

Abstract
Transforming growth factor β (TGFβ) signaling plays an important role in regulating tumor malignancy, including in non–small cell lung cancer (NSCLC). The major biological responses of TGFβ signaling are determined by the effector proteins SMAD2 and SMAD3. However, the regulators of TGFβ–SMAD signaling are not completely revealed yet. Here, we showed that the scaffolding protein PDLIM5 (PDZ and LIM domain protein 5, ENH) critically promotes TGFβ signaling by maintaining SMAD3 stability in NSCLC. First, PDLIM5 was highly expressed in NSCLC compared with that in adjacent normal tissues, and high PDLIM5 expression was associated with poor outcome. Knockdown of PDLIM5 in NSCLC cells decreased migration and invasion in vitro and lung metastasis in vivo. In addition, TGFβ signaling and TGFβ-induced epithelial–mesenchymal transition was repressed by PDLIM5 knockdown. Mechanistically, PDLIM5 knockdown resulted in a reduction of SMAD3 protein levels. Overexpression of SMAD3 reversed the TGFβ-signaling-repressing and anti-migration effects induced by PDLIM5 knockdown. Notably, PDLIM5 interacted with SMAD3 but not SMAD2 and competitively suppressed the interaction between SMAD3 and its E3 ubiquitin ligase STUB1. Therefore, PDLIM5 protected SMAD3 from STUB1-mediated proteasome degradation. STUB1 knockdown restored SMAD3 protein levels, cell migration, and invasion in PDLIM5-knockdown cells. Collectively, our findings indicate that PDLIM5 is a novel regulator of basal SMAD3 stability, with implications for controlling TGFβ signaling and NSCLC progression.
Keywords: lung cancer, proteasome degradation, ENH, STUB1, E3 ubiquitin ligase, transforming growth factor beta (TGF-B), lung cancer, cell invasion, cell motility, invasion, protein degradation, signal transduction, tumor metastasis, ubiquitin
Transforming growth factor β (TGFβ) signaling plays essential and multifaceted roles in tumor malignancy (1). At the initial stage of tumor formation, TGFβ acts as a tumor suppressor by inhibiting cell proliferation; however, at the later stage, TGFβ plays prometastatic roles (2, 3). The prometastatic effects of TGFβ signaling include accelerated tumor infiltration, cell adhesion, angiogenesis, cell epithelial–mesenchymal transition (EMT), and extracellular matrix degradation.
In previous decades, lung cancer constituted the leading cause of cancer mortality worldwide (4, 5). Clinical data indicate that non–small cell lung cancer (NSCLC) accounts for most lung cancer cases. Nevertheless, although numerous therapeutic approaches are available for treating NSCLC, its prognosis remains poor (6). As with most cancers, metastasis represents the dominant reason of NSCLC-related death (7). According to clinical studies, TGFβ signaling is frequently enhanced in NSCLC, promoting EMT and tumor metastasis (8, 9). SMAD2 and SMAD3 are two key downstream regulators of TGFβ signaling and play critical roles in TGFβ-mediated biologic functions (10). SMAD3 inhibition diminishes TGFβ-induced EMT in cancer cells (8). In addition, the overexpression of SMAD3 accelerates TGFβ-mediated metastasis in NSCLC cells (11). Notably, the post-translational modification of SMAD3 protein is crucial for regulation of TGFβ signaling. The E3 ligase DEAR has been reported to interact with SMAD3, thus resulting in SMAD3 polyubiquitination and degradation (12). Moreover, phosphorylation at Thr66 in SMAD3, mediated by GSK3β, primes the protein for phosphorylation-directed ubiquitination (13). Hence, it is important to fully understand SMAD3 regulatory mechanisms and how their regulation influences the TGFβ signaling pathway.
The scaffolding protein PDLIM5 (also termed ENH), a member of the PDZ–LIM domain protein family, was initially identified as a PKC kinase–binding protein (14). PDLIM5 contains one PDZ and three LIM domains that mediate protein–protein interactions (15). In addition to PKC, PDLIM5 binds multiple protein kinases, including PKD, AMPK, and PKA, making it a versatile scaffolding protein in signaling transduction pathways (16, 17). PDLIM5 itself is a substrate of AMPK; its phosphorylation represses the migration of vascular smooth muscle cells (18). Furthermore, PDLIM5 functions as a cytoplasmic sequestration protein for transcription factors ID2 and YAP (19, 20). PDLIM5 is up-regulated in various cancers (21, 22). It has been suggested that PDLIM5 promotes EMT and migration of prostate cancer cells (23). In addition, PDLIM5 regulates TGFβ3 signaling in vascular smooth muscle cells and hypoxia-induced pulmonary hypertension in mice (24). Moreover, silencing of PDLIM5 suppresses TGFβ signaling and SMAD3 expression in alveolar epithelial cells (25). However, the role of PDLIM5 in NSCLC and in regulation TGFβ signaling remains largely unknown.
In the current study, we found that PDLIM5 was up-regulated in NSCLC tissues. PDLIM5 knockdown in NSCLC cells inhibited cellular migration and invasion and TGFβ signaling. PDLIM5 interacted with SMAD3, and PDLIM5 knockdown enhanced the interaction between SMAD3 and its E3 ligase STUB1, resulting in SMAD3 degradation. Together, these results demonstrated that PDLIM5 is a novel SMAD3 regulator linking to SMAD3-mediated TGFβ signaling and lung cancer progression.
Results
PDLIM5 is up-regulated in NSCLC and associated with poor outcome
To evaluate the importance of PDLIM5 in NSCLC, PDLIM5 expression was analyzed using data sets available in the Oncomine database (RRID:SCR_007834). PDLIM5 was highly expressed in three different cohorts of NSCLC patients (Fig. 1, A–C). We confirmed the up-regulation of PDLIM5 in NSCLC samples by Western blotting analysis (Fig. 1D). Moreover, PDLIM5 were significantly increased in NSCLC tissues compared with paired adjacent normal lung tissues as evaluated by immunohistochemistry assay in 40 NSCLC subjects (Fig. 1, E and F). Finally, survival analysis was performed to estimate the association between PDLIM5 expression and lung cancer patient survival using the Kaplan–Meier plotter online bioinformatics resource (RRID:SCR_018753), which revealed that elevated PDLIM5 expression in lung cancer patients was correlated with poor prognosis (Fig. 1G). Thus, PDLIM5 is up-regulated in lung cancer and associated with poor outcome.
Figure 1.
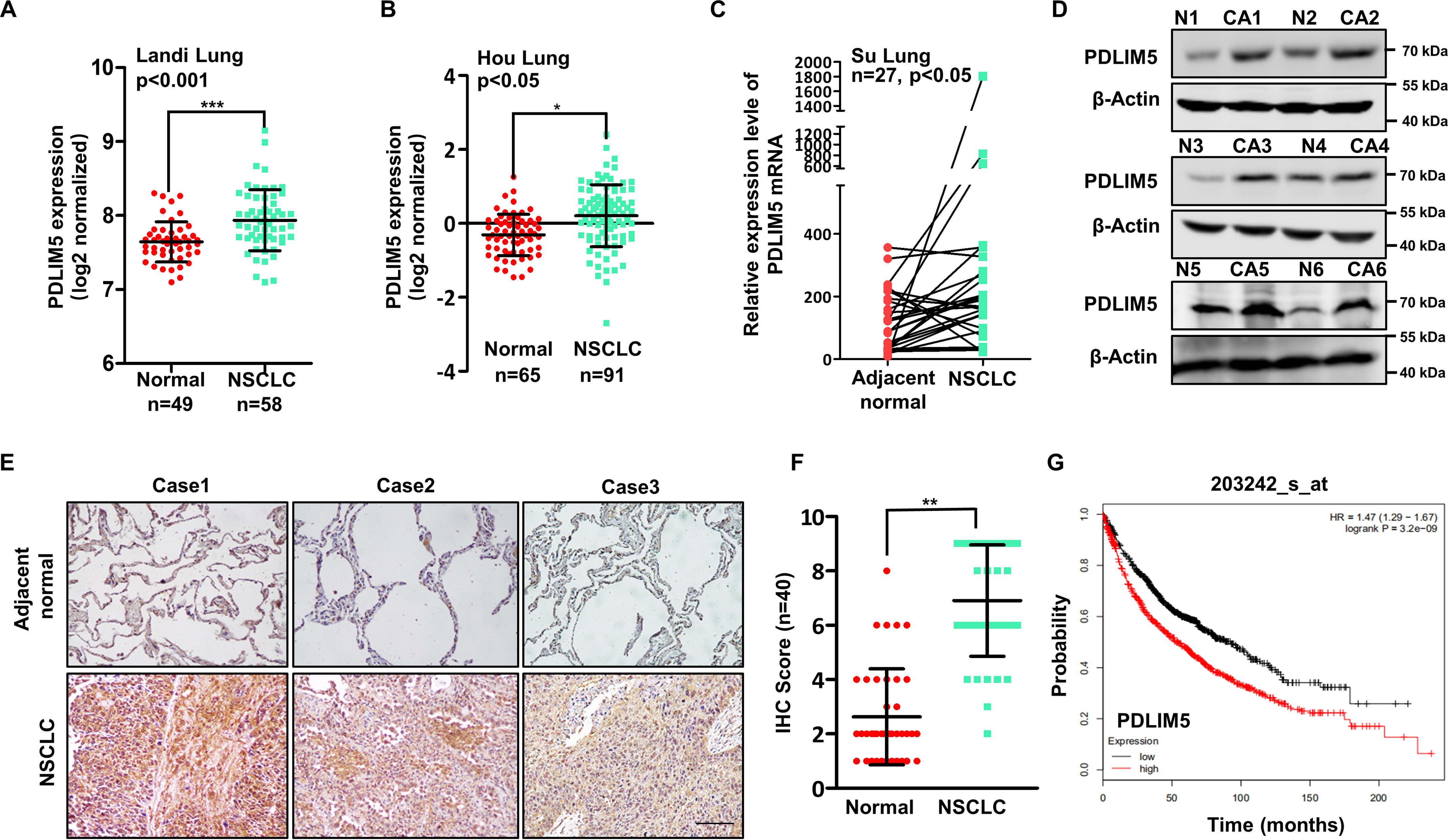
PDLIM5 is up-regulated in NSCLC and associated with poor outcome. A and B, expression of PDLIM5 in lung cancer tissues compared with normal tissues from the Oncomine database. The data are shown as log2 median-centered intensity. C, expression of PDLIM5 in 27 NSCLC tissues compared with paired adjacent normal tissues from the Oncomine database. D, Western blotting analysis of PDLIM5 levels in six cases of NSCLC tissues (CA) and paired adjacent normal tissues (N). β-Actin was used as a loading control. E and F, immunohistochemical analysis of PDLIM5 levels in 40 cases of NSCLC tissues and paired adjacent normal tissues (E). Scale bar, 50 μm. Quantitative analysis was shown in F. Analysis was performed using Wilcoxon matched-pairs signed-rank test. G, overall survival rate analysis of 1926 patients with lung cancer according to the PDLIM5 levels. The analysis was performed by using the Kaplan–Meier Plotter database data. The data are shown as the means ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001. IHC, immunohistochemistry.
