

Abstract
Neurofibrillary tangles of abnormally hyperphosphorylated Tau are a hallmark of Alzheimer's disease (AD) and related tauopathies. Tau is truncated at multiple sites by various proteases in AD brain. Although many studies have reported the effect of truncation on the aggregation of Tau, these studies mostly employed highly artificial conditions, using heparin sulfate or arachidonic acid to induce aggregation. Here, we report for the first time the pathological activities of various truncations of Tau, including site-specific phosphorylation, self-aggregation, binding to hyperphosphorylated and oligomeric Tau isolated from AD brain tissue (AD O-Tau), and aggregation seeded by AD O-Tau. We found that deletion of the first 150 or 230 amino acids (aa) enhanced Tau's site-specific phosphorylation, self-aggregation, and binding to AD O-Tau and aggregation seeded by AD O-Tau, but deletion of the first 50 aa did not produce a significant effect. Deletion of the last 50 aa was found to modulate Tau's site-specific phosphorylation, promote its self-aggregation, and cause it to be captured by and aggregation seeded by AD O-Tau, whereas deletion of the last 20 aa had no such effects. Among the truncated Taus, Tau151–391 showed the highest pathological activities. AD O-Tau induced aggregation of Tau151–391 in vitro and in cultured cells. These findings suggest that the first 150 aa and the last 50 aa protect Tau from pathological characteristics and that their deletions facilitate pathological activities. Thus, inhibition of Tau truncation may represent a potential therapeutic approach to suppress Tau pathology in AD and related tauopathies.
Keywords: Alzheimer's disease, Tau protein (Tau), truncation, phosphorylation, aggregation, prion-like activity, protein aggregation, pathogenesis
Tau is a major neuronal microtubule-associated protein. Aggregation of Tau into neurofibrillary tangles (NFTs) is a hallmark brain lesion of Alzheimer's disease (AD) and related tauopathies (1–4). The number of NFTs correlates with the severity of dementia (5–7), and the regional distribution of Tau pathology is apparently associated with the progression of AD (8, 9).
In AD brain, Tau is abnormally hyperphosphorylated at many sites (10–12). Hyperphosphorylation of Tau leads to its loss of normal function, gain of neurotoxicity, and aggregation into NFTs (13–15). Tau pathology can be induced in the mouse brain by injection of Tau aggregates isolated from AD or Tau transgenic mouse brains or produced in vitro (16–20). Abnormally hyperphosphorylated and cytosolic Tau isolated from AD brain (AD P-Tau) by sedimentation and ion-exchange chromatography (10) can sequester/capture normal Tau and template it into filaments in vitro (13). We recently found that like AD P-Tau, oligomeric Tau isolated from AD brain (AD O-Tau) by sedimentation also effectively induces Tau aggregation in cultured cells (21) and templates Tau pathology in vivo (22–24) in a prion-like fashion, which may underlie the amplification and propagation of Tau pathology throughout AD brain.
In addition to hyperphosphorylation, Tau is abnormally truncated at multiple sites in AD brain (25, 26). Many proteases, including calpains and caspases, proteolyze Tau in vitro and in vivo (27–29). Tau can be cleaved by caspase 6 at Asp13 and Asp402, by caspase 2 at Asp314, by caspase 3 at Asp25 and Asp421, by chymotrypsin at Tyr197, by an unknown thrombin-like cytosolic protease at Lys257, by asparaginyl endopeptidase at Asn255 and Asn368, and by calpain at Lys44 and Arg230 (30–32). At Glu391, Tau is cleaved by an unknown protease (30). Puromycin-specific aminopeptidase proteolyzes residues stepwise from the N terminus of Tau (4, 30). In AD brain, various Tau fragments have been identified (29, 33, 34). In addition to these protease-mediated cleavages, 21 novel proteolytic fragments of Tau have been identified (35), which, as of yet, have not had a protease identified for their generation (33, 35, 36). Among all truncations of Tau, truncations at Glu391 and Asp421 were the most reported in AD brain. We recently found that SDS- and reducing agent–resistant high-molecular-weight (HMW) aggregates of Tau from AD brain lack the N-terminal portion (21, 37). Many studies showed that truncation of Tau promotes its aggregation (30, 38), implying that Tau truncation may play a critical role in Tau pathogenesis (25, 33). However, these studies were carried out employing mostly heparin or arachidonic acid for induction of aggregation, which are highly artificial conditions. The impact of Tau truncation on its pathological activities, including hyperphosphorylation, self-aggregation, and binding to and aggregation seeded by AD O-Tau, is not well-documented. Based on the terminal acidic regions of Tau and the truncations reported in AD brain (25, 26, 39, 40), in the present study, we generated Tau truncations with deletions from both the N and C termini and determined their pathological activities. We found that Tau truncation from either the N- or C-terminal toward microtubule-binding repeat region (MTBR) modulated the site-specific phosphorylation and enhanced its self-aggregation, its binding to AD O-Tau, and aggregation seeded by AD O-Tau. Among these truncations, Tau151–391 showed the highest pathological activities and aggregation induced by AD O-Tau, both in vitro and in cultured cells.
Results
Construction and expression of various truncation forms of Tau
In AD brain, Tau is truncated at multiple sites by various proteases (25, 33). Normally, the acidic termini of Tau interact with the microtubule-binding repeats to prevent its aggregation (41). Based on the truncation sites reported in AD brain and on the acidic terminal regions of Tau (Fig. 1A), we constructed 11 truncation forms of Tau from both the N and C termini of the protein (Fig. 1A), expressed them in HEK-293FT cells, and analyzed the expression by Western blots developed with a battery of monoclonal Tau antibodies (Fig. 1B). Western blots showed that the truncation forms with deletion of the first 50 amino acids (aa)—Tau51–441, Tau51–421, and Tau51–391—were not recognized by antibody 43D (6–18 aa). Truncation with the deletion of the first 150 aa—Tau151–441, Tau151–421, and Tau151–391—was not recognized by antibodies 43D or 63B (74–103 aa). Truncation with the deletion of the first 230 aa—Tau231–441, Tau231–421, and Tau231–391—was not recognized by antibodies 43D, 63B, or Tau 5 (210–230 aa). Deletions of the last 20 aa—Tau1–421, Tau51–421, Tau151–421—or the last 50 aa—Tau1–391, Tau51–391, Tau151–391, and Tau231–391—were not recognized by antibody Tau46.1 (428–441 aa) (Fig. 1C). mAb 77G7 against MTBR was able to detect all truncation forms (Fig. 1C). All truncation forms were tagged with HA at the N terminus and were detected by anti-HA (Fig. 1C). These results indicate that the 12 truncation forms of Tau were constructed and expressed as expected.
Figure 1.

Truncation forms of Tau show expected size and immunolabeling in Western blots. A, truncation forms of Tau used in the present study. pI of every 10-aa fragment is presented with the color code as indicated. B, schematic showing the position of epitopes of Tau antibodies used in this study. C, truncation forms of Tau in A were overexpressed in HEK-293FT cells and analyzed with Western blots developed with the indicated antibodies. D, various truncates of Tau in A were overexpressed in HEK-293T cells for 48 h, and then the relative cell cytotoxicity and viability were measured by LDH activity in cultured medium and by Cell Counting Kit 8, respectively. The data are presented as means ± S.D.
To learn the toxicity of these Taus, we overexpressed Tau1–441 and 11 truncates of Tau in HEK-293T cells and analyzed lactate dehydrogenase (LDH) activity in the medium leaked from cells 48 h after transfection. We found similar levels of released LDH activity from cells overexpressed with the above 12 forms of Tau (Fig. 1D). We further analyzed cell viability in HEK-293T cells by Cell Counting Kit 8 and found similar cell viability in cells with overexpression of the 12 Taus (Fig. 1D). Thus, Tau truncates showed similar effect in cytotoxicity and cell viability as the full-length protein.
N- and C-terminal truncations modulate Tau phosphorylation
Abnormal hyperphosphorylation is central to Tau pathogenesis (3). To determine the effect of Tau truncation on its phosphorylation, we overexpressed the above truncated Taus in HEK-293FT cells and analyzed phosphorylation by Western blots developed with site-specific and phosphorylation-dependent Tau antibodies. We found no detectable Tau phosphorylation at Ser199, Thr205, Thr212, Ser214, or Thr217 in Tau231– (Fig. 2, A–E); at Ser396 or Ser404 in Tau–391 (Fig. 2, I and J); and at Ser422 in Tau–421 and Tau–391 truncation forms (Fig. 2K), confirming the site-specific phosphorylation of Tau.
Figure 2.
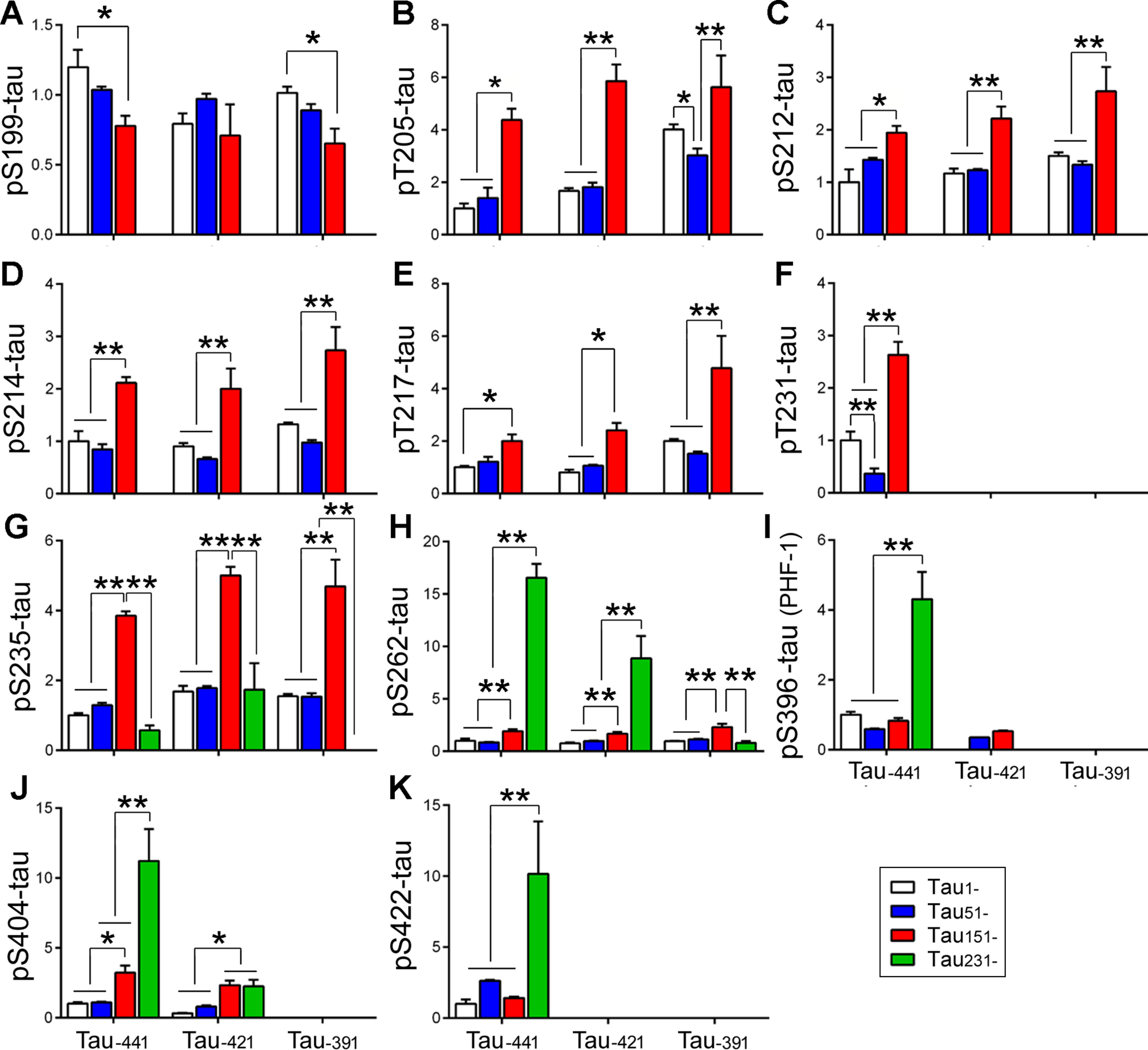
N-terminal truncation of Tau enhances its phosphorylation. Truncated Taus were expressed in HEK-293FT cells and analyzed by Western blots developed with site-specific and phosphorylation-dependent Tau antibodies. The phosphorylation of Tau at individual sites was calculated and statistically analyzed by one-way ANOVA. The data are presented as means ± S.D. (A–K). *, p < 0.05; **, p < 0.01.
