

Abstract
Aim:
L-2-hydroxyglutaric aciduria is a slowly progressive neurometabolic disorder caused by an enzymatic deficiency of L-2-hydroxyglutarate dehydrogenase. Here, we aimed to evaluate the clinical, neuroradiologic, and genotypic characteristics of patients with L-2-hydroxyglutaric aciduria who were followed in our outpatient clinic.
Material and Methods:
Twenty-five patients with L-2-hydroxyglutaric aciduria were enrolled in the study. Data regarding demographic, clinical, and neuroradiologic findings and molecular analysis were evaluated retrospectively.
Results:
The mean age of patients at the time of diagnosis was 12.09±8.02 years, whereas the mean age at the time of the first symptoms was 39.47±29.96 months. Diagnostic delay was found as 9.95±7.78 years. Developmental delay, decrease in school success, and seizures were the most common initial symptoms; however, behavioral problems and seizures became more prominent in the disease course. At the time of diagnosis, mental retardation and at least one pathologic cerebellar finding were detected in all symptomatic patients. Three patients developed brain tumors. The most common neuroimaging findings were subcortical white matter changes and cerebellar dentate nucleus involvement. In one patient, there was only isolated basal ganglia involvement without white matter lesions. Patients with similar genotypic features exhibited different clinical and radiologic findings.
Conclusion:
Although clinical symptoms appear early in L-2-hydroxyglutaric aciduria, there is approximately a ten-year delay in diagnosis. In subjects in whom brain tumor is detected in early childhood, L-2-hydroxyglutaric aciduria should be considered in the differential diagnosis in the presence of mental retardation accompanied by developmental delay, cerebellar and pyramidal findings, and behavior disorders in a wide spectrum ranging from autism spectrum disorder to psychosis. In patients with L-2-hydroxyglutaric aciduria, incipient headache, tinnitus, altered consciousness, and seizures can be indicative of brain tumors.
Keywords: Brain magnetic resonance imaging, L2HGDH, L-2-hydroxyglutaric aciduria, subcortical white matter
Öz
Amaç:
L-2-hidroksiglutarik asidüri; L-2-hidroksiglutarat dehidrogenaz enzimindeki yetersizlik sonucu gelişen, yavaş seyirli bir nörometabolik hastalıktır. Çalışmamızda kliniğimizde izlenen L-2-hidroksiglutarik asidüri olgularının, klinik, nöroradyolojik ve genotipik özelliklerinin tanımlanması amaçlanmıştır.
Gereç ve Yöntemler:
Çalışmaya L-2-hidroksiglutarik asidüri tanılı 25 olgu alındı. Olgulara ait demografik veriler, klinik belirti ve bulgular, görüntüleme bulguları ile genetik analiz sonuçları geriye dönük değerlendirildi.
Bulgular:
Olguların ortalama tanı yaşı 12,09±8,02 idi. İlk yakınmaları ortalama 39,47±29,96 ayda başlamıştı ve ortalama 9,95±7,78 yıllık tanısal gecikme saptandı. Gelişimsel gerilik, okul başarısında azalma ve nöbetler en sık görülen ilk başvuru yakınmaları iken hastalığın seyrinde davranış sorunları ve nöbetlerin öne çıktığı görüldü. Belirtili olguların tümünde, tanı anında zekâ geriliği ve en az bir patolojik serebellar fizik bakı bulgusu saptandı. Üç olguda beyin tümörü gelişti. Subkortikal ak madde değişiklikleri ile serebellar dentat nükleus tutulumu en sık görülen görüntüleme bulgularıydı. Bir olguda ak madde tutulumu olmayıp, izole bazal gangliyon tutulumu vardı. Benzer genotipik özelliklere sahip olan olgular, farklı klinik ve radyolojik bulgulara sahipti.
Çıkarımlar:
Klinik bulguların erken yaşlarda ortaya çıkmasına karşın, L-2-hidroksiglutarik asidüri olgularında ortalama 10 yıllık tanısal gecikme yaşanmaktadır. Erken yaşta beyin tümörü saptanan olgularda, zekâ geriliğine eşlik eden gelişimsel gecikme, serebellar ya/ya da piramidal bulguların varlığında ve otizm spektrumu bozukluğundan psikoza uzanan geniş spektrumdaki davranış bulgularının varlığında L-2-hidroksiglutarik asidüri ayırıcı tanıda akılda tutulmalıdır. Tanılı olgularda yeni başlayan baş ağrısı, kulak çınlaması, bilinç düzeyi değişikliği ve karakter değiştiren nöbet, malignite gelişimi açısından uyarıcı olmalıdır. Özgül nöroradyolojik bulguların tanınması, olguların daha erken tanı almasını sağlayabilir.
