

Abstract
The human chemokine family consists of 46 protein ligands that induce chemotactic cell migration by activating a family of 23 G protein–coupled receptors. The two major chemokine subfamilies, CC and CXC, bind distinct receptor subsets. A sequence motif defining these families, the X position in the CXC motif, is not predicted to make significant contacts with the receptor, but instead links structural elements associated with binding and activation. Here, we use comparative analysis of chemokine NMR structures, structural modeling, and molecular dynamic simulations that suggested the X position reorients the chemokine N terminus. Using CXCL12 as a model CXC chemokine, deletion of the X residue (Pro-10) had little to no impact on the folded chemokine structure but diminished CXCR4 agonist activity as measured by ERK phosphorylation, chemotaxis, and Gi/o-mediated cAMP inhibition. Functional impairment was attributed to over 100-fold loss of CXCR4 binding affinity. Binding to the other CXCL12 receptor, ACKR3, was diminished nearly 500-fold. Deletion of Pro-10 had little effect on CXCL12 binding to the CXCR4 N terminus, a major component of the chemokine-GPCR interface. Replacement of the X residue with the most frequent amino acid at this position (P10Q) had an intermediate effect between WT and P10del in each assay, with ACKR3 having a higher tolerance for this mutation. This work shows that the X residue helps to position the CXCL12 N terminus for optimal docking into the orthosteric pocket of CXCR4 and suggests that the CC/CXC motif contributes directly to receptor selectivity by orienting the chemokine N terminus in a subfamily-specific direction.
Keywords: G protein-coupled receptor (GPCR), cell migration, nuclear magnetic resonance (NMR), signal transduction, structure-function, chemokine, CC/CXC motif, chemokine network
Multicellular organisms orchestrate the migration of many cell types (1). In humans, this essential function is carried out by the chemokine signaling network, a family of 46 secreted proteins that orchestrate directed cell migration through interactions with 23 chemokine-receptive G protein–coupled receptors (GPCRs) (2). With twice the number of ligands as receptors, it is not surprising that most receptors bind multiple chemokines (3). Some chemokine ligands also bind promiscuously to more than one receptor, although monogamous or nearly monogamous pairs do exist (4). Initially, this redundancy was considered a result of evolutionary pressure to organize the immune system against multiple pathogens and avoid immune resistance (5). However, studies of chemokine signaling reveal unique functions for each chemokine-receptor pair (6–8). An improved description of the chemokine interaction network is as selectively promiscuous, neither fully specific nor fully promiscuous, where overlapping high affinity chemokine-receptor pairs exist as a subset of all possible combinations (e.g. CCR3 binds many but not all chemokines, and these ligands bind several but not all receptors).
Unraveling the principles underlying the selectively promiscuous chemokine network has proven challenging despite determined structures of chemokines, chemokine receptors (CKRs), and chemokine-receptor co-crystal complexes (9–11). These structural studies have revealed a common tertiary fold for chemokines and validated that CKRs adopt the GPCR seven-transmembrane architecture. The highly conserved structures of chemokines and CKRs yield a conserved interface, with the chemokine N terminus binding the orthosteric pocket of the receptor. This binding pose was predicted by an initial model of the interaction that separates the chemokine interface into two distinct sites (12). In this two-site model, the CKR N terminus binds the globular chemokine core but does not lead to receptor activation (site-1). Activation is achieved through interactions between the chemokine N terminus and the receptor extracellular loop and transmembrane residues (site-2). Determination of the co-crystal structures added to this model by revealing a large protein-protein interface that included regions, such as the chemokine loop linking the β1- and β2-strands near residue 30 (i.e. the 30s loop), not previously predicted to significantly interact (13–16). Thus, although the simplistic two-site model fails to capture the full complexity of chemokine-CKR recognition, it remains a useful framework for structure-function analyses.
Contrary to the chemokine conserved structure, chemokine sequence is highly variable. The most conserved residues are four characteristic cysteines that form two disulfide bonds necessary for function in some chemokines (17). The first two cysteine residues separate the N terminus from the N-loop and classify chemokines into two major families where the cysteines are adjacent (CC-family) or separated by a single residue (CXC-family) (18).
CKRs lack a CC/CXC motif and thus adopt the family of its chemokine ligand. In a truly promiscuous system, this nomenclature would break down. However, receptors rarely bind chemokines from multiple families. Thus, CXC receptors are activated by CXC chemokines, and CC receptors are activated by CC chemokines. There has been little evidence to support a family-wide explanation for this familial selectivity. As the deterministic residue for family classification is the X residue in the CXC motif, this X-factor likely plays a functional role in chemokine discrimination.
Remarkably, many chemokine genes segregate into two major clusters corresponding to their family (19). In humans, chromosome 4 houses a majority of CXC chemokines, and chromosome 17 contains most CC chemokines. This implies that selectivity has been maintained through the successive gene duplications that rapidly expanded the chemokine family and that this selectivity may be as old as the molecules themselves. Phylogenetic analysis suggests that CXCL12 was a founding member of the CXC family. Duplication of a primordial chemokine gene followed by a permissive mutation that removed the X residue yielded the CC family progenitor (20–22). Subsequent duplication events and transmutation occurred throughout chemokine evolution, thus driving recognition complexity and allowing the expansion of the chemokine system to orchestrate the inducible migration and constitutive homing of many cell types.
The CC/CXC motif is not contained in the binding (site-1) or activation (site-2) regions of the chemokine; however, it is poised directly between these regions, allowing for structural modulation that could alter receptor binding or activation. In co-crystal structures of chemokine-bound GPCRs, the CC motif is above the pocket near the top of TM1 and the receptor N terminus. In the absence of a structure of a CXC chemokine bound to its receptor, it is unknown how the X residue interacts with the GPCR. Thus, the chemokine X-factor could determine selectivity between CC and CXC chemokines, and this effect could be achieved through structural perturbation of the chemokine N-loop (site-1), the N terminus (site-2), or a more subtle allosteric effect.
To shed light on selectivity determinants between CC and CXC chemokines, we interrogated the structural impact and functional consequences of mutation and deletion of the X residue in the prototypical homeostatic chemokine CXCL12. Using NMR, cell-based functional assays, and radioligand binding, we observed structural perturbation as well as reduced receptor binding and activation upon mutation or deletion of the X residue. Interactions with the CKR N terminus (site-1) were largely unchanged, localizing the binding defect to the chemokine interface with the CKR extracellular loops or transmembrane regions (site-2). Analysis of every chemokine with an NMR solution-state structure suggested preferred N-terminal states between CC and CXC chemokines. Homology modeling and subsequent molecular dynamics (MD) simulations of CXCL12-P10del support the hypothesis that the CC/CXC motif directly impacts chemokine N-terminal orientation important for receptor recognition.
Results
The chemokine network segregates CC and CXC chemokines
Analysis of the chemokine interaction network revealed that each chemokine subfamily activates a distinct subset of receptors (Fig. 1A). In other words, aside from the atypical (e.g. ACKR1) or viral (e.g. US28) receptors, chemokine receptors exhibit subfamily (e.g. CC versus CXC) selectivity. The presence or absence of the X position is the only absolutely conserved sequence difference that distinguishes CC and CXC chemokines. Although there is no consensus X residue among the 17 CXC chemokines (Fig. 1B), this amino acid may contribute to receptor preference within the CXC family. Of the 11 different amino acids found at the X position, polar and aliphatic side chains are found in the ligands for the more promiscuous CXCR1, CXCR2, and CXCR3 receptors, whereas charged (Arg and Glu), aromatic (Tyr), or constrained (Pro) amino acids correspond to selective or monogamous chemokine-receptor interactions. We hypothesized that deletion of the intervening X residue would alter chemokine structure in a way that significantly impairs recognition by CXC family receptors, opening the door to interactions with other GPCRs.