To simplify the comparison, we first analyzed the impact of N-terminal truncation on Tau phosphorylation. We found that deletion of the first 50 aa did not affect Tau phosphorylation at Ser199 (Fig. 2A), Thr212 (Fig. 2C), Ser214 (Fig. 2D), Thr217 (Fig. 2E), Ser235 (Fig. 2G), Ser262 (Fig. 2H), Ser396 (Fig. 2I), Ser404 (Fig. 2J), and Ser422 (Fig. 2K), regardless of C-terminal truncations but slightly suppressed Tau phosphorylation at Thr205 (Fig. 2B) and Thr231 (Fig. 2F). Deletion of the first 150 aa suppressed Tau phosphorylation at Ser199 but significantly increased it at most sites, except at Ser396 and Ser422, independent of the C-terminal truncation (Fig. 2), suggesting that deletion of the first 150 aa enhances Tau phosphorylation at most sites. Deletion of the first 230 aa did not change (Tau–441 and Tau–421) or decreased (Tau-391) Ser235 phosphorylation (Fig. 2G) but significantly increased Tau phosphorylation at Ser262 (Fig. 2H), Ser396 (Fig. 2I), Ser404 (Fig. 2J), and Ser422 (Fig. 2K) in Tau–441 and at Ser262 (Fig. 2H) in Tau–421 (Fig. 2G). However, there was no increase of Ser262 phosphorylation (Fig. 2H) in Tau-391 and of Ser396 phosphorylation (Fig. 2I) in Tau–421. Thus, deletion of the first 50 aa did not affect, but deletion of the first 150 aa significantly enhanced, Tau phosphorylation regardless of the C-terminal truncation. The increase in phosphorylation in Tau with deletion of the first 230 aa depended on intact or partially intact C terminus.
Then we determined the effect of the C-terminal truncations on Tau phosphorylation. We found that the C-terminal truncation also modulated Tau phosphorylation site-specifically (Fig. 3). Deletion of the last 20 aa decreased Ser199 phosphorylation in Tau1–, but not in the Tau51– and Tau151– truncation forms (Fig. 3A). Deletion of the last 20 aa did not significantly affect Tau phosphorylation at Thr205, Thr212, Ser214, Thr217, or Ser262 (Fig. 3, B–E), increased at Ser235 (Fig. 3G), and decreased at Ser396 and Ser404 (Fig. 3, I and J) in most truncation forms. Deletion of the last 50 aa increased or tended to increase Tau phosphorylation at Thr205, Thr212, Ser214, Thr217, and Ser235 but did not significantly affect phosphorylation at Ser262 in the most N-terminal truncates (Fig. 3, B–E). Interestingly, deletion of the last 20 or 50 aa suppressed Ser262 phosphorylation in Tau231– truncation forms (Fig. 3H), and deletion of either the last 20 or 50 aa almost abolished or dramatically suppressed Tau phosphorylation at Thr231, Ser396, and Ser404 regardless of N-terminal truncation (Fig. 3, F, I, and J), suggesting that the last 20 aa play a critical role in the regulation of phosphorylation at these sites. These findings suggest that generally, C-terminal truncation can increase the phosphorylation of Tau at the sites located in proline-rich domain (Thr205, Thr212, Ser214, and Thr217) but decrease at sites in microtubule-binding repeat (Ser262) and C-terminal domains (Ser396 and Ser404).
Figure 3.

C-terminal truncation of Tau increases its phosphorylation at the sites in proline-rich domain and decreases at the sites in and downstream of the microtubule-binding repeats region. The phosphorylation of Tau at individual sites was normalized by total Tau and statistically analyzed by one-way ANOVA. The data are presented as means ± S.D. (A–K). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Truncation of Tau promotes its aggregation
Tau is aggregated into NFTs in AD brain. To learn the impact of truncation in Tau aggregation, we overexpressed various HA-tagged truncated Taus in HEK-293FT cells for 48 h, separated RIPA buffer–soluble and –insoluble fractions, and analyzed Tau levels by Western blots developed with anti-HA. We found a significant amount of RIPA buffer–insoluble Tau in cells expressed with Tau truncations, except Tau231–391 (Fig. 4B). Interestingly, we found ∼70-kDa HMW-Tau only in the RIPA buffer–insoluble fraction of Tau151–391 (Fig. 4B), suggesting that Tau151–391 formed the AD-like SDS- and reducing agent–resistant aggregates described previously (21, 37).
Figure 4.
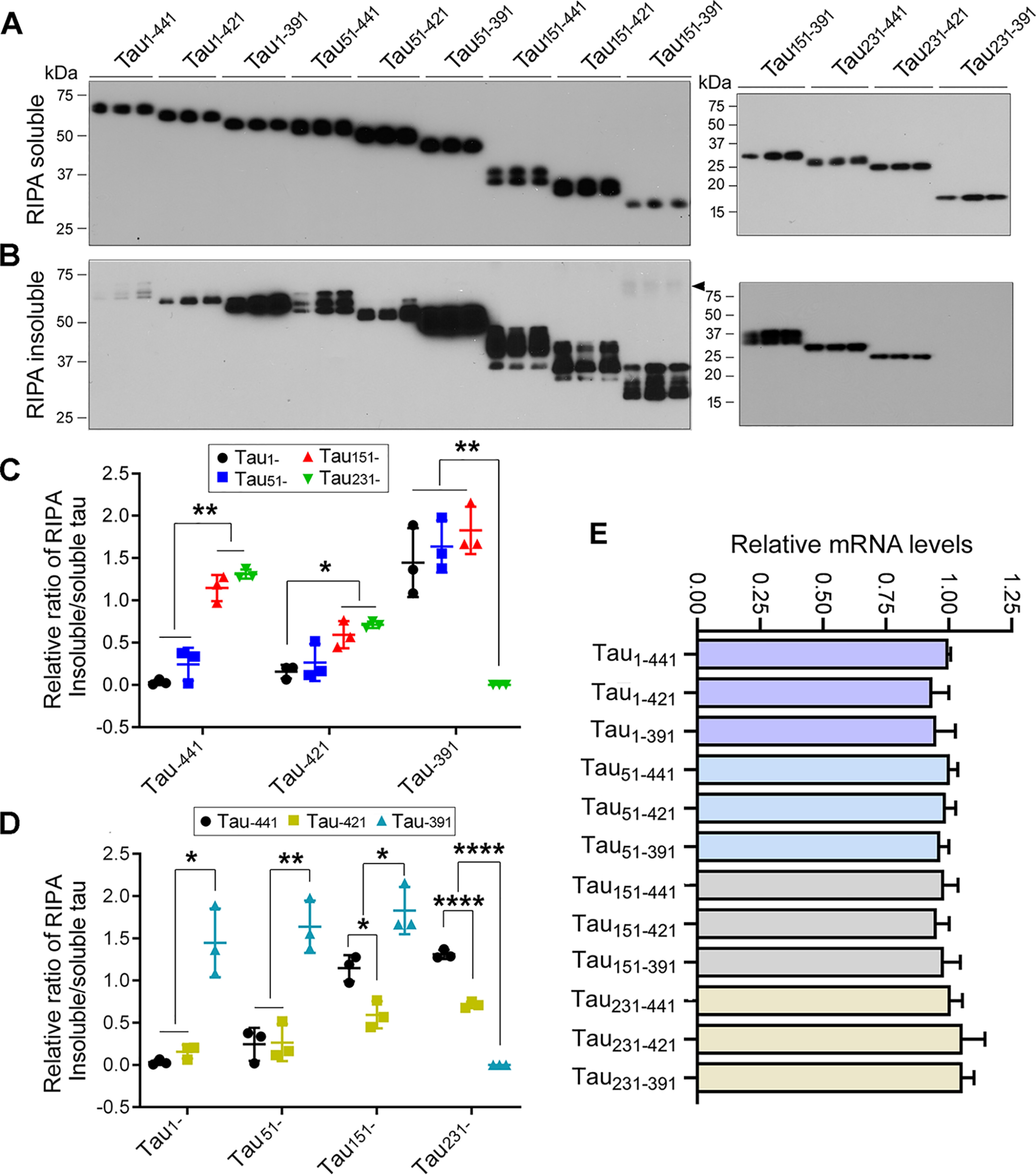
N- and C-terminal truncations enhance Tau's aggregation. A and B, HA-tagged truncated Taus were expressed in HEK-293FT cells for 48 h. The cells were lysed in RIPA buffer and centrifuged at 130,000 × g for 45 min. RIPA buffer-soluble (A) and -insoluble (B) Taus were analyzed by Western blots developed with anti-HA. The arrowhead indicates SDS- and reducing agent–resistant high molecular Tau aggregates. C and D, the ratio of RIPA buffer–insoluble/soluble Tau was calculated. The data are presented as means ± S.D. *, p < 0.05; **, p < 0.01; ****, p < 0.0001. E, various truncates of Tau were overexpressed in HEK-293T cells for 48 h. The levels of Tau mRNA were detected by quantitative RT-PCR and normalized by GAPDH. The relative levels of Tau mRNA are presented as means ± S.D.
We determined the impact of N-terminal truncations on Tau aggregation by analyzing the ratio of RIPA buffer–insoluble/soluble Tau. We found that in general, deletion from the N terminus toward MTBR increased the ratio of RIPA buffer–insoluble/soluble Tau (Fig. 4, A and B), suggesting that N-terminal truncation increases Tau's capacity to aggregate. Deletion of the first 50 aa did not significantly affect the ratios of RIPA buffer–insoluble/soluble Tau, regardless of the C-terminal truncations (Fig. 4, A and B). However, deletion of the first 150 aa significantly increased the ratio of RIPA buffer-insoluble/soluble Tau in Tau–441 and Tau–421, but not in Tau–391 truncation forms (Fig. 4, A and B), suggesting that the first 150 aa inhibit Tau aggregation, but this inhibition depends on the intact or partially intact C terminus. Compared with Tau151–, Tau231– forms did not further enhance the ratios of RIPA buffer–insoluble/soluble Tau (Fig. 4, A and B), suggesting that the first 150 aa are essential to inhibit Tau self-aggregation. The expression level of Tau231–391 was very low, which may lead to undetectable aggregation of Tau231–391. These findings suggest that N-terminal truncation enhances Tau aggregation, and the first 150 aa are critical to prevent Tau from self-aggregation.
