

Abstract
Prader-Willi syndrome (PWS) is caused by the loss of function of the paternally inherited 15q11-q13 locus. This region is governed by genomic imprinting, a phenomenon in which genes are expressed exclusively from one parental allele. The genomic imprinting of the 15q11-q13 locus is established in the germline and is largely controlled by a bipartite imprinting centre. One part, termed the Prader-Willi syndrome imprinting center (PWS-IC), comprises a CpG island that is unmethylated on the paternal allele and methylated on the maternal allele. The second part, termed the Angelman syndrome imprinting centre, is required to silence the PWS_IC in the maternal germline. The loss of the paternal contribution of the imprinted 15q11-q13 locus most frequently occurs owing to a large deletion of the entire imprinted region but can also occur through maternal uniparental disomy or an imprinting defect. While PWS is considered a contiguous gene syndrome based on large-deletion and uniparental disomy patients, the lack of expression of only non-coding RNA transcripts from the SNURF-SNRPN/SNHG14 may be the primary cause of PWS. Patients with small atypical deletions of the paternal SNORD116 cluster alone appear to have most of the PWS related clinical phenotypes. The loss of the maternal contribution of the 15q11-q13 locus causes a separate and distinct condition called Angelman syndrome. Importantly, while much has been learned about the regulation and expression of genes and transcripts deriving from the 15q11-q13 locus, there remains much to be learned about how these genes and transcripts contribute at the molecular level to the clinical traits and developmental aspects of PWS that have been observed.
Keywords: Prader-Willi syndrome, imprinting, non-coding RNA, neurodevelopmental disorder, snoRNA, epigenetics
1. Overview
Prader-Willi Syndrome (PWS) is a neurodevelopmental disorder with hallmark traits of hypotonia, hypogonadism and hyperphagia/obesity. It affects approximately 1 out of 15 000 live births and currently has no known cure [1–3]. Although patients universally possess the hallmark traits, they can also demonstrate growth hormone deficiency, characteristic facial features, developmental delay and behavioural problems. The onset of various neuroendocrine phenotypes suggests that PWS primarily impacts the hypothalamus although other organs might still be affected. Patients are diagnosed at birth and are followed by endocrinologists throughout their lives. Despite several advancements in clinical care, the majority of individuals with PWS have a life expectancy of 29.5 years with the most common causes of mortality being from respiratory, cardiac and gastrointestinal failures [4].
PWS is caused by the loss of function of the paternally inherited 15q11-q13 locus. This region is governed by genomic imprinting, a phenomenon in which genes are expressed exclusively from one parental allele. The genomic imprinting of the 15q11-q13 locus is established in the germline and is largely controlled by a bipartite imprinting centre. One part, termed the Prader-Willi syndrome imprinting centre (PWS-IC) [5–7], comprises a CpG island that is unmethylated on the paternal allele and methylated on the maternal allele. The second part, termed the Angelman syndrome imprinting centre (AS-IC), is required to silence the PWS_IC in the maternal germline [8,9]. The loss of the paternal contribution of the imprinted 15q11-q13 locus most frequently occurs owing to a large deletion (LD) of the entire imprinted region (LD approx. 5 Mb, 65%-75%) [10,11] but can also occur through maternal uniparental disomy (UPD, 20%–30%) [12–14] or an imprinting defect (ID) (1%–3%) [15,16] (figure 1). Large deletions typically occur between five common breakpoint regions illustrated in figure 2. While PWS is considered a contiguous gene syndrome based on LD and UPD patients, the lack of expression of only non-coding RNA transcripts from the SNURF-SNRPN/SNHG14 may be the primary cause of PWS. Patients with small atypical deletions (SD) of the paternal SNORD116 cluster alone appear to have most of the PWS related clinical phenotypes [17–22]. However, the milder phenotypes present in these patients probably indicate that concurrent absence of other regions in the locus may contribute to greater severity of PWS phenotype. The loss of the maternal contribution of the 15q11-q13 locus causes a separate and distinct condition called Angelman syndrome [23]. Importantly, while much has been learned about the regulation and expression of genes and transcripts deriving from the 15q11-q13 locus, there remains much to be learned about how these genes and transcripts contribute at the molecular level to the clinical traits and developmental aspects of PWS that have been observed.
Figure 1.

The chr15q11-q13 region is genomically imprinted, with some genes only expressed from the paternal chromosome and some only from the maternal chromosome. Prader-Willi syndrome is associated with loss of expression from the paternal chromosome. This can occur either through paternal deletion, uniparental disomy (UPD) or an imprinting defect (ID).
Figure 2.

Overview of the chr15q11-q13 region. Genes in blue are expressed only from the paternal chromosome (PAT) and genes in red are expressed only from the maternal chromosome (MAT). Genes in grey are expressed biallelically and those in black are silent imprinted genes. ATP10A is partially imprinted (hatched boxes). Expressed non-coding regions are denoted in green. The imprinting centre (PWS-IC) that controls paternal chromosome expression is noted. The Angelman syndrome imprinting centre (AS-IC) lies within the U-exons on the paternal chromosome. Unfilled circles represent unmethylated DMRs and filled circles represent methylated DMRs. PWS patients often contain genetic deletions between breakpoints 1 or 2 (BP1, BP2) and breakpoints 3, 4 or 5 (BP3–5). Transcription is noted by horizontal arrows and is discussed in detail in the text. Not drawn to scale.
2. Clinical features
2.1. Diagnosis
Patients suspected of having PWS are often first screened using a DNA methylation assay for the PWS-IC [24–26] (figure 3). Unaffected individuals will show one unmethylated allele and one methylated allele. However, the vast majority of PWS patients will show only a methylated allele. The PWS subtype can be differentiated further through additional assays. The deletion subtype can be identified by chromosomal microarray or DNA fluorescence in situ hybridization (FISH). The former assay can also identify the precise deletion breakpoints as well as microdeletions involving the IC and SNORD116 cluster, depending on the size limitations of the microarray analysis. The UPD and IC defect subtypes are determined by interrogating the parental inheritance of the two chromosome 15 alleles [27]. The parents of the proband are typed to identify their specific microsatellite marker alleles. If the proband has microsatellite marker contribution from one parent, the proband has UPD, whereas contributions from both parents indicate ID. Rare cases of chromosomal rearrangements such as translocations and inversions are detected by using both FISH and chromosomal karyotyping. A very rare cohort of patients with PWS owing to deletions involving the SNORD116 cluster may not test positive using the DNA methylation test. Therefore, if PWS is suspected and the DNA methylation test is negative, a chromosomal microarray may still be warranted.
Figure 3.

Workflow for PWS diagnosis. See text for details.
Advanced diagnostics tools such as methylation specific-multiplex ligation-dependent probe amplification allow for simultaneous evaluation of DNA methylation and the presence of deletions [28]. The probe targets five different differentially methylated regions (DMR) in the locus and can help identify both IC and SNORD116 cluster microdeletions. If no deletions are detected, DNA polymorphism assay still must be conducted to differentiate between UPD and ID subtypes.
2.2. Nutritional phases
The different stages of PWS can be divided into phases 0–4 based on the onset of specific nutritional phenotypes [29] (figure 4). Seven distinct phases were identified by Miller et al. Individuals with PWS demonstrate decreased fetal movements and present at birth with unexplained failure to thrive and severe hypotonia (phase 0). These traits continue in the newborn phase (phase 1a: 0–9 months) and are followed by approximately a 17-month period of normal development (phase 1b: 9–25 months). In the childhood years, PWS patients begin to develop metabolic syndrome with weight gain despite the absence of additional food consumption (phase 2a: 2.1–4.5 years). The patients then begin to develop hyperphagia with some satiety (phase 2b: 4.5–8 years). Through adolescence and into adulthood, the patients' metabolic syndrome and hyperphagia continue to worsen and peaks around this time (phase 3: 8 years–adulthood). The appetite is reported to be impossible to satiate for some patients during this period. The hyperphagic drive begins to decrease and becomes satiable again for some patients (phase 4: adulthood). Many behavioural and cognitive problems that are present through the clinical phases are heavily correlated to the degree of patients’ hyperphagia [30].
Figure 4.

PWS clinical phenotypes, their prevalence and age of onset.
2.3. Hallmark traits
Hypotonia is most prominent in the neonatal phase. Patients demonstrate severe weakness, poor reflexes, decreased arousal and poor suck/appetite [29,31]. These traits lead to failure to thrive and often require patients to be placed on feeding tubes for various amounts of time. The cause of hypotonia is central in nature (deficiencies in growth hormone (GH), thyroid stimulating hormone, and cortisol) [32] as neuromuscular studies yield insignificant findings [33]. Hypotonia begins to improve once the patients are able to feed themselves and becomes mild in adulthood [29,31]. The lower muscle tone in patients leads to decreased energy expenditure and lower overall caloric requirements.
Hypogonadism is noted at birth and is present throughout the patient's lifetime. Patients of both sexes demonstrate genital hypoplasia, incomplete puberty, underdevelopment of secondary sexual characteristics and infertility later in life [34,35]. Hypogonadism was initially believed to be entirely owing to hypothalamic deficits. However, decrease in levels of hormones directly synthesized by the gonads in non-hypogonadotropic patients indicate that both the hypothalamus and the primary gonads may be involved [35–38].
Obesity accompanied by hyperphagia is perhaps the most recognizable feature of PWS. The degree and onset of hyperphagia and obesity depend on the nutritional phases mentioned in the previous section. PWS patients' food-seeking behaviour is thought to be lack of satiety owing to hypothalamic dysfunction. Both children and adults with PWS have been found to have significantly elevated levels of the orexigneic hormone ghrelin [39–41]. However, short and long-term pharmacological interventions that lowered ghrelin to physiological levels failed to improve hyperphagia and obesity in children (age 11–14) and adults (age 25) [42–44]. Thus it is unlikely that a single pathway controls appetite in PWS patients. The lack of satiety, metabolic syndrome in phase 2a and lower caloric requirements (see hypotonia) are believed to be primary contributors to the observed obesity in patients.
2.4. Other endocrinologic traits
PWS is also associated with a number of other traits, including hypothalamic dysfunction, respiratory distress, sleep disturbance, type 2 diabetes, musculoskeletal issues and behavioural problems (figure 4). PWS patients suffer from growth hormone deficiency and demonstrate reduced growth hormone secretion both in childhood and adulthood [32,45]. Patients have short statures in childhood and the absence of a growth spurt during puberty results in more pronounced phenotype in adulthood [46]. Central hypothyroidism is reported in a quarter of children with PWS. The lower circulating levels of T3 and T4 [47,48] are believed to compound patients' symptoms such as hypotonia and obesity. Later in life, the incidence of hypothyroidism drops to comparable levels to that of unaffected populations [49]. Central adrenal insufficiency (CAI) is also noted in some PWS patients. The lower levels of cortisol are believed to disturb the metabolism of carbohydrates, proteins and fats in PWS patients. The precise prevalence of CAI is to be determined as one study reported prevalence up to 60% [50] while others reported much lower prevalence [45,51]. type 2 diabetes is observed in a quarter of PWS patients and is a secondary complication of obesity [52]. However, it is rarely observed in the absence of obesity in PWS patients [47]. Maintenance of an appropriate caloric diet, growth hormone therapy (GHT), and counselling can dramatically reduce the incidence of obesity and type 2 diabetes in PWS patients.
2.4.1. Respiratory distress
Respiratory distress in PWS patients is multifactorial in origin. During a normal physiological response, the hypothalamus helps adjust the respiratory rate to compensate for increase in carbon dioxide and decrease in oxygen levels. However, PWS patients often show an imbalance in this response owing to hypothalamic dysfunction and do not compensate adequately to hypercapnic states [53,54]. The hypotonia associated with PWS can also lead to poor respiratory muscle tone and depressed respiratory response [55]. This feature can lead to increased aspiration and respiratory infection owing to weaker respiratory musculature. Lastly, obesity can lead to obstructive sleep apnea [56].