Figure 1.

Selectivity in the chemokine network between CC and CXC chemokines. A, the chemokine network is illustrated with chemokine receptors labeled inside the membrane and the chemokines that bind that receptor labeled outside. The inner phylogenetic tree describes the evolutionary relationships between chemokine receptors. Colors represent each chemokine family: CC (blue), CXC (green), CX3C (red), XC (orange), and atypical (purple). B, the CXC receptors are shown linked with the X residues of their cognate ligands. Each one-letter code is colored by amino acid property: hydrophilic (green), hydrophobic (pink), positive charge (blue), conformationally special (purple), aromatic (yellow), or negative charge (red). The 17 CXC chemokines are represented below the corresponding X residue. C, unique solution-state NMR structures of CXC chemokines were analyzed to determine intramolecular contacts with the side chain of the X residue. These data are illustrated on the structure of CXCL12 (PDB entry 2KED). D, the average lengths of the β1- and β2-strands are shorter in CC chemokines than CXC chemokines, as calculated from 27 solution-state NMR structures. This is demonstrated by structures of CXCL12 (PDB entry 2KED) and CCL20 (PDB entry 2JYO). The X residue in CXCL12 (Pro-10) makes contacts with Leu-29, Thr-31, and Gln-37 to extend secondary structure into the 30s loop. E, each conformer in CC and CXC NMR structures were separated and aligned via the conserved chemokine core. Vectors describing the N terminus of each structure were averaged by family to construct the CC or CXC mean axes, which are separated by 45°. F, the structure of vMIP-II bound to CXCR4 (PDB entry 4RWS) is overlaid with the coordinate system of D. The CXC mean axis is near the N terminus of vMIP-II in the orthosteric pocket, whereas the CC mean axis is outside of the receptor.
Chemokine structural characteristics
To investigate the structural role of the X position, we tabulated all contacts between that side chain and other amino acids in the NMR structures of 11 different CXC chemokines (Fig. 1C). Most X interactions involved residues in the β1- or β2-strands, with fewer contacts with the end of the 30s loop or the beginning of the β3-strand. Although there were contacts with the N terminus, this region is unstructured and flexible in solution, and they likely represent transient interactions (23). Inspection of the NMR structures of 16 different CC chemokines revealed that the β1-β2 hairpin is longer in CXC chemokines, with an average of two additional residues that participate in β-strand hydrogen bonding (Fig. 1D). This is most apparent in CXCL12, where the proline at the X position packs tightly with residues flanking the 30s loop (Leu-29, Thr-31, and Gln-37), resulting in both β1- and β2-strands extending an additional two residues into the 30s loop. In chemokines with bulkier X residues, such as glutamine in CXCL8, this region protrudes away from the N terminus into solution.
In addition to secondary structure characteristics, several N-terminal preferences became apparent after alignment of CC and CXC chemokines by the conserved core. To quantify this, each conformer in the NMR structure was separated (520 total models), and an average vector was calculated for each N terminus (Fig. 1E). These vectors were averaged by family to calculate the difference in the mean orientations for CC and CXC ligands of 45°. This preference in solution is influenced by the CC/CXC motif that bridges the N terminus to the chemokine body. Upon overlaying the CXCR4 co-crystal structure (4RWS) with the aligned coordinate system, the mean axis for CXC chemokine N termini was oriented in an appropriate direction for receptor docking and activation (Fig. 1F). In contrast, the mean axis of the CC ligand family extended past TM1 in a direction incompatible with binding the orthosteric pocket. This analysis extended our hypothesis that deletion of the X residue would impair binding to CXC receptors by altering the preferred orientation of the receptor-activating N terminus.
Chemokine sequence characteristics
Although the CC/CXC motif is used to separate chemokines into easily classified families, it is not necessarily the sole factor contributing to familial differences. Chemokine N-terminal sequences were analyzed in an attempt to identify consistent differences in the activation domain of CC and CXC chemokines (Fig. 2A, left). Sequences are displayed in a structural format due to the high sequence variance with the conserved N-terminal cysteines aligned. This format best represents the N-terminal length and depth of contact in the orthosteric pocket. The ELR motif is found directly preceding the first cysteine in 7 of 17 CXC chemokines. This region has the highest conservation yet is found in less than half of the family. However, the arginine in this motif is the most conserved position, with a basic residue found in 13 of 17 CXC chemokines. Among the other four CXC chemokines, CXCL4 and CXCL14 have not been shown to bind or activate one of the CXC receptors, and CXCL17 is a newly discovered ligand with an unconventional sequence that may adopt a different structure. Only one of the 26 CC chemokines has a basic residue directly preceding the cysteine. The CC family N termini have low conservation and no apparent family-spanning trends.
Figure 2.

Sequence and structure variation between CC and CXC chemokines. A, the sequences of the activation (site-1) and binding (site-2) motifs were compared between CC and CXC chemokines. Both the chemokine and receptor N termini are unstructured in the unbound state and thus are aligned to conserved cysteine residues. The X residue in the CXC motif unequivocally divides the CC and CXC chemokines. The next deterministic position directly precedes the first cysteine and is negatively charged in 76% CXC chemokines and only 4% of CC chemokines. No difference in average N-terminal length was observed. B, constructs of CXCL12-WT, -P10Q, and -P10del are shown. Glutamine was chosen for substitution as it is the most common X residue. C, structural models of CXCL12-P10del were constructed using the solution-state NMR structure 2KED. Deviation from the WT structure was observed at the N terminus and 30s loop. D, to understand the dynamic impact, 100-ns MD simulations were performed with CXCL12-WT and CXCL12-P10del in explicit solvent. Orientation of the proximal N terminus consistently differs between CC and CXC final states shown overlaid with the CXCR4 co-crystal structure (PDB entry 4RWS). The position of Arg-8 in CXCL12-WT is near the expected binding partner CXCR4:Asp-262, whereas Arg-8 in CXCL12-P10del is facing the opposite side of the pocket. E, each construct was examined by NMR HSQC. Uniform peak distribution supports tertiary folding. Unequal peak intensities in CXCL12-P10del result from an increase in dynamics. Overlays with CXCL12-WT demonstrate altered peak position. F, the intrinsic fluorescence of each chemokine was measured throughout thermal denaturation. The plotted first derivative illustrates the Tm of CXCL12-WT (91.1 °C), CXCL12-P10Q (88.3 °C), or CXCL12-P10del (86.3 °C) performed in triplicate.
Familial selectivity could also be encoded at the level of binding rather than activation. CKR N-terminal sequences were analyzed to search for conserved binding epitopes between CC and CXC receptors (Fig. 2A, right). Like the chemokine, the unbound CKR N terminus is unstructured with high sequence variability and is displayed continuously with the conserved N-terminal cysteine aligned. The most conserved position directly precedes the aligned cysteine and is a proline in half of the receptors with no preference between families.
The presence of the X position unequivocally divides CC and CXC chemokines, implying a direct functional role. In the absence of compelling sequence differences in binding or activation domains, the CC/CXC motif that bridges these two sites was the most likely factor to determine familial selectivity. To interrogate the role of the X residue in structure and function, a CC version of CXCL12 was engineered by deletion of the X residue (Fig. 2B, CXCL12-P10del). We selected CXCL12 for this study due to its high conservation across species and its specific binding to CXCR4 (one known chemokine ligand) and ACKR3 (two known chemokine ligands). As a candidate for the primordial chemokine, it is plausible that a CXC-to-CC transition occurred near the beginning of chemokine evolution (20, 21). Another construct was designed to probe the role of the X residue identity by mutating the X in CXCL12 to glutamine, the most common X residue (Fig. 2B, CXCL12-P10Q).