Introduction
L-2-hydroxyglutaric aciduria (L2HGA) is an autosomal recessive neurometabolic disorder caused by an enzymatic deficiency of L-2-hydroxyglutarate dehydrogenase due to homozygous or compound heterozygous mutations in the L2HGDH gene (1, 2). The main clinical findings of the disease include developmental and cognitive retardation, febrile and/or afebrile seizures, gait problems, and behavioral problems. Pathologic physical examination findings include progressive cerebellar ataxia, pyramidal and extrapyramidal system findings, and macrocephaly. Acute metabolic decompensation is not expected and rapid neurologic deterioration is generally not reported. The fact that the initial clinical findings are nonspecific, causes the diagnosis to be made frequently in adolescence or adulthood (2, 3). Development of malignant brain tumors has also been reported in the clinical course of L-2-hydroxyglutaric aciduria (4). Neuroradiologic findings specific to the disease facilitate the diagnosis. Symmetrical white matter involvement predominant in the subcortical area, and cerebellar dentate nucleus involvement constitute the frequently observed brain magnetic resonance imaging (MRI) findings. In different case series, different rates of globus pallidus, putamen, and caudate nucleus involvement have been reported. In the advancing stages of the disease, cerebral and cerebellar atrophy may be added to the radiologic findings (5, 6). The definite diagnosis is made by demonstrating increased L-2-hydroxyglutaric acid in body fluids and/or by molecular analysis of the L2HGDH gene (1, 7). Although patients who benefited partially from riboflavin, carnitine, and flavin adenine dinucleotide (FAD) therapies have been reported in a small number of studies, there is still no efficient treatment (8, 9). In this study, it was aimed to define the clinical, neuroradiologic, and genotypic findings in patients with L2HGA who were followed up by our clinic.
Material and Methods
Twenty-five patients who were being followed up with a diagnosis of L2HGA in Istanbul University-Cerrahpaşa, Cerrahpaşa Medical Faculty, Division of Pediatric Nutrition and Metabolism, were included in the study. In all patients included in the study, the diagnosis was confirmed by demonstrating increased L-2-hydroxyglutaric acid in urine using capillary electrophoresis and/or through molecular analysis of the L2HGDH gene. The subjects who did not have regular follow-up and who had deficient data were not included in the study.
Demographic data such as the age at the time of diagnosis, sex, and consanguinity in the family, the age at the time of the first symptom, the symptom at the time of the first presentation, clinical findings, brain tumors that developed during the disease, brain MRI findings, and molecular analysis results belonging to the L2HGDH gene, were recorded retrospectively.
Our study was conducted in accordance with the principles of the Helsinki Declaration. Approval was obtained from our hospital’s local ethics committee for our study (June 2017- Decision no: 214159). Written consent was obtained from the families of all subjects.
Results
Twenty-five patients who were being followed up with a diagnosis of L-2-hydroxyglutaric aciduria were included in the study. Fifteen subjects were female and ten were male. Twenty-three subjects had a history of consanguinity in the family. In eight families, more than one sibling with a diagnosis of L2HGA were included in the study. Two subjects had a diagnosis of spondylo-ocular syndrome in addition to L2HGA (Case 12, 13).
The mean age at the time of diagnosis was found as 12.09±8.02 years in the subjects who were included in the study. The mean age at the time of the first presentation symptom associated with L-2-hydroxyglutaric aciduria was found as 39.47±29.96 months and the mean diagnostic delay was found as 9.95±7.78 years.
Clinical findings
The most common symptoms at the time of presentation included developmental retardation (n=9), decrease in academic success (n=5), and febrile and/or afebrile seizure (n=5). Two subjects were asymptomatic at the time of diagnosis. The first of the asymptomatic subjects was diagnosed at the age of six months as a result of family screening performed because the first child in the family was diagnosed as having L2HGA (Case 12). The other subject had a history of premature birth and the presence of suspicious radiologic findings in terms of L2HGA on routine brain MRI ordered by developmental neurology at the age of one year, which led to investigations for L2HGA, and early diagnosis of the disease (Case 14). The presence of seizures (48%) and behavioral problems (36%) during the disease period from the onset of the first symptoms to the time of diagnosis were noted as predominant clinical findings. Behavioral findings were found in nine patients at the time of diagnosis and during the course of the disease. One subject was referred for investigations for autism spectrum disorder (ASD) (Case 21). One subject was diagnosed while receiving medical treatment for atypical psychosis (Case 11) and one subject was diagnosed while being followed up because of catatonia (Case 20). When the subjects’ neurologic physical examination findings at the time of diagnosis were evaluated, it was observed that all symptomatic subjects had different degrees of mental retardation and at least one cerebellar examination finding including mainly ataxia and/or intentional tremor. Pyramidal system findings such as spasticity, increased deep tendon reflexes, and pathological reflex accompanied the picture in thirteen patients, and extrapyramidal findings such as athetosis and dystonia were found in only four subjects.