2.4.2. Sleep disturbances
Alterations in sleep patterns are well reported and can be caused by both hypothalamic dysfunction and respiratory distress [56–59]. Disturbances in crucial hormones such as orexin and acetyl cholinergic neurons in the pedunculo-pontine tegmental nucleus lead to abnormalities in the circadian rhythm, sleep/wake cycles and sleep architecture [57,60,61]. Abnormalities in respiratory response and illnesses such as obstructive sleep apnea can further compound sleep disturbances in patients [57,58].
2.4.3. Behavioural problems
PWS patients demonstrate both hyperphagia related and non-hyperphagia related behavioural problems. Non-hyperphagia related problems include tantrums, stubbornness, obsessive compulsive disorder and skin picking [62–66]. These behaviours are heavily correlated with the patient's degree of obesity and hyperphagia [63]. Food seeking behavioural problems such as stealing, manipulative behaviour and self-injury are also well documented [62]. A subset of PWS patients are also diagnosed with autism spectrum disorder (ASD), attention deficit hyperactivity disorder, and psychosis which can further compound behavioural problems [67–70].
2.4.4. Prader-Willi syndrome facial features
PWS patients present with distinct facial features such as narrow temple and nasal bridge, almond shaped eyes, thin upper lip and downturned mouth (collectively referred to as PWS facial features). It is reported that PWS facial features may not be present at birth and may develop over a patient's life. In addition, PWS patients have small hands and feet from GH deficiency [46] Osteoporosis leading to fractures and scoliosis is also a concern for some patients [71].
2.4.5. Life expectancy
The average life expectancy for PWS patients is currently 29.5 years and the causes of mortality differ greatly between adult and child patients [4]. Cardiac, pulmonary and gastrointestinal failures are the leading causes of death. However, complications from type 2 diabetes and infections are also reported. In pediatric patients, the most common cause of mortality is respiratory failure and infections [72]. PWS patients are also vulnerable to sudden and unexpected death (SED). A myriad of studies have attempted to identify the cause of SED in PWS patients. Although no direct cause was identified, cardiovascular disease, respiratory illness and thrombosis were identified as potential risk factors for increased SED [4]. The role of CAI in SED is less certain as a few studies have found lower incidences of CAI in PWS patients [45,51] than previously reported [50].
2.5. Genotype–phenotype correlations
Several correlations between different PWS genetic etiologies and clinical phenotypes have been noted. More studies are required to truly elucidate the extent and significance of these correlations (figure 5).
Figure 5.
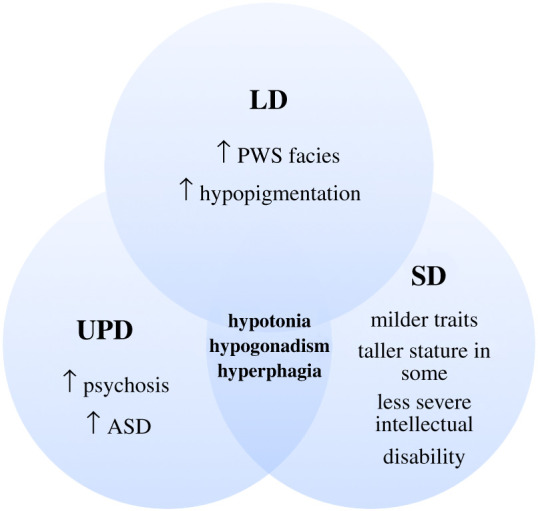
Features shared and distinct among patients with the three PWS genotypes. The hallmark traits are still shared by all three groups.
2.5.1. Effects of large deletions
Deletion patients are reported to have increased occurrence of PWS facial features [12,13], hypopigmentation (owing to deletion of one copy of Oculocutaneous Albinism II gene (OCA2)) [13,73,74] and intellectual disability [74]. Some studies report that type I deletion patients (BP1 to BP3) have exacerbated cases of intellectual disability and compulsory behaviour compared to type II deletion patients (BP2 to BP3) [75,76].
2.5.2. Uniparental disomy
UPD patients are reported to have less PWS facial features [12,13] and little to no hypopigmentation [13,73,74]. However, they are reported to have increased risk for psychosis [67,70] and ASD [68,69]. Interestingly, PWS UPD patients are also reported to have less compulsory and behavioural problems despite these increased risks [76,77]. The differences observed between UPD and LD patients may potentially be owing to expression of two copies of UBE3A in UPD patients.
2.5.3. Small deletions
PWS patients with SD in the proximal SNHG14 transcript encompassing the SNORD116 cluster are reported to demonstrate milder phenotype and absence of some clinical traits associated with PWS [17–22]. These patients still possess the hallmark traits of hypotonia, hypogonadism and hyperphagia/obesity albeit in milder forms. Some patients had normal to tall stature and absence of PWS facial features [19–22]. These findings might indicate that the absence of SNORD116 plays a crucial role in the development of PWS phenotype. However, the absence of other surrounding regions may play a role in the severity of the phenotypes as seen in LD and UPD patients.
2.6. Current clinical interventions
Current treatments address a particular phenotype and are not aimed to cure the disorder.
2.6.1. Hypotonia: growth hormone therapy
The natural history of PWS can be significantly improved with clinical interventions. Endocrinologists in almost all instances administer GHT for PWS patients from infancy. GHT improves the body composition by increasing muscle mass, reducing body fat while normalizing height [78,79]. It is also reported that GHT improves cognitive function and IQ scores for patients [80–82]. In adults, GHT was also shown to be beneficial by improving body composition, muscle mass and sleep-disordered breathing [83]. The impact of GHT on body mass index, hyperphagia and food-seeking behaviour is reported to be minimal but more future studies are warranted. Along with GHT, physical therapy can greatly aid PWS patients develop and maintain muscle tone in childhood and adulthood [84].
Although GHT provides tremendous benefits, each patient must be monitored carefully for potential side effects. PWS patients with CAI may experience an adrenal crisis owing to increased metabolism of cortisol [85]. Those with respiratory illness may risk hypoxemia and sleep disturbance owing to increased basal metabolism in the absence of respiratory compensation [86]. Patients with diabetes may also experience worsening of their symptoms owing to antagonism of insulin [87]. Thus endocrinologists must closely monitor each individual PWS patient based on their response to treatment. Despite these potential adverse effects, the long-term benefits of GHT are still considered to outweigh the potential risks [88,89].
2.6.2. Hypogonadism
Sex hormone therapy is administered and often helps patients develop secondary sex characteristics [34]. The dosage of testosterone and oestrogen has to be carefully monitored with each patient to avoid any negative side effects. With testosterone treatment, exacerbation of behavioural problems such as aggression may be noted. With oestrogen treatment, osteoporosis and possible fertility must be considered. Fertility was reported in some female PWS patients and thus sex education has to be emphasized [90,91].
2.6.3. Hyperphagia/obesity
Pharmaceutical interventions are currently unavailable to address hyperphagia. PWS patients are followed by both endocrinologists and dietitians. Caloric requirements are set at 60% to 80% of the age-appropriate regular daily allowance with vitamin supplementation (Miller diet) [92]. Additionally, parents and carers are coached on exercise programmes and ways to limit alternate food access by PWS patients.
2.6.4. Sleep disturbances
A sleep study is recommended for most PWS patients to monitor for both potential respiratory distress and effects of GHT [86]. Interventions such as tonsillectomy and continuous positive airway pressure/bilevel positive airway pressure are available for patients with obstructive sleeping difficulties [93].
2.6.5. Behavioural therapy
Speech therapy and special courses are often used to help supplement any learning difficulties [94]. Serotonin reuptake inhibitors have been reported to be effective for obsessive compulsive disorder [95] and selective serotonin reuptake inhibitors have been reported to be effective for psychosis [96].
2.6.6. Experimental interventions in progress
Several experimental interventions are currently being evaluated. These interventions are often designed to address molecular deficits found in PWS. For example, PWS patients are reported to have reduced oxytocin secreting neurons which may help decrease appetite and promote satiety [97]. As a result, oxytocin therapy was developed to address this deficiency. Upon treatment, patients were reported to have reduced appetite and improved behaviour [97,98]. For hypotonia, a myostatin inhibitor may promote muscle growth and tone, and may improve metabolism [99] for hyperphagia/obesity. There are a number of other experimental interventions to treat hyperphagia/obesity as well [98,100–104].
3. Prader-Willi syndrome molecular genetics
The genomic imprinting of the 15q11-q13 locus is established in the germline and is controlled by the bipartite imprinting centre. PWS-IC [5–7] (figure 2) comprises a CpG island that is unmethylated on the paternal allele and methylated on the maternal allele and includes the first exon of the SNURF-SNRPN gene. The other part, the AS-IC represses the PWS-IC in the maternal germline and silences the maternal allele of chromosome 15q11-q13 in somatic tissues. The PWS-IC serves as a promoter for the approximately 600 kb long SNRPN transcript that serves not only as a pre-messenger RNA (mRNA) for SNURF and SNRPN but also encodes a non-coding RNA, SNHG14, which is a host transcript for the production of a number of both long and short non-coding RNAs such as SPA1, SPA2, sno-lncRNAs 1–5, SNORD116, IPW, SNORD115 and UBE3A-ATS. The proximal portion of SNHG14, between SNRPN and IPW is expressed in virtually all cell types in humans. The distal portion comprised of SNORD115 and UBE3A-ATS is only expressed in neurons. UBE3A-ATS, silences paternal UBE3A in neurons. Thus, UBE3A is biallelically expressed in many tissues but is expressed exclusively from the maternal allele in neurons [105,106].
3.1. The SNRPN transcript
The bicistronic SNRPN transcript encodes two protein-coding genes SNURF and SNRPN as well as a approximately 600 kb long non-coding RNA (lncRNA) termed SNHG14 [107–109]. SNURF is encoded by the first three exons and has unknown function while SNRPN is encoded by exons 4 through to 10 and produces SMN, a non-essential protein which may be involved in mRNA splicing [24,25]. The SNHG14 non-coding RNA initiates at the upstream exons of SNRPN and hosts multiple RNA species with a diverse range of putative functions. For example, two clusters of small nucleolar RNAs (snoRNAs) are processed from the introns of SNHG14. The SNORD116 cluster of 30 box C/D snoRNAs is closer to the PWS-IC and is expressed in most tissues while the SNORD115 cluster of 45 box C/D snoRNAs are more distal and are expressed almost exclusively in neurons [105,106]. snoRNA ended, polyadenylated RNAs and IPW are processed lncRNAs with incompletely understood functions. As mentioned earlier, the distal-most portion of SNHG14 encodes UBE3A-ATS, which silences paternal UBE3A via transcriptional interference [110]. By RNA FISH, the SNHG14 host gene appears within nuclei as a large RNA cloud with unknown function that localizes near its site of transcription on the paternally inherited allele of chromosome 15 [109].
3.2. Box C/D small nucleolar RNAs
Of more than 100 reported post-transcriptional RNA modifications [111,112], most are found in ribosomal RNAs (rRNAs), transfer RNAs (tRNAs) and other small RNAs. However, an increasing number are becoming identified in mRNAs and lncRNAs. The most abundant RNA modifications are 2′-O methylation and pseudouridylation. These are mostly directed by snoRNAs that are usually concentrated in Cajal bodies or nucleoli where they modify either small nuclear RNAs (snRNAs) or rRNA, or participate in the processing of rRNA during ribosome biogenesis [113–115]. There are several hundred known snoRNAs, the majority of which are encoded in introns of protein-coding genes [116]. Box C/D snoRNAs are processed from excised and debranched introns by exonucleolytic trimming (figure 6b) [117,118] and carry out their functions in complex with specific protein components, forming ribonucleoprotein complexes (snoRNPs) consisting of the proteins NOP56, NOP58, Fibrillarin and 15.5 KD/NHPX (figure 6a) [113]. Box C/D snoRNAs harbour antisense sequences that basepair with target RNA substrates and guide the placement of 2′-O-methylation modification on the fifth basepair upstream of the D and D' box (figure 6a, blue star). 2′-O methylations directed by box C/D snoRNAs are biologically important and so far verified to exist internally only in rRNAs and snRNAs [119] and a substantial portion of known methylated sites in rRNA lie in close proximity to functional sites such as the peptidyltransferase centre, suggesting potential involvement in rRNA folding, stability and translation [119]. Interestingly, 2′-O-methylation within coding regions of artificial mRNAs has recently been reported to disrupt key steps in codon reading during cognate tRNA selection [120]. Furthermore, snoRNAs may be involved in brain development or function [121].