Molecular dynamics analysis of CXCL12-P10del
To determine whether the X deletion of CXCL12 would be tolerated or if additional mutations would be needed to stabilize the CC disulfide arrangement, models of CXCL12-P10del were constructed and were similar to CXCL12-WT with a core Cα root mean square deviation of 0.6 Å (Fig. 2C). Deviations were noted at the 30s loop and N terminus, but the cysteines adopted normal disulfide stereochemistry, suggesting that tertiary structure would be maintained. This modeling did not sample alternative conformations. To assess the impact of the X-factor on conformational dynamics, five 100-ns MD simulations were performed with CXCL12-WT or CXCL12-P10del in explicit solvent. Over the course of each run, the N terminus fluctuated through many states; however, residues proximal to the CC/CXC motif, particularly Arg-8, consistently differed between CXCL12-WT and CXCL12-P10del. With the CXCR4 co-crystal structure overlaid, Arg-8 in CXCL12-WT was near its expected binding partner, CXCR4:Asp-262 (13) (Fig. 2D). Recent modeling and mutations support this contact as important for receptor activation (24, 25). In CXCL12-P10del, Arg-8 is facing the opposing side of the pocket and unlikely to establish this interaction. This reorientation of the proximal N terminus mimicked the differences between the mean N-terminal states found in NMR solution structures and may be a defining characteristic between CC and CXC chemokines.
Structure and stability of X position mutants
We expressed and purified [U-15N]CXCL12-WT, -P10Q, and -P10del proteins for experimental comparisons of folding, stability, and receptor binding. Protein identity and folding were confirmed by MS and reverse-phase HPLC. NMR heteronuclear single quantum coherence (HSQC) spectra verified that each protein formed a stable, folded tertiary structure similar to CXCL12-WT (Fig. 2E). Larger peak shifts in the CXCL12-P10del spectrum relative to CXCL12-P10Q and variable peak intensities suggest that the deletion of the X residue caused greater structural perturbation and altered the internal dynamics of the protein. This increase in dynamics did not destabilize the protein as assessed by thermal denaturation, with Tm > 85 °C for each of the three constructs (Fig. 2F). We attempted to acquire three-dimensional NMR data to determine the structure of CXCL12-P10del but were prevented by extensive line broadening that reduced the quality of the spectra.
Receptor binding and activation at CXCR4
To further examine the structural effects of X residue deletion suggested by NMR HSQC spectra and MD simulations, receptor binding was assessed by radioligand displacement of [125I]CXCL12 from CXCR4 (Fig. 3A and Table 1). After incubation with HEK-293T membranes overexpressing CXCR4, CXCL12-WT displaced the radioligand (Table 2) (Ki = 0.14 nm). CXCL12-P10del showed displacement, but with a 100-fold decrease in affinity (Ki = 14.8 nm). Intermediate between WT and X deletion, CXCL12-P10Q had a loss of affinity of ∼10-fold (Ki = 1.5 nm). Although these data suggested that X residue deletion disrupts chemokine-GPCR binding, it was unclear whether this disruption prevented chemokine contacts with the receptor N terminus (site-1) or the receptor body (site-2) and whether biological function was maintained.
Figure 3.

Modification of the CXC X-factor reduces signaling and binding to CXCR4. A, displacement of [125I]CXCL12-WT from CXCR4 was measured for each construct via scintillation after a 4-h equilibration with membrane suspensions at room temperature. Measurements were performed in duplicate from three independent experiments. B, inhibition of cAMP was measured via the GloSensor assay after stimulation with forskolin. Measurements were performed in quadruplicate from at least three independent experiments. C, HEK-293 cells were stimulated with 10 nm chemokine for 5 min, and ERK phosphorylation was determined by Western blotting. Quantification of ERK phosphorylation relative to vehicle is shown to the right. D, transwell chemotaxis assays were performed over 2 h with THP1 cells. Raw counts of migrated cells display the full chemotactic curve (left). The first half of the biphasic curve (right) shows a loss in efficacy and potency in CXCL12-P10del. Measurements were performed in quadruplicate from at least three independent experiments.
Table 1.
[125I]CXCL12 saturation binding parameter estimates
CXCR4 and ACKR3 Bmax and Kd values were estimated using [125I]CXCL12 saturation assays performed in duplicate from at least two independent experiments.
| [125I]CXCL12 | Bmax | Kd (pKd ± S.E.) |
|---|---|---|
| fmol/mg protein | nm | |
| CXCR4 | 224 | 0.05 (10.27 ± 0.17) |
| ACKR3 | 264 | 0.01 (10.95 ± 0.05) |
Table 2.
[125I]CXCL12 competition binding estimates
Chemokine binding was fit to a one-site model from data obtained in duplicate from at least three independent experiments.
| [125I]CXCL12 Ki |
|||||
|---|---|---|---|---|---|
| CXCL12-WT, Ki (pKi ± S.E.) | CXCL12-P10Q |
CXCL12-P10del |
|||
| Ki (pKi ± S.E.) | ΔpKi (mutant − WT) | Ki (pKi ± S.E.) | ΔpKi (mutant − WT) | ||
| nm | nm | nm | |||
| CXCR4 | 0.14 (9.85 ± 0.28) | 1.51 (8.82 ± 0.16) | −1.03 | 14.8 (7.83 ± 0.36) | −2.02 |
| ACKR3 | 0.09 (10.04 ± 0.37) | 0.69 (9.16 ± 0.14) | −0.88 | 42.7 (7.37 ± 0.48) | −2.67 |
In preliminary studies on receptor activation, CXCL12-P10del failed to activate CXCR4 as assessed by ERK phosphorylation at 10 nm (Fig. 3C). Conversely, CXCL12-P10Q maintained robust ERK phosphorylation, demonstrating an X identity change being less impactful than familial exchange through X deletion. A result of balanced chemokine signaling is cell migration; thus, transwell chemotaxis assays were used to characterize the X mutants' ability to accomplish this main role. The biphasic response to increasing chemokine concentration was noted (Fig. 3D, left). Analyzing the increasing slope of this curve results in similar trends to ERK phosphorylation with CXCL12-P10Q having an intermediate impact compared with CXCL12-P10del (Fig. 3D, right).
As signaling through endogenous receptors was consistently perturbed, direct Gi/o function was monitored via cAMP inhibition to gain detailed signaling insight (Table 3). Chemokine receptors are primarily coupled to Gαi/o proteins, which act directly to inhibit cAMP production. After stimulation of CXCR4-expressing HEK-293T cells with forskolin, CXCR4 exhibited robust cAMP inhibition (Fig. 3B) with CXCL12-WT, demonstrating nanomolar potency (EC50 = 0.26 nm, Emax = 100%). Compared with CXCL12-WT, CXCL12-P10del was the most deficient, with a 100-fold decrease in potency (EC50 = 25 nm, Emax = 103%), and again CXCL12-P10Q was intermediate, with a 20-fold decrease in potency (EC50 = 4.90 nm, Emax = 90%).
Table 3.
