., Villanueva R. A., Thomas D. L., Wakita T., and Lemon S. M. (2006) Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103, 2310–2315 10.1073/pnas.0510727103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krishnan P., Schnell G., Tripathi R., Ng T., Reisch T., Beyer J., Dekhtyar T., Irvin M., Xie W., Larsen L., Mensa F., Pilot-Matias T., and Collins C. (2017) Pooled resistance analysis in HCV genotype 1–6-infected patients treated with glecaprevir/pibrentasvir in phase 2 and 3 clinical trials. J. Hepatol. 66, S500 10.1016/S0168-8278(17)31399-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Welzel T. M., Bhardwaj N., Hedskog C., Chodavarapu K., Camus G., McNally J., Brainard D., Miller M. D., Mo H., Svarovskaia E., Jacobson I., Zeuzem S., and Agarwal K. (2017) Global epidemiology of HCV subtypes and resistance-associated substitutions evaluated by sequencing-based subtype analyses. J. Hepatol. 67, 224–236 10.1016/j.jhep.2017.03.014 [DOI] [PubMed] [Google Scholar]
- 24. Seth R. B., Sun L., Ea C. K., and Chen Z. J. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 25. Choi H. J., Park A., Kang S., Lee E., Lee T. A., Ra E. A., Lee J., Lee S., and Park B. (2018) Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun. 9, 125 10.1038/s41467-017-02624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang B., Xi X., Lei X., Zhang X., Cui S., Wang J., Jin Q., and Zhao Z. (2013) Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9, e1003231 10.1371/journal.ppat.1003231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feng H., Sander A. L., Moreira-Soto A., Yamane D., Drexler J. F., and Lemon S. M. (2019) Hepatovirus 3ABC proteases and evolution of mitochondrial antiviral signaling protein (MAVS). J. Hepatol. 71, 25–34 10.1016/j.jhep.2019.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patel M. R., Loo Y. M., Horner S. M., Gale M. Jr and Malik H. S. (2012) Convergent evolution of escape from hepaciviral antagonism in primates. PLos Biol. 10, e1001282 10.1371/journal.pbio.1001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sumpter R., Loo Y.-M., Foy E., Li K., Yoneyama M., Fujita T., Lemon S. M., and Gale M. Jr (2005) Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79, 2689–2699 10.1128/JVI.79.5.2689-2699.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yi M., Hu F., Joyce M., Saxena V., Welsch C., Chavez D., Guerra B., Yamane D., Veselenak R., Pyles R., Walker C. M., Tyrrell L., Bourne N., Lanford R. E., and Lemon S. M. (2014) Evolution of a cell culture-derived genotype 1a hepatitis C virus (H77S.2) during persistent infection with chronic hepatitis in a chimpanzee. J. Virol. 88, 3678–3694 10.1128/JVI.03540-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kolykhalov A. A., Agapov E. V., Blight K. J., Mihalik K., Feinstone S. M., and Rice C. M. (1997) Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277, 570–574 10.1126/science.277.5325.570 [DOI] [PubMed] [Google Scholar]
- 32. Yanagi M., St Claire M., Shapiro M., Emerson S. U., Purcell R. H., and Bukh J. (1998) Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology 244, 161–172 10.1006/viro.1998.9092 [DOI] [PubMed] [Google Scholar]
- 33. Meylan E., Martinon F., Thome M., Gschwendt M., and Tschopp J. (2002) RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-κ B and is processed during apoptosis. EMBO Rep. 3, 1201–1208 10.1093/embo-reports/kvf236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taremi S. S., Beyer B., Maher M., Yao N., Prosise W., Weber P. C., and Malcolm B. A. (1998) Construction, expression, and characterization of a novel fully activated recombinant single-chain hepatitis C virus protease. Protein Sci. 7, 2143–2149 10.1002/pro.5560071011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 36. Copeland R. A. (2013) Evaluation of enzyme inhibitors in drug discovery: A guide for medicinal chemists and pharmacologists, 2nd Ed., Wiley-Interscience, New Jersey: [PubMed] [Google Scholar]
- 37. Beran R. K., and Pyle A. M. (2008) Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J. Biol. Chem. 283, 29929–29937 10.1074/jbc.M804065200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ferreira A. R., Magalhães A. C., Camões F., Gouveia A., Vieira M., Kagan J. C., and Ribeiro D. (2016) Hepatitis C virus NS3-4A inhibits the peroxisomal MAVS-dependent antiviral signalling response. J. Cell. Mol. Med. 20, 750–757 10.1111/jcmm.12801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Romano K. P., Laine J. M., Deveau L. M., Cao H., Massi F., and Schiffer C. A. (2011) Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J. Virol. 85, 6106–6116 10.1128/JVI.00377-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frigerio F., Coda A., Pugliese L., Lionetti C., Menegatti E., Amiconi G., Schnebli H. P., Ascenzi P., and Bolognesi M. (1992) Crystal and molecular structure of the bovine α-chymotrypsin-eglin c complex at 2.0 A resolution. J. Mol. Biol. 225, 107–123 10.1016/0022-2836(92)91029-O [DOI] [PubMed] [Google Scholar]