Figure 6.

(a) A typical box C/D snoRNP complex including the snoRNA and associated proteins. Typical snoRNPs associate via RNA complementarity with rRNAs. Fibrillarin (Fib) catalyses the 2′-O methylation of targeted rRNA regions at the 5th bp upstream of D and D′ box as indicated by the blue star. (b) Box C/D snoRNAs are processed from excised introns following debranching and exonucleolytic trimming. (c) sno-lncRNAs are produced from introns that contain two snoRNAs. Thus, they lack 5′ cap structures and 3′ poly(A) tails but are stabilized by their terminal snoRNP components. (d) SPA RNAs are processed similarly to sno-lncRNAs but can be spliced and have 5′ snoRNP structures and 3′ poly(A) tails.
3.3. The SNORD116 (HBII-85) cluster
The SNORD116 cluster of box C/D snoRNAs is expressed in most tissues but their expression is much higher in the brain. The importance of this cluster to PWS pathology is very high, because all reported deletions and mutations associated with PWS lead to loss of expression from this region. A number of atypical PWS deletions have narrowed the putative PWS critical region to approximately 80 kB primarily spanning the SNORD116 cluster (figure 4) [17–22]. While most known snoRNAs have been shown to target the modification of rRNA, the SNORD116 snoRNAs are classified as ‘orphans' because no known targets have been identified and their sequences show no significant complementarity to rRNAs. Thus, it is crucial to identify the targets and functions of SNORD116 snoRNAs. Mapping sites of 2′-O methylation on RNA molecules is challenging but several groups have recently developed genome-wide methods to chemically isolate and map the positions of sites of 2′-OMe [122,123]. With this technology, it will be important to identify SNORD116 targets by comparing results from human and animal models that do or do not express SNORD116s, although such models may not accurately reflect the full spectrum of human phenotypes. While snoRNAs are present across a vast spectrum of organisms and often share ancient and conserved elements [113], SNORD116 and SNORD115, represent blossoming groups of RNAs that may have lineage-specific molecular functions. Zhang et al. used a computational approach, snoSeeker, to investigate the evolution of imprinted snoRNAs across 12 placental mammalian species [124]. They discovered that the number of copies of the SNORD116 and SNORD115 varied widely with human and rodent lineages demonstrating the highest gains in snoRNA copies. For example, primates and rodents all possess greater than 20 copies of the SNORD116 family, while some species such as cows and elephants possess as few as 12 and 1 copies, respectively. In addition, the birth of new snoRNA copies revealed that nucleotide substitutions occurred the most within the snoRNA sequence and not in the flanking regions.
The SNORD116 snoRNA cluster has been further divided into groups 1–3 (SNOG1, SNOG2, SNOG3) based on sequence and expression heterogeneity [125]. In the hypothalamus, SNOG1 (SNORD116-1 to SNORD116-9) is expressed most highly compared to SNOG2 (SNORD116-10 to SNORD116-24) and SNOG3 (SNORD116-25 to SNORD116-29). The higher reported levels of expression may actually be owing to increased stability of individual snoRNAs mediated by snoRNP complexes. In Fibrillarin RIP-Seq experiments in ovarian teratocarcinoma PA1 cells, enrichment was observed over the first third of the SNORD116 cluster but not the latter two thirds [126]. Thus, the absence of SNOG1 may play an important role in PWS. Kocher et al. investigated the sequence similarity of the SNORD116 family across different primates and rodents [127]. Between humans and mouse, the authors noted that SNOG1 and SNOG2 shared greater homology, while SNOG3 possessed smaller overlap in homology. Thus the authors proposed that the variance phenotype that is observed in murine models may be explained by the differences in SNOG3. As primates such as chimpanzees and macaques also share significant homology in the SNORD116 sequence, primates may ultimately offer better models for PWS than mice.
Along with studies involving PWS SD patients, Burnett et al. demonstrated the importance of SNORD116 in neurons derived from both PWS patient induced pluripotent stem cells (iPSCs) and Snord116 knockout (KO) murine models [128]. These models showed reduced levels of nescient helix loop helix2 (NHLH2) and the prohormone convertase PC1 enzyme (PCSK1). Nhlh2 is reported to promote Pcsk1 expression which in turn promotes the conversion of prohormones into mature hormones. The failure of proper hormone maturation may explain the various neuroendocrine phenotypes seen in PWS.
Several murine models of PWS have been generated to study the impact of genes relevant to the disorder. These include large deletions of the locus as well as individual deletion of Snord116 cluster. Each model demonstrates slightly different phenotypes and severity based on genetic background of the mouse strain. However, both the LD and Snord116 deletion models demonstrated the same phenotype to one another [129–131]. These findings further indicate that Snord116 may be the critical gene and that its absence can be responsible for causing the majority of the PWS phenotype. Other murine models such as those with Magel2 deletions also shared some similarities with Snord116 deletion models and thus the influence of other genes in the locus cannot entirely be ruled out [132,133].
3.4. Short nucleolar-long non-coding RNAs
The SNORD116 cluster also harbours five sno-lncRNAs. These unusual RNAs have snoRNA sequences at their 5′ and 3′ caps but lack 5′-cap structures and poly(A) tails [134] (figure 6c). The SNHG14 transcript hosts 5 sno-lncRNAs within the SNORD116 cluster. sno-lncRNA1 spans from SNORD116-6 to SNORD116-7, sno-lncRNA2 spans from SNORD116-13 to SNORD116-14, sno-lncRNA3 spans from SNORD116-18 to SNORD116-19, sno-lncRNA4 spans from SNORD116-20 to SNORD116-21 and sno-lncRNA5 spans from SNORD116-26 to SNORD116-27. sno-lncRNAs are strictly retained in the nucleus and accumulate at or near their site of synthesis [134]. While the complete functions of the sno-lncRNAs are not known, they harbour multiple consensus binding sites for the Fox family of splicing regulators, and have been shown to bind RBFOX2 in nuclei and together promote specific alternative splicing patterns [134]. This and the fact that all sno-lncRNAs derive from the minimal region associated with PWS (figure 7) have led to the suggestion that changes in alternative splicing may underlie at least some of the PWS clinical features. It should be noted that while sno-lncRNAs are highly expressed in human and rhesus monkeys, they are undetectable in mouse [135], perhaps partly explaining the difference in phenotypes between SNORD116 region deletions in the different species.
Figure 7.

Comparison of reported microdeletions from patients with PWS features narrows the critical region to that spanning the SNORD116 cluster, SPA2 and the sno-lncRNAs. Not drawn to scale.
3.5. snoRNA ended, polyadenylated RNAs
SPA RNAs are a newly described class of lncRNA that possess 5′ snoRNA cap and 3′ poly(A) tails [126]. Thus, these novel RNAs, like sno-lncRNAs, lack typical 5′-cap structures that are associated with most RNA polymerase II generated spliced transcripts. The long SNHG14 primary transcript houses two SPAs (figure 6d). SPA1 is approximately 34 000 bp in length and has seven exons. The 5′ end corresponds to SNORD107 and the poly(A) tail forming the 3′ end is located upstream of SNORD109A. SPA2 is approximately 16 000 bp in length and contains 30 exons. The 5′ cap corresponds to SNORD109A while the 3′ end aligns to the 3′ end of IPW. SPA1 was shown to bind to the RNA binding protein TDP43, while SPA2 binds RBFOX2 and HNRNPM [126]. Like the SNORD116 snoRNAs and sno-lncRNAs, functions of SPA RNAs are currently not completely understood. It should be noted that while sno-lncRNAs are highly expressed in both stem cells and neurons [134], the expression of SPA1 and SPA2 is much greater in neurons than in stem cells [126]. SPA1 and particularly SPA2 are not expressed in PWS patients.
3.6. The SNORD115 (HBII-52) cluster
The SNORD115 cluster of box C/D snoRNAs consist of 45 copies and is expressed almost exclusively in neurons [107]. Unlike SNORD116, these snoRNAs are almost identical to one another and have an 18 bp complementarity to the serotonin 2c receptor (HTR2C) mRNA. This interaction has been reported to promote alternative splicing and production of mature HTR2c spliceform [107].
The HTR2C transcript has been reported to undergo three possible post transcriptional modifications. First, exclusion of exon 5b results in a shorter protein that is retained in the endoplasmic reticulum Second, the incorporation of exon 5b produces a fully functional receptor [136]. Third, the HTR2C transcript can undergo A to I editing to generate a product that includes exon 5b, but confers lower receptor activity [137,138]. It has been proposed that SNORD115 promotes the generation of the fully functional HTR2C receptor transcript by blocking of splicing silencing factors and competing with deaminases for the binding of the HTR2C transcript [139]. Because they are expressed in some PWS patients, these snoRNAs are not likely a major cause of PWS clinical manifestations but still may worsen symptoms.
3.7. The partial short nucleolar RNA debate
Through RNA-Seq, several studies identified new RNA species that are produced from further processing of snoRNAs (psnoRNAs) [140,141]. The psnoRNAs are reported to have miRNA-like properties and can affect the mRNA abundance of specific transcripts. Kishore et al. used MBII-52 (Snord115) overexpression construct in a mouse neuroblastoma cell line and observed that psnoRNAs are preferentially generated over traditional canonical snoRNAs [142]. The group also report that their overexpression construct led to the expression of snoRNAs that fail to associate with classic snoRNP proteins and instead associate with splicing factors. The same group also reported that Snord116 may undergo the same processing as Snord115 [143].
In contrast to these above studies, other groups have failed to find the presence of abundant psnoRNAs. Bortolin-Cavaillé et al. found that the majority of SNORD115 RNA species are the full length variant [144]. A smaller truncated species (larger size than psnoRNAs) was found in small abundance in mouse brain samples but completely absent in human brain samples. Both the truncated and full length SNORD115s were found to associate with the canonical snoRNP complex member fibrillarin. Thus these authors argued that the psnoRNAs observed in the Kishore studies were probably degradation products stemming from snoRNA overexpression. Galiveti et al. also failed to observe psnoRNAs across several human samples in their Northern blot experiments [145]. Consequently, the discrepancy in results from these experiments drives uncertainty about the abundance of psnoRNAs and their role in PWS.
3.8. SNORD107, SNORD64, SNORD108, SNORD109A, SNORD109B and IPW
Several single-copy Box C/D snoRNAs lie within the SNHG14 transcript. These snoRNAs are SNORD107, SNORD64, SNORD108, SNORD109A and SNORD109B. Like the SNORD116 snoRNAs, these appear to be orphans with no known targets in identified rRNA and other RNAs.
IPW is annotated as a lncRNA that was initially thought to have no functional role. In one study, however, IPW was reported to have a trans-regulatory role on the DLK1-DIO3 imprinted region on chromosome 14 [146]. Upon overexpression of IPW in PWS iPSCs, the maternally expressed genes in the DLK1-DIO3 imprinted locus was significantly downregulated. Of interest, this element also harbours a poly(A) site which serves as the termination site for the SNHG14 transcript in stem cells [105] as well as for SPA2 [126,147].
3.9. UBE3A
UBE3A encodes an E3 ubiquitin ligase that places a ubiquitin mark on proteins targeted for degradation by the proteasome [148,149]. It is known to target itself and RING1B, as well as several other putative proteins in vitro, but other targets, including bona fide in vivo targets, remain unknown. UBE3A is biallelically expressed in most tissues. However, it is imprinted in neurons by the expression of UBE3A-ATS from the paternal allele. It is expressed exclusively from the maternal allele in most neurons in the central nervous system [108]. The loss of function of UBE3A results in a separate disorder called Angelman syndrome [23].
3.10. ‘The left field genes' - MKRN3, MAGEL2 and NDN
Three genomically imprinted and paternally expressed intronless genes MKRN3, MAGEL2 and NDN lie approximately 1.3 Mb upstream of SNPRN. These genes may play a role in PWS based on the phenotypes that are observed upon their loss. However, some studies also report that the deletion of MKRN3, MAGEL2 and NDN alone do not cause a PWS phenotype [150].