PDLIM5 knockdown impairs migration and invasion of lung cancer cells
Next, we evaluated the expression of PDLIM5 in various NSCLC cell lines. PDLIM5 was abundant in A549, H1975, and MSTO cells but not in PC9 cells (Fig. 2A). A549 and H1975 cells were selected for further loss-of-function studies. PDLIM5 was reduced by PDLIM5 knockdown in A549 and H1975 cells using viral-mediated RNA inference (shPDLIM5) (Fig. 2B). Among the three different targeting sequences, shPDLIM5-1 and shPDLIM5-3 resulted in high knockdown efficiency (Fig. 2, B and C). We next performed wound-healing and Transwell migration assays to investigate the function of PDLIM5. PDLIM5 knockdown significantly inhibited the wound-healing capacity (Fig. 2, D and E) and cellular migration (Fig. 2, F and G) of A549 and H1975 cells. Cell invasion was also greatly halted in PDLIM5-knockdown lung cancer cells (Fig. 2, H and I). In addition, PDLIM5 was overexpressed in PC9 cells. PDLIM5 overexpression increased wound-healing capacity, cellular migration, and invasion (Fig. S1, A–D). To explore the role of PDLIM5 in lung metastasis in vivo, we intravenously injected Pdlim5-knockdown Lewis lung carcinoma (LLC) cells (Fig. 2J) into BALB/c nude mice via tail vein. Lung metastatic nodules were significantly reduced in mice injected with Pdlim5-knockdown cells compared with those in mice receiving control LLC cells (Fig. 2, K and L). Together, these observations suggest that PDLIM5 promotes cell migration, invasion, and lung metastasis of lung cancer cells.
Figure 2.
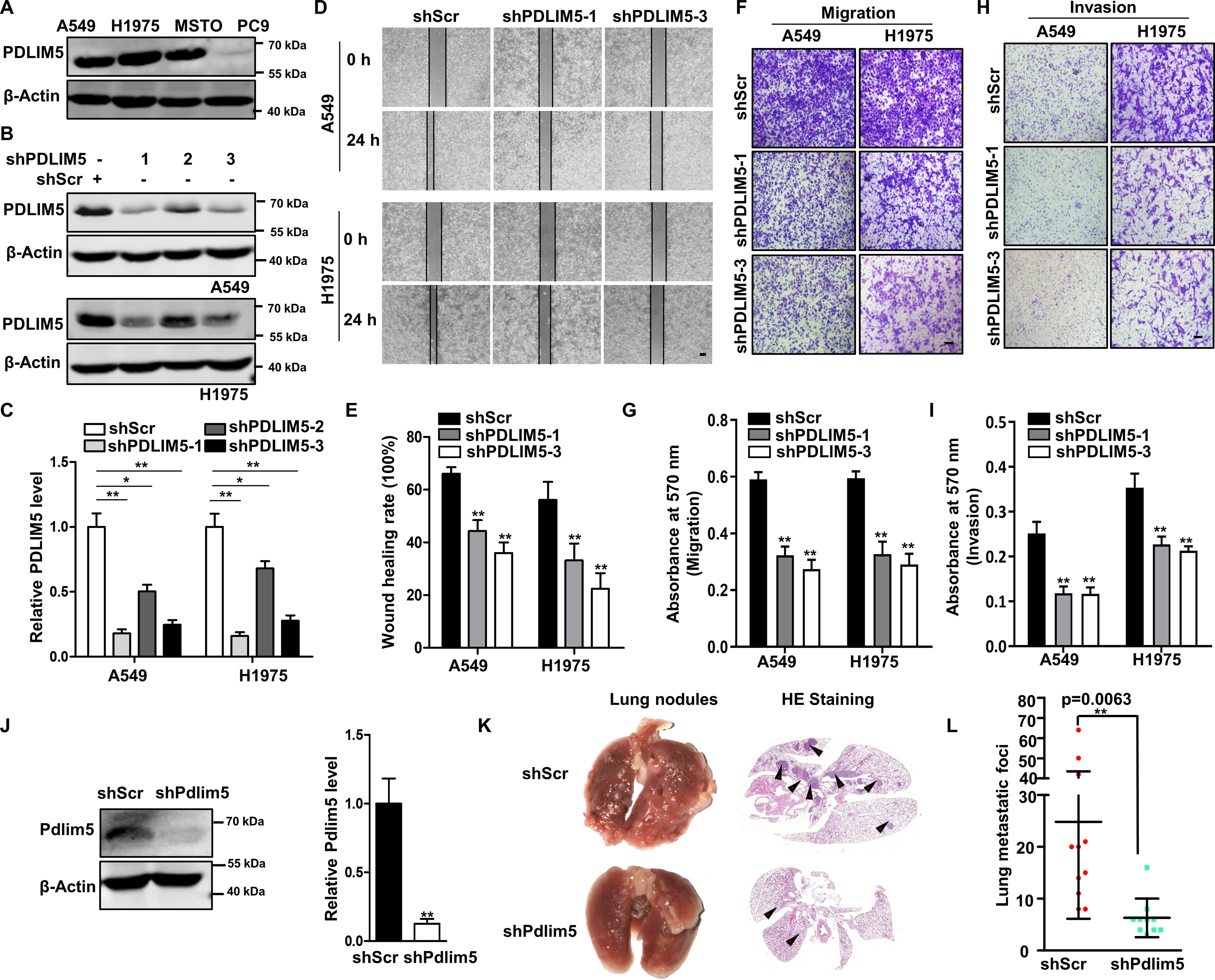
PDLIM5 knockdown impairs migration and invasion of lung cancer cells. A, Western blotting analysis of PDLIM5 in various NSCLC cell lines. β-Actin was used as a loading control. B, Western blotting analysis of PDLIM5 knockdown efficiency in A549 and H1975 cells achieved by using three different shRNA constructs. C, the knockdown efficiency of PDLIM5 was quantified and normalized to cells infected with control shRNA (shScr) (n = 3). β-Actin was used as a loading control. D, representative images of the wound-healing assay of PDLIM5 knockdown A549 and H1975 cells. The images were captured at 0 and 24 h after scratching. Scale bar, 200 μm. E, the wound-healing rate was analyzed by ImageJ software (n = 4). F and H, representative images of the Transwell migration (F) and Transwell invasion assay (H) of PDLIM5 knockdown A549 and H1975 cells. Scale bar, 200 μm. G and I, the migration (G) and invasion (I) index were quantified (n = 3). J, Western blotting analysis of Pdlim5 levels in Pdlim5-knockdown LLC cells. β-Actin was used as a loading control. K and L, representative bright-field images, and hematoxylin and eosin staining of the mouse lung with metastatic lesions (K). The lungs were collected 28 days after the injection of Pdlim5-knockdown LLC cells. The lesions were quantified (L). Arrows indicate the metastatic lesions. The data were shown as the means ± S.D. Analysis was performed using two-tailed Student's t test for C, E, G, I, J, and L. *, p < 0.05; **, p < 0.01.
Next, PDLIM5-knockdown in A549 and H1975 cells reduced cell spreading (Fig. S2A) and adhesion to vascular endothelial cells (Fig. S2, B and C). Using a real-time cell analyzer to record cell adhesion (26), we found that PDLIM5 knockdown reduced cell adhesion to fibronectin (Fig. S2D). Thus, PDLIM5 is required for cell spreading and cell–ECM and cell–cell interactions. PDLIM5 knockdown in A549 and H1975 cells also significantly attenuated vasculogenic mimicry (Fig. S2, E and F), a process associated with tumor malignancy whereby tumor cells mimic the vascular network (27). Moreover, PDLIM5 knockdown did not affect cell proliferation, colony formation, and the expression of cell cycle markers PCNA and cyclin D1 (Fig. S1, F–H). Consistently, cell proliferation was not altered in PC9 cells with PDLIM5 overexpression (Fig. S1E).
Furthermore, we isolated mouse embryonic fibroblasts from Pdlim5 knockout and WT mouse embryos to evaluate the universality of the findings. As in lung cancer cells, Pdlim5 deletion decreased wound-healing capacity without affecting cell proliferation (Fig. S1, I–K).
PDLIM5 knockdown attenuates TGFβ signaling
Cell migration and invasion are regulated by TGFβ signaling, and PDLIM5 has been reported to be related to this signaling (24, 25). To examine the role of PDLIM5 in regulating TGFβ signaling in lung cancer, gene set enrichment analysis was used with The Cancer Genome Atlas database of lung cancer to analyze the potential association between PDLIM5 and TGFβ pathway. PDLIM5 was strongly co-expressed with genes belonging to the TGFβ pathway in lung cancer (Fig. 3A). TGFβ1 markedly induced SBE-Luc activity in A549 cells, which was significantly repressed by PDLIM5 knockdown (Fig. 3B). PAI1 and JUNB are two well-known downstream targeting genes of TGFβ signaling, whose expression was notably decreased in PDLIM5-knockdown A549 and H1975 cells (Fig. 3C) and in embryonic fibroblasts isolated from Pdlim5-knockout mice (Fig. S3A). Consistent with this, forced expression of PDLIM5 in PC9 cells increased the expression of PAI1 and JUNB (Fig. S3B). EMT is typically induced in lung cancer cells treated with TGFβ1 (9). Accordingly, upon TGFβ1 stimulation, A549 and H1975 cells exhibited a fibroblast-like morphology, whereas this morphological change was blocked by PDLIM5 knockdown (Fig. 3D). Furthermore, RT-PCR and Western blotting analysis revealed that PDLIM5 knockdown in A549 and H1975 cells significantly decreased TGFβ1-responsive and mesenchymal-related gene expression following TGFβ1 treatment (Fig. 3, E and F).
Figure 3.