Gi/o-mediated cAMP inhibition and β-arrestin-2 recruitment
Best fits were calculated with a variable slope from data performed in triplicate or quadruplicate from at least three independent experiments.
| Functional activity |
||||||
|---|---|---|---|---|---|---|
| CXCL12-WT |
CXCL12-P10Q |
CXCL12-P10del |
||||
| EC50 (pEC50 ± S.E.) | Emax | EC50 (pEC50 ± S.E.) | Emax | EC50 (pEC50 ± S.E.) | Emax | |
| nm | % CXCL12 | nm | % CXCL12 | nm | % CXCL12 | |
| CXCR4 | 0.03 (9.59 ± 0.20) | 100 | 4.90 (8.31 ± 0.18) | 90 ± 9 | 25.1 (7.60 ± 0.26) | 103 ± 16 |
| ACKR3 | 3.00 (8.52 ± 0.07) | 100 | 5.40 (8.27 ± 0.04) | 89 ± 2 | 38.1 (7.42 ± 0.09) | 80 ± 5 |
Receptor binding and activation at ACKR3
As CXC receptors appeared to be sensitive to mutation or deletion of the X position, we sought to test binding and function at another family of chemokine receptors. Atypical chemokine receptor 3 (ACKR3) is a scavenger receptor for CXCL12 and CXCL11 and is an example of a GPCR with inherent arrestin bias (26). Compared with CXCR4, CXCL12-WT displaced the radioligand bound to ACKR3 at a lower concentration (Ki = 0.09 nm) (Fig. 4A). Whereas mutation of the X residue moderately reduced affinity (Ki = 0.69 nm), deletion at this position reduced affinity over 450-fold (Ki = 42.7 nm).
Figure 4.

Effects of CXC mutants on another chemokine receptor, ACKR3. A, displacement of [125I]CXCL12-WT from ACKR3 was measured for each construct via scintillation after a 4- h equilibration with membrane suspensions at room temperature. Measurements were performed in duplicate from at least three independent experiments. B, recruitment of β-arrestin was measured via the Tango assay performed in quadruplicate from at least three independent experiments. C, summary of the X-factor impact in each signaling and binding experiment. CXCL12-P10del requires a significantly higher concentration to reach 50% effectiveness. CXCL12-P10Q is intermediate between WT and P10del.
ACKR3 activation was assessed by β-arrestin-2 recruitment measured using the Tango assay (Fig. 4B and Table 3) as ACKR3 has no known G protein activity. Signaling trended with radioligand binding, with CXCL12-WT being the most potent (EC50 = 3.0 nm), followed by CXCL12-P10Q (EC50 = 5.4 nm) and then CXCL12-P10del (EC50 = 38.1 nm). This trend with effective concentration was consistent among all assays (Fig. 4C).
Binding affinity to the CXCR4 N-terminal domain
To determine the source of the 100-fold reduction in CXCR4 binding affinity caused by X residue deletion, a peptide consisting of the first 38 residues of CXCR4 (P38) was titrated into each chemokine and observed by NMR HSQC (Fig. 5A). The chemokine-P38 interaction corresponds to site-1 in the two-site model and is thought to contribute mainly to chemokine binding (Fig. 5C). Throughout the 16-point titration, peak shifts were consistent between each chemokine, supporting a common binding mode. Nonlinear fitting of total chemical shift perturbations for each titration point yielded an apparent binding affinity of 2 μm for CXCL12-WT and CXCL12-P10Q (Fig. 5B). Several peaks broadened beyond detection in the CXCL12-P10del/P38 titration, but consistent shifts calculated an apparent affinity of 7 μm. To obtain more precise estimates of the P38 binding affinity, we used microscale thermophoresis (MST) to measure binding of each chemokine to P38 peptide that had been conjugated to a C-terminal Cy5 dye. Increasing concentrations of each CXCL12 variant were mixed with 25 nm P38-Cy5, and the resulting fluorescence after excitation indicated that no aggregation or adsorption occurred in CXCL12-WT (Fig. 5D). Similar traces were observed for CXCL12-P10Q and CXCL12-P10del. Dissociation coefficients were calculated for CXCL12-WT (2.0 ± 0.5 μm), CXCL12-P10Q (5.0 ± 1.0 μm), and CXCL12-P10del (0.9 ± 0.4 μm) and broadly support the low micromolar range calculated from the NMR titrations (Fig. 5E). However, the MST data suggest a slight increase in affinity for CXCL12-P10del compared with WT. Thus, rather than distorting the site-1 binding site on the CXCL12 surface, deletion of the X residue has no negative impact on binding to the CXCR4 N-terminal domain P38 peptide.
Figure 5.
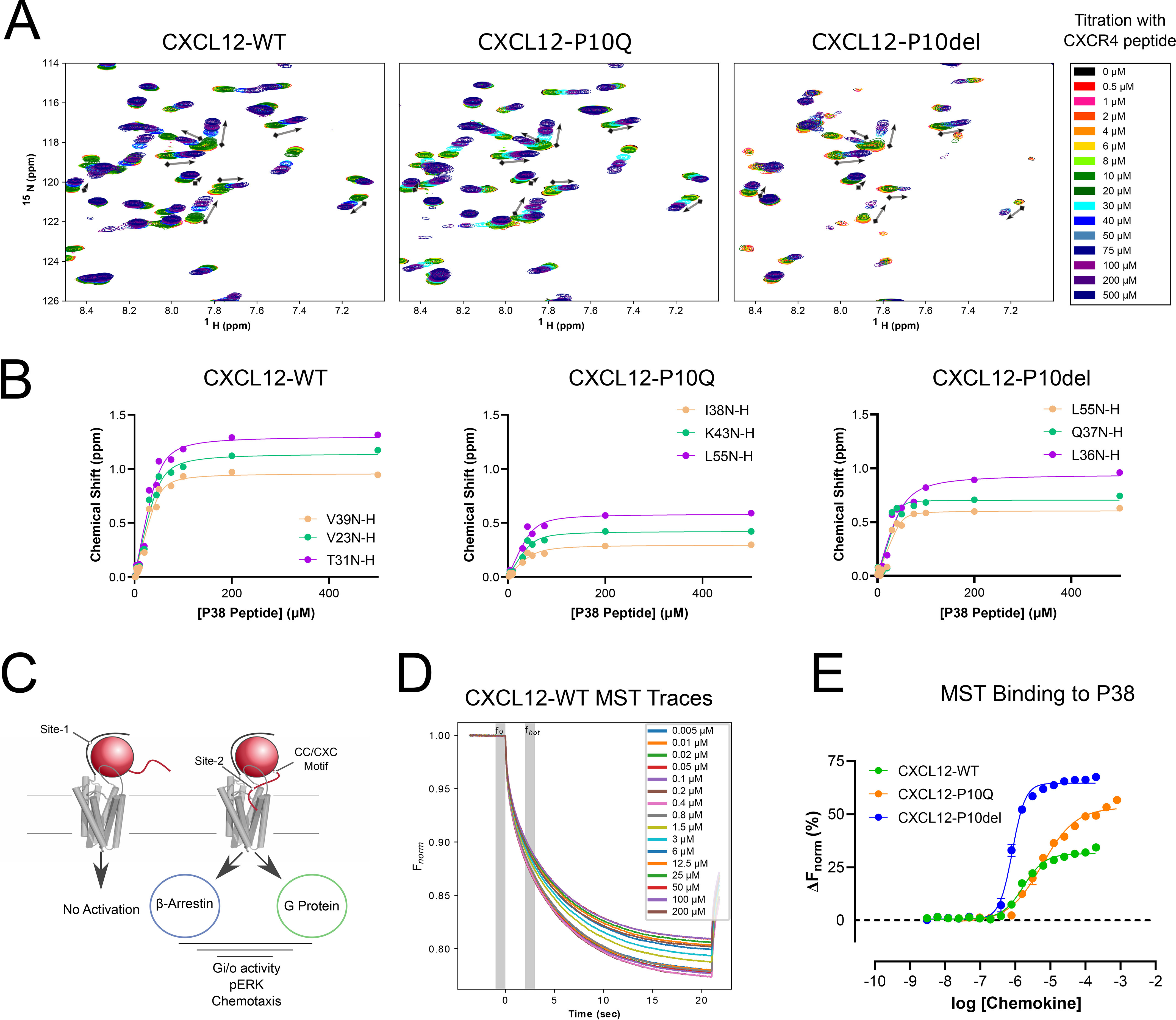
Receptor N-terminal binding is maintained through X-mutation and deletion. A, the NMR HSQC spectra over a 16-point titration series shows specific peak movement with the addition of a 38-residue CXCR4 peptide. Directionality of peak movement is consistent for each protein. B, example nonlinear fits for three residues in each titration series. Apparent affinity was calculated for CXCL12-WT (2 μm), CXCL12-P10Q (2 μm), and CXCL12-P10del (7 μm) from averaging the 15 largest shifting residues. C, a cartoon demonstrates the two-site model of chemokine interaction. The peptide used in the NMR titration series corresponds to site-1 binding. D, MST traces plot the resulting fluorescence (Fnorm) at each concentration (0.006–200 μm) of chemokine mixed with 25 nm P38-Cy5 normalized to the 1.0-s equilibration f0. Time region fhot corresponds to the 1.0-s data collection interval (1.5–2.5 s). Sample conditions were optimized to prevent aggregation, adsorption, photobleaching, and insufficient fluorescence (signal/noise > 20.0). E, dose-response curve plot calculated ΔFnorm (%), with error bars showing S.E. with n > 5 runs for each chemokine. Dissociation constants were calculated for CXCL12-WT (2.0 ± 0.5 μm), CXCL12-P10Q (5.0 ± 1.0 μm), and CXCL12-P10del (0.9 ± 0.4 μm).