MKRN3 encodes a zinc finger protein and is believed to play a role in puberty. Three frameshift mutations leading to truncation of the protein as well as a missense mutation have been reported in patients with central precocious puberty [151]. The precise function of MKRN3 is yet to be elucidated.
MAGEL2 encodes a protein that enhances the activity of an E3 ubiquitin ligase complex [152]. It is a part of the MUST complex composed of MAGEL2, USP7 and TRIM27 [153]. The complex activates the WASH complex by ubiquitination and promotes retrograde and endosomal transport of target proteins. In addition, MAGEL2 has also been reported to interact with proteins involved in the regulation of circadian rhythm [133].
Patients harbouring truncating mutations of MAGEL2 have a distinct disorder called Schaaf-Yang syndrome (SYS) [154,155]. The majority of truncating mutations arises in the region of nucleotides 1990–1996 which is described as a mutational hotspot. SYS patients have some features, such as intellectual disability and hypotonia that overlap with PWS. However, they also present with ASD, contractures and other dysmorphic features which are not common to PWS. Surprisingly, deletion of MAGEL2 has been reported to cause little to no phenotype [154,155]. These findings indicate that the truncating mutations of MAGEL2 may encode a defective protein which acts in a dominant negative fashion [155].
The potential role of Necdin (NDN) has been most studied in murine models. It encodes a DNA binding protein reported to be involved in neuronal maturation by promoting cessation of cell division and promoting axonal outgrowth [156,157]. NDN has several known interacting partners. It binds the intracellular domain of the nerve growth factor receptor along with MAGEH1 and it interacts with MAGEL2 to prevent the degradation of FEZ1, an important promoter of axonal outgrowth.
Ndn may play an especially important role in gonadotropin releasing hormone (GnRH) neurons [158]. Overexpressed NDN protein in murine models was shown to co-immunoprecipitate with a known GnRH repressor MSX. In addition, NDN was also shown to be crucial for the generation of all subtypes of GnRH neurons and their correct projections. For this reason, NDN is thought to play a role in hypogonadotropic hypogonadism seen in PWS patients.
3.11. Other genes of unknown significance
Several genes of unknown significance lie in the 15q11-q13 locus. These genes are also imprinted and expressed from the paternal allele. NPAP1/C15ORF2 is an imprinted and intronless gene that lies upstream of SNRPN and may encode a protein [159]. Two additional loci, PWRN1 and PWRN2 are annotated and lie upstream of NPAP1 and the SNRPN. These genes appear to be non-coding [160].
4. Prader-Willi syndrome epigenetics
4.1. Epigenetic regulation of the 15q11-q13 locus
Understanding the mechanism of repression of the maternal 15q11-q13 locus is critical from a therapeutic point of view. PWS patients lack paternal contribution of the 15q11-q13 locus but possess an intact but epigenetically silent set of genes on the maternal chromosome. The regulation of gene expression at the 15q11-q13 locus is largely controlled by the PWS-IC. The PWS-IC is the master regulator and also influences DMRs at MKRN3, MAGEL2 and NDN, which are unmethylated on the paternal allele and methylated on the maternal allele. The differential methylation at the PWS-IC is established in the germline. The paternal allele remains unmethylated, perhaps owing to the maintained expression from the major SNRPN promoter (PWS-IC) in sperm [160]. The maternal allele becomes methylated as the result of transcriptional activation of an oocyte-specific promoter(s) upstream of SNRPN. This transcription leads to gene body methylation as it transcribes across the PWS-IC [9,161]. The PWS-IC, and therefore the major SNRPN promoter is then repressed in somatic cells. How the PWS-IC influences methylation at the DMRs in MKRN3, MAGEL2 and NDN is not known. This mechanism establishes the initial silencing of the maternal chromosome. In recent studies, however, the existence of separate somatic imprints in silencing the maternal allele has been discovered [162].
One of the first therapeutic proof of principle studies used the global DNA methyltransferase inhibitor 5-azadeoxycytidine (5-aza-dC) in PWS patient derived lymphoblastoid cells [163]. This compound was shown to be able to demethylate the PWS-IC leading to activation of the maternal genes. Thus the activation of the normally silent maternal genes may offer a potential therapeutic option for PWS patients.
4.2. The role of ZNF274 in repressing the maternal 15q11-q13 allele
Zinc Finger Protein 274 (ZNF274) is composed of a SCAN leucine rich domain, one or two Krüppel associated box (KRAB) domains, and five C2H2 zinc finger domains and also has four isoforms (a-d) using different polyadenylation signals [164]. Isoforms b and d are shorter and possess one KRAB domain. Isoforms a and c possess two KRAB domains and are longer. The DNA sequence specificity is conferred by the five zinc finger domains. ZNF274 forms a silencing complex with SET domain bifurcated 1 (SETDB1) to deposit the repressive histone mark, H3K9me3 [165]. It is reported to co-bind to multiple genomic loci with ZNF75D but also has approximately 1000 independent binding sites across the genome [166].
Cruvinel et al. previously discovered that ZNF274 binds to six sites within the maternal allele of SNORD116 [167]. Along with the enrichment of ZNF274 on the maternal allele, the enrichment of H3K9me3 was also observed on the maternal allele. After knockdown of SETDB1, activation of maternal SNORD116 in iPSCs from patients with PWS was seen. These results led to the proposal of a model in which ZNF274 recruits SETDB1 to maternal SNORD116, where it deposits H3K9me3 and contributes to repression of the maternal allele.
In subsequent studies, ZNF274 was knocked out in iPSCs from PWS patients. Although the activation of the maternal genes was modest in iPSCs, SNORD116 was fully activated in neural precursors and neurons differentiated from them [162]. This finding suggests that ZNF274 mediated repression of maternal SNORD116 may represent a role for ZNF274 in maintaining a neuron-specific somatic imprint rather than a germline imprint. Interestingly, the activation of SNORD116 initiated from the upstream, neuron-specific exons and not the PWS-IC, which remained fully methylated. These findings also reinforce the idea that this additional somatic imprint is required to maintain the repression of the maternal genes in the neural lineage.
4.3. G9a and GLP
G9a (Euchromatic histone lysine N-methyltransferase-2) and GLP (Euchromatic histone lysine N-methyltransferase-1) may also play an important role in establishing imprinting at the 15q11-q13 locus. G9a was previously reported to be crucial in establishing CpG methylation at the PWS-IC and catalysing the placement of H3K9me2 marks on histones in mouse embryonic stem cells [168]. Interestingly, DNA methylation was unchanged at the PWS-IC at E9.5 upon G9a KO. The precise interplay between H3K9me2 and DNA methylations is not known. However, it is believed that H3K9me2 marks help recruit DNA methyltransferases to the target locus and catalyse the methylation of CpG islands [169].
More recently, Kim et al. identified two inhibitors of G9a in a screen to identify activators of maternal Snrpn in mouse fibroblasts [170]. They observed partial activation of the normally silent maternal genes such as SNORD116 in both mouse and human fibroblasts PWS models and demonstrated improved survival and growth compared to the untreated when administered by intraperitoneal injection into PWS mice. These studies revealed that the inhibition of G9a leads to decrease in H3K9me2 marks but no changes to DNA methylation at the PWS-IC. A preprint from Wu et al. confirmed the ability of G9a to activate maternal SNORD116 in neural progenitors and neurons derived from human PWS iPSCs [171]. These results probably indicate that G9a plays an important role in the establishment of DNA methylation at the PWS-IC but the activation of the maternal genes are dependent on the status of H3K9 methylation, independent of the PWS-IC DNA methylation.
The relationship between G9a/GLP and ZNF274/SETDB1 and whether they share any common pathways is currently unknown. For example, SETDB1 may depend on G9a/GLP to establish H3K9me2 before it can catalyse the placement of H3K9me3. The inhibition of G9a/GLP would thus also indirectly inhibit SETDB1 by limiting the amount of available H3K9me2 substrates. It is also possible that the G9a inhibitors also directly inhibit SETDB1. Additionally, experiments to evaluate the recruitment of G9a and H3K9me2 to the human 15q11-q13 locus are necessary. It is possible that ZNF274 may co-recruit both G9a/Glp1 and SETDB1 to the locus. However, the presence of other transcription factors and recruiters for G9a/GLP cannot be ruled out.
4.4. SMCHD1
Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) encodes an epigenetic repressor. Its loss is associated with hypometyhlation and an increase in active histone modifications in its target region through manipulation of the chromatin architecture [172–175]. Both human and murine SMCHD1 are critical in the process of X-inactivation [176,177] while murine Smchd1 is also reported to act on other loci such as the region upstream of Snrpn and Igf2r cluster [178]. SMCHD1 is believed to be involved in the embryonic development of several structures in the head such as the eyes and the nose. However, its exact involvement in these developmental pathways is unknown. Mutations of SMCHD1 have been implicated in disorders such as Facioscapulohumeral muscular dystrophy [179,180] and Bosma arhinia microphthalmia syndrome [181,182].
In the PWS locus, SMCHD1 was shown to be responsible for establishing the methylation imprint at Mkrn3, Magel2 and Ndn [183]. Smchd1 KO cells showed upregulation of these genes as well as loss of DNA methylation at the respective CpG sites. ChIP-Seq experiments also showed enrichment of H3K4me3, indicating active promoters, for Ndn and Mkrn3. SMCHD1 itself showed enrichment over the three genes as well as four other distal sites. The authors interestingly noted that four of the sites of SMCHD1 enrichment overlapped with CTCF binding sites. Thus the authors propose that SMCHD1 may antagonize CTCF mediated chromatin interactions and help establish imprinted repression of the maternal region.
The role of SMCHD1 has not been as extensively explored in human systems. It is possible that SMCHD1 may help establish imprinting for MKRN3, MAGEL2 and NDN in human cells. However, the PWS-IC is clearly the master regulator controlling the imprinting of MKRN3, MAGEL2 and NDN along with SNRPN because it has long been known that the deletion of the paternal PWS-IC is sufficient to cause the loss of expression of these upstream genes [164–166,184–186]. This begs the question of how DNA methylation at the PWS-IC, DNA methylation and/or histone modifications influenced by SMCHD1, and histone modifications regulated by ZNF274/SETDB1/G9A work together to repress maternal 15q11-q13.
The role of PWS-IC as the master regulator is also seen in murine models. Bressler et al. generated a deletion removing most of the IC in a murine model [187]. These models exhibited a similar phenotype of hypotonia and failure to thrive at birth followed by early death in 40% of the mice. The surviving mice showed developmental delay but did not develop infertility or hyperphagia/obesity. The models with a Snrpn segment deletion that left the IC intact did not demonstrate any phenotypes [188,189]. These studies also noted that deletion of the IC led to the loss of other paternally imprinted genes while Snrpn deletions sparing the IC failed to show this phenotype.
4.5. Implications for future therapeutics
The studies involving epigenetic regulators on the maternal chromosome, such as ZNF274 and G9a, may serve as therapeutic strategies for PWS (figure 8). The activation of the maternal allele may provide a permanent solution for PWS patients missing critical genes and can serve as a potential cure for the disorder. These approaches need to be further fine-tuned in the future to avoid potential genome-wide off target effects from inhibiting these epigenetic regulators. Thus future studies must explore methods to specifically exert epigenetic effects within the locus while minimizing impact at other loci. In addition, 15q11-q13 locus specific effects such as the potential silencing of UBE3A from over-activation of UBE3A-ATS also need to be monitored.
Figure 8.

The maternal allele might serve as a therapeutic target for PWS. (a) The maternal allele is normally silenced owing to epigenetic silencing, some of which involves SMCHD1, ZNF274 and H3K9me3. Colour scheme is as in figure 2. (b) Removal of repressors such as ZNF274 and G9a have been shown to be able to activate the genes on the silent maternal chromosome, although we still do not know the full extent of activation.