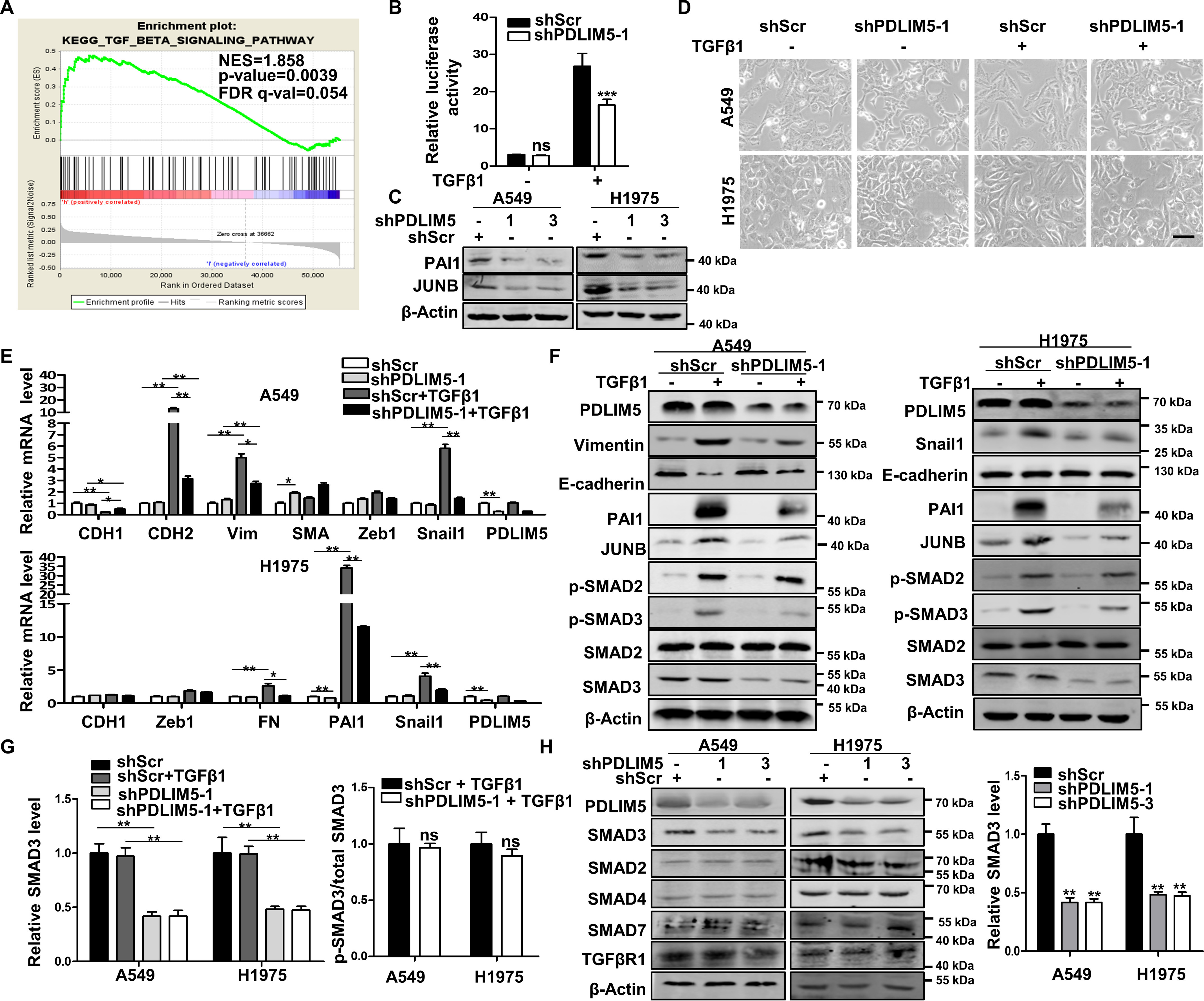
PDLIM5 knockdown attenuates TGFβ signaling. A, GSEA of The Cancer Genome Atlas lung cancer data sets revealed the association between TGFβ-responsive genes and PDLIM5. B, luciferase reporter assay with PDLIM5 knockdown A549 cells. The cells were stimulated, or not, with TGFβ1 (10 ng/ml). The data were normalized to the Renilla luciferase activity. C, Western blotting analysis of TGFβ signaling-related proteins PAI1 and JUNB in PDLIM5 knockdown A549 and H1975 cells. β-Actin was used as a loading control. D, morphology of PDLIM5 knockdown A549 and H1975 cells treated, or not, with TGFβ1 (5 ng/ml) for 24 h. Scale bar, 50 μm. E, RT-PCR analysis of the expression of genes for EMT markers in PDLIM5 knockdown A549 and H1975 cells treated, or not, with TGFβ1 (5 ng/ml) for 24 h. The data were normalized to 18S RNA (n = 3). F, Western blotting analysis of TGFβ signaling proteins and EMT markers in PDLIM5 knockdown A549 and H1975 cells treated, or not, with TGFβ1 (5 ng/ml) for 24 h. β-Actin was used as a loading control. G, quantitative analysis of SMAD3 and p-SMAD3 in PDLIM5 knockdown NSCLC cells. SMAD3 value was normalized to the levels in shScr cells, and β-actin was used as a loading control. p-SMAD3 value was normalized to the shScr + TGFβ1 levels (n = 3). H, Western blotting analysis of TGFβ signaling proteins in PDLIM5 knockdown A549 and H1975 cells. SMAD3 value was quantified and normalized to shScr value. β-Actin was used as a loading control. The data are shown as the means ± S.D. Analysis was performed using one-way ANOVA with Tukey post hoc test for B, E, and G and two-tailed Student's t test for H. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Interestingly, SMAD3 but not SMAD2 expression was decreased in PDLIM5-knockdown cells (Fig. 3, F–H) and in Pdlim5-knockout mouse embryonic fibroblasts (Fig. S3C). p-Smad3/Smad3 was unaltered in TGFβ1-treated PDLIM5-knockdown cells (Fig. 3G). PDLIM5 knockdown did not affect the expression of SMAD4, SMAD7, and TGFβR1 (Fig. 3H). In addition, PC9 cells with PDLIM5 overexpression increased the SMAD3 level (Fig. S3D). Together, these results indicated that PDLIM5 regulates SMAD3-mediated TGFβ signaling.
SMAD3 mediates PDLIM5 function
To explore whether PDLIM5 function in lung cancer cells was mediated by SMAD3, we first explored the role of SMAD3 in cell migration and invasion. SMAD3 knockdown in A549 cells inhibited cell wound-healing capacity, cellular migration, and invasion (Fig. S1, L–P). The expression of SMAD3 was reduced in Pdlim5-knockout mouse lungs (Fig. S3E). Pdlim5 deletion in mice resulted in alveolarization retardation, which was similar to Smad3-knockout mice (28) (Fig. S3, F and G). Therefore, similar to PDLIM5, SMAD3 promotes cancer cell migration and invasion and lung development.
Next, we restored SMAD3 in PDLIM5-knockdown A549 cells. The decreased expression of PAI1 and JUNB in PDLIM5-knockdown A549 cells was recovered upon SMAD3 overexpression (Fig. 4A).
Figure 4.
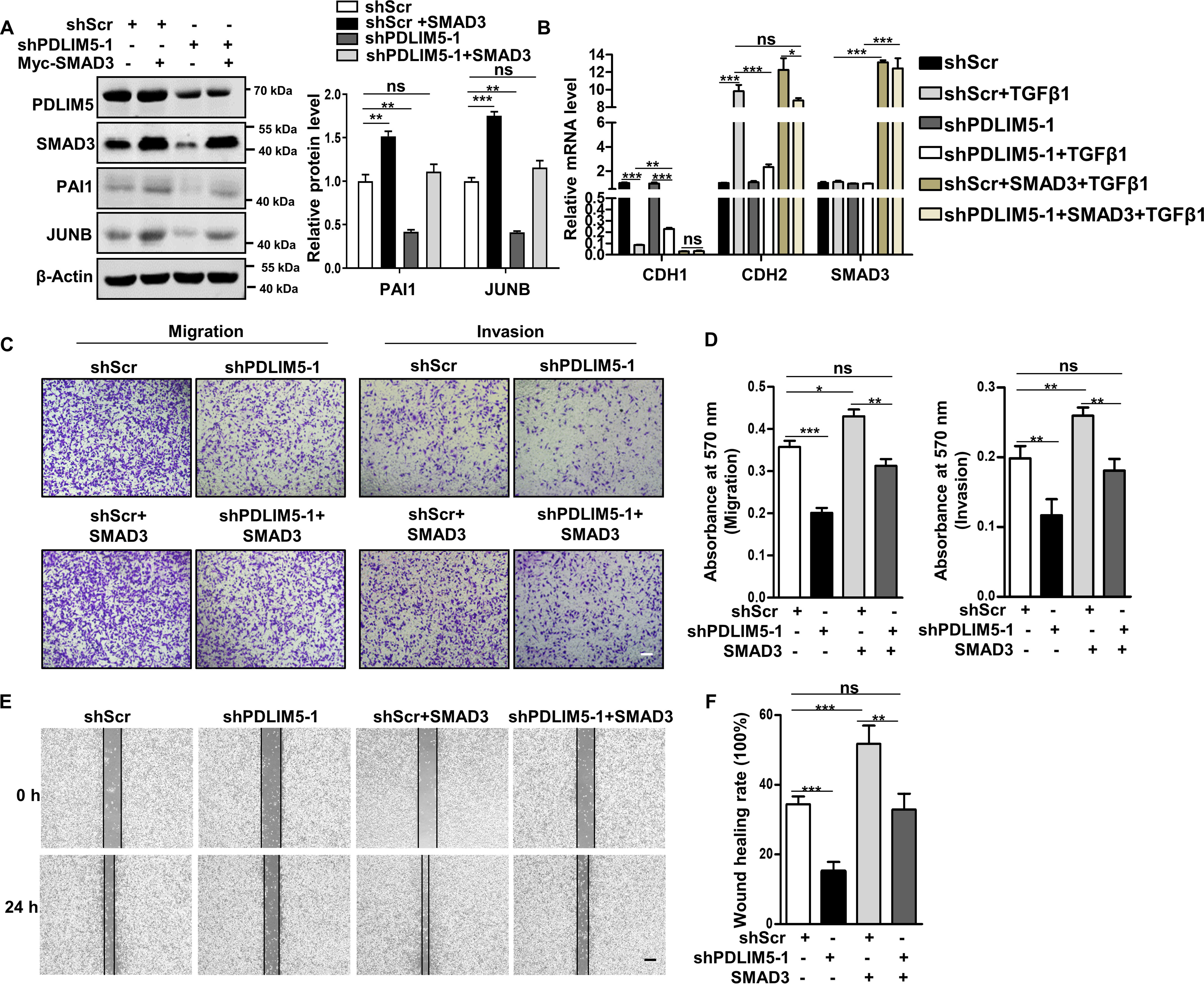
SMAD3 mediates PDLIM5 function in cancer cells. A, Western blotting analysis of PAI1 and JUNB in PDLIM5 knockdown A549 cells, with or without SMAD3 overexpression. PAI1 and JUNB were quantified and normalized to shScr value (n = 3). β-Actin was used as a loading control. B, RT-PCR analysis of EMT markers in PDLIM5 knockdown A549 cells, with or without SMAD3 overexpression. The cells were treated, or not, with TGFβ1 (5 ng/ml) for 24 h. The data were normalized to 18S RNA (n = 3). C, representative images of the Transwell migration and Transwell invasion assay of PDLIM5 knockdown A549 cells with or without SMAD3 overexpression. Scale bar, 200 μm. D, the migration and invasion index were quantified (n = 3). E, representative images of the wound-healing assay of PDLIM5 knockdown A549 cells with or without SMAD3 overexpression. The images were captured at 0 and 24 h after wounding. Scale bar, 200 μm. F, the wound-healing rates were analyzed by ImageJ software (n = 3). The data are shown as means ± S.D. Analysis was performed using one-way ANOVA with Tukey post hoc test for A, B, D, and F. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
The repression of EMT induced by PDLIM5 knockdown was also attenuated by SMAD3 overexpression (Fig. 4B). Furthermore, SMAD3 ectopic expression rescued PDLIM5 knockdown–induced defects in cellular attachment to fibronectin (Fig. S2G) and vasculogenic mimicry (Fig. S2H). Finally, the decreased wound-healing capacity, cellular migration, and invasion in PDLIM5-knockdown A549 cells were all restored by SMAD3 overexpression (Fig. 4, C–F). Taken together, these results clearly support our assessment that the defects in PDLIM5-knockdown cells were due to the decrease of SMAD3.