Deletion of C-terminal 20 aa did not affect the ratio of RIPA buffer–insoluble/soluble Tau in Tau1– and Tau51– but decreased in Tau151– and Tau231– truncation forms (Fig. 4, A and C), suggesting that the last 20 aa may not protect Tau from the self-aggregation. However, deletion of the last 50 aa significantly increased the aggregation regardless of N-terminal truncations except Tau231– truncation forms (Fig. 4, A and C), suggesting that the last 50 aa are also critical to prevent Tau from self-aggregation. Taken together, these findings indicate that both the first 150 aa and the last 50 aa protect Tau from self-aggregation, and deletion of either the first 150 aa or the last 50 aa promotes Tau aggregation. Tau151–391 showed the highest self-aggregation capacity (Fig. 4).
To investigate the cause of low expression of Tau231–391, we determined mRNA levels of Taus by real-time quantitative PCR 48 h after transfection. We found similar mRNA levels in these 12 Tau forms expressing cells (Fig. 4E), suggesting that a lower level of Tau231–391 could have resulted from instability of this protein fragment.
Truncation of Tau enhances its capture by AD O-Tau
Isolated AD O-Tau was previously found to sequester/capture normal Tau (13, 14), which is a basis of Tau aggregation induced by pathological Tau. To learn the effect of Tau truncation on its capture by AD O-Tau, we performed an overlay assay. We overexpressed HA-tagged truncated Taus in HEK-293FT cells. The cell extracts containing similar levels of truncated Taus in PBS were incubated with nitrocellulose membrane predotted with various amounts of AD O-Tau overnight. AD O-Tau–captured truncated Taus were detected by development with anti-HA followed by HRP–anti-IgG and ECL. We found that Tau with various truncations was captured differentially by AD O-Tau (Fig. 5A). The levels of truncated Taus captured by AD O-Tau were increased with truncation from the N terminus toward MTBR (Fig. 5A). Deletion of the first 50 aa did not have an effect, but deletion of the first 150 aa significantly enhanced the level of Tau captured by AD O-Tau, regardless of C-terminal truncations (Fig. 5, A and B). Deletion of the first 230 aa enhanced the level of Tau captured by AD O-Tau in Tau–441 and Tau–421, but not in Tau–391 truncation forms (Fig. 5, A and B). In the C-terminal truncations, deletion of the last 20 aa did not affect the capture of Tau by AD O-Tau (Fig. 5, A and C). Although deletion of the last 50 aa did not enhance Tau capture by AD O-Tau in Tau1– and Tau51– truncations, it enhanced it in Tau151– and decreased it in Tau231– forms (Fig. 5, A and C). These results suggest that the first 150 aa and the last 50 aa protect Tau from being captured by AD O-Tau, and their deletions enhance Tau's capture by AD O-Tau; Tau151–391 is the Tau most potently captured by AD O-Tau.
Figure 5.
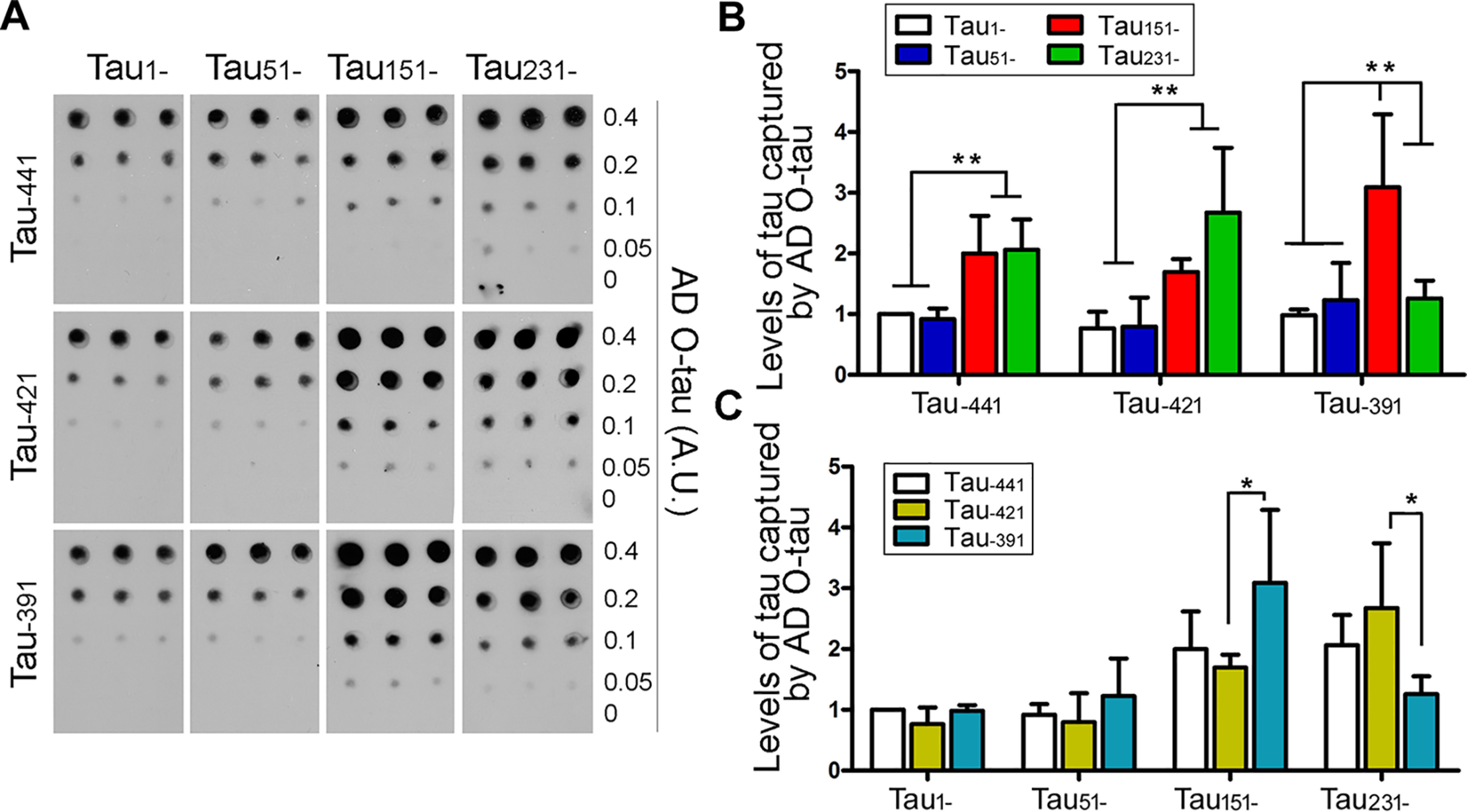
N- and C-terminal truncations enhance Tau's binding to AD O-Tau. HA-tagged truncated Taus were expressed in HEK-293FT cells for 48 h. The cells were lysed in PBS by probe sonication and centrifuged to yield extracts. Various amounts of AD O-Tau were dotted on nitrocellulose membrane. The membrane was incubated with cell extracts containing Tau truncations. Captured Tau was analyzed by anti-HA, followed with HRP-anti-IgG and ECL (A). The data are presented as means ± S.D. (B and C). *, p < 0.05; **, p < 0.01.
Truncation of Tau enhances its aggregation seeded by AD O-Tau
Misfolded Tau templates Tau aggregation in vitro and in vivo, which is a basis of propagation of Tau pathology (14, 42). To determine the effect of Tau truncations on AD O-Tau–seeded aggregation, we overexpressed Tau truncations in HEK-293FT cells and treated these cells with AD O-Tau for 42 h after 6 h of transfection. RIPA buffer–soluble and –insoluble Taus from these cells were analyzed by Western blots. We found that the AD O-Tau treatment caused the reduction of RIPA buffer–soluble Tau (Fig. 6A, upper panel) and increased levels of RIPA buffer–insoluble Tau in all truncation forms (Fig. 6A, lower panel). In addition to the HA-Tau, AD O-Tau was detected by R134d and 92e in RIPA buffer–insoluble factions of cells treated with AD O-Tau (Fig. 6B). We also found HMW-Tau in the RIPA buffer–insoluble fraction of cells transfected with Tau151–391, Tau231–441, and Tau231–421 (Fig. 6A). The expression level of Tau231–391 was extremely low, and the RIPA buffer–insoluble Tau was undetectable in the cells treated with AD O-Tau at this condition (Fig. 6A). Thus, collectively these findings suggest that AD O-Tau induces Tau aggregation, which is modulated by truncation.
Figure 6.
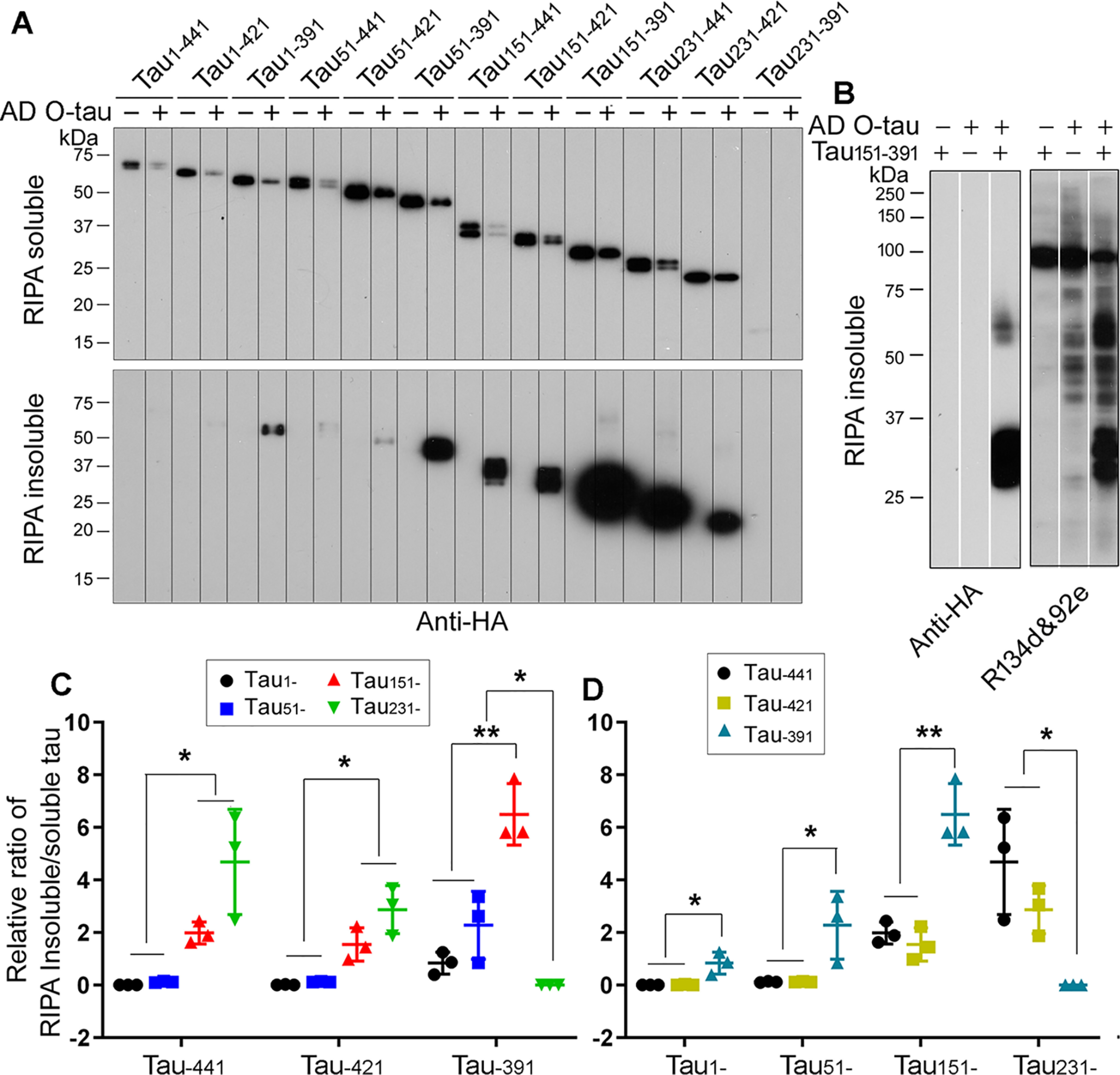
N- and C-terminal truncations enhance Tau aggregation seeded by AD O-Tau. A, HA-tagged truncated Taus were expressed in HEK-293FT cells for 6 h, and then the cells were treated with AD O-Tau for 42 h. The cells were lysed in RIPA buffer and centrifuged at 130,000 × g for 45 min. RIPA buffer–soluble (upper panel) and –insoluble (lower panel) Taus were analyzed by Western blots developed with anti-HA. B, HA-Tau151–391 was expressed in HEK-293FT cells. RIPA buffer–insoluble Tau was analyzed by Western blots developed with anti-HA and mixture of R134d and 92e. C and D, the ratios of RIPA buffer–insoluble/soluble Tau were calculated, and the data are presented as means ± S.D. (n = 3). *, p < 0.05; **, p < 0.01.