5. Concluding remarks
The field of PWS is constantly evolving with new discoveries driving improved outcomes for patients. Understanding the molecular underpinnings behind the clinical presentation and usage of model systems have led to the discovery of treatments that drastically improved the natural history of the disorder. For example, GHT and oxytocin therapy have greatly improved the quality of life for patients by alleviating hyperphagia and behavioural problems. However, these therapies address specific phenotypes and are not permanent cures for PWS. Recent advancements in genetic diagnosis tools have helped further pinpoint critical genes in the 15q11-q13 locus. Patients harbouring SD encompassing the SNORD116 cluster present a majority of the PWS clinical phenotypes. Thus understanding the function of SNORD116 including the roles of snoRNAs, sno-lncRNAs and SPA RNAs may be crucial for discovering molecular deficits that may exist between PWS and unaffected cells. In addition, the silent maternal 15q11-q13 allele may serve as an intriguing treatment option for PWS. Patients possess an intact set of genes on the maternal 15q11-q13 allele but are unable to express them owing to epigenetic silencing. Two promising silencing factors, ZNF274 and G9a, were identified in previous studies. The knockout of ZNF274 showed robust activation of maternal SNORD116 in neurons derived from PWS patient iPSCs. The inhibition of G9a also demonstrated activation of the maternal SNORD116 in PWS patient fibroblasts. These promising studies provide both molecular precedence and hope that innovative therapeutics involving SNORD116 and the maternal allele can be developed in the future.
Acknowledgements
We thank members of the Carmichael and Chamberlain laboratories for helpful discussions.
Data accessibility
This article has no additional data.
Authors' contributions
This article was primarily written by M.S.C. with significant input from the other authors.
Competing interests
We declare we have no competing interests.
Funding
This work was supported by grant HD099975 (to G.G.C. and S.J.C.) from the NIH and by a grant to G.G.C. from the Foundation for Prader-Willi Research.
References
- 1.McCandless SE. 2011. Clinical report-health supervision for children with Prader-Willi syndrome. Pediatrics 127, 195–204. ( 10.1542/peds.2010-2820) [DOI] [PubMed] [Google Scholar]
- 2.Vogels A, Van Den Ende J, Keymolen K, Mortier G, Devriendt K, Legius E, Fryns J-P, 2004. Minimum prevalence, birth incidence and cause of death for Prader–Willi syndrome in Flanders. Eur. J. Hum. Genet. 12, 238–240. ( 10.1038/sj.ejhg.5201135) [DOI] [PubMed] [Google Scholar]
- 3.Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H. 2001. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J. Med. Genet. 38, 792–798. ( 10.1136/jmg.38.11.792) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler MG, Manzardo AM, Heinemann J, Loker C, Loker J. 2017. Causes of death in Prader-Willi syndrome: Prader-Willi Syndrome Association (USA) 40-year mortality survey. Genet. Med. 19, 635–642. ( 10.1038/gim.2016.178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DuBose AJ, Smith EY, Johnstone KA, Resnick JL. 2012. Temporal and developmental requirements for the Prader–Willi imprinting center. Proc. Natl Acad. Sci. USA 109, 3446–3450. ( 10.1073/pnas.1115057109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnstone KA, DuBose AJ, Futtner CR, Elmore MD, Brannan CI, Resnick JL. 2006. A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Hum. Mol. Genet. 15, 393–404. ( 10.1093/hmg/ddi456) [DOI] [PubMed] [Google Scholar]
- 7.Shemer R, Hershko AY, Perk J, Mostoslavsky R, Tsuberi B-Z, Cedar H, Buiting K, Razin A. 2000. The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat. Genet. 26, 440–443. ( 10.1038/82571) [DOI] [PubMed] [Google Scholar]
- 8.Brannan CI, Bartolomei MS. 1999. Mechanisms of genomic imprinting. Curr. Opin Genet. Dev. 9, 164–170. ( 10.1016/S0959-437X(99)80025-2) [DOI] [PubMed] [Google Scholar]
- 9.Smith EY, Futtner CR, Chamberlain SJ, Johnstone KA, Resnick JL. 2011. Transcription is required to establish maternal imprinting at the Prader-Willi syndrome and Angelman syndrome locus. PLoS Genet. 7, e1002422 ( 10.1371/journal.pgen.1002422) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amos-Landgraf JM, et al. 1999. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am. J. Hum. Genet. 65, 370–386. ( 10.1086/302510) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR, Nakao M, Surti U, Chakravarti A, Ledbetter DH. 1995. Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am. J. Hum. Genet. 57, 40–48. [PMC free article] [PubMed] [Google Scholar]
- 12.Cassidy SB, Forsythe M, Heeger S, Nicholls RD, Schork N, Benn P, Schwartz S. 1997. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am. J. Med. Genet. 68, 433–440. ( 10.1002/(SICI)1096-8628(19970211)68:4<433::AID-AJMG12>3.0.CO;2-T) [DOI] [PubMed] [Google Scholar]
- 13.Gillessen-Kaesbach G, Robinson W, Lohmann D, Kaya-Westerloh S, Passarge E, Horsthemke B. 1995. Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum. Genet. 96, 638–643. ( 10.1007/BF00210291) [DOI] [PubMed] [Google Scholar]
- 14.Nicholls RD, Knoll JH, Butler MG, Karam S, Lalande M. 1989. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 342, 281–285. ( 10.1038/342281a0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horsthemke B, Buiting K. 2008. Genomic imprinting and imprinting defects in humans. Adv. Genet. 61, 225–246. ( 10.1016/S0065-2660(07)00008-9) [DOI] [PubMed] [Google Scholar]
- 16.Ohta T, et al. 1999. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am. J. Hum. Genet. 64, 397–413. ( 10.1086/302233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bieth E, et al. 2015. Highly restricted deletion of the SNORD116 region is implicated in Prader-Willi syndrome. Eur. J. Hum. Genet. 23, 252–255. ( 10.1038/ejhg.2014.103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butler MG, Christian SL, Kubota T, Ledbetter DH. 1996. A 5-year-old white girl with Prader-Willi syndrome and a submicroscopic deletion of chromosome 15q11q13. Am. J. Med. Genet. 65, 137–141. ( 10.1002/(SICI)1096-8628(19961016)65:2<137::AID-AJMG11>3.0.CO;2-R) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Smith AJ, et al. 2009. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 18, 3257–3265. ( 10.1093/hmg/ddp263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duker AL, et al. 2010. Paternally inherited microdeletion at 15q11. 2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader–Willi syndrome. Eur. J. Hum. Genet. 18, 1196–1201. ( 10.1038/ejhg.2010.102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL. 2008. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 40, 719–721. ( 10.1038/ng.158) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan Q, Potter KJ, Burnett LC, Orsso CE, Inman M, Ryman DC, Haqq AM. 2020. Prader–Willi-like phenotype caused by an atypical 15q11. 2 microdeletion. Genes 11, 128 ( 10.3390/genes11020128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chamberlain SJ, Lalande M. 2010. Angelman syndrome, a genomic imprinting disorder of the brain. J. Neurosci. 30, 9958–9963. ( 10.1523/JNEUROSCI.1728-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glenn CC, Saitoh S, Jong MT, Filbrandt MM, Surti U, Driscoll DJ, Nicholls RD. 1996. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 58, 335–346. [PMC free article] [PubMed] [Google Scholar]
- 25.Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. 1997. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol. Hum. Reprod. 3, 321–332. ( 10.1093/molehr/3.4.321) [DOI] [PubMed] [Google Scholar]
- 26.Kubota T, Sutcliffe JS, Aradhya S, Gillessen-Kaesbach G, Christian SL, Horsthemke B, Beaudet AL, Ledbetter DH. 1996. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am. J. Med. Genet. 66, 77–80. ( 10.1002/(SICI)1096-8628(19961202)66:1<77::AID-AJMG18>3.0.CO;2-N) [DOI] [PubMed] [Google Scholar]
- 27.Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB. 2001. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet. Med. 3, 206–211. ( 10.1097/00125817-200105000-00011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Procter M, Chou LS, Tang W, Jama M, Mao R. 2006. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin. Chem. 52, 1276–1283. ( 10.1373/clinchem.2006.067603) [DOI] [PubMed] [Google Scholar]
- 29.Miller JL, et al. 2011. Nutritional phases in Prader-Willi syndrome. Am. J. Med. Genet. A 155a, 1040–1049. ( 10.1002/ajmg.a.33951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dykens EM, Maxwell MA, Pantino E, Kossler R, Roof E. 2007. Assessment of hyperphagia in Prader-Willi syndrome. Obesity 15, 1816–1826. ( 10.1038/oby.2007.216) [DOI] [PubMed] [Google Scholar]
- 31.Gunay-Aygun M, Schwartz S, Heeger S, O'Riordan MA, Cassidy SB. 2001. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 108, E92 ( 10.1542/peds.108.5.e92) [DOI] [PubMed] [Google Scholar]
- 32.Burman P, Ritzen EM, Lindgren AC. 2001. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr Rev. 22, 787–799. ( 10.1210/edrv.22.6.0447) [DOI] [PubMed] [Google Scholar]
- 33.Reus L, Zwarts M, van Vlimmeren LA, Willemsen MA, Otten BJ, Nijhuis-van der Sanden MW. 2011. Motor problems in Prader-Willi syndrome: a systematic review on body composition and neuromuscular functioning. Neurosci. Biobehav. Rev. 35, 956–969. ( 10.1016/j.neubiorev.2010.10.015) [DOI] [PubMed] [Google Scholar]
- 34.Crino A, et al. 2003. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur. J. Pediatr. 162, 327–333. ( 10.1007/s00431-002-1132-4) [DOI] [PubMed] [Google Scholar]
- 35.Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O, Gross-Tsur V. 2010. Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur. J. Endocrinol. 162, 377–384. ( 10.1530/EJE-09-0901) [DOI] [PubMed] [Google Scholar]
- 36.Gross-Tsur V, Hirsch HJ, Benarroch F, Eldar-Geva T. 2012. The FSH-inhibin axis in Prader-Willi syndrome: heterogeneity of gonadal dysfunction. Reprod. Biol. Endocrinol. 10, 39 ( 10.1186/1477-7827-10-39) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirsch HJ, Eldar-Geva T, Benarroch F, Rubinstein O, Gross-Tsur V. 2009. Primary testicular dysfunction is a major contributor to abnormal pubertal development in males with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 94, 2262–2268. ( 10.1210/jc.2008-2760) [DOI] [PubMed] [Google Scholar]
- 38.Radicioni A, et al. 2012. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader–Willi syndrome. Clin. Endocrinol. 76, 72–77. ( 10.1111/j.1365-2265.2011.04161.x) [DOI] [PubMed] [Google Scholar]
- 39.Bizzarri C, Rigamonti AE, Luce A, Cappa M, Cella SG, Berini J, Sartorio A, Müller EE, Salvatoni A. 2010. Children with Prader-Willi syndrome exhibit more evident meal-induced responses in plasma ghrelin and peptide YY levels than obese and lean children. Eur. J. Endocrinol. 162, 499 ( 10.1530/EJE-09-1033) [DOI] [PubMed] [Google Scholar]
- 40.DelParigi A, Tschöp M, Heiman ML, Salbe AD, Vozarova B, Sell SM, Bunt JC, Tataranni PA. 2002. High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 87, 5461–5464. ( 10.1210/jc.2002-020871) [DOI] [PubMed] [Google Scholar]
- 41.Purtell L, Sze L, Loughnan G, Smith E, Herzog H, Sainsbury A, Steinbeck K, Campbell LV, Viardot A. 2011. In adults with Prader–Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-1 levels. Neuropeptides 45, 301–307. ( 10.1016/j.npep.2011.06.001) [DOI] [PubMed] [Google Scholar]