PDLIM5 suppresses proteasome-dependent degradation of SMAD3
To explore the regulation between PDLIM5 and SMAD3, first, we analyzed PDLIM5 and SMAD3 mRNA levels in NSCLC cell lines available in the Expression Atlas database. No relationship between PDLIM5 and SMAD3 mRNA levels was suggested (r = 0.03, p = 0.77) (Fig. 5A). In contrast, PDLIM5 and SMAD3 protein levels were highly correlated in NSCLC cell lines and NSCLC tissues (Fig. 5, B and C). Further, RT-PCR analysis indicated that PDLIM5 knockdown did not alter SMAD3 mRNA level in A549 and H1975 cells (Fig. 5D). These observations indicated that in the lung cancer cells, PDLIM5 specifically regulates SMAD3 stability.
Figure 5.

PDLIM5 suppresses proteasome-dependent degradation of SMAD3. A, the correlation between PDLIM5 and SMAD3 mRNA levels in NSCLC cells. B, Western blotting analysis of PDLIM5 and SMAD3 in different NSCLC cells. The correlation between PDLIM5 and SMAD3 protein levels was shown. β-Actin was used as a loading control. C, Western blotting analysis of PDLIM5 and SMAD3 in six NSCLC tissues and paired adjacent normal tissues. The correlation between PDLIM5 and SMAD3 protein levels was shown. β-Actin was used as a loading control. D, mRNA levels of SMAD2, SMAD3, and SMAD5 in PDLIM5 knockdown A549 and H1975 cells. The data were normalized to 18S RNA levels (n = 3). E and F, Western blotting analysis of SMAD3 in PDLIM5 knockdown (E) and PDLIM5 overexpressed (F) A549 cells treated with CHX (50 µg/ml) for 4 or 8 h. SMAD3 levels at the indicated times after CHX treatment were quantified (n = 3). β-Actin was used as a loading control. G, ubiquitination assay of exogenously overexpressed SMAD3 in PDLIM5 overexpressed and PDLIM5 knockdown A549 cells. H, Western blotting analysis of SMAD3 in PDLIM5 knockdown A549 cells treated with 20 μm MG132 for 8 h. SMAD3 levels were quantified and normalized to shScr values. β-Actin was used as loading control. The data are shown as the means ± S.D. Analysis was performed using two-tailed Student's t test for D and one-way ANOVA with Tukey post hoc test for E, F, and H. *, p < 0.05; **, p < 0.01; ***, p < 0.001. IB, immunoblotting.
Next, to evaluate SMAD3 turnover, we treated A549 cells with cycloheximide (CHX) to inhibit protein synthesis. PDLIM5 knockdown promoted SMAD3 turnover (Fig. 5E), whereas this was markedly inhibited upon PDLIM5 overexpression in A549 cells (Fig. 5F). Because ubiquitination is an important mechanism for regulating SMAD3 stability, we co-expressed FLAG–SMAD3 and ubiquitin constructs in A549 cells to evaluate whether PDLIM5 regulates SMAD3 ubiquitination. We found that SMAD3 ubiquitination was enhanced upon PDLIM5 knockdown (Fig. 5G) and repressed after PDLIM5 overexpression (Fig. 5G). Importantly, MG132 (an inhibitor of proteasome degradation) effectively inhibited SMAD3 decrease in PDLIM5-knockdown cells (Fig. 5H). Collectively, these findings indicate that PDLIM5 protects SMAD3 from proteasome-mediated degradation.
PDLIM5 interacts with SMAD3
It has been reported that the serine/threonine kinase GSK3β is responsible for SMAD3 degradation (13). However, GSK3β activity, as indicated by its phosphorylated state, was not altered in PDLIM5 knockdown A549 cells (Fig. S4A). Moreover, GSK3β knockdown (Fig. S4B) and a GSK3β inhibitor (LiCl) (Fig. S4C) all failed to restore SMAD3 levels in PDLIM5-knockdown A549 cells. Then we questioned whether PDLIM5 stabilizes SMAD3 by direct binding. To test this, FLAG-tagged SMAD3 and HA-tagged PDLIM5 were ectopically expressed in HEK293T cells. Protein pellets precipitated using the anti-FLAG antibody contained the HA-tagged PDLIM5 protein. Specifically, no interaction between SMAD2 and PDLIM5 was detected (Fig. 6A). Immunoprecipitation of endogenous proteins in A549 cells confirmed the interaction between PDLIM5 and SMAD3 (Fig. 6B). We therefore proceeded to map the interacting domains in PDLIM5 and SMAD3. LIM domains, the PDZ domain, and PDLIM5 without LIM domains (ΔLIM) were individually expressed in HEK293T cells. SMAD3 was found to interact with the LIM but not the PDZ domain of PDLIM5 (Fig. 6C). In comparison, either the SMAD3 MH1 or MH2 domain interacted with PDLIM5 (Fig. 6D). Last, PDLIM5 co-localization with SMAD3 as revealed by immunofluorescence assay further confirmed this interaction (Fig. 6E). Therefore, PDLIM5 interacts with SMAD3 but not SMAD2.
Figure 6.
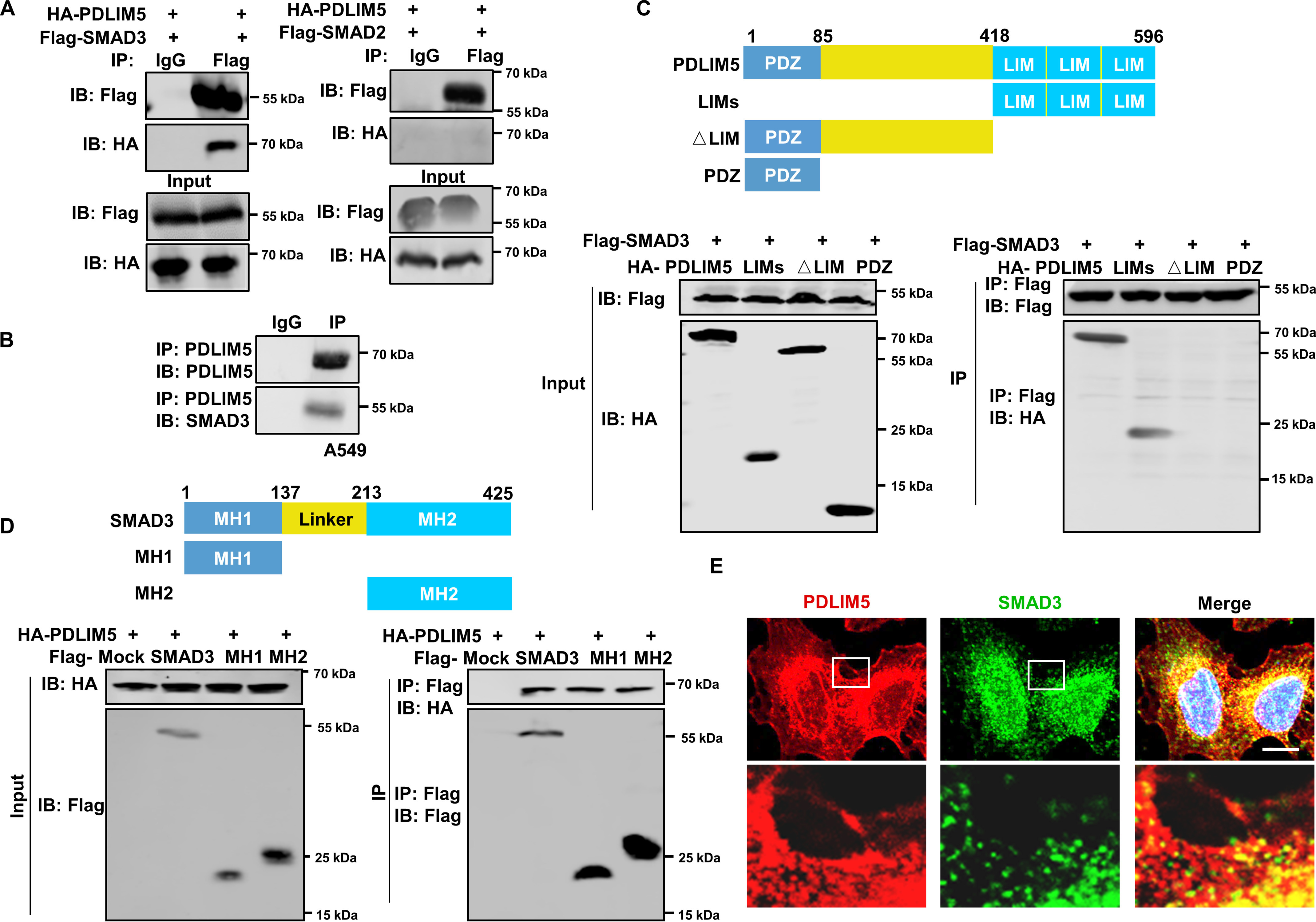
PDLIM5 interacts with SMAD3. A, co-immunoprecipitation analysis of the interaction between PDLIM5 and SMAD3 or SMAD2 in HEK293T cells. B, co-immunoprecipitation analysis of the endogenous interaction between PDLIM5 and SMAD3 in A549 cells. C, mapping PDLIM5 fragment that interacted with SMAD3. HEK293T cells were co-transfected with FLAG–SMAD3 and PDLIM5 truncated fragment (HA-PDLIM5, 1–596 amino acids; LIMs, 418–596 amino acids; △LIM, 1–417 amino acids; PDZ, 1–85 amino acids) for immunoprecipitation assays. D, mapping SMAD3 fragment that interacted with PDLIM5. HEK293T cells were co-transfected with HA-PDLIM5 and SMAD3 truncated fragment (FLAG–SMAD3, 1–425 amino acids; MH1, 1–137 amino acids; MH2, 231–425 amino acids) for immunoprecipitation assays. E, immunofluorescence analysis to show co-localization of SMAD3 (green) and PDLIM5 (red) in A549 cells. The cell nucleus is stained blue. Scale bar, 50 μm. IB, immunoblotting.
To answer whether the interaction between PDLIM5 and SMAD3 plays a role in stabilizing SMAD3 by PDLIM5, we first transfected truncated PDLIM5 without LIM domains (ΔLIM) in PC9 cells to detect whether it promotes SMAD3 expression. As expected, PDLIM5 mutation, destroying the PDLIM5–SMAD3 interaction, failed to up-regulate SMAD3 in PC9 cells (Fig. S3D). Similarly, PDLIM5 re-expression restored the wound-healing capacity, cellular migration, and invasion in PDLIM5-knockdown cells. However, expression of PDLIM5 mutation (ΔLIM) did not restore the defects (Fig. S3, H–L). Thus, the interaction is required for PDLIM5 to stabilize SMAD3.