Discussion
Human CXCL12, one of the best-characterized chemokines, is necessary for development and has ancient origins indicating that it may be the primordial chemokine (21, 22, 27, 28). A CXC-to-CC transition was simulated in this protein by deletion of the X residue to create CXCL12-P10del. We predicted that this mutation would significantly impair receptor recognition via a structural change in the chemokine.
Modeling and NMR data suggested that P10del was a structurally tolerated mutation, and thermal shift assay confirmed stability (Fig. 2F), suggesting that reduced receptor binding was not due to a change in the bulk tertiary structure. However, NMR HSQC peak position differed from WT with variable peak intensity, suggesting an increase in protein dynamics. These altered spectra were not seen in X deletion of CXCL8, possibly due to CXCL12 having a unique proline at the X position (29). Most NMR structures of CXC chemokines have the X position packed tightly with β1- and β2-strand residues to extend these secondary structures into the 30s loop, and X deletion likely disrupts this stability and leads to increased motion in this region and the adjacent N terminus.
MD simulations demonstrate that the CC/CXC motif may also constrain the preferred states of the N terminus in solution. This was most consistently seen in the residues proximal to the CC/CXC motif, particularly in the arginine immediately preceding the first cysteine in most CXC chemokines. In MD simulations of CXCL12, Arg-8 is reoriented upon deletion of the X residue, as shown in Fig. 2D. This residue has previously been identified as a potential determinant of subfamily selectivity, and disrupting its interaction with the receptor via reorientation of the N terminus may be deleterious to receptor binding (30). Comparing the solution-state NMR structures of chemokines supports this hypothesis, with the average CC N-terminal axis oriented 45° away from the average CXC axis. These preferred N-terminal ranges would limit CC chemokines ability to bind the orthosteric pocket and activate a CXC receptor.
CXCL12-P10del retains signaling ability through CXCR4, albeit reduced up to 100-fold in cAMP inhibition. With endogenous receptors, no ERK phosphorylation or chemotaxis was detected until 10 nm. At this concentration, CXCL12-WT reached a maximal effect. From an evolutionary perspective, this newly established CC chemokine would retain minimalistic function to preserve gene existence but be mostly free of selective pressure to undergo neofunctionalization (31). CXCL12-P10Q was designed to investigate the role of the identity of the X residue. In every aspect tested, this construct was intermediate between WT and P10del, suggesting that the familial difference in the CC/CXC motif is a larger factor in selectivity than X identity. However, the X identity change did reduce signaling ability and thus likely plays a role in discrimination between CXC chemokines.
At high concentrations, a peptide corresponding to the N terminus of CXCL12 can activate CXCR4 (32). Similar studies were the basis for the two-site model of chemokine-receptor interaction (Fig. 5C), which separates regions involved in binding (CKR N terminus) and receptor activation (chemokine N terminus). This model overly simplifies the protein-protein interaction yet remains useful as a broad framework to understanding CKR activation. Thus, it is unlikely that modifying the X residue would eliminate the activation ability of CXCL12 as the N terminus is intact. The more probable defect in these mutants is the ability to bind the receptor. This is supported by radioligand displacement as the affinity compared with CXCL12-WT is decreased by 10- or 100-fold for CXCL12-P10Q and CXCL12-P10del, respectively. Interestingly, ACKR3 binds CXCL12-P10del relatively worse than CXCR4, with a decreased affinity of over 450-fold. As this receptor encodes selectivity across two chemokines, it may be more reliant on the CXC motif to discriminate binding partners than the monogamous CXCL12-CXCR4 relationship.
To localize the defect in binding, a peptide consisting of the first 38 residues of CXCR4 was titrated and observed through NMR HSQC experiments. The affinities of CXCL12-WT and CXCL12-P10Q with P38 were comparable, whereas CXCL12-P10del showed a 3-fold loss of affinity. In the NMR structure of CXCL12 bound to this peptide, the X residue makes minimal contacts with P38. Thus, it was expected that CXCL12-P10Q would not disturb peptide-binding affinity. The MST experiments more precisely measured the binding affinity and confirmed the low micromolar range of binding. However, these experiments calculate CXCL12-P10del as binding tighter than WT to P38.
The relatively small change in affinity for CXCL12-P10del binding to P38 cannot account for the larger affinity loss when binding the intact receptor. This implicates the chemokine N terminus or 30s loop as the main contributor to reduced binding affinity, as these regions directly contact the CKR body. As the chemical matter of these regions is unchanged, a conformational explanation is required. The co-crystal structures of CXC and CC receptors have unique binding modes with differing chemokine tilts and binding depths (Fig. 6A). Particularly, the 30s loop of the CC chemokine penetrates into the orthosteric pocket of the receptor and may be facilitated by an increased chemokine tilt in this direction. Compared with the CXCR4-bound chemokine, this tilt is 36° and may act to counter the 45° difference in average N-terminal position in solution.
Figure 6.

Impact of the chemokine X-factor. A, the two determined co-crystal structures of CC and CXC receptors display unique chemokine binding orientations. The CC chemokine body interacts closely with the receptor core and includes penetration of the 30s loop into the orthosteric pocket. The CXCR4 co-crystal complex has vMIP-II above the extracellular vestibule with few interactions between the receptor and the 30s loop. B, a proposed model describing the potential influence of the X-factor between CC and CXC chemokines. The X residue directly interacts with the β1-β2 hairpin to stabilize this region in an extended conformation. The loss of the X position pulls the 30s loop into a narrow conformation that favors the increased CC binding depth. Additionally, the presence of the X position constrains the orientation of the chemokine N terminus in solution. The more linear N terminus found in CXC structures matches with the chemokine position above the receptor pocket in the co-crystal structure.
On the basis of these data and previous observations of N-terminal orientation and 30s loop dynamics (7, 13), we propose a model of chemokine familial selectivity based on the X-factor in the CC/CXC motif (Fig. 6B). In this model, the X-factor is best described as a fulcrum that orients the N terminus into preferential binding positions, predominantly the proximal residues, including the arginine in the ELR motif found in most CXC chemokines. Chemokines with improperly aligned N termini have reduced binding affinity, as this region would be unable to establish ideal contacts with the receptor and may be unable to reach the proper orientation in the pocket to achieve efficient receptor activation. Additionally, the X residue is a stabilizing factor in CXC chemokines and adds rigidity by extending the β1- and β2-strands through direct interactions. This sterically applies a pushing force on the 30s loop and results in a wide profile. In CC chemokines that lack this extended secondary structure and the steric burden of the X residue, the 30s loop is pulled into a narrow position. This would facilitate the unique binding mode found in CC chemokines while sterically inhibiting the 30s loop of CXC chemokines from penetrating the orthosteric pocket.