Brain tumor developed during the course of the disease in three patients. The pathologic findings of these patients were as follows: grade 3 anaplastic ependymoma in one subject (Case 6), grade 2 astrocytoma in one subject (Case 11), and grade 3 anaplastic oligodendroglioma in one subject (Case 22). In one subject, no specific neurologic findings were related to the present brain tumor; the mass was detected on brain MRI performed during investigations for mental retardation and cerebellar findings (Case 6). One subject had not been diagnosed despite the presence of all clinical findings and pathologic physical examination findings; this subject was diagnosed with a picture of acutely developing encephalopathy and status epilepticus (Case 11). In the final subject who developed brain tumor, new-onset severe headache developed while being followed up with a diagnosis of L2HGA, which was a warning sign in terms of the development of malignancy (Case 22). The pathologic brain MRI images of the two patients who developed brain tumor are shown in Figure 1, and the subjects’ demographic, clinical, and genotypic findings are summarized in Table 1.
Figure 1.
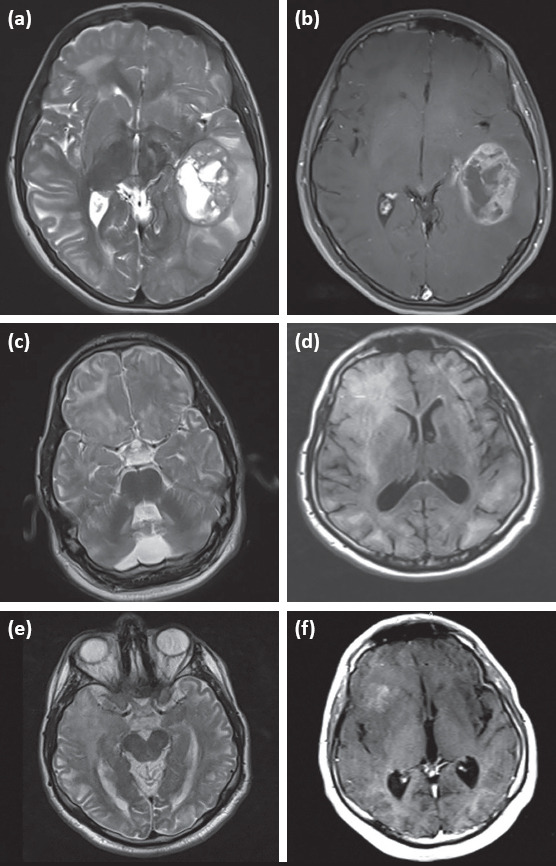
Brain MRI findings belonging to two patients with L2HGA who had brain tumor; 1 (a, b): brain MRI findings of subject 6; mass compressing the surrounding tissues with heterogeneous contrast uptake and cystic component in the temporoparietal region in the left hemisphere in T2-weighted and contrast-enhanced axial sections 1 (c–e): brain MRI findings of subject 22; 1 (c, d): asymmetrical increased signal intensity in the subcortical and deep white matter in the right frontal region in T2 and FLAIR axial sections dated 2011, 1 (e-f): mass with heterogeneous contrast uptake in the right temporal region in T2 axial section and the frontal regions in the contrast-enhanced section extending to the inferior temporal area dated 2013
Table 1.
Demographic, clinical and genotypic findings of the subjects with L2HGA
| Patient | Family | Sex | Age at the time of onset of the findings (months) | Age at diagnosis (years) | Symptom at presentation | Mental retardation | Cerebellar findings | Pyramidal findings | Extrapyramidal findings | Seizure | Speech disorder | Delay in developmental stages1 | Behavioral problems F1 | Genetic analysis M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F1 | M | 84 | 17.3 | Decrease in academic success | + | + | – | – | – | – | – | – | p.Tyr123Cys (c.368A>G) |
| 2 | F1 | F | 84 | 14.6 | Decrease in academic success | + | + | + | – | – | – | – | – | p.Tyr123Cys (c.368A>G) |
| 3 | F1 | M | 24 | 21.4 | Developmental delay | + | + | + | – | – | – | + | – | p.Tyr123Cys (c.368A>G) |
| 4 | F2 | F | 84 | 16.6 | Decrease in academic success | + | + | – | – | +ϖ | + | – | – | p.Gly55Asp (c.164G>A) |
| 5 | F3 | F | 48 | 17.4 | Unsteady gait | + | + | + | – | – | – | – | + | p.Gly55Asp (c.164G> A) |
| 6 | F3 | F | 12 | 14.6 | Afebrile seizure | + | + | – | – | +ϖ | – | – | – | p.Gly55Asp (c.164G> A) |
| 7 | F4 | M | 6 | 23.3 | Developmental delay | + | + | + | + | +ϖ | + | + | – | p.Gly55Asp (c.164G> A) |
| 8 | F4 | F | 6 | 21.1 | Developmental delay | + | + | + | + | +ϖ | + | + | – | p.Gly55Asp (c.164G>A) |
| 9 | F5 | F | 30 | 5.0 | Febrile seizure | + | + | – | – | +ϖν | + | – | + | p.Arg335Ter (c.1003C >T) |