PDLIM5 stabilizes SMAD3 by counteracting the interaction between SMAD3 and STUB1
Because several E3 ligases reportedly regulate SMAD3 degradation, we hypothesized that PDLIM5 may modulate SMAD3 degradation by regulating the activity of certain E3 ligase. An immunoprecipitation (IP)–MS experiment revealed that PDLIM5 bound multiple E3 ligases, among which STUB1 is a E3 ligase for SMAD3 (Fig. 7A). The interaction was confirmed in HEK293T cells expressing HA-PDLIM5 and FLAG-STUB1 by a co-immunoprecipitation assay (Fig. S4D). Overexpression of STUB1 decreased SMAD3 without altering SMAD2 in A549 cells (Fig. S4E). Moreover, PAI1 and JUNB expression was reduced in STUB1 overexpressed cells (Fig. S4F). Importantly, STUB1 knockdown restored SMAD3 level in PDLIM5 knockdown cells (Fig. 7B). Specifically, STUB1 interacted with the MH2 domain of SMAD3 (Fig. S4G and Fig. 7C). Because both PDLIM5 and STUB1 interacted with the MH2 domain of SMAD3, we next tested whether PDLIM5 affected the interaction between STUB1 and SMAD3. Immunoprecipitation analysis revealed that reduced PDLIM5 expression enhanced the interaction between STUB1 and SMAD3, whereas PDLIM5 overexpression perturbed this interaction (Fig. 7, D and E). Next, acceptor bleach FRET experiments were carried out with CFP-SMAD3 as the donor and YFP-STUB1 as the acceptor to verify the results. Photobleaching of YFP-STUB1 significantly increased the postbleaching fluorescence intensity of CFP-SMAD3, indicating significant FRET between these two protein. The FRET between CFP-SMAD3 and YFP-STUB1 was disrupted upon PDLIM5 overexpression, suggesting that PDLIM5 repressed STUB1 interacting with SMAD3 (Fig. 7F). Moreover, the decrease of SMAD3 in STUB1-overexpression A549 cells was reversed by the expression of PDLIM5 but not LIM-domain deletion PDLIM5 mutation (Fig. 7G). Notably, STUB1 overexpression in A549 cells reduced cell migration, invasion, cell adhesion, and vasculogenic mimicry (Fig. 8, A–D, and Fig. S2, I and J), similar to those in PDLIM5-knockdown cells. Furthermore, knockdown STUB1 restored the wound-healing capacity, cellular migration, and invasion of PDLIM5-knockdown cells (Fig. 8, E–H). All of these observations indicate that PDLIM5 and STUB1 competitively bind to SMAD3, and PDLIM5 stabilizes SMAD3 by protecting it from STUB1-mediated proteasome degradation.
Figure 7.
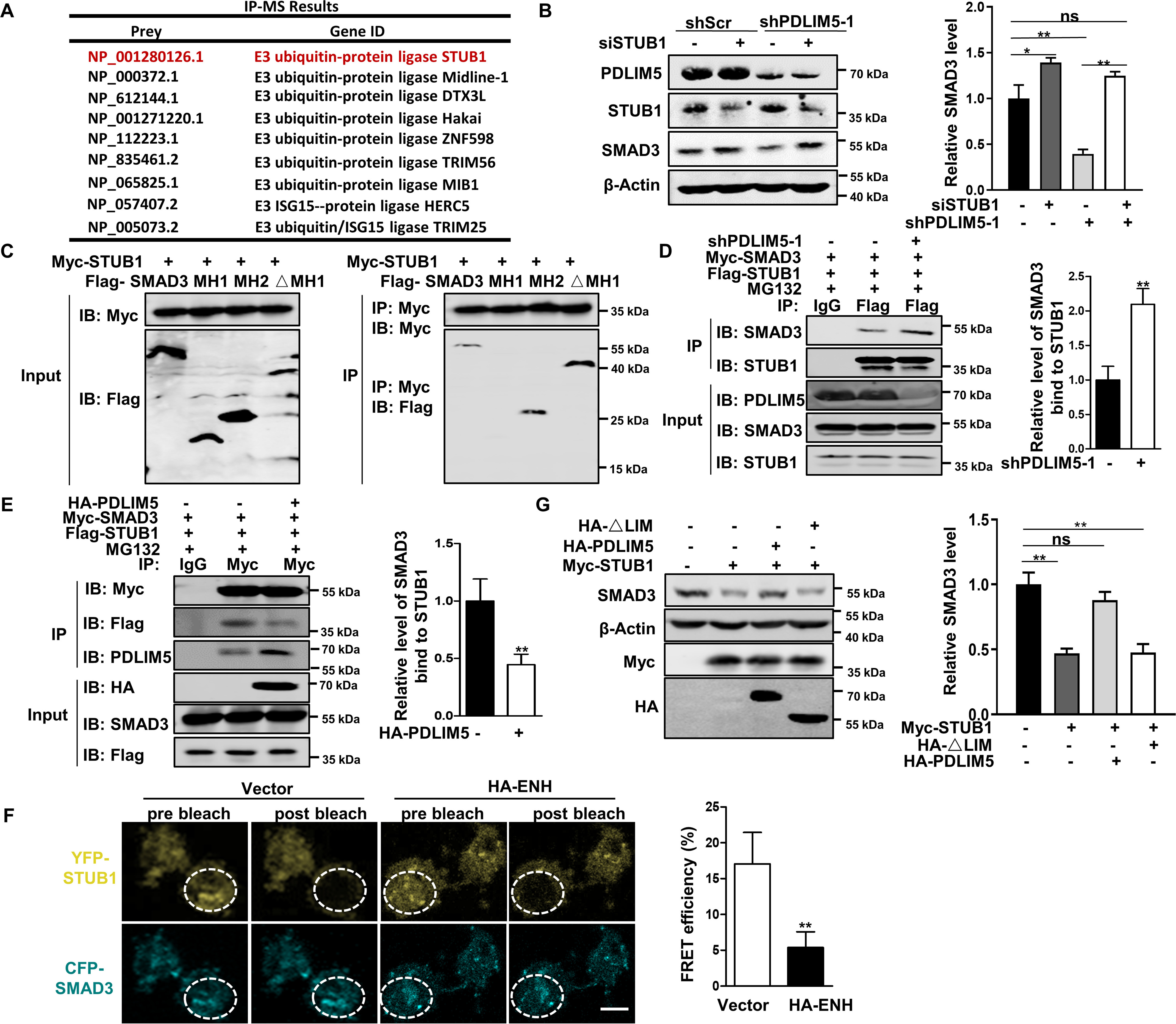
PDLIM5 stabilizes SMAD3 by counteracting the interaction between SMAD3 and STUB1. A, IP-MS analysis of candidate proteins interacting with PDLIM5 in A549 cells. B, Western blotting analysis of SMAD3 in PDLIM5 knockdown, alone or combination with STUB1 knockdown in A549 cells. SMAD3 value was quantified and normalized to the control (n = 3). β-Actin was used as a loading control. C, mapping SMAD3 fragment that interacted with STUB1. HEK293T cells were co-transfected with Myc-STUB1 and SMAD3 truncated fragment (FLAG–SMAD3, 1–425 amino acids; MH1, 1–137 amino acids; MH2, 231–425 amino acids; ΔMH1, 137–425 amino acids) for immunoprecipitation assays. D and E, co-immunoprecipitation analysis of the interaction between STUB1 and SMAD3 in PDLIM5 knockdown (D) or PDLIM5 overexpressed (E) A549 cells. F, FRET experiments were performed using PDLIM5 overexpressed A549 cells co-transfected with YFP-STUB1 (YFP; top row) and CFP-SMAD3 (CFP; bottom row). Representative images for pre- and postbleaching were shown. Quantification of FRET efficiency was calculated with following formula: %FRET = 100 × (CFPpost CFPpre)/CFPpost. G, Western blotting analysis of SMAD3 in A549 cells transfected with STUB1 expressing construct, alone or together with PDLIM5 full-length or LIM-domain deletion mutant. SMAD3 were quantified and normalized to the control (n = 3). β-Actin was used as a loading control. The data are shown as the means ± S.D. Analysis was performed using two-tailed Student's t test for D–F and one-way ANOVA with Tukey post hoc test for B and G. *, p < 0.05; **, p < 0.01. IB, immunoblotting.
Figure 8.
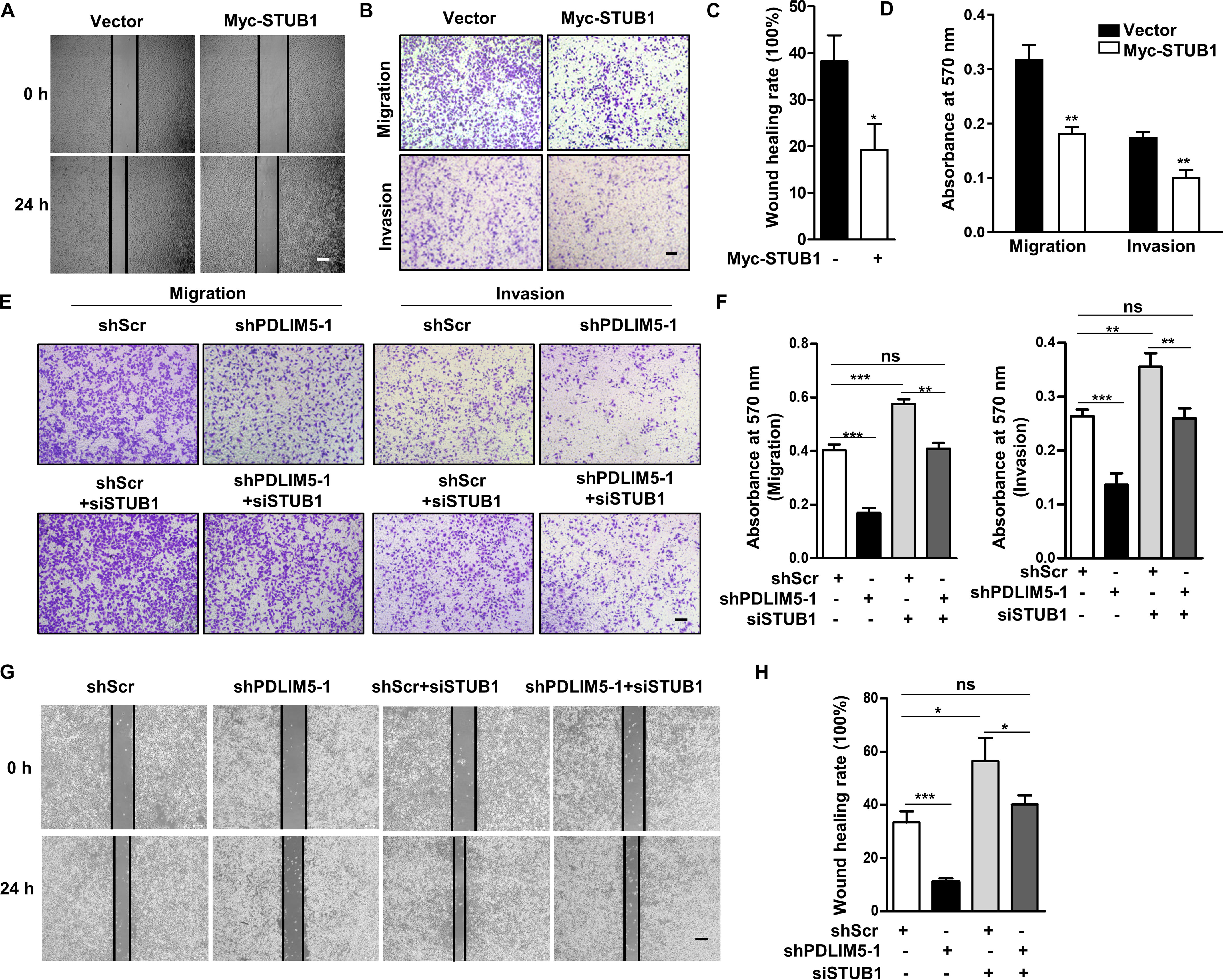
STUB1 knockdown reverses the defects in cell migration and invasion upon PDLIM5 knockdown. A, representative images of the wound-healing assay for STUB1 overexpressed cells. The images were captured at 0 and 24 h after scratching. Scale bar, 200 μm. C, the wound-healing rate was analyzed by ImageJ software (n = 3). B, representative images of Transwell migration and Transwell invasion assay for STUB1 overexpressed A549 cells. Scale bar, 200 μm. D, the migration and invasion index were quantified (n = 3). E, representative images of the Transwell migration and Transwell invasion assay of PDLIM5-knockdown A549 cells with or without STUB1 knockdown. Scale bar, 200 μm. F, the migration and invasion index were quantified (n = 3). G, representative images of the wound-healing assay of PDLIM5 knockdown A549 cells with or without STUB1 knockdown. The images were captured at 0 and 24 h after scratching. Scale bar, 200 μm. H, the wound-healing rate was analyzed by ImageJ software (n = 3). The data are shown as means ± S.D. Analysis was performed using two-tailed Student's t test for C and D and one-way ANOVA with Tukey post hoc test for F and H. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Discussion
TGFβ signaling has a prominent role in NSCLC tumorigenesis and metastases (3). SMAD3, as a key mediator in the TGFβ signaling pathway, plays an essential role in tumor invasiveness. In this study, we found that PDLIM5 is a new interacting protein to SMAD3 but not SMAD2. The interaction disrupts the binding between SMAD3 and its E3 ligase STUB1 to prevent SMAD3 degradation, thus promoting TGFβ signaling and cancer cell migration, invasion, and NSCLC progression. Our findings therefore demonstrated a novel mechanism for PDLIM5 in regulating TGFβ–SMAD3 signaling and NSCLC progression.