The role of the CC/CXC motif in receptor binding and activation has now been investigated in CXCL8 and CXCL12; however, knowledge of this motif in CC chemokines has not been adequately pursued. Based on the results of this study, it is plausible that insertion of an intervening X residue in a CC chemokine would substantially reduce binding and activation to CC receptors. Furthermore, addition, deletion, or modification of the X residue could be a step toward engineered chemokines with novel receptor-binding profiles. Chemokines interact with their receptors via two divergent, unstructured N-terminal domains (i.e. site-1 and site-2), suggesting that family-wide specificity is encoded in the structural scaffold. The CC/CXC motif couples the chemokine body and N terminus and plays an important role in determining binding affinity. We speculate that the orientation of the chemokine N terminus is influenced by the presence or absence of the X residue and is a defining factor for familial selectivity.
Experimental procedures
Chemokine purification
CXCL12-WT, -P10Q, and -P10del were expressed and purified as described previously (33). Briefly, chemokines (construct 6xHIS-SUMO-chemokine) were expressed recombinantly in Escherichia coli at 37 °C for 5 h before harvest by centrifugation. Pellets were resuspended and lysed by French press at 4 °C and centrifuged. The remaining insoluble pellet was resuspended with 6 m guanidine and partially purified by nickel column. Elution fractions were refolded by infinite dilution (100 mm Tris, 10 mm cysteine, 0.5 mm cystine, pH 8) with agitation overnight. After concentration, the fusion protein was cleaved by the addition of the protease ULP1 for at least 4 h. Additional purification was accomplished by cation-exchange chromatography and reverse-phase HPLC. Final elution fractions were pooled and lyophilized, and identity was confirmed by MS.
Sequence alignments
Sequences for the 46 human chemokines were taken from the UniProt database with the signal sequences removed. These sequences were visualized in Jalview and sent for alignment via MUSCLE with default settings. Jalview was used to separate CXC and CC chemokines and produce figures with continuous sequences aligned by conserved cysteines.
Structural analysis
NMR solution-state structures were taken from the Protein Data Bank for each available chemokine (CCL1, 1EL0; CCL3, 1B50; CCL4, 2FIN; CCL5, 5YAM; CCL5, 6FGP; CCL7, 4ZKC; CCL11, 2MPM; CCL15, 2HCC; CCL19, 2MP1; CCL20, 2JYO; CCL21, 2L4N; CCL23, 1G91; CCL24, 1EIH; CCL26, 1G2T; CCL27, 2KUM; CCL28, 6CWS; CX3CL1, 1B2T; XCL1, 1J8I; CXCL1, 1MGS; CXCL2, 1QNK; CXCL5, 2MGS; CXCL8, 1ILP; CXCL8, 5WDZ; CXCL10, 1LV9; CXCL11, 1RJT; CXCL12, 2KED; CXCL12, 2N55; CXCL13, 5L7M; CXCL14, 2HDL). Conformer structures were separated, and all structures were aligned in PyMOL by the conserved three-stranded β-sheet. All but the N termini were removed to determine the N-terminal axis in UCSF Chimera. Resulting vectors were averaged by family to construct a mean axis for CC, CXC, XC, and CX3C chemokines. Figures overlaying co-crystal complexes were generated in UCSF Chimera. PyMOL was used to determine residues contacting the X position with a cutoff of 4 Å. Positions were separated based on contact with the main chain or side chain of the X residue. The interdisulfide distance was measured in PyMOL between the second and third cysteines.
Molecular dynamics simulation
A model of CXCL12-P10del was constructed using RosettaCM as described previously (34, 35). The input template used was CXCL12-WT (PDB entry 2KED). After relaxation, the final models were compared with the NMR structure via Cα deviation using PyMOL. MD simulations were performed following previous protocols (36). Briefly, CXCL12-WT and CXCL12-P10del were simulated for 100 ns in Gromacs 2018 using the OPLS-AA/L force field and the SPC/E water model (n = 5). Simulations were overlaid with the CXCR4 co-crystal structure (PDB entry 4RWS), and differential positions for arginine 8 were illustrated in PyMOL.
Chemotaxis transwells
Chemotaxis assays were performed as described previously (37). THP1 cells were washed twice with RPMI 1640 with HEPES and 0.2% BSA. The lower chamber of Corning HTS transwell plates was loaded with varying chemokine dilutions. The upper chamber was seeded with 75,000 cells. Plates were incubated at 37 °C at 5% CO2 for 2 h. The migrated cells in the lower well were quantified via flow cytometry.
Western blotting analysis
HEK-293 cells (Microbix, Toronto, Canada) were cultured in 6-well plates and transfected with 5 μg of FLAG-CXCR4 48 h before the assay, following transfection protocols as described previously (38). Cells were serum-starved in Dulbecco's modified Eagle's medium with 20 mm HEPES for 5 h and then stimulated for 5 min with 10 nm chemokine and immediately harvested for Western blotting analysis for pERK-1/2 (Sigma, catalog no. M8159) and total ERK-1/2 (Sigma, catalog no. M5670). Blots were subjected to densitometric analysis, and pERK-1/2 levels were normalized to total ERK-1/2 and quantified relative to vehicle.
NMR spectroscopy
For NMR studies, recombinant chemokines were grown in M9 minimal medium supplemented with 15NH4Cl and purified as described previously (39). Samples were prepared in a 3-mm tube at 50 μm chemokine concentration in 25 mm MES, pH 7.6, containing 10% D2O and 0.02% sodium azide. NMR HSQC experiments were carried out on a 600-MHz Bruker spectrometer at 25 °C. Titration series added buffer-matched peptide (CXCR4 residues 1–38, C28A) at specified concentrations.
Radioligand binding
HEK-293T cells were transfected with CXCR4 or ACKR3 for membrane purification. After 48 h, cells were resuspended in hypotonic lysis buffer (1 mm HEPES, 2 mm EDTA, pH 7.4) on ice and centrifuged at 30,000 × g for 30 min. The membrane pellet was resuspended in binding buffer (20 mm HEPES, 10 mm MgCl2, 0.1 mm EDTA, pH 7.4) and homogenized via Polytron. 1-ml membrane aliquots were frozen at −80 °C. For the competition binding assay 15–50 pm [125I]CXCL12 (PerkinElmer Life Sciences, specific activity = 2200 Ci/mmol) was incubated with diluted membrane and chemokine for 4 h at room temperature with 1 mm CaCl2 0.1% BSA, and 100 mm NaCl added to the binding buffer. Membranes were harvested via vacuum filtration onto a 96-well filtermat-A presoaked in 0.3% polyethyleneimine and washed three times with binding buffer. After drying, MeltiLex was applied to each filter, and radioactivity was assessed by luminescence via a MicroBeta plate counter. Results were analyzed in GraphPad Prism 8. Saturation experiments were performed similarly to those above, with the radioligand titrated from 4 to 130 pm in the presence or absence of 320 nm unlabeled CXCL12-WT. Protein concentrations of membrane preparations were measured via Bradford assay.
GloSensor Gi/o-mediated cAMP inhibition assay
HEK-293T cells were transfected 48 h before assay with CXCR4 and cAMP GloSensor-22F (Promega) plasmids overnight. Cells were transferred to a poly-l-lysine–coated 384-well white clear-bottom plate 24 h before the assay. The assay was started upon the addition of chemokine and GloSensor reagent in assay buffer (HBSS, 20 mm HEPES, 0.1% BSA, pH 7.4) using a FLIPR. After 15 min at room temperature, cAMP accumulation was initiated by forskolin (1 μm). After an additional 15 min, luminescence was recorded via a MicroBeta plate counter. Results were analyzed in GraphPad Prism 8.