- 42.De Waele K, Ishkanian SL, Bogarin R, Miranda CA, Ghatei MA, Bloom SR, Pacaud D, Chanoine JP.. 2008. Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader-Willi syndrome. Eur. J. Endocrinol. 159, 381–388. ( 10.1530/EJE-08-0462) [DOI] [PubMed] [Google Scholar]
- 43.Haqq AM, Stadler DD, Rosenfeld RG, Pratt KL, Weigle DS, Frayo RS, LaFranchi SH, Cummings DE, Purnell JQ. 2003. Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 88, 3573–3576. ( 10.1210/jc.2003-030205) [DOI] [PubMed] [Google Scholar]
- 44.Tan TM, Vanderpump M, Khoo B, Patterson M, Ghatei MA, Goldstone AP. 2004. Somatostatin infusion lowers plasma ghrelin without reducing appetite in adults with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 89, 4162–4165. ( 10.1210/jc.2004-0835) [DOI] [PubMed] [Google Scholar]
- 45.Grugni G, et al. 2013. Central adrenal insufficiency in young adults with Prader-Willi syndrome. Clin. Endocrinol. 79, 371–378. ( 10.1111/cen.12150) [DOI] [PubMed] [Google Scholar]
- 46.Butler MG, Lee J, Manzardo AM, Gold J.-A., Miller JL, Kimonis V, Driscoll DJ. 2015. Growth charts for non-growth hormone treated Prader-Willi syndrome. Pediatrics 135, e126-e135. ( 10.1542/peds.2014-1711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diene G, Mimoun E, Feigerlova E, Caula S, Molinas C, Grandjean H, Tauber M. 2010. Endocrine disorders in children with Prader-Willi syndrome–data from 142 children of the French database. Horm. Res. Paediatr. 74, 121–128. ( 10.1159/000313377) [DOI] [PubMed] [Google Scholar]
- 48.Miller JL, Goldstone AP, Couch JA, Shuster J, He G, Driscoll DJ, Liu Y, Schmalfuss IM. 2008. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am. J. Med. Genet. A 146a, 570–577. ( 10.1002/ajmg.a.31677) [DOI] [PubMed] [Google Scholar]
- 49.Butler MG, Theodoro M, Skouse JD. 2007. Thyroid function studies in Prader–Willi syndrome. Am. J. Med. Genet. A 143, 488 ( 10.1002/ajmg.a.31683) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Lind van Wijngaarden RF, Otten BJ, Festen DA, Joosten KF, de Jong FH, Sweep FC, Hokken-Koelega AC. 2008. High prevalence of central adrenal insufficiency in patients with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 93, 1649–1654. ( 10.1210/jc.2007-2294) [DOI] [PubMed] [Google Scholar]
- 51.Corrias A, et al. 2012. Assessment of central adrenal insufficiency in children and adolescents with Prader–Willi syndrome. Clin. Endocrinol. 76, 843–850. ( 10.1111/j.1365-2265.2011.04313.x) [DOI] [PubMed] [Google Scholar]
- 52.Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. 2002. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev. Med. Child Neurol. 44, 248–255. ( 10.1017/S001216220100202X) [DOI] [PubMed] [Google Scholar]
- 53.Cielo C, Marcus CL. 2014. Central hypoventilation syndromes. Sleep Med. Clin. 9, 105–118. ( 10.1016/j.jsmc.2013.10.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gozal D, Arens R, Omlin KJ, Ward SL, Keens TG. 1994. Absent peripheral chemosensitivity in Prader-Willi syndrome. J. Appl. Physiol. 77, 2231–2236. ( 10.1152/jappl.1994.77.5.2231) [DOI] [PubMed] [Google Scholar]
- 55.Hákonarson H, Moskovitz J, Daigle KL, Cassidy SB, Cloutier MM. 1995. Pulmonary function abnormalities in Prader-Willi syndrome. J. Pediatr. 126, 565–570. ( 10.1016/S0022-3476(95)70350-0) [DOI] [PubMed] [Google Scholar]
- 56.Festen DA, de Weerd AW, van den Bossche RA, Joosten K, Hoeve H, Hokken-Koelega AC. 2006. Sleep-related breathing disorders in prepubertal children with Prader-Willi syndrome and effects of growth hormone treatment. J. Clin. Endocrinol. Metab. 91, 4911–4915. ( 10.1210/jc.2006-0765) [DOI] [PubMed] [Google Scholar]
- 57.Bruni O, Verrillo E, Novelli L, Ferri R. 2010. Prader-Willi syndrome: sorting out the relationships between obesity, hypersomnia, and sleep apnea. Curr. Opin Pulm. Med. 16, 568–573. ( 10.1097/MCP.0b013e32833ef547) [DOI] [PubMed] [Google Scholar]
- 58.Miller J, Wagner M. 2013. Prader-Willi syndrome and sleep-disordered breathing. Pediatr. Ann. 42, e210-e214. ( 10.3928/00904481-20130924-10) [DOI] [PubMed] [Google Scholar]
- 59.Priano L, Grugni G, Miscio G, Guastamacchia G, Toffolet L, Sartorio A, Mauro A. 2006. Sleep cycling alternating pattern (CAP) expression is associated with hypersomnia and GH secretory pattern in Prader-Willi syndrome. Sleep Med. 7, 627–633. ( 10.1016/j.sleep.2005.12.004) [DOI] [PubMed] [Google Scholar]
- 60.Hayashi M, Miyata R, Tanuma N. 2011. Decrease in acetylcholinergic neurons in the pedunculopontine tegmental nucleus in a patient with Prader-Willi syndrome. Neuropathology 31, 280–285. ( 10.1111/j.1440-1789.2010.01157.x) [DOI] [PubMed] [Google Scholar]
- 61.Nevsimalova S, Vankova J, Stepanova I, Seemanova E, Mignot E, Nishino S. 2005. Hypocretin deficiency in Prader-Willi syndrome. Eur. J. Neurol. 12, 70–72. ( 10.1111/j.1468-1331.2004.00969.x) [DOI] [PubMed] [Google Scholar]
- 62.Dimitropoulos A, Feurer I, Butler M, Thompson T. 2001. Emergence of compulsive behavior and tantrums in children with Prader-Willi syndrome. Am. J. Ment. Retard. 106, 39–51. ( 10.1352/0895-8017(2001)106<0039:EOCBAT>2.0.CO;2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dykens EM, Kasari C. 1997. Maladaptive behavior in children with Prader-Willi syndrome, Down syndrome, and nonspecific mental retardation. Am. J. Ment. Retard. 102, 228–237. ( 10.1352/0895-8017(1997)102<0228:MBICWP>2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 64.Dykens EM, Rosner BA. 1999. Refining behavioral phenotypes: personality—motivation in Williams and Prader-Willi syndromes. Am. J. Ment. Retard. 104, 158–169. ( 10.1352/0895-8017(1999)104<0158:RBPPIW>2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 65.Dykens EM. 2004. Maladaptive and compulsive behavior in Prader-Willi syndrome: new insights from older adults. Am. J. Ment. Retard. 109, 142–153. ( 10.1352/0895-8017(2004)109<142:MACBIP>2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 66.Ho AY, Dimitropoulos A. 2010. Clinical management of behavioral characteristics of Prader–Willi syndrome. Neuropsychiatr. Dis. Treat. 6, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holland AJ, Whittington JE, Butler J, Webb T, Boer H, Clarke D. 2003. Behavioural phenotypes associated with specific genetic disorders: evidence from a population-based study of people with Prader-Willi syndrome. Psychol. Med. 33, 141–153. ( 10.1017/S0033291702006736) [DOI] [PubMed] [Google Scholar]
- 68.Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF. 2004. Prader-Willi syndrome: a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur. Child Adolesc. Psychiatry 13, 42–50. ( 10.1007/s00787-004-0354-6) [DOI] [PubMed] [Google Scholar]
- 69.Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H. 2004. Cognitive abilities and genotype in a population-based sample of people with Prader–Willi syndrome. J. Intellect. Disabil. Res. 48, 172–187. ( 10.1111/j.1365-2788.2004.00556.x) [DOI] [PubMed] [Google Scholar]
- 70.Yang L, Zhan GD, Ding JJ, Wang HJ, Ma D, Huang GY, Zhou WH. 2013. Psychiatric illness and intellectual disability in the Prader-Willi syndrome with different molecular defects: a meta analysis. PLoS ONE 8, e72640 ( 10.1371/journal.pone.0072640) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shim JS, Lee SH, Seo SW, Koo KH, Jin DK. 2010. The musculoskeletal manifestations of Prader-Willi syndrome. J. Pediatr. Orthop. 30, 390–395. ( 10.1097/BPO.0b013e3181da857d) [DOI] [PubMed] [Google Scholar]
- 72.Tauber M, Diene G, Molinas C, Hebert M. 2008. Review of 64 cases of death in children with Prader-Willi syndrome (PWS). Am. J. Med. Genet. A 146a, 881–887. ( 10.1002/ajmg.a.32131) [DOI] [PubMed] [Google Scholar]
- 73.Butler MG. 1989. Hypopigmentation: a common feature of Prader-Labhart-Willi syndrome. Am. J. Hum. Genet. 45, 140–146. [PMC free article] [PubMed] [Google Scholar]
- 74.Torrado M, et al. 2007. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. Am. J. Med. Genet. A 143a, 460–468. ( 10.1002/ajmg.a.31520) [DOI] [PubMed] [Google Scholar]
- 75.Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. 2004. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 113, 565–573. ( 10.1542/peds.113.3.565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hartley SL, Maclean WE Jr, Butler MG, Zarcone J, Thompson T. 2005. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am. J. Med. Genet. A 136, 140–145. ( 10.1002/ajmg.a.30771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG. 2000. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J. Intellect. Disabil. Res. 44, 25–30. ( 10.1046/j.1365-2788.2000.00250.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Craig ME, Cowell CT, Larsson P, Zipf WB, Reiter EO, Albertsson Wikland K, Ranke MB, Price DA. 2006. Growth hormone treatment and adverse events in Prader-Willi syndrome: data from KIGS (the Pfizer International Growth Database). Clin. Endocrinol. 65, 178–185. ( 10.1111/j.1365-2265.2006.02570.x) [DOI] [PubMed] [Google Scholar]
- 79.Deal CL, Tony M, Höybye C, Allen DB, Tauber M, Christiansen JS, Participants GH.i.P.-W.S.C.C.G.W. 2013. Growth Hormone Research Society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 98, E1072-E1087. ( 10.1210/jc.2012-3888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Butler MG, Matthews NA, Patel N, Surampalli A, Gold JA, Khare M, Thompson T, Cassidy SB, Kimonis VE. 2019. Impact of genetic subtypes of Prader–Willi syndrome with growth hormone therapy on intelligence and body mass index. Am. J. Med. Genet. A 179, 1826–1835. ( 10.1002/ajmg.a.61263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Osorio J. 2012. Growth and development: growth hormone therapy improves cognition in children with Prader-Willi syndrome. Nat. Rev. Endocrinol. 8, 382 ( 10.1038/nrendo.2012.73) [DOI] [PubMed] [Google Scholar]
- 82.Siemensma EP, et al. 2012. Beneficial effects of growth hormone treatment on cognition in children with Prader-Willi syndrome: a randomized controlled trial and longitudinal study. J. Clin. Endocrinol. Metab. 97, 2307–2314. ( 10.1210/jc.2012-1182) [DOI] [PubMed] [Google Scholar]
- 83.Sode-Carlsen R, Farholt S, Rabben KF, Bollerslev J, Schreiner T, Jurik AG, Christiansen JS, Hoybye C. 2012. Growth hormone treatment in adults with Prader-Willi syndrome: the Scandinavian study. Endocrine 41, 191–199. ( 10.1007/s12020-011-9560-4) [DOI] [PubMed] [Google Scholar]
- 84.Schlumpf M, Eiholzer U, Gygax M, Schmid S, van der Sluis I, l'Allemand D. 2006. A daily comprehensive muscle training programme increases lean mass and spontaneous activity in children with Prader-Willi syndrome after 6 months. J. Pediatr. Endocrinol. Metab. 19, 65–74. ( 10.1515/JPEM.2006.19.1.65) [DOI] [PubMed] [Google Scholar]
- 85.Scaroni C, Ceccato F, Rizzati S, Mantero F. 2008. Concomitant therapies (glucocorticoids and sex hormones) in adult patients with growth hormone deficiency. J. Endocrinol. Invest. 31, 61–65. ( 10.1007/BF03345609) [DOI] [PubMed] [Google Scholar]
- 86.Miller J, Silverstein J, Shuster J, Driscoll DJ, Wagner M. 2006. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 91, 413–417. ( 10.1210/jc.2005-1279) [DOI] [PubMed] [Google Scholar]
- 87.Cutfield WS, Wilton P, Bennmarker H, Albertsson-Wikland K, Chatelain P, Ranke MB, Price DA. 2000. Incidence of diabetes mellitus and impaired glucose tolerance in children and adolescents receiving growth-hormone treatment. Lancet 355, 610–613. ( 10.1016/S0140-6736(99)04055-6) [DOI] [PubMed] [Google Scholar]