PDLIM5 belongs to the PDZ–LIM protein family, featured as containing one PDZ domain and one or three LIM domains. The most studied PDZ–LIM domain proteins in cancers are PDLIM1, PDLIM2, and PDLIM4. PDLIM1/CLP36 is up-regulated to mediate breast cancer metastasis by interacting with α-actinin (29). In contrast, PDLIM1 is down-regulated to inhibit cellular migration and invasion in highly metastatic colorectal cancer cells and metastatic hepatocellular cancer (30, 31). PDLIM2 controls COP9 signalosome activity and promotes the degradation of multiple tumor inducers including NF-κB and STAT3 in colorectal cancer (32, 33). A similar anti-tumor role of PDLIM2 was recently reported in lung cancer (34). PDLIM4 is a tumor suppressor and reportedly down-regulated in various cancers (35). Here, we showed that PDLIM5 is up-regulated in NSCLC tissues, which is associated with poor outcome. Furthermore, our data revealed that PDLIM5 is required for NSCLC cell adhesion, migration, invasion, and lung metastasis in mice. Although it has been reported that PDLIM5 knockdown inhibits prostate cancer and thyroid carcinoma cell proliferation (22, 23), we showed that PDLIM5 insufficiency in NSCLC cells and mouse embryonic fibroblasts does not inhibit cell proliferation, indicating that the function of PDLIM5 is context-dependent. Therefore, the present results revealed a protumor role of PDLIM5 in NSCLC.
Upon TGFβ stimulation, SMAD2 and SMAD3 are phosphorylated at their C termini by the activated TGFβRI and shuttle the signal to nucleus, which is the main events in TGFβ signaling (36). Despite their similarity in sequence, SMAD2 and SMAD3 have different roles in tumor progression and are differentially regulated (37). In current study, we showed that PDLIM5 interacts with SMAD3 but not SMAD2 and stabilizes basal SMAD3. Several studies have demonstrated that SMAD3 knockdown remarkably inhibits TGFβ-induced EMT and tumor metastasis in multiple cancers including NSCLC (11, 38). In addition, the expression of SMAD3 target genes, including PAI1 and JUNB, are positively correlated with tumor progression (39, 40). We found that both PAI1 and JUNB were down-regulated in PDLIM5 knockdown NSCLC cells, and there were similar defects in cellular migration and invasion in both PDLIM5 and SMAD3 knockdown cells. Importantly, ectopic expression of SMAD3 prominently restored the deficiency in TGFβ signaling, migration, and invasion caused by PDLIM5 loss. These observations demonstrated that PDLIM5 specifically regulates SMAD3-mediating TGFβ signaling.
The TGFβ–SMAD pathway is regulated by ubiquitin-mediated degradation (41). Various E3 ligases have been reported to mediate the degradation of multiple components in TGFβ signaling, including SMAD3 and SMAD2. PDLIM5 contains three LIM domains that mediate protein–protein interactions. Notably, LIM domain proteins have recently emerged as vital regulators that tightly control the activity of E3 ubiquitin ligase (42, 43). Consistent with this, we found that PDLIM5 binds multiple E3 ligases, including STUB1. STUB1 ubiquitinates and degrades SMAD3 in a basal level, thus desensitizing the cells to TGFβ signaling (44). We showed that PDLIM5 stabilizes basal SMAD3 by repressing SMAD3 binding to STUB1. Whether other PDLIM5-interacting E3 ligases mediates the malignance of lung cancer requires more exploration.
The dichotomous roles of TGFβ in cancers complicate the development of TGFβ-targeted anticancer drugs. However, proteins that interact with SMAD3, such as PDLIM5, bring new light to this field. The reason is that targeting PDLIM5 does not promote the proliferation of NSCLC cells, while it inhibits TGFβ signaling and tumor malignance.
In conclusion, we find that PDLIM5 specifically interacts with SMAD3 and prevents its degradation, and its expression is up-regulated to promote TGFβ signaling and malignance of lung cancer. These findings pinpoint PDLIM5 as a potential prognostic biomarker and intervention target for NSCLC.
Experimental procedures
Plasmids and reagents
SBE-Luc was purchased from Addgene (#16500, Watertown, MA, USA). pRL-TK was from Promega (Madison, WI, USA). FLAG–SMAD3 and FLAG-STUB1 were purchased from Youbio (Changsha, China). CFP-SMAD3, YFP-STUB1, FLAG-PDLIM5, HA-PDLIM5, Myc-SMAD3, Myc-STUB1, and all the truncated protein-encoding plasmids were constructed as described previously (24). FLAG–SMAD2 was a generous gift from Dr. Lin-Long Lu (Zhejiang University School of Medicine, Hangzhou, China). siRNA (target sequences are shown in Table S1) was from Hanbio (Shanghai, China). MG132 and CHX were obtained from MedChemExpress (Shanghai, China). Active recombinant human TGFβ1 was obtained from PeproTech (Rocky Hill, NJ, USA).
NSCLC tissues
Forty-six NSCLC tissues and matched tumor-adjacent normal lung tissues were obtained from the First Affiliated Hospital, College of Medicine, Zhejiang University (Hangzhou, China) and confirmed by pathological diagnosis. The protein levels of PDLIM5 and SMAD3 were analyzed in fresh specimens from six cases. The other 40 paraffin-embedded tissues were analyzed by immunohistochemical staining. This study was approved by the Research Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University (Hangzhou, China, No. 2019-1393) and abided by the Declaration of Helsinki principles. Written informed consent was obtained from all patients prior to the study.
Bioinformatics analysis
The Oncomine (RRID:SCR_007834) database was utilized to analyze the expression profiles of PDLIM5 in normal and NSCLC tissues. The survival Kaplan–Meier estimates of patients with NSCLC exhibiting different PDLIM5 expression levels was assessed using a Kaplan–Meier plotter (RRID:SCR_018753).
Cell culture and transient transfection
Mouse lung cancer cell line LLC, human HEK293T cells, and NSCLC cell lines A549, MSTO, H1975, H1299, H358, and PC9 were from the American Type Culture Collection (Manassas, VA, USA). WT and Pdlim5-knockout mouse embryonic fibroblasts were isolated and cultured as described previously (45). The cells were grown in RPMI 1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum.
Mice
Pdlim5 global knockout (Pdlim5−/−) mice were bred from heterozygous mice, as described previously (46). The left lungs of Pdlim5 WT and knockout mice were embedded by paraffin. The tissues were cut into 5 μm sections and stained with hematoxylin and eosin for histological examination. 6–8-week-old nude mice were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, China) for in vivo metastasis assay. The mice were injected with LLC lung cancer cells suspended in PBS (2.5 × 106 cells/mouse) via the tail vein. 28 days after the injection, the mice were euthanized by pentobarbital injection, and the lungs were fixed in 4% paraformaldehyde (PFA) to prepare paraffin sections. The lung metastasis regions were counted under a light microscope. The total number of metastatic nodules in each mouse was determined. All animal experiments were reviewed and approved by the Animal Care and Use Committee of the Zhejiang University School of Medicine.
Lentivirus infection
To produce lentiviruses, HEK293T cells were transfected with plKO-puro shPDLIM5 (the target sequences are provided in Table S1) or control shRNA (shScr) and packaging plasmids (pMD2.G and psPAX2; Sigma–Aldrich). Lentivirus-containing supernatants were collected 48 h after the transfection and filtered through a 0.22 μm filter. Prior to infection, lentiviruses at same virus multiplicity of infection (multiplicity of infection of 30) were incubated with Polybrene (Beyotime, Shanghai, China) to increase the infection efficiency.
Transwell assay
For the migration assay, 2 × 104 cells suspended in 200 μl of RPMI 1640 serum-free medium containing 0.5% BSA were seeded into 8-μm Boyden chambers (Corning, Armonk, NY, USA). For the invasion assay, 2 × 105 cells suspended in 200 μl of RPMI 1640 serum-free medium containing 0.5% BSA were plated in the upper chamber precoated with 50 μl of 2 μg/ml Matrigel (Corning). Then 600 μl of the complete medium was added to the lower chamber to encourage cell migration and invasion. The incubation time for migration was 24 h. The invasion required an additional 24 h. After incubation, the upper chambers were collected and fixed in methanol, and nonmigrating or noninvading cells were removed. The migrating and invading cells were stained with crystal violet solution (Beyotime), and representative images were acquired using a light microscope (Olympus, Tokyo, Japan). The crystal violet-stained cells were then washed with 33% acetic acid, and absorbance was measured at 570 nm (Molecular Devices, Sunnyvale, CA, USA).