To compare the efficiency of aggregation of various truncated Taus seeded by AD O-Tau, we analyzed the ratio of RIPA buffer–insoluble/soluble Tau. We found that deletion of the N terminus toward MTBR increased AD O-Tau–seeded aggregation regardless of C-terminal truncation (Fig. 6, A and C). Similar to in the self-aggregation, deletion of the first 50 aa did not significantly affect Tau aggregation templated by AD O-Tau, but deletion of the first 150 aa enhanced AD O-Tau–seeded aggregation in all C-terminal truncations (Fig. 6, A and C). Deletion of the N-terminal 230 aa enhanced AD O-Tau–seeded aggregation in Tau–441 and Tau–421, but not in Tau–391 truncation forms (Fig. 6, A and C). Tau151–391 was the most potent truncation to the aggregation induced by AD O-Tau (Fig. 6, A–D). Deletion of the last 20 aa did not affect AD O-Tau–induced aggregation but suppressed it in Tau231- truncations (Fig. 6, A and D). Deletion of the last 50 aa enhanced Tau aggregation templated by AD O-Tau, except in Tau231– truncations (Fig. 6, A and D). These data suggest that consistently, the first 150 aa and the last 50 aa of Tau prevent AD O-Tau–seeded aggregation. Collectively, these findings reveal that deletions of either the first 150 aa or the last 50 aa can enhance AD O-Tau–seeded Tau aggregation and that Tau151–391 is the most prone to be seeded to aggregate by AD O-Tau.
AD O-Tau induces Tau151–391 aggregation in vitro and in cultured cells
Our studies described above indicated that Tau151–391 appears to have the highest activity to self-aggregate and bind to AD O-Tau. Tau aggregation can be induced in vitro by polyanionic molecules, such as heparin, heparan sulfate, and RNA (43–45). We further studied its aggregation induced in vitro and in cultured cells. We first determined the aggregation of Tau151–391 induced by heparin. We incubated recombinant Tau1–441 (rTau1–441) or rTau151–391 with heparin in the presence of thioflavin T for various time periods at room temperature. Aggregated Tau was detected by fluorescence density and plotted against the incubation time. We found that heparin induced aggregation much faster and to a much higher extent in Tau151–391 than in Tau1–441 (Fig. 7A). Then we studied Tau151–391 aggregation induced by AD O-Tau. We incubated rTau151–391 or rTau1–441 with AD O-Tau for various time periods at room temperature and detected aggregation by thioflavin T fluorescence. We found that AD O-Tau induced aggregation of Tau151–391, but not Tau1–441, in a time-dependent manner (Fig. 7B). These results reveal that Tau151–391 has a higher capacity than Tau1–441 to be induced to aggregate by either heparin or AD O-Tau in vitro.
Figure 7.

Aggregation of Tau151–391 induced by AD O-Tau. A and B, recombinant Tau1–441 and Tau151–391 were incubated with heparin (A) or AD O-Tau (B) at room temperature for up to 180 min or 87 hr. The aggregation of Tau was measured by thioflavin T fluorescence. The data are presented as means ± S.D. (n = 3) and plotted to the incubation time. C–F, Tau1–441 (C and E) and Tau151–391 (D and F) tagged with HA were overexpressed in HeLa cells for 6 h and then treated without (C and D) or with (E and F) AD O-Tau for 42 h. The cells were immunostained with anti-HA followed by fluorescence-conjugated second antibody. G, the number of cells with aggregates was calculated, and the data are presented as means ± S.D. (n = 3). **, p < 0.01; ****, p < 0.001. H, Tau151–391 and Tau1–441 were overexpressed in HeLa cells and treated with AD O-Tau for 42 h. The cells were immunostained with anti-HA and co-stained with Thioflavin T.
To further verify AD O-Tau–induced aggregation, we overexpressed HA-tagged Tau1–441 or Tau151–391 in HeLa cells and treated the cells with AD O-Tau for 42 h after 6 h of transfection. The cells were then immunofluorescence-stained with anti-HA. Although we found <1% of Tau1–441 cells with aggregates (Fig. 7, C and G), ∼6% cells with atypical aggregates were seen in the Tau151–391 cells (Fig. 7, D and G). However, we found that the treatment with AD O-Tau markedly increased the number (∼22%) of Tau151–391 cells with aggregates in the cytoplasm and the nucleus (Fig. 7, F and G). This increase was weak in Tau1–441 cells (Fig. 7, E and G). The aggregates of Tau151–391, but not Tau1–441, seeded by AD O-Tau were thioflavin T–positive (Fig. 7H). Thus, Tau151–391 was found to display a high potency for templation and aggregation by AD O-Tau.
Discussion
Since the first report on Tau truncation in AD paired helical filament in 1993 (36), more than 50 fragments of Tau have been identified, and over 30 were found present in AD brain (33). However, the role of truncation in Tau pathogenesis in AD and related tauopathies was not well-understood. Based on several reported cleavage sites, in the present study we studied effects of different Tau truncations on pathological activities. We found that truncation at both the N and C termini enhanced pathological activities of Tau, including an increase of site-specific hyperphosphorylation, self-aggregation, capture by AD O-Tau, and AD O-Tau–seeded aggregation (Fig. 8). Among the truncated fragments, Tau151–391 was found to be the most pathogenic.
Figure 8.

Proposed Tau truncation and its pathological activities. Deletion of the first 150 aa and the last 50 aa enhances Tau hyperphosphorylation, aggregation, and binding to and aggregation seeded by AD O-Tau. Tau151–391 is prone to aggregation.
Prion-like activity of pathological Tau may underlie the progression of AD and related tauopathies. Our group previously found that recombinant Tau can be sequestered by AD P-Tau by overlay assay (13). In the present study, we developed a novel assay with AD O-Tau as capture to quantitively validate the ability of Tau “seeds” to capture pro-aggregation truncated Tau fragments, which is a significant advance over artificial conditions using heparan or other methods to induce aggregation. Here we found that N-terminal truncation is particularly significant to pathogenesis, whereas prior work focused on C-terminal truncation. Our present study also challenges the dogma of the importance of caspase cleavage at Asp421 because we did not find any increase in pathogenic activity in Tau truncated at this site. The capture assay identified pathological cores for assembly, which mimic the early steps of Tau aggregation. Similarly, the method also can be used to detect prion-like activity of pathological Tau by overlay with crude extract of HEK-293FT/HA-Tau151–391.
Tau RD P301S FRET Biosensor, in which a HEK-293 cell line that stably expresses the repeat domain (amino acids 244–372) of 2N4R Tau with two disease-associated mutations (P301L and V337M), is widely used to assess prion-like seeding activity (46). We here expressed various forms of HA-Tau in HEK-293FT cells, treated the cells with AD O-Tau for 42 h after 6 h transfection, and harvested the cells with RIPA buffer. Aggregated Tau induced by AD O-Tau was yielded in RIPA buffer–insoluble fraction by 130,000 × g for 45 min and quantified by immunoblots. By using this method, we found different efficacies of various Tau truncates in AD O-Tau seeded aggregation in cultured cells, and Tau151–391 was found to be the most prone to be induced to aggregate. Consistently, we found that N-terminal truncation is more significant to pathogenesis than C-terminal truncation at Asp421. This method also can be used to evaluate the seeding activity of pathological Tau from AD and 3xTg-AD mouse brains (21).
Heparin and other polyanionic molecules had been shown to induce Tau aggregation in vitro (44, 45, 47). Therefore, heparin-induced Tau aggregation assay combined with thioflavin T fluorescence detection was most commonly used to access Tau aggregation (48, 49). Nevertheless, heparin-induced filament core is structurally different from that seen in the AD brain (49). In this study, Tau151–391 aggregation in vitro was induced by incubating with AD O-Tau. However, the prion-like activity of AD O-Tau–induced Tau aggregates in vivo remains to be investigated.
Tau molecule shows a preference to form a paperclip-like conformation by folding the acidic N- and C-terminal portions back on the basic MTBR region (50) (Fig. 8), which might protect Tau from self-aggregation. It was reported that two sequences, 275VQIINK280 and 306VQIVYK311, within repeats (R) 1 and 2, respectively, of the MTBR are required for self-aggregation of Tau protein (51, 52). In the present study, we found that deletion of either the first 50 aa or the last 20 aa, which removes part of the acidic N and C termini, did not significantly affect Tau's self-aggregation and its binding to and aggregation seeded by AD O-Tau. However, deletion of either the first 150 aa or the last 50 aa, which removes the acidic portions of N or C terminus completely, markedly increased the pathological activities of Tau (Fig. 8). Thus, both the N- and C-terminal acidic portions of Tau appear to protect Tau from aggregation. Partial deletion of the N- or C-terminal acidic regions may not be able to disrupt the paperclip structure, but complete removal of these acidic regions probably disrupts the paperclip structure and increases the aggregation tendency. Thus, Tau151–391 is the most prone to (a) self-aggregation, (b) binding to AD O-Tau, and (c) AD O-Tau–seeded aggregation. Interestingly, deletion of 230 aa increased self-aggregation, its capture by AD O-Tau, and AD O-Tau-seeded aggregation in Tau–441 and Tau–421 truncation forms as compared with deletion of the first 150 aa. However, Although Tau231–391 was captured by AD O-Tau efficiently, no self-aggregation and AD O-Tau–induced aggregation of Tau were detected in Tau231–391 cells. We found a very low expression level of Tau231–391 in cells, which could be a reason why we could not find any detectable Tau aggregation with or without AD O-Tau induction. However, mRNA level of Tau231–391 was similar to other Tau truncates in cells, suggesting a lower stability of the protein.
Aggregation of hyperphosphorylated Tau into NFTs is apparently central to the pathogenesis of AD and related tauopathies (3). In AD brain, Tau is hyperphosphorylated and truncated. Caspase cleavage of Tau may precede the hyperphosphorylation, where especially caspase cleavage at Asp421 has been shown to initiate the cascade leading to Tau aggregation (29, 53, 54). In the present study, we found that deletion of the first 50 aa did not affect significantly Tau phosphorylation. However, deletion of the first 150 aa increased Tau phosphorylation at Thr205, Thr212, Ser214, Thr217, Thr231, and Ser235, but not at Ser262, Ser396, Ser404, and Ser422, suggesting that deletion of the 150 aa makes the region upstream of MTBR, but not MTBR and downstream of MTBR, a favorable substrate for Tau kinases and/or an unfavorable substrate for Tau phosphatases. It is also possible that facilitation of Tau phosphorylation by the truncation may result from the molecule opening or/and from reduction of acidic nature of the molecule by deletion of the N-terminal portion. Similarly, deletion of N-terminal 230 aa enhanced Tau phosphorylation at the sites in MTBR and downstream of it. However, deletion of C-terminal either 20 aa or 50 aa did not increase or weakly increased Tau phosphorylation at the majority of phospho-sites but suppressed that at Thr231, Ser262, Ser396, and Ser404. Thus, N-terminal truncation generally enhanced, but C-terminal truncation suppressed, Tau phosphorylation. Hyperphosphorylation leads to Tau aggregation (55). Pseudo-phosphorylation at Ser396 and Ser404 stimulates the rate of Tau polymerization (56). Thus, truncation at the N terminus may also enhance Tau aggregation via hyperphosphorylation.