| 10 | F5 | M | 12 | 8.1 | Febrile seizure | + | + | – | – | +ν | – | – | – | p.Arg335Ter (c.1003C>T) |
| 11 | F6 | M | 24 | 32.0 | Developmental delay | + | + | + | – | +ν | + | + | + | not performed |
| 12 | F7 | F | Asymptomatic | 0.6 | None | – | – | – | – | – | – | – | – | p.Ile278Ser (c.883T>G) |
| 13 | F7 | F | 36 | 4.2 | Developmental delay | + | + | + | – | – | – | + | + | p.Ile278Ser (c.883T>G) |
| 14 | F8 | M | Asymptomatic | 1.0 | None | – | – | – | – | – | – | – | – | p.Lys246= (c.738G>A) |
| 15 | F9 | F | 11 | 4.1 | Unsteady gait | + | + | – | + | – | – | – | + | p.Arg335Ter (c.1003C> T) |
| 16 | F10 | F | 7 | 20.0 | Afebrile seizure | + | + | + | – | +ϖ | + | – | – | – |
| 17 | F11 | M | 60 | 8.8 | Behavior problems | + | + | – | – | – | + | – | + | p.Met372Serfs (c.1115delT) |
| 18 | F12 | F | 30 | 7.9 | Developmental delay | + | + | + | – | – | + | + | – | p.Gly55Asp (c.164G> A) |
| 19 | F12 | F | 10 | 7.4 | Developmental delay | + | + | + | – | +ϖν | + | + | – | p.Gly55Asp (c.164G>A) |
| 20 | F13 | M | 6 | 6.5 | Developmental delay | + | + | + | + | +ϖ | + | + | + | p.Met372Serfs (c.1115delT) |
| 21 | F13 | M | 48 | 0.1 | Behavior problems | + | + | – | – | – | + | – | + | p.Met372Serfs (c.1115delT) |
| 22 | F14 | M | 48 | 12.6 | Febrile seizure | + | + | + | – | +ϖν | + | + | – | p.His98Tyr (c.292C>T) |
| 23 | F14 | M | 84 | 14.8 | Decrease in academic success | + | + | + | – | – | + | – | – | p.His98Tyr (c.292C>T) |
| 24 | F15 | F | 70 | 7.9 | Unsteady gait | + | + | – | – | – | + | – | – | p.Arg277X (c.829C> T) |
| 25 | F16 | F | 84 | 15.0 | Decrease in academic success | + | + | – | – | +ϖ | – | – | + | p.Gly55Asp (c.164G>A) |
ϖ: Afebrile seizure; ν: Febrile seizure
Radiologic findings
All subjects included in the study had brain MRI examinations. It was observed that the most common radiologic findings included subcortical white matter changes and cerebellar dentate nucleus involvement. When the extension of the white matter involvement was examined, subcortical involvement was found in 96% of the subjects and deep and periventricular white matter involvement was found in 85% and 32% of the subjects, respectively. When the gray matter was examined, it was found that 23 patients had involvement of the dentate nucleus, 20 patients had involvement of the globus pallidus, 10 patients had involvement of the putamen, 10 patients had involvement of the caudate nucleus, and one patient had involvement of the thalamus. Brain stem involvement was reported in only two subjects, and cerebellar white matter was intact in all subject. Cerebellar atrophy was observed with a rate of 72% and cerebellar atrophy with a predominance of vermis atrophy was observed with a rate of 68%.
White matter involvement was absent in one patient, though symptoms and pathologic physical examination findings were present at the time of diagnosis. Brain MRI findings included isolated globus pallidus, caudate nucleus, putamen, and dentate nucleus involvement (Case 13).
When the MRI of Case 22, who was reported to develop brain tumor, performed 2 years ago was examined, it was noted that malignancy originated from the area of asymmetrical white matter involvement, and no such characteristics were noted in the other two patients who had brain tumor.
Different brain MRI images belonging to the subjects are shown in Figure 2 and radiologic findings are summarized in Table 2 as titles.
Figure 2.