Tango β-arrestin recruitment assay
Tango assays were performed as described previously with minor modifications (40). Briefly, HEK-293 HTLA cells were transfected with ACKR3 48 h before assay using the polyethyleneimine transfection method. Cells were transferred to a poly-l-lysine–coated 384-well white clear-bottom plate 24 h before the assay. Cells were stimulated with varying concentrations of chemokine prepared in drug buffer (HBSS, 20 mm HEPES, 0.1% BSA, pH 7.4) using a FLIPR (Molecular Devices). Assay plates were incubated overnight at room temperature and monitored using a MicroBeta (PerkinElmer Life Sciences) luminescence counter. Results were analyzed in GraphPad Prism 8.
Microscale thermophoresis
MST assays were performed using a Monolith® NT.115 (NanoTemper) system. Binding affinity was evaluated between CXCL12-WT, -P10Q, and -P10del chemokines and C-terminally Cy5-labeled P38 CXCR4 peptide. The P38-Cy5 peptide was purified by reverse-phase HPLC with identity confirmed by linear trap quadrupole MS. For MST measurement, CXCL12-WT, -P10Q, and -P10del were dialyzed with P38-Cy5 into assay buffer (20 mm MES, 0.05% Tween 20, pH 6.5). Optimization for sufficient fluorescent signal and checking for sample aggregation or capillary adsorption was performed. Monolith® NT.115 premium capillaries were loaded from low-binding 200-μl fast reaction tubes containing chemokine at a maximum concentration of 200 μm for CXCL12-WT and -P10del or 800 μm for CXCL12-P10Q and with the instrument set with an LED excitation power of 20% and using MST power at 40%. Resulting dose response (ΔFnorm) obtained from normalized fluorescence was analyzed by least-squares curve fit according to a 1:1 binding model (Nanotemper) to calculate Kd values. Figures were created using GraphPad Prism 8.
Differential scanning fluorimetry
Thermal protein denaturation was monitored via differential scanning fluorimetry in a Prometheus NT.48 (Nanotemper) using the intrinsic fluorescence of native Tryptophan residues. Each chemokine was prepared at 0.5 mg/ml in assay buffer (20mM MES, pH 6.5) and loaded into capillary tubes. Thermal unfolding was initiated by a linear thermal ramp from 20°C–95°C (1°C/minute) with an excitation power of 50%. Unfolding transition points were determined from wavelengths of Tryptophan fluorescence at 330 and 350 nm, and the first derivative of the 330/350 ratio was plotted as a function of temperature.
Data availability
The data supporting the findings of this study are contained within the article. The data sets generated during this study are available from the corresponding author (Brian F. Volkman, bvolkman@mcw.edu) upon request.
Acknowledgments
This research was completed in part with computational resources and technical support provided by the Research Computing Center at the Medical College of Wisconsin.
Author contributions—M. J. W. conceptualization; M. J. W., S. A. M., A. E. G., K. S. C., F. C. P., J. D. M., and B. F. V. data curation; M. J. W., S. A. M., A. E. G., K. S. C., F. C. P., J. D. M., and B. F. V. formal analysis; M. J. W., A. M., J. D. M., and B. F. V. validation; M. J. W., A. M., J. D. M., and B. F. V. investigation; M. J. W., K. S. C., and B. F. V. visualization; M. J. W., S. A. M., A. E. G., K. S. C., F. C. P., A. M., J. D. M., and B. F. V. methodology; M. J. W. writing-original draft; M. J. W., J. D. M., and B. F. V. project administration; M. J. W., S. A. M., A. E. G., K. S. C., F. C. P., A. M., J. D. M., and B. F. V. writing-review and editing; S. A. M., A. E. G., F. C. P., A. M., and J. D. M. supervision; A. E. G., A. M., J. D. M., and B. F. V. funding acquisition; F. C. P., J. D. M., and B. F. V. resources; J. D. M. software.
Funding and additional information—This work was supported by National Institutes of Health Grants R01 AI058072 (to B. F. V.), GM106727 (to A. M.), and R35 GM133421 (to J. D. M.) and Shared Instrumentation Grant S10 OD020000 and a Early Career Research Grant from the National Psoriasis Foundation (to A. E. G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—B. F. V. and F. C. P. have ownership interests in Protein Foundry, LLC.
- GPCR
- G protein–coupled receptor
- HSQC
- heteronuclear single quantum coherence
- CKR
- chemokine receptor
- PDB
- Protein Data Bank
- ERK
- extracellular signal–regulated kinase
- MST
- microscale thermophoresis
- MD
- molecular dynamics
- HBSS
- Hanks' balanced salt solution.
References
- 1. Thomas M. A., Kleist A. B., and Volkman B. F. (2018) Decoding the chemotactic signal. J. Leukoc. Biol. 104, 359–374 10.1002/JLB.1MR0218-044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bachelerie F., Ben-Baruch A., Burkhardt A. M., Combadiere C., Farber J. M., Graham G. J., Horuk R., Sparre-Ulrich A. H., Locati M., Luster A. D., Mantovani A., Matsushima K., Murphy P. M., Nibbs R., Nomiyama H., et al. (2014) International Union of Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 66, 1–79 10.1124/pr.113.007724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander S. P. H., Mathie A., and Peters J. A. (2011) Guide to receptors and channels (GRAC), 5th Ed. Br. J. Pharmacol. 164, S1–S2 10.1111/j.1476-5381.2011.01649_1.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stone M. J., Hayward J. A., Huang C., Huma Z. E., and Sanchez J. (2017) Mechanisms of regulation of the chemokine-receptor network. Int. J. Mol. Sci. 18, 342 10.3390/ijms18020342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mantovani A. (1999) The chemokine system: redundancy for robust outputs. Immunol. Today 20, 254–257 10.1016/s0167-5699(99)01469-3 [DOI] [PubMed] [Google Scholar]
- 6. Smith J. S., Nicholson L. T., Suwanpradid J., Glenn R. A., Knape N. M., Alagesan P., Gundry J. N., Wehrman T. S., Atwater A. R., Gunn M. D., MacLeod A. S., and Rajagopal S. (2018) Biased agonists of the chemokine receptor CXCR3 differentially control chemotaxis and inflammation. Sci. Signal. 11, eaaq1075 10.1126/scisignal.aaq1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zlotnik A., and Yoshie O. (2012) The chemokine superfamily revisited. Immunity 36, 705–716 10.1016/j.immuni.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Proudfoot A. E. I. (2002) Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2, 106–115 10.1038/nri722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arimont M., Hoffmann C., de Graaf C., and Leurs R. (2019) Chemokine receptor crystal structures: what can be learned from them? Mol. Pharmacol. 96, 765–777 10.1124/mol.119.117168 [DOI] [PubMed] [Google Scholar]