Many proteases are expressed in the infant brain, including A disintegrin and metalloproteinase 10 (ADAM10), thrombin, caspase, and calpain. Under physiological circumstances, they are involved in the control of brain development via regulating multiple substrates in the signal cascades, brain inflammation, apoptosis, and so on. In AD, up-regulation and/or mislocalization of these proteases could lead to increased levels and/or inappropriate positioning of truncated Tau. Unlike in infant or normal adult brain, in AD brain hyperphosphorylation probably opens Tau's paper clip structure, which makes its cleavage at aa 150 by thrombin and/or ADAM 10 possible (57). Furthermore, an elevated level of Tau in AD brain provides abundant substrate for the proteases and aggravates the pathological accumulation of truncated fragments.
Two proteases, thrombin and ADAM10, which cleave Tau at Arg155 and Ala152, respectively, might be responsible for N-terminal truncation of Tau near Lys150 in vivo (33, 58, 59). Both enzymes are expressed in adult human brain (58, 60). ADAM10, first identified as the primary α-secretase of β-amyloid protein precursor (61), showed a 2-fold increase of mRNA levels in AD hippocampus (62). It is mainly localized to cell membrane and functions as a transmembrane protease, but its activity may not be limited to the membrane. For example, ADAM10 might exert proteolytic activity in its trafficking route to the plasma membrane, and ADAM10 still functions at the early stages after endocytosis when it is not fully degraded (63, 64). Although Tau is mostly cytoplasmic, it is released in the extracellular space through multiple pathways including direct leakage, exocytosis, exosomes, synaptic vesicles, or translocation across the plasma membrane (65). In addition, we cannot exclude the possibility that Tau might be cleaved extracellularly and then internalized. It was reported that ADAM10-cleaved Tau fragment (Tau-A) was elevated 10-fold high in the brains of the Tg4510 mouse model of Tau deposition (66). Although high levels of serum Tau-A were associated with lower risk of AD and dementia in a prospective study (67), serum Tau-A levels did show a significant increase in AD and mild cognitive impairment (68). In AD patients, Tau-A level in serum correlated inversely with the cognitive assessment score (Mattis dementia rating scale) (65) and was related to the change of cognitive function over time in early AD patients (69), supporting the possibility that Tau-A could indicate the rate of clinical progression of AD. Thrombin stains extra- and intracellular NFTs, implying that the enzyme could function both outside and inside cells (58). Nevertheless, the current evidence is not sufficient to reveal whether ADAM10 and thrombin cleave Tau intracellularly or extracellularly or both.
NFTs in AD contains full-length Tau (70). The truncations probably occur after NFTs are formed and after cell death become extracellular. However, we recently reported that HMW-Tau in AD brains lacked the N-terminal portion (37). The truncations could play a significant role as aggregation-promoting seeds and thus in this way become important in promoting and accelerating secondary spread of Tau pathology. Thus, it remains to be investigated whether these truncations occur prior to aggregation of Tau in one or more tauopathies other than AD.
Truncations of Tau at Asp421 (TauD421) and Glu391 (TauE391) are the most studied truncation forms in AD brain. Tau is cleaved at Asp421 by caspase-3 and at Glu391 by an unknown protease. TauD421 and TauE391 aggregate more rapidly and to a greater degree than full-length Tau in the presence of heparin or arachidonic acid (38, 56). TauD421 and TauE391 detected by the mAb C3 and mAb423 are also present at early stages of Tau aggregation (71, 72). Immunohistochemical studies indicate that cleavage at Asp421 occurs after formation of the Alz50 epitope but prior to truncation at Glu391. Thus, TauD421 appears to occur relatively early in the disease state, contemporaneous with the initial Alz50 folding event that heralds the appearance of filamentous Tau in NFTs, neuropil threads, and the dystrophic neurites surrounding amyloid plaques (73). The levels of Tau fragment cleaved at Asp421 (Tau-C) in serum were not significantly related to the change of cognitive function over time in early AD patients (69). In AD brain, the level of TauD421 is increased significantly (29, 74, 75) but was not found in HMW-Tau (37). In the present study, we found that truncation of Tau at Asp421, a partial deletion of the acidic C-terminal, did not enhance the self-aggregation in cultured cells and AD O-Tau–seeded aggregation in cultured cells, which may be due to the reduction of Tau phosphorylation at Thr231, Ser262, Ser396, and Ser404. In a previous study we showed that hyperphosphorylation of Tau at Thr231 and Ser262 are required for its self-aggregation into paired helical filaments (PHFs) in vitro (76). In contrast to TauD421 and TauE391, the deletion of the whole acidic C-terminal region dramatically enhanced Tau self-aggregation, capture by AD O-Tau, and AD O-Tau–seeded aggregation. Further deletion of N-terminal 150 aa, which results in Tau151–391, the Tau fragment of the protease-resistant core of PHFs isolated from AD brain (77), showed the highest pathological activities. Rats transgenic for Tau151–391 develop neurofibrillary pathology and also display hyperphosphorylation and production of HMW-Tau species (25). In addition, SDS- and β-mercaptoethanol–resistant HMW-Tau in AD brain homogenates was immunoreactive with anti–pSer422-Tau strongly. Thus, these findings argue against the impact of Tau truncation at Asp421 in Tau pathogenesis and suggest that N-terminal truncation is particularly significant to pathogenesis of AD.
Our previous studies showed that hyperphosphorylation of Tau inhibits its binding to tubulin and promotion of the assembly and stabilization of microtubules and increases its self-aggregation and its capture of normal Tau, the characteristics that suggest the opening of the paperclip structure of Tau (13–15). The present study suggests that truncation of Tau generating Tau151–391 results in similar characteristics. Furthermore, truncation of Tau at Lys150 enhances Tau phosphorylation. Thus, in addition to the inhibition of the hyperphosphorylation, inhibition of the proteases responsible for the truncation of Tau at Lys150 and/or Glu391 could be required to effectively inhibit Tau pathology.
Although similar cytotoxicity and cell viability were found in cells overexpressing various Tau truncates, truncation may generate toxic Tau fragments. N-terminal Tau fragments including Tau 1–44 aa, 26–44 aa, 26–230 aa, 1–156 aa, and 45–230 aa exert powerful toxicity (78–80), but Tau 1–230 aa protects neuron from apoptosis (78). Activation of calpain by aggregated Aβ generated a neurotoxic 17-kDa fragment (81, 82). Deletion of the microtubule-binding domain facilitates the spine localization of Tau (82). Phosphorylation of Tau at distinct sites increases Tau targeting spine, which may regulate the spine localization of Src kinase Fyn (82, 83). Thus, truncation of Tau may have broad roles in AD pathogenesis.
We analyzed here Tau truncation and its pathological changes in vitro and in cultured HEK-293FT cells that do not contain high concentration of assembled microtubules or axonal processes. Also, unique Tau kinase-sensitive phosphorylated sites and chaperones like HSP90 influence normal folding and what is exposed to proteases. Thus, the role of Tau truncation and its phosphorylation, interaction with, and aggregation seeded by AD O-Tau remain to be studied in neuronal cells and in vivo.
In summary, the acidic N and C termini of Tau suppress its pathological activities. Truncation of Tau from the N or C terminus toward MTBR enhances its site-specific phosphorylation, self-aggregation, capture by AD O-Tau, and AD O-Tau–seeded aggregation. Deletion of either the first 150 aa or the last 50 aa, but not the first 50 aa or the last 20 aa, markedly increases the pathological activities, which may initiate Tau pathogenesis in AD brain and related tauopathies. Tau151–391 is the most potent Tau fragment for pathological activities, which can be used for seeding activity in vitro and in vivo. Inhibition of Tau truncation may be a therapeutic approach to prevent or inhibit Tau pathogenesis.
Materials and methods
Plasmids, antibodies, and other reagents
pCI/HA-Tau truncations were generated by PCR amplification with the primers listed in Table 1 by using pCI/HA-Tau as a template and were constructed into pCI-neo. All constructs were verified by DNA sequence analysis. The primary antibodies used in the present study are listed in Table 2. Peroxidase-conjugated anti-mouse and anti-rabbit IgGs were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Alexa Fluor 555–conjugated goat anti-mouse IgG was from Life Technologies Inc. Heparin and thioflavin T were from Sigma–Aldrich. CyQUANT™ LDH cytotoxicity assay kit and the ECL kit were from Thermo Fisher Scientific. Cell Counting Kit 8 was from Dojindo Molecular Technologies (Rockville, MD).
Table 1.
Primers used in this study
| Truncations | Primer |
|
|---|---|---|
| Forward | Reverse | |
| Tau1–441 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTatggctgagccccgccaggagttcg | ataagaatgcggccgctcacaaaccctgcttggccagggagg |
| Tau1–421 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTatggctgagccccgccaggagttcg | ataagaat gcggccgctcactccgccccgtggtctgtcttgg |
| Tau1–391 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTatggctgagccccgccaggagttcg | ataagaatgcggccgctcaGTCtaccatgtcgatgctgccgg |
| Tau51–441 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTATGacccccactgaggacggatctga | ataagaatgcggccgctcacaaaccctgcttggccagggagg |
| Tau51–421 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTATGacccccactgaggacggatctga | ataagaat gcggccgctcactccgccccgtggtctgtcttgg |
| Tau51–391 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTATGacccccactgaggacggatctga | ataagaatgcggccgctcaGTCtaccatgtcgatgctgccgg |
| Tau151–441 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTatcgccacaccgcggggagcag | ataagaatgcggccgctcacaaaccctgcttggccagggagg |
| Tau151–421 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTatcgccacaccgcggggagcag | ataagaat gcggccgctcactccgccccgtggtctgtcttgg |
| Tau151–391 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTatcgccacaccgcggggagcag | ataagaatgcggccgctcaGTCtaccatgtcgatgctgccgg |
| Tau231–441 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTATGactccacccaagtcgccgtcttccgc | ataagaatgcggccgctcacaaaccctgcttggccagggagg |
| Tau231–421 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTATGactccacccaagtcgccgtcttccgc | ataagaatgcggccgctcactccgccccgtggtctgtcttgg |
| Tau231–391 (SalI, NotI) | ACGCGTCGACATGTACCCATACGATGTTCCAGATTACGCTATGactccacccaagtcgccgtcttccgc | ataagaatgcggccgctcaGTCtaccatgtcgatgctgccgg |
Table 2.