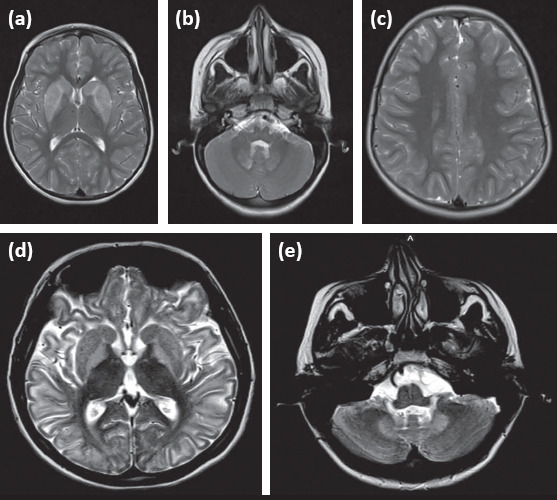
Different brain MRI findings of patients with L2HGA; 2 (a–c): brain MRI findings of subject 13; it is observed that there is increased signal intensity in bilateral caudate nuclei, putamens and dentate nuclei in T2 axial sections, while no white matter change is observed; 2 (d, e): brain MRI findings of subject 16; 2 (d): marked increased signal intensity in bilateral basal ganglia predominant in bilateral symmetrical subcortical white matter and globus pallidus in T2 axial section, 2 (e): increased signal intensity in bilateral dentate nuclei in T2 axial section
Table 2.
Changes found in brain MR imagings in patients with L-2-hydroxyglutaric aciduria and their frequencies (n=25)
| n | % | |
|---|---|---|
| White matter changes | ||
| Subcortical white matter | 24 | 96 |
| Deep white mattera | 21 | 84 |
| Periventricular white matter | 8 | 32 |
| Cerebellar white matter | – | – |
| Capsula interna | 10 | 40 |
| Capsula externa | 19 | 76 |
| Capsula extrema | 22 | 88 |
| Corpus callosum | 3 | 12 |
| Brain stem | 2 | 8 |
| Subcortical cystsb | 19 | 76 |
| Gray matter changes | ||
| Globus pallidus | 20 | 80 |
| Caudate nucleus | 10 | 40 |
| Putamen | 10 | 40 |
| Dentate nucleus | 23 | 92 |
| Thalamus | 1 | 4 |
| Presence of atrophy | ||
| Cerebral | 18 | 72 |
| Cerebellar | 17 | 68 |
a: White matter of the cerebral hemisphere lobes; b: Hypointense white matter rarefaction areas in T1 and FLAIR sections in subcortical white matter
Genotypic findings
Molecular analysis of the L2HGDH gene was performed in 24 patients. No mutation was found and no polymerase chain reaction (PCR) product belonging to exome 1 was observed in one of the subjects in whom molecular analysis was performed. In the patient in whom markedly increased L-2-hydroxyglutaric acid excretion was demonstrated with capillary electrophoresis in accompaniment with clinical and radiologic findings compatible with the disease, it was thought that a homozygous deletion could be responsible for the clinical picture, but multiple ligation-dependent probe amplification (MLPA) could not be performed (Case 16).
Homozygous p.Gly55Asp (c.164G>A) was the most common variation in the subjects in whom molecular analysis was performed. The presence of sibling cases with different clinical and radiologic findings despite similar genotypic findings in our study, in which consanguineous marriage was found with a rate of 92%, was interpreted such that there was no genotype-phenotype relationship.
Discussion
L-2-hydroxyglutaric aciduria is a congenital metabolism disorder characterized by slowly progressive neurodegenerative findings. Although developmental delay and cognitive retardation begin in the early period of life, symptomatic pyramidal and extrapyramidal findings and cerebellar findings are frequently manifested after the first ten years of life (3). The fact that initial findings are nonspecific leads to diagnostic delay, and the diagnosis is made in advancing years of life only after brain tumor develops in some cases. In the present study, the clinical findings that could guide physicians are listed by the stages of life to prevent diagnostic delay in L2HGA, and the specific neuroradiologic findings related to the disease are emphasized. We investigated whether different mutations belonging to the L2HGDH gene influenced the clinical course of the disease by presenting the genotypic data belonging to a single center.