- 88.Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB. 2010. Long-term growth hormone therapy changes the natural history of body composition and motor function in children with Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 95, 1131–1136. ( 10.1210/jc.2009-1389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wolfgram PM, Carrel AL, Allen DB. 2013. Long-term effects of recombinant human growth hormone therapy in children with Prader-Willi syndrome. Curr. Opin. Pediatr. 25, 509–514. ( 10.1097/MOP.0b013e328362c7a2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Åkefeldt A, Törnhage C.-J, Gillberg C. 1999. A woman with Prader–Willi syndrome gives birth to a healthy baby girl. Dev. Med. Child Neurol. 41, 789–790. ( 10.1017/S0012162299221573) [DOI] [PubMed] [Google Scholar]
- 91.Schulze A, Mogensen H, Hamborg-Petersen B, Graem N, Østergaard J, Brøndum-Nielsen K. 2001. Fertility in Prader-Willi syndrome: a case report with Angelman syndrome in the offspring. Acta Paediatr. 90, 455–459. ( 10.1111/j.1651-2227.2001.tb00451.x) [DOI] [PubMed] [Google Scholar]
- 92.Miller JL, Lynn CH, Shuster J, Driscoll DJ. 2013. A reduced-energy intake, well-balanced diet improves weight control in children with Prader-Willi syndrome. J. Hum. Nutr. Diet 26, 2–9. ( 10.1111/j.1365-277X.2012.01275.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pavone M, Paglietti M, Petrone A, Crino A, De Vincentiis G, Cutrera R.. 2006. Adenotonsillectomy for obstructive sleep apnea in children with Prader-Willi syndrome. Pediatr. Pulmonol. 41, 74–79. ( 10.1002/ppul.20334) [DOI] [PubMed] [Google Scholar]
- 94.Goldstone A, Holland A, Hauffa B, Hokken-Koelega A, Tauber M, PWS SC.a.t.S.E.M.o.t.C.C.o.P.W. 2008. Recommendations for the diagnosis and management of Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 93, 4183–4197. ( 10.1210/jc.2008-0649) [DOI] [PubMed] [Google Scholar]
- 95.Dykens E, Shah B. 2003. Psychiatric disorders in Prader-Willi syndrome: epidemiology and management. CNS Drugs 17, 167–178. ( 10.2165/00023210-200317030-00003) [DOI] [PubMed] [Google Scholar]
- 96.Soni S, Whittington J, Holland AJ, Webb T, Maina E, Boer H, Clarke D. 2007. The course and outcome of psychiatric illness in people with Prader-Willi syndrome: implications for management and treatment. J. Intellect. Disabil. Res. 51, 32–42. ( 10.1111/j.1365-2788.2006.00895.x) [DOI] [PubMed] [Google Scholar]
- 97.Atasoy D, Betley JN, Su HH, Sternson SM. 2012. Deconstruction of a neural circuit for hunger. Nature 488, 172–177. ( 10.1038/nature11270) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tauber M, et al. 2011. Oxytocin may be useful to increase trust in others and decrease disruptive behaviours in patients with Prader-Willi syndrome: a randomised placebo-controlled trial in 24 patients. Orphanet J. Rare Dis. 6, 47 ( 10.1186/1750-1172-6-47) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haidet AM, et al. 2008. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl Acad. Sci. USA 105, 4318–4322. ( 10.1073/pnas.0709144105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Allas S, Abribat T. 2013. Clinical perspectives for ghrelin-derived therapeutic products. In The Ghrelin system (eds Benso A, Casanueva FF, Ghigo E, Granata R), pp. 157–166. Basel, Switzerland: Karger Publishers. [DOI] [PubMed] [Google Scholar]
- 101.Fintini D, Grugni G, Brufani C, Bocchini S, Cappa M, Crinò A. 2014. Use of GLP-1 receptor agonists in Prader-Willi syndrome: report of six cases. Diabetes Care 37, e76-e77. ( 10.2337/dc13-2575) [DOI] [PubMed] [Google Scholar]
- 102.Chen KY, et al. 2015. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J. Clin. Endocrinol. Metab. 100, 1639–1645. ( 10.1210/jc.2014-4024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Knani I, et al. 2016. Targeting the endocannabinoid/CB1 receptor system for treating obesity in Prader-Willi syndrome. Mol. Metab. 5, 1187–1199. ( 10.1016/j.molmet.2016.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kimonis V, Surampalli A, Wencel M, Gold JA, Cowen NM. 2019. A randomized pilot efficacy and safety trial of diazoxide choline controlled-release in patients with Prader-Willi syndrome. PLoS ONE 14, e0221615 ( 10.1371/journal.pone.0221615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martins-Taylor K, Hsiao JS, Chen P-F, Glatt-Deeley H, De Smith AJ, Blakemore AI, Lalande M, Chamberlain SJ.. 2014. Imprinted expression of UBE3A in non-neuronal cells from a Prader-Willi syndrome patient with an atypical deletion. Hum. Mol. Genet. 23, ddt628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Numata K, Kohama C, Abe K, Kiyosawa H. 2010. Highly parallel SNP genotyping reveals high-resolution landscape of mono-allelic Ube3a expression associated with locus-wide antisense transcription. Nucleic Acids Res. 39, 2649–2657. ( 10.1093/nar/gkq1201) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cavaillé J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie J-P, Brosius J, Hüttenhofer A. 2000. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl Acad. Sci. USA 97, 14 311–14 316. ( 10.1073/pnas.250426397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Runte M, Hüttenhofer A, Groß S, Kiefmann M, Horsthemke B, Buiting K. 2001. The IC-SNURF–SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 10, 2687–2700. ( 10.1093/hmg/10.23.2687) [DOI] [PubMed] [Google Scholar]
- 109.Vitali P, Royo H, Marty V, Bortolin-Cavaillé M-L, Cavaillé J. 2010. Long nuclear-retained non-coding RNAs and allele-specific higher-order chromatin organization at imprinted snoRNA gene arrays. J. Cell Sci. 123, 70–83. ( 10.1242/jcs.054957) [DOI] [PubMed] [Google Scholar]
- 110.Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. 2010. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl Acad. Sci. USA 107, 17 668–17 673. ( 10.1073/pnas.1004487107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Machnicka MA, et al. 2013. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 41, D262–D267. ( 10.1093/nar/gks1007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lewis CJ, Pan T, Kalsotra A. 2017. RNA modifications and structures cooperate to guide RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 18, 202–210. ( 10.1038/nrm.2016.163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kiss T. 2001. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 20, 3617–3622. ( 10.1093/emboj/20.14.3617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI.. 2007. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8, 574–585. ( 10.1038/nrm2184) [DOI] [PubMed] [Google Scholar]
- 115.Matera AG, Terns RM, Terns MP. 2007. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 8, 209–220. ( 10.1038/nrm2124) [DOI] [PubMed] [Google Scholar]
- 116.Filipowicz W, Pogacic V. 2002. Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol. 14, 319–327. ( 10.1016/S0955-0674(02)00334-4) [DOI] [PubMed] [Google Scholar]
- 117.Tycowski KT, Shu MD, Steitz JA. 1993. A small nucleolar RNA is processed from an intron of the human gene encoding ribosomal protein S3. Genes Dev. 7, 1176–1190. ( 10.1101/gad.7.7a.1176) [DOI] [PubMed] [Google Scholar]
- 118.Kiss T, Filipowicz W. 1995. Exonucleolytic processing of small nucleolar RNAs from pre-mRNA introns. Genes Dev. 9, 1411–1424. ( 10.1101/gad.9.11.1411) [DOI] [PubMed] [Google Scholar]
- 119.Decatur WA, Fournier MJ. 2002. rRNA modifications and ribosome function. Trends Biochem. Sci. 27, 344–351. ( 10.1016/S0968-0004(02)02109-6) [DOI] [PubMed] [Google Scholar]
- 120.Choi J, et al. 2018. 2'-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat. Struct. Mol. Biol. 25, 208–216. ( 10.1038/s41594-018-0030-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stepanov GA, Filippova JA, Komissarov AB, Kuligina EV, Richter VA, Semenov DV. 2015. Regulatory role of small nucleolar RNAs in human diseases. Bio. Med. Res. Int. 2015, 206 849 ( 10.1155/2015/206849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dai Q, Moshitch-Moshkovitz S, Han D, Kol N, Amariglio N, Rechavi G, Dominissini D, He C. 2017. Nm-seq maps 2'-O-methylation sites in human mRNA with base precision. Nat. Methods 14, 695–698. ( 10.1038/nmeth.4294) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu Y, Pirnie SP, Carmichael GG. 2017. High-throughput and site-specific identification of 2'-O-methylation sites using ribose oxidation sequencing (RibOxi-seq). RNA 23, 1303–1314. ( 10.1261/rna.061549.117) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang YJ, Yang JH, Shi QS, Zheng LL, Liu J, Zhou H, Zhang H, Qu LH. 2014. Rapid birth-and-death evolution of imprinted snoRNAs in the Prader-Willi syndrome locus: implications for neural development in Euarchontoglires. PLoS ONE 9, e100329 ( 10.1371/journal.pone.0100329) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Castle JC, et al. 2010. Digital genome-wide ncRNA expression, including SnoRNAs, across 11 human tissues using polyA-neutral amplification. PLoS ONE 5, e11779 ( 10.1371/journal.pone.0011779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wu H, Yin QF, Luo Z, Yao RW, Zheng CC, Zhang J, Xiang JF, Yang L, Chen LL. 2016. Unusual processing generates SPA LncRNAs that sequester multiple RNA binding proteins. Mol. Cell. 64, 534–548. ( 10.1016/j.molcel.2016.10.007) [DOI] [PubMed] [Google Scholar]
- 127.Kocher MA, Good DJ. 2017. Phylogenetic analysis of the SNORD116 locus. Genes 8, 358 ( 10.3390/genes8120358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Burnett LC, et al. 2017. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Invest. 127, 293–305. ( 10.1172/JCI88648) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cattanach BM, et al. 1992. A candidate mouse model for Prader–Willi syndrome which shows an absence of Snrpn expression. Nat. Genet. 2, 270–274. ( 10.1038/ng1292-270) [DOI] [PubMed] [Google Scholar]
- 130.Gabriel J, Merchant M, Ohta T, Ji Y, Caldwell R, Ramsey M, Tucker J, Longnecker R, Nicholls R. 1999. A transgene insertion creating a heritable chromosome deletion mouse model of Prader-Willi and Angelman syndromes. Proc. Natl Acad. Sci. USA 96, 9258–9263. ( 10.1073/pnas.96.16.9258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Skryabin BV, Gubar LV, Seeger B, Pfeiffer J, Handel S, Robeck T, Karpova E, Rozhdestvensky TS, Brosius J. 2007. Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet. 3, e235 ( 10.1371/journal.pgen.0030235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bischof JM, Stewart CL, Wevrick R. 2007. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 16, 2713–2719. ( 10.1093/hmg/ddm225) [DOI] [PubMed] [Google Scholar]
- 133.Kozlov SV, et al. 2007. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 39, 1266–1272. ( 10.1038/ng2114) [DOI] [PubMed] [Google Scholar]
- 134.Yin Q-F, Yang L, Zhang Y, Xiang J-F, Wu Y-W, Carmichael GG, Chen L-L. 2012. Long noncoding RNAs with snoRNA ends. Mol. Cell 48, 219–230. ( 10.1016/j.molcel.2012.07.033) [DOI] [PubMed] [Google Scholar]
- 135.Zhang XO, Yin QF, Wang HB, Zhang Y, Chen T, Zheng P, Lu X, Chen LL, Yang L. 2014. Species-specific alternative splicing leads to unique expression of sno-lncRNAs. BMC Genom. 15, 287 ( 10.1186/1471-2164-15-287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang Z, Shen M, Gresch PJ, Ghamari-Langroudi M, Rabchevsky AG, Emeson RB, Stamm S. 2016. Oligonucleotide-induced alternative splicing of serotonin 2C receptor reduces food intake. EMBO Mol. Med. 8, 878–894. ( 10.15252/emmm.201506030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Flomen R, Knight J, Sham P, Kerwin R, Makoff A. 2004. Evidence that RNA editing modulates splice site selection in the 5-HT2C receptor gene. Nucleic Acids Res. 32, 2113–2122. ( 10.1093/nar/gkh536) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martin C, et al. 2013. RNA splicing and editing modulation of 5-HT 2C receptor function: relevance to anxiety and aggression in VGV mice. Mol. Psychiatry 18, 656–665. ( 10.1038/mp.2012.171) [DOI] [PubMed] [Google Scholar]