- 10. Miller M. C., and Mayo K. H. (2017) Chemokines from a structural perspective. Int. J. Mol. Sci. 18, 2088 10.3390/ijms18102088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kufareva I., Gustavsson M., Zheng Y., Stephens B. S., and Handel T. M. (2017) What do structures tell us about chemokine receptor function and antagonism?. Annu. Rev. Biophys. 46, 175–198 10.1146/annurev-biophys-051013-022942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajagopalan L., and Rajarathnam K. (2006) Structural basis of chemokine receptor function: a model for binding affinity and ligand selectivity. Biosci. Rep. 26, 325–339 10.1007/s10540-006-9025-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qin L., Kufareva I., Holden L. G., Wang C., Zheng Y., Zhao C., Fenalti G., Wu H., Han G. W., Cherezov V., Abagyan R., Stevens R. C., and Handel T. M. (2015) Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 10.1126/science.1261064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burg J. S., Ingram J. R., Venkatakrishnan A. J., Jude K. M., Dukkipati A., Feinberg E. N., Angelini A., Waghray D., Dror R. O., Ploegh H. L., and Garcia K. C. (2015) Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 10.1126/science.aaa5026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng Y., Han G. W., Abagyan R., Wu B., Stevens R. C., Cherezov V., Kufareva I., and Handel T. M. (2017) Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46, 1005–1017.e5 10.1016/j.immuni.2017.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleist A. B., Getschman A. E., Ziarek J. J., Nevins A. M., Gauthier P. A., Chevigné A., Szpakowska M., and Volkman B. F. (2016) New paradigms in chemokine receptor signal transduction: moving beyond the two-site model. Biochem. Pharmacol. 114, 53–68 10.1016/j.bcp.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rajarathnam K., Sykes B. D., Dewald B., Baggiolini M., and Clark-Lewis I. (1999) Disulfide bridges in interleukin-8 probed using non-natural disulfide analogues: dissociation of roles in structure from function. Biochemistry 38, 7653–7658 10.1021/bi990033v [DOI] [PubMed] [Google Scholar]
- 18. Graves D. T., and Jiang Y. (1995) Chemokines, a family of chemotactic cytokines. Crit. Rev. Oral. Biol. Med. 6, 109–118 10.1177/10454411950060020101 [DOI] [PubMed] [Google Scholar]
- 19. Zlotnik A., and Yoshie O. (2000) Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 10.1016/S1074-7613(00)80165-X [DOI] [PubMed] [Google Scholar]
- 20. Nomiyama H., Osada N., and Yoshie O. (2010) The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 21, 253–262 10.1016/j.cytogfr.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 21. DeVries M. E., Kelvin A. A., Xu L., Ran L., Robinson J., and Kelvin D. J. (2006) Defining the origins and evolution of the chemokine/chemokine receptor system. J. Immunol. 176, 401–415 10.4049/jimmunol.176.1.401 [DOI] [PubMed] [Google Scholar]
- 22. Najakshin A. M., Mechetina L. V., Alabyev B. Y., and Taranin A. V. (1999) Identification of an IL-8 homolog in lamprey (Lampetra fluviatilis): early evolutionary divergence of chemokines. Eur. J. Immunol. 29, 375–382 [DOI] [PubMed] [Google Scholar]
- 23. Baryshnikova O. K., and Sykes B. D. (2006) Backbone dynamics of SDF-1α determined by NMR: interpretation in the presence of monomer-dimer equilibrium. Protein Sci. 15, 2568–2578 10.1110/ps.062255806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ziarek J. J., Kleist A. B., London N., Raveh B., Montpas N., Bonneterre J., St-Onge G., DiCosmo-Ponticello C. J., Koplinski C. A., Roy I., Stephens B., Thelen S., Veldkamp C. T., Coffman F. D., Cohen M. C., et al. (2017) Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation. Sci. Signal. 10, eaah5756 10.1126/scisignal.aah5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ngo T., Stephens B. S., Gustavsson M., Holden L. G., Abagyan R., Handel T. M., and Kufareva I. (2020) Crosslinking-guided geometry of a complete CXC receptor-chemokine complex and the basis of chemokine subfamily selectivity. PLoS Biol. 18, e3000656 10.1371/journal.pbio.3000656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajagopal S., Kim J., Ahn S., Craig S., Lam C. M., Gerard N. P., Gerard C., and Lefkowitz R. J. (2010) β-Arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. U. S. A. 107, 628–632 10.1073/pnas.0912852107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Doitsidou M., Reichman-Fried M., Stebler J., Köprunner M., Dörries J., Meyer D., Esguerra C. V., Leung T. C., and Raz E. (2002) Guidance of primordial germ cell migration by the chemokine SDF-1. Cell 111, 647–659 10.1016/S0092-8674(02)01135-2 [DOI] [PubMed] [Google Scholar]
- 28. Ma Q., Jones D., Borghesani P. R., Segal R. A., Nagasawa T., Kishimoto T., Bronson R. T., and Springer T. A. (1998) Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. U. S. A. 95, 9448–9453 10.1073/pnas.95.16.9448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joseph P. R. B., Sarmiento J. M., Mishra A. K., Das S. T., Garofalo R. P., Navarro J., and Rajarathnam K. (2010) Probing the role of CXC motif in chemokine CXCL8 for high affinity binding and activation of CXCR1 and CXCR2 receptors. J. Biol. Chem. 285, 29262–29269 10.1074/jbc.M110.146555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stephens B. S., Ngo T., Kufareva I., and Handel T. M. (2020) Functional anatomy of the full-length CXCR4-CXCL12 complex systematically dissected by quantitative model-guided mutagenesis. Sci. Signal. 13, eaay5024 10.1126/scisignal.aay5024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gangele K., Jamsandekar M., Mishra A., and Poluri K. M. (2019) Unraveling the evolutionary origin of ELR motif using fish CXC chemokine CXCL8. Fish Shellfish Immunol. 93, 17–27 10.1016/j.fsi.2019.07.034 [DOI] [PubMed] [Google Scholar]
- 32. Filippo T. R. M., Galindo L. T., Barnabe G. F., Ariza C. B., Mello L. E., Juliano M. A., Juliano L., and Porcionatto M. A. (2013) CXCL12 N-terminal end is sufficient to induce chemotaxis and proliferation of neural stem/progenitor cells. Stem Cell Res. 11, 913–925 10.1016/j.scr.2013.06.003 [DOI] [PubMed] [Google Scholar]
- 33. Takekoshi T., Ziarek J. J., Volkman B. F., and Hwang S. T. (2012) A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol. Cancer Ther. 11, 2516–2525 10.1158/1535-7163.MCT-12-0494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song Y., Dimaio F., Wang R. Y. R., Kim D., Miles C., Brunette T., Thompson J., and Baker D. (2013) High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 10.1016/j.str.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wedemeyer M. J., Mueller B. K., Bender B. J., Meiler J., and Volkman B. F. (2019) Modeling the complete chemokine–receptor interaction. Methods Cell Biol. 149, 289–314 10.1016/bs.mcb.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lemkul J. (2019) From proteins to perturbed Hamiltonians: a suite of tutorials for the GROMACS-2018 molecular simulation package. Living J. Comput. Mol. Sci. 1, 5068 10.33011/livecoms.1.1.5068 [DOI] [Google Scholar]
- 37. Getschman A. E., Imai Y., Larsen O., Peterson F. C., Wu X., Rosenkilde M. M., Hwang S. T., and Volkman B. F. (2017) Protein engineering of the chemokine CCL20 prevents psoriasiform dermatitis in an IL-23–dependent murine model. Proc. Natl. Acad. Sci. U. S. A. 114, 12460–12465 10.1073/pnas.1704958114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alekhina O., and Marchese A. (2016) β-Arrestin1 and signal-transducing adaptor molecule 1 (STAM1) cooperate to promote focal adhesion kinase autophosphorylation and chemotaxis via the chemokine receptor CXCR4. J. Biol. Chem. 291, 26083–26097 10.1074/jbc.M116.757138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Veldkamp C. T., Peterson F. C., Pelzek A. J., and Volkman B. F. (2005) The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 14, 1071–1081 10.1110/ps.041219505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kroeze W. K., Sassano M. F., Huang X. P., Lansu K., McCorvy J. D., Giguère P. M., Sciaky N., and Roth B. L. (2015) PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 10.1038/nsmb.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data supporting the findings of this study are contained within the article. The data sets generated during this study are available from the corresponding author (Brian F. Volkman, bvolkman@mcw.edu) upon request.