In a study conducted with 16 subjects in which the mean age at the time of enrollment was 16.2 years, the clinical findings included developmental delay, presence of a history of seizures, speech disorder, and balance disorder in order of frequency (10). Macrocephaly was found in four patients and development of brain tumor during the course of the disease was recorded in four patients. In another study conducted with eight Turkish subjects in which the mean age at the time of diagnosis was 8 years, development of brain tumor or macrocephaly was not reported in any subject; it was reported that all subjects had psychomotor retardation (11). When the subjects were evaluated according to neurologic physical examination findings, it was observed that seven subjects had extrapyramidal findings and five had cerebellar ataxia. Fifty percent of the subjects had a history of seizure. In a study in which 61 patients with L2HGA were evaluated, the mean age at the time of the onset of the first symptom related to the disease was 2 (range, 0-7) years and the first symptoms recorded included developmental delay (52%), epilepsy (42%), and ataxia (20%). Macrocephaly was found in 48% of the subjects and no subject who developed brain tumor was reported (7). In another study conducted with 29 patients with L2HGA, the mean age at the time of symptom onset was reported as 8.2 years, and the mean age at the time of diagnosis was reported as 13.4 years; the mean diagnostic delay time was 5.2 years (12). Having seizures was a common clinical finding and it was shown that cerebellar findings accompanying mental retardation predominated among pathologic physical examination findings. In this study, which also gave information about follow-up processes, it was reported that the subjects started to display cerebellar findings at about the age of 12 years, and dystonia developed in one patient. Medulloblastoma was observed in one patient. Behavioral disorders have been reported with different frequencies in patients with L2HGA (7, 10, 11); it was observed that findings such as attention-deficit and hyperactivity before adolescence were replaced by mood changes such as depression after adolescence (12). In recent years, it has been emphasized that L2HGA should be considered in the differential diagnosis in subjects presenting with ASD (13, 14). In our study, which was compatible with the literature in terms of the variability of clinical findings, it was shown that there was a mean delay time of 9.95±7.78 years from the onset of the first symptoms to diagnosis. In one subject, the diagnosis was delayed up to the age of 32 years when the picture of encephalopathy secondary to the development of brain tumor and status epilepticus developed, although all clinical symptoms and physical examination findings were recognized before. In contrast to the literature, pyramidal system findings were also included in predominant pathologic physical examination findings in addition to cerebellar findings. It is known that the psychiatric findings related to the disease may show variability ranging from ASD to atypical psychosis. Therefore, it was emphasized in our study that suspecting L2HGA disease in the presence of behavioral findings accompanying mental retardation, especially in early childhood period, could prevent diagnostic delay.
Brain tumors in cases of L-2-hydroxyglutaric aciduria have been previously reported in the literature, though the underlying pathophysiologic mechanisms could not be elucidated fully (15). The effect of L-2-hydroxyglutaric acid level on oncogenesis has been frequently discussed, but this view could not maintain validity considering similar urine acid levels in subjects who did and did not develop a malignancy. Also, it is thought that the acid level in body fluid is not equivalent to the acid level in the brain tissue. The presence of mitogenic factors with increased production in the demyelination and remyelination processes that develop during the course of the disease was excluded because similar tumors have not been observed in other white matter disorders. Finally, it is thought that increased L-2-hydroxyglutarate accumulation in some mutations of the L2HDGH gene could have oncogenic action (15). In a literature data review involving 295 patients, it was reported that brain tumor developed between the ages of 3 and 36 years in 14 patients, and it was shown that the frequencies of different neoplasms varied by different age ranges. In the early periods of life, development of medulloblastoma was predominant, whereas the frequency of low-grade supratentorial astrocytoma increased between the ages of 9 and 12 years, and the frequency of high-grade glioma increased in the advanced age period (9). In subjects aged over 9 years, the frequency of development of glioma is increased, and it is generally localized in the frontal or temporal areas (4). In the literature, development of Wilms’ tumor has been reported only in one subject during the course of L2HGA disease, and it was proposed that the disease could also be associated with malignancies arising from the tissues other than the central nervous system (16). In our study, brain tumor developed in three patients; severe headache in one of these subjects while being followed up with a diagnosis of L2HGA was a warning sign in terms of malignancy, and imaging was repeated. Considering that the white matter involvement on the past imaging belonging to this patient was asymmetrical, and the neoplasm that developed later originated from this area, new symptoms should be considered carefully in patients with a diagnosis of L2HGA, and it should be kept in mind that asymmetrical white matter involvement areas may carry risk in terms of the development of malignancy later in life.
Brain MRI findings are quite specific in terms of L2HGA disease and enable making a diagnosis as a result of imaging obtained because of different symptoms. In a study conducted with 17 subjects, frontotemporal white matter involvement was reported in all subjects; parietal white matter involvement was found in 94% subjects and occipital white matter involvement was found in 88% (6). When the extension of white matter involvement was examined, it was observed that subcortical involvement predominated (82.3%) and central and periventricular white matter involvement were found in 11.7% and 5.9% of the subjects, respectively. When the gray matter was evaluated, dentate nucleus involvement was reported in all subjects, putamen involvement was reported in 70.6%, globus pallidus involvement was reported in 58.8%, caudate nucleus involvement was reported in 35.2%, and thalamic involvement was reported in 17.6%. Brain stem involvement was reported in only one subject and corpus callosum and cerebellar white matter were preserved in all subjects. In the study in which cerebral atrophy was observed with a rate of 47.1%, vermis atrophy (11.8%) predominated in subjects who had cerebellar atrophy. Only one subject had atrophy in the cerebellar hemispheres. In the literature, there are different studies in which similar MRI findings have been reported (10, 12). In contrast to the data reported, the mean age at the time of imaging was found as 11.9±8.5 years and the mean disease duration was found as 9.2±8.1 in another study in which the relationship between disease duration and progression of radiological findings was evaluated. Similar to the literature, subcortical white matter involvement was found in all subjects (more predominant in the frontal area) and dentate nucleus (98%), caudate nucleus (100%) and putamen (100%) involvement were noted in the gray matter. When the effect of increasing disease duration on neuroradiologic findings was evaluated, it was observed that involvement predominant in the outer border of the caudate nucleus and putamen in short-term disease showed a homogeneous extension in the whole nucleus as the disease duration prolonged. Again, white matter involvement predominant in the frontal area with prolonged disease duration was replaced by a global white matter involvement. Globus pallidus and dentate nucleus involvement are present even in the early periods of the disease and are not correlated with disease progression. A statistically significant correlation was found between cerebral and cerebellar atrophy and disease duration (5). When the data of our study were evaluated in terms of the diversity of radiologic findings, they were found to be compatible with the literature. However, white matter involvement was present in all cases reported up to the present time. In one of the subjects in our study, isolated basal ganglion involvement was found without white matter involvement. This radiologic appearance has not been reported in cases of L2HGA in previous studies.