Cell adhesion assay
To detect cell adhesion to endothelial cells, H1975 and A549 cells were labeled with calcein AM. The labeled cells (1 × 104/100 μl/well) were co-cultured with an endothelial monolayer of human umbilical vein endothelial cells (isolated as described (47)) in 96-well plates for 1 h. After the incubation, the detached cells were washed off with PBS, and the adherent cells were counted under a fluorescence microscope (Nikon Eclipse TE2000U-E, Tokyo, Japan). The xCELLigence real-time cell analyzer instrument (Acea Biosciences, San Diego, CA, USA) was used to evaluate the adhesion capacity of A549 and H1975 cells to fibronectin. For the experiment, 1 × 104 cells suspended in 200 μl of RPMI 1640 medium containing 0.5% BSA were seeded into 96-well E-plates precoated with 2.5 μg fibronectin/well. The attachment was monitored by recording the cell index every minute for 2 h.
Vasculogenic mimicry assay
Vasculogenic mimicry assay was used to evaluate the vasculogenic mimicry of cancer cells in vitro. For the experiment, 10 μl of Matrigel (Corning) was placed in an u-Slide well (Ibidi, Martinsried, Germany) and incubated at 37 °C for 30 min to allow it to solidify. A549 and H1975 cells suspended in complete medium were seeded into the u-Slide at a density of 2 × 104 cells/well and incubated for 6 h. The formation of tubes was captured using an inverted light microscope at 100× magnification in five random fields. Tube length was also measured in five random fields, using ImageJ software.
Immunofluorescence staining
The cells cultured on glass coverslips were fixed in 4% PFA and permeabilized using 0.5% Triton X-100 in PBS before incubation with an appropriate primary antibody (Table S2). The signal was visualized by incubating with goat anti-mouse antibodies conjugated with Alexa Fluor 594 or goat anti-rabbit antibodies conjugated with Alexa Fluor 488 at room temperature for 1 h. The nucleus was labeled using 4′,6-diamidino-2-phenylindole. The images were acquired by using the FV1000 confocal microscope (Olympus, Tokyo, Japan).
Immunohistochemical staining
Paraffin-embedded tissue samples were stained with anti-PDLIM5 antibody (Table S2). Then each sample was scored based on the percentage of positively stained cells (0, 0%; 1, 1–25%; 2, 26–50%; 3, 51–75%; 4, 76%–100%) and the staining intensity (0, negative; 1, weak; 2, moderate; 3, strong). The two scores were multiplied for the final score (48).
Western blotting
All tissue and cellular proteins were extracted by RIPA lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Nonidet P-40) following the manufacturer's instructions (Beyotime). The lysate was boiled at 100 °C for 8 min, and the proteins were separated on 10 and 13% SDS-PAGE gels. They were then transferred onto nitrocellulose membrane (Pall, Port Washington, NY, USA), probed with primary antibody at 4 °C overnight. The primary antibodies used herein were listed in Table S2. The membranes were then washed three times with TBST (150 mm NaCl, 50 mm Tris, 0.05% Tween 20, pH 7.6) and probed with a secondary antibody (Table S2) at room temperature for 1 h. Finally, the target protein was visualized by using the Odyssey system (LI-COR Biosciences, Lincoln, NE, USA).
Co-immunoprecipitation
The cells were lysed in Nonidet P-40 buffer (20 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm EDTA, and 1% Nonidet P-40) freshly supplemented with a protease inhibitor (Roche) and a phosphatase inhibitor (Roche). Then 30 μl of magnetic beads (Bio-Rad) were incubated with 3 μg of the appropriate antibody with agitation for 10 min at 37 °C. Cell lysates were incubated with antibody-conjugated magnetic beads at 4 °C overnight. The immunoprecipitates were then eluted by boiling in SDS sample buffer (Beyotime) for 5 min. For the ubiquitination assay, the cells transfected with FLAG–SMAD3 and ubiquitin constructs were treated with 10 μm MG132 for 8 h and then lysed for a subsequent co-immunoprecipitation assay.
IP-MS
A549 cells were transduced with HA-PDLIM5 plasmids for 24 h, then lysed, and incubated with anti-HA antibody–conjugated beads. The immunoprecipitate was subjected to SDS-PAGE. After electrophoresis, the protein gel was stained with Coomassie Brilliant Blue and then analyzed by MS by Qinglian Bio (Beijing, China).
Double luciferase reporter assay
The SBE reporter assay was performed as previously described (49). Reporter gene activity was detected according to the protocol for the Dual-Luciferase reporter gene assay kit (Promega) and measured by using SpectraMax M5 (Molecular Devices).
Quantitative RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen). For cDNA synthesis, 1 μg of total RNA was transcribed using the ReverTraAce quantitative RT-PCR kit (Toyobo Inc., Osaka, Japan). Quantitative PCR was performed using the LightCycler Roche480 (Roche) according to the manufacturer's instructions of the SYBR Green dye (cwbio, Beijing, China). Gene expression was normalized to the endogenous control (18S RNA) and calculated using 2 – ΔΔCt. All primers used for the analysis are listed in Table S3.
Acceptor photobleaching FRET
CFP-SMAD3 (donor) and YFP-STUB1 (acceptor) together with HA-PDLIM5 or empty vector were co-transfected in A549 cell. After 24 h of transfection, the cells were fixed by 4% PFA. Acceptor photobleaching FRET were performed using the FV1000 confocal microscope (Olympus, Tokyo, Japan) as described previously (50). The CFP and YFP fluorophores were excited with excitation wavelengths of 458 and 514 nm, respectively. After acquired the emission images, the cells marked by a region of interest and this region was bleached by a high laser power (20 iterations, 100% laser power, 514 nm). The FRET efficiency was measured as the percentage increase of donor after photobleaching the acceptor: %FRET = 100 × (CFPpost − CFPpre)/CFPpost. CFPpost and CFPpre indicate CFP emission after and before bleaching, respectively.
Statistical analysis
Statistical analysis was performed by GraphPad Prism 5. The data are presented as the means ± S.D. We performed unpaired t tests to determine the significance of differences between two groups. For the comparison of more than two groups, one-way ANOVA was used. A Wilcoxon matched-pair signed-rank test was used to determine the expression difference between the cancer tissue and adjacent normal tissue. A p value < 0.05 was considered statistically significant.
Data Availability
All the data are included in the article.
Supplementary Material
Acknowledgments
We thank Shuangshuang Liu for helping with confocal microscopy. We thank Dr. Lin-Long Lu (Zhejiang University School of Medicine, Hangzhou, China) for the SMAD2 plasmid. We also thank Editage for language editing.
This article contains supporting information.
Author contributions—Y. S. and H. C. conceptualization; Y. S., Y. K., and H. C. data curation; Y. S., X. W., Z. X., C. G., and H. C. validation; Y. S., X. W., Y. H., J. H., J. Z., Y. K., and H. C. investigation; Y. S., Z. X., and C. G. visualization; Y. S., X. W., Y. H., J. H., J. Z., and H. C. methodology; Y. S., X. W., Z. X., YL, and H. C. writing-original draft; Y. S., X. Z., and Y. K. project administration; Y. S. and Y. L. writing-review and editing; Z. X. software; L. H., C. H., C. C., C. Z., L. W., and Y. K. resources; X. Z., Y. K., and H. C. supervision; Y. K. and H. C. funding acquisition.
Funding and additional information—This work was supported Zhejiang Provincial Natural Science Foundation of China Grant LZ18H020001 (to H. C.) and National Natural Science Foundation of China Grants 81673512 (to L. W.), 31871399 (to H. C.), 81530001 (to Y. K.), and 81700064 (to C. H.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- TGFβ
- transforming growth factor β
- EMT
- epithelial–mesenchymal transition
- NSCLC
- non–small cell lung cancer
- CHX
- cycloheximide
- IP
- immunoprecipitation
- ANOVA
- analysis of variance
- LLC
- Lewis lung carcinoma
- HA
- hemagglutinin
- PFA
- paraformaldehyde
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein.
References
- 1. Massagué J., Blain S. W., and Lo R. S. (2000) TGFβ signaling in growth control, cancer, and heritable disorders. Cell 103, 295–309 10.1016/S0092-8674(00)00121-5 [DOI] [PubMed] [Google Scholar]
- 2. Massagué J. (2008) TGFβ in cancer. Cell 134, 215–230 10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miyazono K., Katsuno Y., Koinuma D., Ehata S., and Morikawa M. (2018) Intracellular and extracellular TGF-β signaling in cancer: some recent topics. Front. Med. 12, 387–411 10.1007/s11684-018-0646-8 [DOI] [PubMed] [Google Scholar]
- 4. Siegel R. L., Miller K. D., and Jemal A. (2019) Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 5. Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., and Jemal A. (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 6. Hirsch F. R., Scagliotti G. V., Mulshine J. L., Kwon R., Curran W. J., Wu Y. L., and Paz-Ares L. (2017) Lung cancer: current therapies and new targeted treatments. Lancet 389, 299–311 10.1016/S0140-6736(16)30958-8 [DOI] [PubMed] [Google Scholar]
- 7. Asselin-Paturel C., Echchakir H., Carayol G., Gay F., Opolon P., Grunenwald D., Chouaib S., and Mami-Chouaib F. (1998) Quantitative analysis of Th1, Th2 and TGF-β1 cytokine expression in tumor, TIL and PBL of non-small cell lung cancer patients. Int. J. Cancer 77, 7–12 [DOI] [PubMed] [Google Scholar]
- 8. Xue J., Lin X., Chiu W. T., Chen Y. H., Yu G., Liu M., Feng X. H., Sawaya R., Medema R. H., Hung M. C., and Huang S. (2014) Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β–dependent cancer metastasis. J. Clin. Invest. 124, 564–579 10.1172/JCI71104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang L., Yang H., Lei Z., Zhao J., Chen Y., Chen P., Li C., Zeng Y., Liu Z., Liu X., and Zhang H. T. (2016) Repression of TIF1γ by SOX2 promotes TGF-β–induced epithelial–mesenchymal transition in non–small-cell lung cancer. Oncogene 35, 867–877 10.1038/onc.2015.141 [DOI] [PubMed] [Google Scholar]
- 10. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ying Z., Tian H., Li Y., Lian R., Li W., Wu S., Zhang H. Z., Wu J., Liu L., Song J., Guan H., Cai J., Zhu X., Li J., and Li M. (2017) CCT6A suppresses SMAD2 and promotes prometastatic TGF-β signaling. J. Clin. Invest. 127, 1725–1740 10.1172/JCI90439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen N., Balasenthil S., Reuther J., and Killary A. M. (2014) DEAR1, a novel tumor suppressor that regulates cell polarity and epithelial plasticity. Cancer Res. 74, 5683–5689 10.1158/0008-5472.CAN-14-1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo X., Ramirez A., Waddell D. S., Li Z., Liu X., and Wang X. F. (2008) Axin and GSK3-control Smad3 protein stability and modulate TGF-signaling. Genes Dev. 22, 106–120 10.1101/gad.1590908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maeno-Hikichi Y., Chang S., Matsumura K., Lai M., Lin H., Nakagawa N., Kuroda S., and Zhang J. F. (2003) A PKCε-ENH-channel complex specifically modulates N-type Ca2+ channels. Nat. Neurosci. 6, 468–475 10.1038/nn1041 [DOI] [PubMed] [Google Scholar]