Primary antibodies used in the present study
Mono, monoclonal; Up, unphosphorylated; Poly, polyclonal; M, mouse; R, rabbit.
| Antibody | Type | Species | Specificity | Source/reference (catalog or lot no.) |
|---|---|---|---|---|
| 43D | Mono | M | Tau8–16 | BioLegend (816601) (21) |
| 63B | Mono | M | Tau74–103 | In house (21) |
| TAU5 | Mono | M | Tau210–241 | Millipore (MAB361/1816394) |
| 77G7 | Mono | M | Tau316–355 | BioLegend (816701) (21) |
| Tau46.1 | Mono | M | Tau428–441 | Upstate (05-838/27088) |
| R134d | Poly | R | Total Tau | In house (21) |
| Anti-pS199 | Poly | R | pSer199 | Invitrogen (44-734G/0300A) |
| Anti-pT205 | Poly | R | pThr205 | Invitrogen (44-738G/RD214239) |
| Anti-pT212 | Poly | R | pThr212 | Invitrogen (44-740G/1709582A) |
| Anti-pS214 | Poly | R | pSer214 | Invitrogen (44-742G/0500B) |
| Anti-pT217 | Poly | R | pSer217 | Invitrogen (44-744/785771A) |
| AT180 | Mono | M | pThr231 | Invitrogen (MN1040/SH2406086) |
| Anti-pS235 | Poly | R | pSer235 | BIOSOURCE (22-3490-0) |
| Anti-pS262 | Poly | R | pSer262 | Invitrogen (44-750G/QK220618) |
| Anti-pS396 | Poly | R | pSer396 | Invitrogen (44752G/567847B) |
| Anti-pS404 | Poly | R | pSer404 | Invitrogen (44-758G/5G255476) |
| R145d | Poly | R | pSer422 | In house (84) |
| PHF-1 | Mono | M | pSer396 | Dr. Peter Davies |
| 92e | Poly | R | Total Tau | In house (21) |
| Anti-HA | Mono | M | HA | Sigma (H9658/101M4776) |
| Anti-HA | Poly | R | HA | Sigma (H6908/110M4845) |
| Anti-GAPDH | Poly | R | GAPDH | Sigma (G9545/015M4824V) |
| Anti–β-actin | Mono | M | β-Actin | Sigma (A1978/046M4789V) |
Cell culture and transfection
HEK-293FT, HEK-293T, and HeLa cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) at 37 °C (5% CO2). Transfections were performed with FuGENE HD (Promega, Madison, WI) according to the manufacturer's instructions.
Western blots and immunodot blots
The samples were diluted with Laemmli SDS sample buffer, followed by boiling for 5 min. Protein concentration was measured using the Pierce™ 660-nm protein assay kit (Thermo Fisher Scientific). The samples were subjected to SDS-PAGE and transferred onto polyvinylidene fluoride membrane (Sigma-Aldrich). The membrane was subsequently blocked with 5% fat-free milk–TBS for 30 min, incubated with primary antibodies (Table 2) in TBS overnight, washed with TBST (TBS with 0.05% Tween 20), incubated with HRP-conjugated secondary antibody for 2 h at room temperature, washed with TBST, incubated with the ECL Western blotting substrate (Thermo Fisher Scientific), and exposed to HyBlot CL® autoradiography film (Denville Scientific Inc., Holliston, MA). Specific immunostaining was quantified by using the Multi Gauge software V3.0 from Fuji Film (Minato, Tokyo, Japan).
Tau level in samples was assayed by immunodot blots as described previously. Briefly, various amounts of a sample were applied onto nitrocellulose membrane (Schleicher and Schuell, Keene, NH) at 5 μl/grid of 7 × 7 mm. The blot was placed in a 37 °C oven for 1 h to allow the protein to bind to the membrane. The membrane was processed as Western blots as described above by using the mixture of R134d and 92e pan Tau antibodies as primary antibodies.
Cytotoxicity and viability assay
Cell cytotoxicity was assessed by measuring LDH release from damaged or dead cells using a CyQUANT LDH cytotoxicity assay kit (Thermo Fisher Scientific) following the manufacturer's instructions. Briefly, HEK-293T cells in 96-well plate were transfected with pCI/HA-Taus with FuGENE HD for 48 h. The culture mediums were collected and incubated with the reaction mixture for 30 min at room temperature. Maximum was yielded by adding lysis buffer to release all LDH to medium. The A490 and A680 were recorded by universal microplate spectrophotometer (BioTek, Winooski, VT) after adding stop solution. Relative cytotoxicity to Tau1–441 was calculated according to the formulas in the manufacturer's instructions.
For cell viability assay, HEK-293T cells cultured in 96-well were transfected with pCI/HA-Taus for 48 h. After adding 10 μl of Cell Counting Kit 8 to each well and incubating at 37 °C for 1 h, A450 was recorded, and relative cell viability to Tau1–441 was calculated.
Quantitative RT-PCR
Total RNA was extracted from cells by using the HP total RNA kit (Omega Bio-tek, Norcross, GA) according to the manufacturer's instructions. Equal amount of RNA was reverse transcribed to cDNA by using SuperScript first-strand cDNA synthesis kit (Biouniquer, Nanjing, China). Real-time PCR was performed with AceQ® quantitative PCR SYBR® Green Master Mix (without ROX) kit (Vazyme Biotech, Nanjing, China) on a LightCycler® 96 real-time PCR system (Roche Diagnostics). The PCR conditions were as follows: 95 °C for 10 min; followed by 45 cycles of 95 °C for 10 s and 60 °C for 30 s; and then 95 °C for 10 s; 65 °C for 60 s; 97 °C for 1 s; and 37 °C for 30 s. The quantitative PCR was performed using these primers: human Tau (forward, 5′-CCAAGTGTGGCTCATTAGGCA-3′; reverse, 5'-CCAATCTTCGACTGGACTCTGT-3′), human GAPDH (forward, 5′-CATGAGAAGTATGACAACAGCCT-3′; reverse, 5′-AGTCCTTCCACGATACCAAAGT-3′). Relative levels of mRNA were quantified by the comparative 2−△△CT method and normalized to GAPDH level.
Preparation of hyperphosphorylated and AD O-Tau
AD O-Tau was isolated from the cerebral cortex of frozen autopsied AD brains as we described previously (10). Briefly, 10% brain homogenate prepared in buffer (20 mm Tris-HCl, pH 8.0, 0.32 M sucrose, 10 mm β-mercaptoethanol, 5 mm MgSO4, 1 mm EDTA, 10 mm glycerophosphate, 1 mm Na3VO4, 50 mm NaF, 1 mm 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), and 10 mg/ml each of aprotinin, leupeptin, and pepstatin) was centrifuged at 27,000 × g for 30 min. The supernatant was further centrifuged at 235,000 × g for 45 min, and the resulting pellet, i.e. AD O-Tau–enriched fractions, was collected, washed three times, and then resuspended in normal saline. The AD O-Tau was probe-sonicated for 10 min at 20% power and stored at −80 °C.
Tau captured by AD O-Tau assay
Various Tau truncations tagged with HA at the N terminus were overexpressed in HEK-293FT cells for 48 h. The cells were lysed in PBS containing 50 mm NaF, 1 mm Na3VO4, 1 mm AEBSF, 5 mm benzamidine, and 10 μg/ml each of aprotinin, leupeptin, and pepstatin by probe sonication at 20% power for 2 min. The cell lysates were centrifuged for 10 min at 10,000 × g. The supernatants containing HA-Tau truncations were stored at −80°C until used.
Various amounts of AD O-Tau isolated from AD brains were dotted on nitrocellulose membrane after serial dilution in TBS and dried at 37 °C for 1 h. The membrane was blocked with 5% fat-free milk in TBS for 2 h and incubated with the above cell extracts containing HA-tagged truncated Taus overnight. After washing three times with TBST, the membrane was incubated with anti-HA in 5% milk in TBST overnight and processed as immuno-dot blot as described above.
AD O-Tau seeded Tau aggregation in cultured cells
HEK-293FT cells were transfected with pCI/HA-Tau truncations with FuGENE HD. AD O-Tau from AD brains was mixed with Lipofectamine 2000 (3.0% in Opti-MEM) (Thermo Fisher Scientific) in 50 μl for 20 min at room temperature. The AD O-Tau/Lipofectamine was added into the cell cultures in 24-well plates after 6 h of transfection and cultured for 42 h. The cells were lysed in RIPA buffer (50 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing 50 mm NaF, 1 mm Na3VO4, 1 mm AEBSF, 5 mm benzamidine, and 10 μg/ml each of aprotinin, leupeptin, and pepstatin for 20 min on ice. The cell lysates were centrifuged at 130,000 × g for 45 min, and the resulting pellet was washed twice with RIPA buffer. The supernatants were pooled together as RIPA buffer–soluble fraction, and the pellet was the RIPA buffer–insoluble fraction. Levels of RIPA buffer–insoluble and -soluble Tau were analyzed by Western blots developed with anti-HA.
To visualize the Tau aggregates in cells, HeLa cells were overexpressed with Tau1–441 and Tau151–391 tagged with HA and treated with AD O-Tau for 42 h as described above. The cells were then fixed for 15 min with 4% paraformaldehyde in phosphate buffer, washed with PBS, and treated with 0.3% Triton in PBS for 15 min at room temperature. After blocking with 5% newborn goat serum, 0.1% Triton X-100, and 0.05% Tween 20 in PBS for 30 min, the cells were incubated with anti-HA in blocking solution overnight at 4 °C, washed with PBS, and incubated with Alexa Fluor 555–conjugated secondary antibody for 2 h at room temperature. After washing with PBS, the cells were mounted with ProLong™ Gold antifade reagent (Thermo Fisher Scientific) and observed with a Nikon confocal microscope.
Expression and purification of recombinant Tau1–441 and Tau151–391
BL21/pGEX-6P1/Tau1–441 and BL21/pGEX-6P1/Tau151–391 cultured in LB medium were treated with 0.5 mm isopropyl β-d-thiogalactopyranoside for 3 h at room temperature to induce the protein expression, after which cells were centrifuged at 5,000 × g for 10 min at 4 °C. The resulting pellets were resuspended in TBS with 1 mm DTT and a mixture of protease inhibitors for bacteria (Sigma) and probe-sonicated on ice with 70% power for 20 min. The bacterial lysate was adjusted to 1% Triton X-100 and centrifuged at 10,000 × g for 15 min. The supernatant was incubated with GSH–Sepharose beads for 1 h at 4 °C. The beads were agitated with PreScissionTM Protease (Sigma–Aldrich) in buffer (50 mm Tris-HCl, pH 7.0, 150 mm NaCl, 1 mm EDTA, and 1 mm DTT) to cleave Tau from GST after extensive washing with TBS. Purified Tau was dialyzed against 5 mm MES, pH 6.8, and 0.5 mm EGTA and lyophilized.
Tau aggregation induced by heparin or AD O-Tau in vitro
Recombinant Tau (25 μm) and heparin (0.5 mg/ml) or AD O-Tau (0.53 mg/ml) in 50 µl of PBS containing 50 μm thioflavin T and 1 mm DTT were incubated by shaking at room temperature. Fluorimetry was performed by using a Spectra Max M5 fluorescence spectrophotometer (set at 440-nm excitation/538-nm emission) every 10 min.
Statistical analysis
The GraphPad Prism 6 software was used for statistical analysis. The results were analyzed by one-way ANOVA followed with Tukey's multiple comparisons test or by two-way ANOVA followed by Sidak's multiple comparisons test for multiple-group analysis.
Data availability
All data are contained within the article.
Acknowledgments
We are thankful to Maureen Marlow for copyediting the manuscript.
Author contributions—J. G., W. X., N. J., L. L., D. C., and F. L. data curation; J. G., W. X., N. J., L. L., and Y. Z. formal analysis; J. G., W. X., N. J., L. L., Y. Z., D. C., and F. L. investigation; C.-X. G., K. I., and F. L. resources; C.-X. G. and K. I. writing-review and editing; F. L. conceptualization; F. L. supervision; F. L. funding acquisition; F. L. validation; F. L. writing-original draft; F. L. project administration.