Missense, nonsense, frameshift, and splice-site mutations belonging to the L2HGDH gene have been reported (7, 17). There is still no proven genotype-phenotype relationship and molecular studies are strongly needed to elucidate the pathophysiology of the disease, especially the mechanisms of the development of brain tumors. The presence of sibling cases with different clinical and radiologic findings despite similar genotypic findings in our study was interpreted as that there was no genotype-phenotype relationship.
In conclusion, L2HGA disease should be kept in mind in the differential diagnosis in subjects who are found to have brain tumor at an early age, in presence of developmental delay, and cerebellar and/or pyramidal findings accompanying mental retardation and in the presence of behavioral findings in a wide spectrum ranging from ASD to psychosis in view of the data presented in our study. In subjects with a diagnosis of L2HGA, new-onset headache, tinnitus, change in consciousness level, and seizure changing characteristics should be warnings in terms of the development of malignancy; one should be careful in terms of tumor development in serial examinations if asymmetrical white matter involvement is noted. The fact that keeping specific neuroradiologic findings in mind could prevent diagnostic delay, and the importance of family screening for the diagnosis of metabolic diseases, especially in countries where the rates of consanguineous marriages are high, were emphasized in our study.
Footnotes
Ethics Committee Approval: The study was conducted in accordance with the principles of the Declaration of Helsinki. Approval was obtained from the local ethics committee of Istanbul University-Cerrahpasa, Cerrahpasa Medical Faculty (June 2017/No: 214159).
Informed Consent: Written consent was obtained from the parents of all patients.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - T.Z., C.Y., Ç.O., E.K., M.Ş.C., A.G., G.Y., E.Ö.E., Ç.A.Z.; Design - T.Z., C.Y., Ç.O., E.K., M.Ş.C., A.G., G.Y., E.Ö.E., Ç.A.Z.; Supervision - T.Z., C.Y., Ç.O., E.K., M.Ş.C., A.G., G.Y., E.Ö.E., Ç.A.Z.; Funding - T.Z., C.Y., E.K., Ç.A.Z.; Analysis and/or Interpretation - T.Z., C.Y., A.G., G.Y.; Literature Review - T.Z., Ç.O., M.Ş.C., E.Ö.E.; Writing - T.Z., C.Y., Ç.O., E.K., Ç.A.Z.; Critical Review - C.Y., Ç.A.Z, E.K.
Conflict of Interest: The authors have no conflicts of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
Etik Kurul Onayı: Çalışma Helsinki deklarasyon prensiplerine uygun olarak gerçekleştirildi. Bu çalışma için etik kurul onayı İstanbul Üniversitesi-Cerrahpaşa, Cerrahpaşa Tıp Fakültesi Lokal Etik Kurulu’ndan alınmıştır (Haziran 2017/No: 214159).
Hasta Onamı: Yazılı hasta onamı hastaların ailelerinden alınmıştır.
Hakem Değerlendirmesi: Dış bağımsız.
Yazar Katkıları: Fikir - T.Z., C.Y., Ç.O., E.K., M.Ş.C., A.G., G.Y., E.Ö.E., Ç.A.Z.; Tasarım - T.Z., C.Y., Ç.O., E.K., M.Ş.C., A.G., G.Y., E.Ö.E., Ç.A.Z.; Denetleme - T.Z., C.Y., Ç.O., E.K., M.Ş.C., A.G., G.Y., E.Ö.E., Ç.A.Z.; Kaynaklar - T.Z., C.Y., E.K., Ç.A.Z.; Analiz ve/veya Yorum - T.Z., C.Y., A.G., G.Y.; Literatür Taraması - T.Z., Ç.O., M.Ş.C., E.Ö.E.; Yazıyı Yazan - T.Z., C.Y., Ç.O., E.K., Ç.A.Z.; Eleştirel İnceleme - C.Y., Ç.A.Z, E.K.