- 139.Kishore S, Stamm S. 2006. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 311, 230–232. ( 10.1126/science.1118265) [DOI] [PubMed] [Google Scholar]
- 140.Brameier M, Herwig A, Reinhardt R, Walter L, Gruber J. 2011. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res. 39, 675–686. ( 10.1093/nar/gkq776) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. 2009. Small RNAs derived from snoRNAs. RNA 15, 1233–1240. ( 10.1261/rna.1528909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kishore S, et al. 2010. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum. Mol. Genet. 19, 1153–1164. ( 10.1093/hmg/ddp585) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shen M, Eyras E, Wu J, Khanna A, Josiah S, Rederstorff M, Zhang MQ, Stamm S. 2011. Direct cloning of double-stranded RNAs from RNase protection analysis reveals processing patterns of C/D box snoRNAs and provides evidence for widespread antisense transcript expression. Nucleic Acids Res. 39, 9720–9730. ( 10.1093/nar/gkr684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bortolin-Cavaillé M-L, Cavaillé J. 2012. The SNORD115 (H/MBII-52) and SNORD116 (H/MBII-85) gene clusters at the imprinted Prader–Willi locus generate canonical box C/D snoRNAs. Nucleic Acids Res. 40, 6800–6807. ( 10.1093/nar/gks321) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Galiveti CR, Raabe CA, Konthur Z, Rozhdestvensky TS. 2014. Differential regulation of non-protein coding RNAs from Prader-Willi syndrome locus. Sci. Rep. 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stelzer Y, Sagi I, Yanuka O, Eiges R, Benvenisty N. 2014. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 46, 551 ( 10.1038/ng.2968) [DOI] [PubMed] [Google Scholar]
- 147.Hsiao JS, Germain ND, Wilderman A, Stoddard C, Wojenski LA, Villafano GJ, Core L, Cotney J, Chamberlain SJ. 2019. A bipartite boundary element restricts UBE3A imprinting to mature neurons. Proc. Natl Acad. Sci. USA 116, 2181–2186. ( 10.1073/pnas.1815279116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Huibregtse JM, Scheffner M, Howley PM. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 10, 4129–4135. ( 10.1002/j.1460-2075.1991.tb04990.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. ( 10.1016/0092-8674(93)90384-3) [DOI] [PubMed] [Google Scholar]
- 150.Kanber D, Giltay J, Wieczorek D, Zogel C, Hochstenbach R, Caliebe A, Kuechler A, Horsthemke B, Buiting K. 2009. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader–Willi syndrome. Eur. J. Hum. Genet. 17, 582–590. ( 10.1038/ejhg.2008.232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Abreu AP, et al. 2013. Central precocious puberty caused by mutations in the imprinted gene MKRN3. New England J. Med. 368, 2467–2475. ( 10.1056/NEJMoa1302160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S.. 2001. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 61, 5544–5551. [PubMed] [Google Scholar]
- 153.Hao Y-H, et al. 2013. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell 152, 1051–1064. ( 10.1016/j.cell.2013.01.051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fountain MD, et al. 2016. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med. 19, 45–52. ( 10.1038/gim.2016.53) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Schaaf CP, et al. 2013. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 45, 1405–1408. ( 10.1038/ng.2776) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lee S, Walker CL, Karten B, Kuny SL, Tennese AA, O'Neill MA, Wevrick R. 2005. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum. Mol. Genet. 14, 627–637. ( 10.1093/hmg/ddi059) [DOI] [PubMed] [Google Scholar]
- 157.Tcherpakov M, Bronfman FC, Conticello SG, Vaskovsky A, Levy Z, Niinobe M, Yoshikawa K, Arenas E, Fainzilber M. 2002. The p75 neurotrophin receptor interacts with multiple MAGE proteins. J. Biol. Chem. 277, 49 101–49 104. ( 10.1074/jbc.C200533200) [DOI] [PubMed] [Google Scholar]
- 158.Miller NL, Wevrick R, Mellon PL. 2009. Necdin, a Prader–Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 18, 248–260. ( 10.1093/hmg/ddn344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Neumann LC, Markaki Y, Mladenov E, Hoffmann D, Buiting K, Horsthemke B. 2012. The imprinted NPAP1/C15orf2 gene in the Prader-Willi syndrome region encodes a nuclear pore complex associated protein. Hum. Mol. Genet. 21, 4038–4048. ( 10.1093/hmg/dds228) [DOI] [PubMed] [Google Scholar]
- 160.Wawrzik M, Spiess AN, Herrmann R, Buiting K, Horsthemke B. 2009. Expression of SNURF-SNRPN upstream transcripts and epigenetic regulatory genes during human spermatogenesis. Eur. J. Hum. Genet. 17, 1463–1470. ( 10.1038/ejhg.2009.83) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lewis MW, Brant JO, Kramer JM, Moss JI, Yang TP, Hansen PJ, Williams RS, Resnick JL. 2015. Angelman syndrome imprinting center encodes a transcriptional promoter. Proc. Natl Acad. Sci. USA 112, 6871–6875. ( 10.1073/pnas.1411261111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Langouët M, Glatt-Deeley HR, Chung MS, Dupont-Thibert CM, Mathieux E, Banda EC, Stoddard CE, Crandall L, Lalande M. 2018. Zinc finger protein 274 regulates imprinted expression of transcripts in Prader-Willi syndrome neurons. Hum. Mol. Genet. 27, 505–515. ( 10.1093/hmg/ddx420) [DOI] [PubMed] [Google Scholar]
- 163.Saitoh S, Wada T. 2000. Parent-of-origin specific histone acetylation and reactivation of a key imprinted gene locus in Prader-Willi syndrome. Am. J. Hum. Genet. 66, 1958–1962. ( 10.1086/302917) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yano K, Ueki N, Oda T, Seki N, Masuho Y, Muramatsu M. 2000. Identification and characterization of human ZNF274 cDNA, which encodes a novel kruppel-type zinc-finger protein having nucleolar targeting ability. Genomics 65, 75–80. ( 10.1006/geno.2000.6140) [DOI] [PubMed] [Google Scholar]
- 165.Frietze S, O'Geen H, Blahnik KR, Jin VX, Farnham PJ. 2010. ZNF274 recruits the histone methyltransferase SETDB1 to the 3′ ends of ZNF genes. PLoS ONE 5, e15082 ( 10.1371/journal.pone.0015082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Imbeault M, Helleboid P.-Y, Trono D. 2017. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554. ( 10.1038/nature21683) [DOI] [PubMed] [Google Scholar]
- 167.Cruvinel E, Budinetz T, Germain N, Chamberlain S, Lalande M, Martins-Taylor K. 2014. Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader-Willi syndrome iPSCs. Hum. Mol. Genet. 23, 4674–4685. ( 10.1093/hmg/ddu187) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xin Z, Tachibana M, Guggiari M, Heard E, Shinkai Y, Wagstaff J. 2003. Role of histone methyltransferase G9a in CpG methylation of the Prader-Willi syndrome imprinting center. J. Biol. Chem. 278, 14 996–15 000. ( 10.1074/jbc.M211753200) [DOI] [PubMed] [Google Scholar]
- 169.Shinkai Y, Tachibana M. 2011. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 25, 781–788. ( 10.1101/gad.2027411) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kim Y, et al. 2016. Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat. Med. 23, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wu H, Ng C, Villegas V, Chamberlain SJ, Cacace A, Wallace G. 2019. Small molecule inhibitors of G9a reactivate the maternal PWS genes in Prader-Willi Syndrome patient derived neural stem cells and differentiated neurons. bioRxiv, 640938.
- 172.Gendrel A-V, Tang YA, Suzuki M, Godwin J, Nesterova TB, Greally JM, Heard E, Brockdorff N. 2013. Epigenetic functions of smchd1 repress gene clusters on the inactive X chromosome and on autosomes. Mol. Cell. Biol. 33, 3150–3165. ( 10.1128/MCB.00145-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jansz N, et al. 2018. Smchd1 regulates long-range chromatin interactions on the inactive X chromosome and at Hox clusters. Nat. Struct. Mol. Biol. 25, 766–777. ( 10.1038/s41594-018-0111-z) [DOI] [PubMed] [Google Scholar]
- 174.Wang CY, Jegu T, Chu HP, Oh HJ, Lee JT. 2018. SMCHD1 merges chromosome compartments and assists formation of super-structures on the inactive X. Cell 174, 406–421. ( 10.1016/j.cell.2018.05.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gdula MR, et al. 2019. The non-canonical SMC protein SmcHD1 antagonises TAD formation and compartmentalisation on the inactive X chromosome. Nat. Commun. 10, 30 ( 10.1038/s41467-018-07907-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Blewitt ME, et al. 2008. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 40, 663 ( 10.1038/ng.142) [DOI] [PubMed] [Google Scholar]
- 177.Nozawa RS, Nagao K, Igami KT, Shibata S, Shirai N, Nozaki N, Sado T, Kimura H, Obuse C. 2013. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat. Struct. Mol. Biol. 20, 566–573. ( 10.1038/nsmb.2532) [DOI] [PubMed] [Google Scholar]
- 178.Mould AW, et al. 2013. Smchd1 regulates a subset of autosomal genes subject to monoallelic expression in addition to being critical for X inactivation. Epigenetics Chromatin 6, 19 ( 10.1186/1756-8935-6-19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lemmers RJ, et al. 2012. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374. ( 10.1038/ng.2454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sacconi S, et al. 2013. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am. J. Hum. Genet. 93, 744–751. ( 10.1016/j.ajhg.2013.08.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gordon CT, et al. 2017. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat. Genet. 49, 249–255. ( 10.1038/ng.3765) [DOI] [PubMed] [Google Scholar]
- 182.Shaw ND, et al. 2017. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 49, 238–248. ( 10.1038/ng.3743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chen K, et al. 2015. Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl Acad. Sci. USA 112, E3535-E3544. ( 10.1073/pnas.1504232112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bielinska B, Blaydes SM, Buiting K, Yang T, Krajewska-Walasek M, Horsthemke B, Brannan CI. 2000. De novo deletions of SNRPN exon 1 in early human and mouse embryos result in a paternal to maternal imprint switch. Nat. Genet. 25, 74–78. ( 10.1038/75629) [DOI] [PubMed] [Google Scholar]
- 185.Rabinovitz S, Kaufman Y, Ludwig G, Razin A, Shemer R. 2012. Mechanisms of activation of the paternally expressed genes by the Prader-Willi imprinting center in the Prader-Willi/Angelman syndromes domains. Proc. Natl Acad. Sci. USA 109, 7403–7408. ( 10.1073/pnas.1116661109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Brant JO, Riva A, Resnick JL, Yang TP. 2014. Influence of the Prader-Willi syndrome imprinting center on the DNA methylation landscape in the mouse brain. Epigenetics 9, 1540–1556. ( 10.4161/15592294.2014.969667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bressler J, Tsai T-F, Wu M-Y, Tsai S-F, Ramirez MA, Armstrong D, Beaudet AL. 2001. The SNRPN promoter is not required for genomic imprinting of the Prader-Willi/Angelman domain in mice. Nat. Genet. 28, 232–240. ( 10.1038/90067) [DOI] [PubMed] [Google Scholar]
- 188.Tsai T-F, Jiang Y-H, Bressler J, Armstrong D, Beaudet AL. 1999. Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum. Mol. Genet. 8, 1357–1364. ( 10.1093/hmg/8.8.1357) [DOI] [PubMed] [Google Scholar]
- 189.Yang T, Adamson TE, Resnick JL, Leff S, Wevrick R, Francke U, Jenkins NA, Copeland NG, Brannan CI. 1998. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat. Genet. 19, 25–31. ( 10.1038/ng0598-25) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.