Funding and additional information—This work was supported in part by funds from New York State Office for People with Developmental Disabilities and Nantong University and by U.S. Alzheimer's Association Grant DSAD-15-363172.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- NFT
- neurofibrillary tangle
- AD
- Alzheimer's disease
- AD O-Tau
- oligomeric Tau isolated from AD brain tissue
- AD P-Tau
- hyperphosphorylated and cytosolic Tau isolated from AD brain
- aa
- amino acid(s)
- MTBR
- microtubule-binding repeat region
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HMW
- high-molecular-weight
- LDH
- lactate dehydrogenase
- RIPA buffer
- radioimmunoprecipitation assay buffer
- HRP
- horseradish peroxidase
- rTau
- recombinant Tau
- HA
- hemagglutinin
- AEBSF
- 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride
- ANOVA
- analysis of variance.
References
- 1. Braak H., Braak E., Grundke-Iqbal I., and Iqbal K. (1986) Occurrence of neuropil threads in the senile human brain and in Alzheimer's disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci. Lett. 65, 351–355 10.1016/0304-3940(86)90288-0 [DOI] [PubMed] [Google Scholar]
- 2. Ballatore C., Lee V. M., and Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- 3. Iqbal K., Liu F., and Gong C. X. (2016) Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15–27 10.1038/nrneurol.2015.225 [DOI] [PubMed] [Google Scholar]
- 4. Wang Y., and Mandelkow E. (2016) Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 5–21 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- 5. Alafuzoff I., Adolfsson R., Grundke-Iqbal I., and Winblad B. (1987) Blood–brain barrier in Alzheimer dementia and in non-demented elderly: an immunocytochemical study. Acta Neuropathol. 73, 160–166 10.1007/BF00693782 [DOI] [PubMed] [Google Scholar]
- 6. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., and Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42, 631–639 10.1212/wnl.42.3.631 [DOI] [PubMed] [Google Scholar]
- 7. Riley K. P., Snowdon D. A., and Markesbery W. R. (2002) Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann. Neurol. 51, 567–577 10.1002/ana.10161 [DOI] [PubMed] [Google Scholar]
- 8. Braak H., and Braak E. (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 9. Braak H., and Braak E. (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–284 10.1016/0197-4580(95)00021-6 [DOI] [PubMed] [Google Scholar]
- 10. Köpke E., Tung Y. C., Shaikh S., Alonso A. C., Iqbal K., and Grundke-Iqbal I. (1993) Microtubule-associated protein Tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384 [PubMed] [Google Scholar]
- 11. Liu F., Shi J., Tanimukai H., Gu J., Gu J., Grundke-Iqbal I., Iqbal K., and Gong C.-X. (2009) Reduced O-GlcNAcylation links lower brain glucose metabolism and Tau pathology in Alzheimer's disease. Brain 132, 1820–1832 10.1093/brain/awp099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., and Binder L. I. (1986) Abnormal phosphorylation of the microtubule-associated protein Tau (Tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 10.1073/pnas.83.13.4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alonso A. C., Zaidi T., Grundke-Iqbal I., and Iqbal K. (1994) Role of abnormally phosphorylated Tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 5562–5566 10.1073/pnas.91.12.5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alonso A. D., Grundke-Iqbal I., and Iqbal K. (1996) Alzheimer's disease hyperphosphorylated Tau sequesters normal Tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787 10.1038/nm0796-783 [DOI] [PubMed] [Google Scholar]
- 15. Alonso A., Zaidi T., Novak M., Grundke-Iqbal I., and Iqbal K. (2001) Hyperphosphorylation induces self-assembly of Tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928 10.1073/pnas.121119298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clavaguera F., Bolmont T., Crowther R. A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A. K., Beibel M., Staufenbiel M., Jucker M., Goedert M., and Tolnay M. (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913 10.1038/ncb1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahmed Z., Cooper J., Murray T. K., Garn K., McNaughton E., Clarke H., Parhizkar S., Ward M. A., Cavallini A., Jackson S., Bose S., Clavaguera F., Tolnay M., Lavenir I., Goedert M., et al. (2014) A novel in vivo model of Tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683 10.1007/s00401-014-1254-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takeda S., Wegmann S., Cho H., DeVos S. L., Commins C., Roe A. D., Nicholls S. B., Carlson G. A., Pitstick R., Nobuhara C. K., Costantino I., Frosch M. P., Müller D. J., Irimia D., and Hyman B. T. (2015) Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight Tau derived from Alzheimer's disease brain. Nat. Commun. 6, 8490 10.1038/ncomms9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clavaguera F., Akatsu H., Fraser G., Crowther R. A., Frank S., Hench J., Probst A., Winkler D. T., Reichwald J., Staufenbiel M., Ghetti B., Goedert M., and Tolnay M. (2013) Brain homogenates from human tauopathies induce Tau inclusions in mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 9535–9540 10.1073/pnas.1301175110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boluda S., Iba M., Zhang B., Raible K. M., Lee V. M., and Trojanowski J. Q. (2015) Differential induction and spread of Tau pathology in young PS19 Tau transgenic mice following intracerebral injections of pathological Tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol. 129, 221–237 10.1007/s00401-014-1373-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li L., Jiang Y., Hu W., Tung Y. C., Dai C., Chu D., Gong C. X., Iqbal K., and Liu F. (2019) Pathological alterations of Tau in Alzheimer's disease and 3xTg-AD mouse brains. Mol. Neurobiol. 56, 6168–6183 10.1007/s12035-019-1507-4 [DOI] [PubMed] [Google Scholar]
- 22. Hu W., Zhang X., Tung Y. C., Xie S., Liu F., and Iqbal K. (2016) Hyperphosphorylation determines both the spread and the morphology of Tau pathology. Alzheimers Dement. 12, 1066–1077 10.1016/j.jalz.2016.01.014 [DOI] [PubMed] [Google Scholar]
- 23. Dai C. L., Hu W., Tung Y. C., Liu F., Gong C. X., and Iqbal K. (2018) Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 x Tg-AD mice. Alzheimers Res Ther 10, 13 10.1186/s13195-018-0341-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miao J., Shi R., Li L., Chen F., Zhou Y., Tung Y. C., Hu W., Gong C. X., Iqbal K., and Liu F. (2019) Pathological Tau from Alzheimer's brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of Tau in vivo. Front. Aging Neurosci. 11, 34 10.3389/fnagi.2019.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zilka N., Filipcik P., Koson P., Fialova L., Skrabana R., Zilkova M., Rolkova G., Kontsekova E., and Novak M. (2006) Truncated Tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 580, 3582–3588 10.1016/j.febslet.2006.05.029 [DOI] [PubMed] [Google Scholar]
- 26. Hasegawa M., Morishima-Kawashima M., Takio K., Suzuki M., Titani K., and Ihara Y. (1992) Protein sequence and mass spectrometric analyses of Tau in the Alzheimer's disease brain. J. Biol. Chem. 267, 17047–17054 [PubMed] [Google Scholar]
- 27. Yang L. S., and Ksiezak-Reding H. (1995) Calpain-induced proteolysis of normal human Tau and Tau associated with paired helical filaments. Eur. J. Biochem. 233, 9–17 10.1111/j.1432-1033.1995.009_1.x [DOI] [PubMed] [Google Scholar]
- 28. Seubert P., Mawal-Dewan M., Barbour R., Jakes R., Goedert M., Johnson G. V., Litersky J. M., Schenk D., Lieberburg I., Trojanowski J. Q., and Lee V. M.-Y. (1995) Detection of phosphorylated Ser262 in fetal Tau, adult Tau, and paired helical filament Tau. J. Biol. Chem. 270, 18917–18922 10.1074/jbc.270.32.18917 [DOI] [PubMed] [Google Scholar]
- 29. Gamblin T. C., Chen F., Zambrano A., Abraha A., Lagalwar S., Guillozet A. L., Lu M., Fu Y., Garcia-Sierra F., LaPointe N., Miller R., Berry R. W., Binder L. I., and Cryns V. L. (2003) Caspase cleavage of Tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 10032–10037 10.1073/pnas.1630428100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y., Garg S., Mandelkow E. M., and Mandelkow E. (2010) Proteolytic processing of Tau. Biochem. Soc. Trans. 38, 955–961 10.1042/BST0380955 [DOI] [PubMed] [Google Scholar]
- 31. Zhao X., Kotilinek L. A., Smith B., Hlynialuk C., Zahs K., Ramsden M., Cleary J., and Ashe K. H. (2016) Caspase-2 cleavage of Tau reversibly impairs memory. Nat. Med. 22, 1268–1276 10.1038/nm.4199 [DOI] [PubMed] [Google Scholar]
- 32. Zhang Z., Song M., Liu X., Kang S. S., Kwon I. S., Duong D. M., Seyfried N. T., Hu W. T., Liu Z., Wang J. Z., Cheng L., Sun Y. E., Yu S. P., Levey A. I., and Ye K. (2014) Cleavage of Tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer's disease. Nat. Med. 20, 1254–1262 10.1038/nm.3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quinn J. P., Corbett N. J., Kellett K. A. B., and Hooper N. M. (2018) Tau proteolysis in the pathogenesis of tauopathies: neurotoxic fragments and novel biomarkers. J. Alzheimers Dis. 63, 13–33 10.3233/JAD-170959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramcharitar J., Albrecht S., Afonso V. M., Kaushal V., Bennett D. A., and Leblanc A. C. (2013) Cerebrospinal fluid Tau cleaved by caspase-6 reflects brain levels and cognition in aging and Alzheimer disease. J. Neuropathol. Exp. Neurol. 72, 824–832 10.1097/NEN.0b013e3182a0a39f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Derisbourg M., Leghay C., Chiappetta G., Fernandez-Gomez F. J., Laurent C., Demeyer D., Carrier S., Buée-Scherrer V., Blum D., Vinh J., Sergeant N., Verdier Y., Buee L., and Hamdane M. (2015) Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. 5, 9659 10.1038/srep09659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Novak M., Kabat J., and Wischik C. M. (1993) Molecular characterization of the minimal protease resistant Tau unit of the Alzheimer's disease paired helical filament. EMBO J. 12, 365–370 10.1002/j.1460-2075.1993.tb05665.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou Y., Shi J., Chu D., Hu W., Guan Z., Gong C. X., Iqbal K., and Liu F. (2018) Relevance of phosphorylation and truncation of Tau to the etiopathogenesis of Alzheimer's disease. Front. Aging Neurosci. 10, 27 10.3389/fnagi.2018.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kovacech B., and Novak M. (2010) Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer's disease. Curr. Alzheimer Res. 7, 708–716 10.2174/156720510793611556 [DOI] [PubMed] [Google Scholar]
- 39. De Strooper B. (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90, 465–494 10.1152/physrev.00023.2009 [DOI] [PubMed] [Google Scholar]
- 40. Drewes G., Trinczek B., Illenberger S., Biernat J., Schmitt-Ulms G., Meyer H. E., Mandelkow E. M., and Mandelkow E. (1995) Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark): a novel protein kinase that regulates Tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J. Biol. Chem. 270, 7679–7688 10.1074/jbc.270.13.7679 [DOI] [PubMed] [Google Scholar]
- 41. Jeganathan S., von Bergen M., Brutlach H., Steinhoff H. J., and Mandelkow E. (2006) Global hairpin folding of Tau in solution. Biochemistry 45, 2283–2293 10.1021/bi0521543 [DOI] [PubMed] [Google Scholar]