Çıkar Çatışması: Yazarlar çıkar çatışması bildirmemişlerdir.
Mali Destek: Yazarlar bu çalışma için mali destek almadıklarını beyan etmişlerdir.
References
- 1.Van Schaftingen E, Rzem R, Veiga-da-Cunha M. L:-2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J Inherit Metab Dis. 2009;32:135–42. doi: 10.1007/s10545-008-1042-3. [DOI] [PubMed] [Google Scholar]
- 2.Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571–87. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffmann GF, Kölker S. Cerebral organic acid disorders and other disorders of lysine catabolism. In: Saudubray JM, Baumgartner MR, Walter J, editors. Inborn Metabolic Diseases:Diagnosis and Treatment. 6th edition. Heidelberg: Springer-Verlag; 2016. pp. 333–48. [Google Scholar]
- 4.Patay Z, Mills JC, Löbel U, Lambert A, Sablauer A, Ellison DW. Cerebral neoplasms in L-2 hydroxyglutaric aciduria:3 new cases and meta-analysis of literature data. AJNR Am J Neuroradiol. 2012;33:940–43. doi: 10.3174/ajnr.A2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steenweg ME, Salomons GS, Yapici Z, et al. L-2-Hydroxyglutaric aciduria:pattern of MR imaging abnormalities in 56 patients. Radiology. 2009;251:856–65. doi: 10.1148/radiol.2513080647. [DOI] [PubMed] [Google Scholar]
- 6.Fourati H, Ellouze E, Ahmadi M, et al. MRI features in 17 patients with l2 hydroxyglutaric aciduria. Eur J Radiol Open. 2016;3:245–50. doi: 10.1016/j.ejro.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steenweg ME, Jakobs C, Errami A, et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants:a genotype-phenotype study. Hum Mutat. 2010;31:380–90. doi: 10.1002/humu.21197. [DOI] [PubMed] [Google Scholar]
- 8.Samuraki M, Komai K, Hasegawa Y, et al. A successfully treated adult patient with L-2-hydroxyglutaric aciduria. Neurology. 2008;70:1051–2. doi: 10.1212/01.wnl.0000287141.90944.95. [DOI] [PubMed] [Google Scholar]
- 9.Yilmaz K. Riboflavin treatment in a case with l-2-hydroxyglutaric aciduria. Eur J Paediatr Neurol. 2009;13:57–60. doi: 10.1016/j.ejpn.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Faiyaz-Ul-Haque M, Al-Sayed MD, Faqeih E, et al. Clinical, neuroimaging, and genetic features of L-2-hydroxyglutaric aciduria in Arab kindreds. Ann Saudi Med. 2014;34:107–14. doi: 10.5144/0256-4947.2014.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canda E, Köse M, Yazıcı H, et al. Clinical, Neuroimaging, and Genetic Features of the Patients with L-2-Hydroxyglutaric Aciduria. J Pediatr Res. 2018;5:39–43. [Google Scholar]
- 12.Topçu M, Aydin OF, Yalçinkaya C, et al. L-2-hydroxyglutaric aciduria:a report of 29 patients. Turk J Pediatr. 2005;47:1–7. [PubMed] [Google Scholar]
- 13.Zafeiriou DI, Ververi A, Salomons GS, et al. L-2-Hydroxyglutaric aciduria presenting with severe autistic features. Brain Dev. 2008;30:305–7. doi: 10.1016/j.braindev.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Kiykim E, Zeybek CA, Zubarioglu T, et al. Inherited metabolic disorders in Turkish patients with autism spectrum disorders. Autism Res. 2016;9:217–23. doi: 10.1002/aur.1507. [DOI] [PubMed] [Google Scholar]
- 15.Haliloglu G, Jobard F, Oguz KK, et al. L-2-hydroxyglutaric aciduria and brain tumors in children with mutations in the L2HGDH gene:neuroimaging findings. Neuropediatrics. 2008;39:119–22. doi: 10.1055/s-2008-1081217. [DOI] [PubMed] [Google Scholar]
- 16.Rogers RE, Deberardinis RJ, Klesse LJ, Boriack RL, Margraf LR, Rakheja D. Wilms tumor in a child with L-2-hydroxyglutaric aciduria. Pediatr Dev Pathol. 2010;13:408–11. doi: 10.2350/09-12-0768-CR.1. [DOI] [PubMed] [Google Scholar]
- 17.Jellouli NK, Hadj Salem I, Ellouz E, et al. Founder effect confirmation of c.241A>G mutation in the L2HGDH gene and characterization of oxidative stress parameters in six Tunisian families with L-2-hydroxyglutaric aciduria. J Hum Genet. 2010;59:216–22. doi: 10.1038/jhg.2014.4. [DOI] [PubMed] [Google Scholar]