- 15. Te V. A., and Bagowski C. P. (2007) PDZ and LIM domain-encoding genes: molecular interactions and their role in development. ScientificWorldJournal 7, 1470–1492 10.1100/tsw.2007.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng M., Cheng H., Banerjee I., and Chen J. (2010) ALP/Enigma PDZ–LIM domain proteins in the heart. J. Mol. Cell Biol. 2, 96–102 10.1093/jmcb/mjp038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin C., Guo X., Lange S., Liu J., Ouyang K., Yin X., Jiang L., Cai Y., Mu Y., Sheikh F., Ye S., Chen J., Ke Y., and Cheng H. (2013) Cypher/ZASP is a novel A-kinase anchoring protein. J. Biol. Chem. 288, 29403–29413 10.1074/jbc.M113.470708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan Y., Tsukamoto O., Nakano A., Kato H., Kioka H., Ito N., Higo S., Yamazaki S., Shintani Y., Matsuoka K., Liao Y., Asanuma H., Asakura M., Takafuji K., Minamino T., et al. (2015) Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat. Commun. 6, 6137 10.1038/ncomms7137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lasorella A., and Iavarone A. (2006) The protein ENH is a cytoplasmic sequestration factor for Id2 in normal and tumor cells from the nervous system. Proc. Natl. Acad. Sci. U.S.A. 103, 4976–4981 10.1073/pnas.0600168103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elbediwy A., Vanyai H., Diaz-de-la-Loza M. D., Frith D., Snijders A. P., and Thompson B. J. (2018) Enigma proteins regulate YAP mechanotransduction. J. Cell Sci. 131, jcs221788 10.1242/jcs.221788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shui I. M., Lindström S., Kibel A. S., Berndt S. I., Campa D., Gerke T., Penney K. L., Albanes D., Berg C., Bueno-de-Mesquita H. B., Chanock S., Crawford E. D., Diver W. R., Gapstur S. M., Gaziano J. M., et al. (2014) Prostate cancer (PCa) risk variants and risk of fatal PCa in the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. Eur. Urol. 65, 1069–1075 10.1016/j.eururo.2013.12.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei X., Zhang Y., Yu S., Li S., Jiang W., Zhu Y., Xu Y., Yang C., Tian G., Mi J., Bergquist J., Zhao M., and Song F. (2018) PDLIM5 identified by label-free quantitative proteomics as a potential novel biomarker of papillary thyroid carcinoma. Biochem. Biophys. Res. Commun. 499, 338–344 10.1016/j.bbrc.2018.03.159 [DOI] [PubMed] [Google Scholar]
- 23. Liu X., Chen L., Huang H., Lv J. M., Chen M., Qu F. J., Pan X. W., Li L., Yin L., Cui X. G., Gao Y., and Xu D. F. (2017) High expression of PDLIM5 facilitates cell tumorigenesis and migration by maintaining AMPK activation in prostate cancer. Oncotarget 8, 98117–98134 10.18632/oncotarget.20981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen T., Zhou G., Zhou Q., Tang H., Ibe J. C., Cheng H., Gou D., Chen J., Yuan J. X., and Raj J. U. (2015) Loss of microRNA-17∼92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am. J. Respir. Crit. Care Med. 191, 678–692 10.1164/rccm.201405-0941OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng H., Chen T., Tor M., Park D., Zhou Q., Huang J. B., Khatib N., Rong L., and Zhou G. (2016) A high-throughput screening platform targeting PDLIM5 for pulmonary hypertension. J. Biomol. Screen. 21, 333–341 10.1177/1087057115625924 [DOI] [PubMed] [Google Scholar]
- 26. Hamidi H., Lilja J., and Ivaska J. (2017) Using xCELLigence RTCA instrument to measure cell adhesion. Bio. Protoc. 7, e2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fernández-Cortés M., Delgado-Bellido D., and Oliver F. J. (2019) Vasculogenic mimicry: become an endothelial cell “but not so much.” Front. Oncol. 9, 803 10.3389/fonc.2019.00803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen H., Sun J., Buckley S., Chen C., Warburton D., Wang X. F., and Shi W. (2005) Abnormal mouse lung alveolarization caused by Smad3 deficiency is a developmental antecedent of centrilobular emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L683–L691 10.1152/ajplung.00298.2004 [DOI] [PubMed] [Google Scholar]
- 29. Liu Z., Zhan Y., Tu Y., Chen K., Liu Z., and Wu C. (2015) PDZ and LIM domain protein 1(PDLIM1)/CLP36 promotes breast cancer cell migration, invasion and metastasis through interaction with α-actinin. Oncogene 34, 1300–1311 10.1038/onc.2014.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen H. N., Yuan K., Xie N., Wang K., Huang Z., Chen Y., Dou Q., Wu M., Nice E. C., Zhou Z. G., and Huang C. (2016) PDLIM1 stabilizes the E-cadherin/β-catenin complex to prevent epithelial–mesenchymal transition and metastatic potential of colorectal cancer cells. Cancer Res. 76, 1122–1134 10.1158/0008-5472.CAN-15-1962 [DOI] [PubMed] [Google Scholar]
- 31. Huang Z., Zhou J. K., Wang K., Chen H., Qin S., Liu J., Luo M., Chen Y., Jiang J., Zhou L., Zhu L., He J., Li J., Pu W., Gong Y., et al. (2020) PDLIM1 inhibits tumor metastasis through activating hippo signaling in hepatocellular carcinoma. Hepatology 71, 1643–1659 10.1002/hep.30930 [DOI] [PubMed] [Google Scholar]
- 32. Bowe R. A., Cox O. T., Ayllón V., Tresse E., Healy N. C., Edmunds S. J., Huigsloot M., and O'Connor R. (2014) PDLIM2 regulates transcription factor activity in epithelial-to-mesenchymal transition via the COP9 signalosome. Mol. Biol. Cell 25, 184–195 10.1091/mbc.E13-06-0306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun F., Xiao Y., and Qu Z. (2015) Oncovirus Kaposi sarcoma herpesvirus (KSHV) represses tumor suppressor PDLIM2 to persistently activate nuclear factor κB (NF-κB) and STAT3 transcription factors for tumorigenesis and tumor maintenance. J. Biol. Chem. 290, 7362–7368 10.1074/jbc.C115.637918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun F., Li L., Yan P., Zhou J., Shapiro S. D., Xiao G., and Qu Z. (2019) Causative role of PDLIM2 epigenetic repression in lung cancer and therapeutic resistance. Nat. Commun. 10, 5324 10.1038/s41467-019-13331-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fu C., Li Q., Zou J., Xing C., Luo M., Yin B., Chu J., Yu J., Liu X., Wang H. Y., and Wang R. F. (2019) JMJD3 regulates CD4 T cell trafficking by targeting actin cytoskeleton regulatory gene Pdlim4. J. Clin. Invest. 129, 4745–4757 10.1172/JCI128293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Massagué J. (2000) How cells read TGF-β signals. Nat. Rev. Mol. Cell. Biol. 1, 169–178 10.1038/35043051 [DOI] [PubMed] [Google Scholar]
- 37. Weinstein M., Yang X., and Deng C. (2000) Functions of mammalian Smad genes as revealed by targeted gene disruption in mice. Cytokine Growth Factor Rev. 11, 49–58 10.1016/S1359-6101(99)00028-3 [DOI] [PubMed] [Google Scholar]
- 38. Bierie B., and Moses H. L. (2006) Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520 10.1038/nrc1926 [DOI] [PubMed] [Google Scholar]
- 39. Pedersen H., Grondahl-Hansen J., Francis D., Osterlind K., Hansen H. H., Dano K., and Brunner N. (1994) Urokinase and plasminogen activator inhibitor type 1 in pulmonary adenocarcinoma. Cancer Res. 54, 120–123 [PubMed] [Google Scholar]
- 40. Sundqvist A., Morikawa M., Ren J., Vasilaki E., Kawasaki N., Kobayashi M., Koinuma D., Aburatani H., Miyazono K., Heldin C. H., van Dam H., and Ten D. P. (2018) JUNB governs a feed-forward network of TGFβ signaling that aggravates breast cancer invasion. Nucleic Acids Res. 46, 1180–1195 10.1093/nar/gkx1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aggarwal K., and Massagué J. (2012) Ubiquitin removal in the TGF-β pathway. Nat. Cell Biol. 14, 656–657 10.1038/ncb2534 [DOI] [PubMed] [Google Scholar]
- 42. Kales S. C., Nau M. M., Merchant A. S., and Lipkowitz S. (2014) Enigma prevents Cbl-c–mediated ubiquitination and degradation of RETMEN2A. PLoS One 9, e87116 10.1371/journal.pone.0087116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xia T., Lévy L., Levillayer F., Jia B., Li G., Neuveut C., Buendia M. A., Lan K., and Wei Y. (2013) The four and a half LIM-only protein 2 (FHL2) activates transforming growth factor β (TGF-β) signaling by regulating ubiquitination of the E3 ligase Arkadia. J. Biol. Chem. 288, 1785–1794 10.1074/jbc.M112.439760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xin H., Xu X., Li L., Ning H., Rong Y., Shang Y., Wang Y., Fu X. Y., and Chang Z. (2005) CHIP controls the sensitivity of transforming growth factor-β signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 280, 20842–20850 10.1074/jbc.M412275200 [DOI] [PubMed] [Google Scholar]
- 45. Sthanam L. K., Barai A., Rastogi A., Mistari V. K., Maria A., Kauthale R., Gatne M., and Sen S. (2017) Biophysical regulation of mouse embryonic stem cell fate and genomic integrity by feeder derived matrices. Biomaterials 119, 9–22 10.1016/j.biomaterials.2016.12.006 [DOI] [PubMed] [Google Scholar]
- 46. Cheng H., Kimura K., Peter A. K., Cui L., Ouyang K., Shen T., Liu Y., Gu Y., Dalton N. D., Evans S. M., Knowlton K. U., Peterson K. L., and Chen J. (2010) Loss of enigma homolog protein results in dilated cardiomyopathy. Circ. Res. 107, 348–356 10.1161/CIRCRESAHA.110.218735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baudin B., Bruneel A., Bosselut N., and Vaubourdolle M. (2007) A protocol for isolation and culture of human umbilical vein endothelial cells. Nat. Protoc. 2, 481–485 10.1038/nprot.2007.54 [DOI] [PubMed] [Google Scholar]
- 48. Jiao J., Zhang R., Li Z., Yin Y., Fang X., Ding X., Cai Y., Yang S., Mu H., Zong D., Chen Y., Zhang Y., Zou J., Shao J., and Huang Z. (2018) Nuclear Smad6 promotes gliomagenesis by negatively regulating PIAS3-mediated STAT3 inhibition. Nat. Commun. 9, 2504 10.1038/s41467-018-04936-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li S., Wang L., Zhao Q., Liu Y., He L., Xu Q., Sun X., Teng L., Cheng H., and Ke Y. (2014) SHP2 positively regulates TGFβ1-induced epithelial–mesenchymal transition modulated by its novel interacting protein Hook1. J. Biol. Chem. 289, 34152–34160 10.1074/jbc.M113.546077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Staruschenko A., Jeske N. A., and Akopian A. N. (2010) Contribution of TRPV1–TRPA1 interaction to the single channel properties of the TRPA1 channel. J. Biol. Chem. 285, 15167–15177 10.1074/jbc.M110.106153 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data are included in the article.