- 41. Rodríguez A., Oliva C., González M., van der Kamp M., and Mulholland A. J. (2007) Comparison of different quantum mechanical/molecular mechanics boundary treatments in the reaction of the hepatitis C virus NS3 protease with the NS5A/5B substrate. J. Phys. Chem. B 111, 12909–12915 10.1021/jp0743469 [DOI] [PubMed] [Google Scholar]
- 42. Case D. A., Berryman J. T., Betz R. M., Cerutti D. S., Cheatham T. E. III, Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Homeyer N., Izadi S., Janowski P., Kaus J., Kovalenko A., et al. (2015) Amber 2015, University of California, San Francisco [Google Scholar]
- 43. Berendsen H. J. C., Grigera J. R., and Straatsma T. P. (1987) The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 10.1021/j100308a038 [DOI] [Google Scholar]
- 44. Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., and Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 10.1002/jcc.10349 [DOI] [PubMed] [Google Scholar]
- 45. Duell E. R., Glaser M., Le Chapelain C., Antes I., Groll M., and Huber E. M. (2016) Sequential inactivation of gliotoxin by the S-methyltransferase TmtA. ACS Chem. Biol. 11, 1082–1089 10.1021/acschembio.5b00905 [DOI] [PubMed] [Google Scholar]
- 46. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., and Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 10.1093/bioinformatics/btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roe D. R., and Cheatham T. E. III (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory. Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 48. Miller B. R. III, McGee T. D. Jr, Swails J. M., Homeyer N., Gohlke H., and Roitberg A. E. (2012) MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory. Comput. 8, 3314–3321 10.1021/ct300418h [DOI] [PubMed] [Google Scholar]
- 49. Grossfield A., and Zuckerman D. M. (2009) Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 5, 23–48 10.1016/S1574-1400(09)00502-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prongay A. J., Guo Z., Yao N., Pichardo J., Fischmann T., Strickland C., Myers J. Jr, Weber P. C., Beyer B. M., Ingram R., Hong Z., Prosise W. W., Ramanathan L., Taremi S. S., Yarosh-Tomaine T., et al. (2007) Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino] carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem. 50, 2310–2318 10.1021/jm060173k [DOI] [PubMed] [Google Scholar]
- 51. Schechter I., and Berger A. (2012) Reprint of “On the size of the active site in proteases. I. Papain. 1967.” Biochem. Biophys. Res. Commun. 425, 497–502 10.1016/j.bbrc.2012.08.015 [DOI] [PubMed] [Google Scholar]
- 52. Li K., Foy E., Ferreon J. C., Nakamura M., Ferreon A. C., Ikeda M., Ray S. C., Gale M. Jr, and Lemon S. M. (2005) Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U. S. A. 102, 2992–2997 10.1073/pnas.0408824102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brenndörfer E. D., Karthe J., Frelin L., Cebula P., Erhardt A., Schulte Am Esch J., Hengel H., Bartenschlager R., Sällberg M., Häussinger D., and Bode J. G. (2009) Nonstructural 3/4A protease of hepatitis C virus activates epithelial growth factor-induced signal transduction by cleavage of the T-cell protein tyrosine phosphatase. Hepatology 49, 1810–1820 10.1002/hep.22857 [DOI] [PubMed] [Google Scholar]
- 54. Morikawa K., Gouttenoire J., Hernandez C., Dao Thi V. L., Tran H. T., Lange C. M., Dill M. T., Heim M. H., Donzé O., Penin F., Quadroni M., and Moradpour D. (2014) Quantitative proteomics identifies the membrane-associated peroxidase GPx8 as a cellular substrate of the hepatitis C virus NS3-4A protease. Hepatology 59, 423–433 10.1002/hep.26671 [DOI] [PubMed] [Google Scholar]
- 55. Kang X., Chen X., He Y., Guo D., Guo L., Zhong J., and Shu H. B. (2013) DDB1 is a cellular substrate of NS3/4A protease and required for hepatitis C virus replication. Virology 435, 385–394 10.1016/j.virol.2012.10.025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data that support the findings of this study are available from the corresponding author upon request.