- 42. Margittai M., and Langen R. (2004) Template-assisted filament growth by parallel stacking of Tau. Proc. Natl. Acad. Sci. U.S.A. 101, 10278–10283 10.1073/pnas.0401911101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gamblin T. C., King M. E., Kuret J., Berry R. W., and Binder L. I. (2000) Oxidative regulation of fatty acid-induced Tau polymerization. Biochemistry 39, 14203–14210 10.1021/bi001876l [DOI] [PubMed] [Google Scholar]
- 44. Kampers T., Friedhoff P., Biernat J., Mandelkow E. M., and Mandelkow E. (1996) RNA stimulates aggregation of microtubule-associated protein Tau into Alzheimer-like paired helical filaments. FEBS Lett. 399, 344–349 10.1016/S0014-5793(96)01386-5 [DOI] [PubMed] [Google Scholar]
- 45. Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., and Crowther R. A. (1996) Assembly of microtubule-associated protein Tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553 10.1038/383550a0 [DOI] [PubMed] [Google Scholar]
- 46. Sanders D. W., Kaufman S. K., DeVos S. L., Sharma A. M., Mirbaha H., Li A., Barker S. J., Foley A. C., Thorpe J. R., Serpell L. C., Miller T. M., Grinberg L. T., Seeley W. W., Diamond M. I. (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 82, 1271–1288 10.1016/j.neuron.2014.04.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilson D. M., and Binder L. I. (1997) Free fatty acids stimulate the polymerization of Tau and amyloid β peptides: in vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Am. J. Pathol. 150, 2181–2195 [PMC free article] [PubMed] [Google Scholar]
- 48. Friedhoff P., Schneider A., Mandelkow E. M., and Mandelkow E. (1998) Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein Tau monitored by fluorescence in solution. Biochemistry 37, 10223–10230 10.1021/bi980537d [DOI] [PubMed] [Google Scholar]
- 49. Zhang W., Falcon B., Murzin A. G., Fan J., Crowther R. A., Goedert M., and Scheres S. H. (2019) Heparin-induced Tau filaments are polymorphic and differ from those in Alzheimer's and Pick's diseases. eLife 8, e43584 10.7554/eLife.43584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jeganathan S., Hascher A., Chinnathambi S., Biernat J., Mandelkow E. M., and Mandelkow E. (2008) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076 10.1074/jbc.M805300200 [DOI] [PubMed] [Google Scholar]
- 51. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., and Mandelkow E. (2000) Assembly of Tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 10.1073/pnas.97.10.5129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mukrasch M. D., Biernat J., von Bergen M., Griesinger C., Mandelkow E., and Zweckstetter M. (2005) Sites of Tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986 10.1074/jbc.M501565200 [DOI] [PubMed] [Google Scholar]
- 53. Rissman R. A., Poon W. W., Blurton-Jones M., Oddo S., Torp R., Vitek M. P., LaFerla F. M., Rohn T. T., and Cotman C. W. (2004) Caspase-cleavage of Tau is an early event in Alzheimer disease tangle pathology. J. Clin. Invest. 114, 121–130 10.1172/JCI200420640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mandelkow E., von Bergen M., Biernat J., and Mandelkow E. M. (2007) Structural principles of Tau and the paired helical filaments of Alzheimer's disease. Brain Pathol. 17, 83–90 10.1111/j.1750-3639.2007.00053.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alonso A. D., Zaidi T., Novak M., Barra H. S., Grundke-Iqbal I., and Iqbal K. (2001) Interaction of Tau isoforms with Alzheimer's disease abnormally hyperphosphorylated Tau and in vitro phosphorylation into the disease-like protein. J. Biol. Chem. 276, 37967–37973 10.1074/jbc.M105365200 [DOI] [PubMed] [Google Scholar]
- 56. Abraha A., Ghoshal N., Gamblin T. C., Cryns V., Berry R. W., Kuret J., and Binder L. I. (2000) C-terminal inhibition of Tau assembly in vitro and in Alzheimer's disease. J. Cell Sci. 113, 3737–3745 [DOI] [PubMed] [Google Scholar]
- 57. Alonso A. D. C., Mederlyova A., Novak M., Grundke-Iqbal I., and Iqbal K. (2004) Promotion of hyperphosphorylation by frontotemporal dementia Tau mutations. J. Biol. Chem. 279, 34873–34881 10.1074/jbc.M405131200 [DOI] [PubMed] [Google Scholar]
- 58. Arai T., Miklossy J., Klegeris A., Guo J. P., and McGeer P. L. (2006) Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 65, 19–25 10.1097/01.jnen.0000196133.74087.cb [DOI] [PubMed] [Google Scholar]
- 59. Arai T., Guo J. P., and McGeer P. L. (2005) Proteolysis of non-phosphorylated and phosphorylated Tau by thrombin. J. Biol. Chem. 280, 5145–5153 10.1074/jbc.M409234200 [DOI] [PubMed] [Google Scholar]
- 60. Marcinkiewicz M., and Seidah N. G. (2000) Coordinated expression of β-amyloid precursor protein and the putative β-secretase BACE and α-secretase ADAM10 in mouse and human brain. J. Neurochem. 75, 2133–2143 10.1046/j.1471-4159.2000.0752133.x [DOI] [PubMed] [Google Scholar]
- 61. Lammich S., Kojro E., Postina R., Gilbert S., Pfeiffer R., Jasionowski M., Haass C., and Fahrenholz F. (1999) Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U.S.A. 96, 3922–3927 10.1073/pnas.96.7.3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gatta L. B., Albertini A., Ravid R., and Finazzi D. (2002) Levels of β-secretase BACE and α-secretase ADAM10 mRNAs in Alzheimer hippocampus. Neuroreport 13, 2031–2033 10.1097/00001756-200211150-00008 [DOI] [PubMed] [Google Scholar]
- 63. Yuan X. Z., Sun S., Tan C. C., Yu J. T., and Tan L. (2017) The role of ADAM10 in Alzheimer's disease. J. Alzheimers Dis. 58, 303–322 10.3233/JAD-170061 [DOI] [PubMed] [Google Scholar]
- 64. Saftig P., and Lichtenthaler S. F. (2015) The α secretase ADAM10: a metalloprotease with multiple functions in the brain. Prog. Neurobiol. 135, 1–20 10.1016/j.pneurobio.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 65. Chu D., and Liu F. (2019) Pathological changes of Tau related to Alzheimer's disease. ACS Chem. Neurosci. 10, 931–944 10.1021/acschemneuro.8b00457 [DOI] [PubMed] [Google Scholar]
- 66. Henriksen K., Wang Y., Sørensen M. G., Barascuk N., Suhy J., Pedersen J. T., Duffin K. L., Dean R. A., Pajak M., Christiansen C., Zheng Q., and Karsdal M. A. (2013) An enzyme-generated fragment of Tau measured in serum shows an inverse correlation to cognitive function. PLoS One 8, e64990 10.1371/journal.pone.0064990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Neergaard J. S., Dragsbæk K., Christiansen C., Karsdal M. A., Brix S., and Henriksen K. (2018) Two novel blood-based biomarker candidates measuring degradation of Tau are associated with dementia: a prospective study. PLoS One 13, e0194802 10.1371/journal.pone.0194802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Inekci D., Henriksen K., Linemann T., Karsdal M. A., Habib A., Bisgaard C., Eriksen F. B., and Vilholm O. J. (2015) Serum fragments of Tau for the differential diagnosis of Alzheimer's disease. Curr. Alzheimer Res. 12, 829–836 10.2174/1567205012666150710111211 [DOI] [PubMed] [Google Scholar]
- 69. Henriksen K., Byrjalsen I., Christiansen C., and Karsdal M. A. (2015) Relationship between serum levels of Tau fragments and clinical progression of Alzheimer's disease. J. Alzheimers Dis. 43, 1331–1341 10.3233/JAD-140984 [DOI] [PubMed] [Google Scholar]
- 70. Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y. C., Zaidi M. S., and Wisniewski H. M. (1986) Microtubule-associated protein Tau: a component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089 [PubMed] [Google Scholar]
- 71. Luna-Muñoz J., Chávez-Macías L., García-Sierra F., and Mena R. (2007) Earliest stages of Tau conformational changes are related to the appearance of a sequence of specific phospho-dependent Tau epitopes in Alzheimer's disease. J. Alzheimers Dis. 12, 365–375 10.3233/jad-2007-12410 [DOI] [PubMed] [Google Scholar]
- 72. Flores-Rodríguez P., Ontiveros-Torres M. A., Cárdenas-Aguayo M. C., Luna-Arias J. P., Meraz-Ríos M. A., Viramontes-Pintos A., Harrington C. R., Wischik C. M., Mena R., Florán-Garduño B., and Luna-Muñoz J. (2015) The relationship between truncation and phosphorylation at the C-terminus of Tau protein in the paired helical filaments of Alzheimer's disease. Front. Neurosci. 9, 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Guillozet-Bongaarts A. L., Garcia-Sierra F., Reynolds M. R., Horowitz P. M., Fu Y., Wang T., Cahill M. E., Bigio E. H., Berry R. W., and Binder L. I. (2005) Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging 26, 1015–1022 10.1016/j.neurobiolaging.2004.09.019 [DOI] [PubMed] [Google Scholar]
- 74. Lyu X. J., Li Z. H., Li X., Zeng W. L., Yang P., Lin Q. X., Zheng J. Y., Du X. L., Gu Y. Z., Zhao Y. Q., Xie R. S., Liu T., Lin H. L., and Ma W. J. (2017) Commuting mode specific exposure to PM(2.5) in urban area of Guangzhou. Zhonghua Liu Xing Bing Xue Za Zhi 38, 309–313 [DOI] [PubMed] [Google Scholar]
- 75. Basurto-Islas G., Luna-Muñoz J., Guillozet-Bongaarts A. L., Binder L. I., Mena R., and García-Sierra F. (2008) Accumulation of aspartic acid 421- and glutamic acid 391-cleaved Tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J. Neuropathol. Exp. Neurol. 67, 470–483 10.1097/NEN.0b013e31817275c7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang J. Z., Grundke-Iqbal I., and Iqbal K. (2007) Kinases and phosphatases and Tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68 10.1111/j.1460-9568.2006.05226.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wischik C. M., Novak M., Thøgersen H. C., Edwards P. C., Runswick M. J., Jakes R., Walker J. E., Milstein C., Roth M., and Klug A. (1988) Isolation of a fragment of Tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 85, 4506–4510 10.1073/pnas.85.12.4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Amadoro G., Serafino A. L., Barbato C., Ciotti M. T., Sacco A., Calissano P., and Canu N. (2004) Role of N-terminal Tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ. 11, 217–230 10.1038/sj.cdd.4401314 [DOI] [PubMed] [Google Scholar]
- 79. Amadoro G., Ciotti M. T., Costanzi M., Cestari V., Calissano P., and Canu N. (2006) NMDA receptor mediates Tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. U.S.A. 103, 2892–2897 10.1073/pnas.0511065103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Corsetti V., Amadoro G., Gentile A., Capsoni S., Ciotti M. T., Cencioni M. T., Atlante A., Canu N., Rohn T. T., Cattaneo A., and Calissano P. (2008) Identification of a caspase-derived N-terminal Tau fragment in cellular and animal Alzheimer's disease models. Mol. Cell Neurosci. 38, 381–392 10.1016/j.mcn.2008.03.011 [DOI] [PubMed] [Google Scholar]
- 81. Park S. Y., and Ferreira A. (2005) The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which Tau mediates β-amyloid–induced neurodegeneration. J. Neurosci. 25, 5365–5375 10.1523/JNEUROSCI.1125-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xia D., Li C., and Götz J. (2015) Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim. Biophys. Acta 1852, 913–924 10.1016/j.bbadis.2014.12.017 [DOI] [PubMed] [Google Scholar]
- 83. Padmanabhan P., Martínez-Mármol R., Xia D., Götz J., and Meunier F. A. (2019) Frontotemporal dementia mutant Tau promotes aberrant Fyn nanoclustering in hippocampal dendritic spines. eLife 8, e45040 10.7554/eLife.45040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pei J. J., Braak E., Braak H., Grundke-Iqbal I., Iqbal K., Winblad B., and Cowburn R. F. (1999) Distribution of active glycogen synthase kinase 3β (GSK-3β) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 58, 1010–1019 10.1097/00005072-199909000-00011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article.