

Abstract
Therapeutic targeting of the immune system in cancer is now a clinical reality and marked successes have been achieved, most notably through the use of checkpoint blockade antibodies and chimeric antigen receptor T cell therapy. However, efforts to develop new immunotherapy agents or combination treatments to increase the proportion of patients who benefit have met with challenges of limited efficacy and/or significant toxicities. Nanomedicines — therapeutics composed of or formulated in carrier materials typically less than 100 nm in size — were originally developed to increase the uptake of chemotherapy agents by tumours and to reduce their off-target toxicities. Here, we discuss how nanomedicine-based treatment strategies are well suited to immunotherapy, based on the ability of nanomaterials to direct immunomodulators to tumours and lymphoid organs, to alter the way biologics engage with target immune cells, and to accumulate in myeloid cells in tumours and systemic compartments. We also discuss early efforts towards clinical translation of nanomedicine-based immunotherapy.
Beginning approximately ten years ago, clinical trials showed significant efficacy of checkpoint blockade and chimeric antigen receptor (CAR) T cell therapy in certain cancers, leading to approval by the U.S. Food and Drug Administration (FDA) of multiple therapies and a major change in the role of immunotherapy in oncology broadly. These treatments have induced prolonged complete remissions in a subset of patients, conclusively demonstrating the potential of immunotherapy in cancer1–3. These successes have fueled a dramatic increase in the number of immunotherapy agents in human testing and an enormous number of clinical trials exploring combination treatments. However, following the initial striking results observed in trials of inhibitors of the programmed cell death 1 (PD1) pathway and CAR T cell therapy in leukaemias and lymphomas, progress has been modest. Combination immunotherapies, such as co-treatment with antibodies that target cytotoxic T lymphocyte antigen 4 (CTLA4) and PD1, have shown moderate enhancements in efficacy while eliciting substantial increases in toxicity4. CAR T cell therapies that have been effective in haematological cancers have so far generally failed to have a major impact on the treatment of much more prevalent solid epithelial cancers. Thus, additional approaches to safely and effectively drive immune responses against cancer remain an important unmet need.
While efforts aiming to further expand our understanding of human immunology in the context of cancer remain critical, broader success with immunotherapy treatments is not limited by a lack of reasonable therapeutic targets. A major challenge lies in safely engaging these targets at the right time and place. Cancer drug development has historically focused on the generation of therapeutics that are systemically administered to ensure access to all sites of metastatic disease. This is an effective strategy for drugs designed to specifically act on pathways that are unique to tumour cells (for example, targeted drugs inhibiting mutant tumour kinases). Applying the same paradigm to immunotherapy agents that act on the host immune system can be problematic owing to our inability to site-specifically stimulate tumour-specific immune cells. For example, the approved checkpoint blockade drugs that interfere with inhibitory pathways in adaptive immunity have shown efficacy but also manageable (albeit significant) toxicities in patients, including gastrointestinal and pulmonary toxicities and autoimmune sequelae5. New therapeutics that instead “turn up” adaptive responses through immune-stimulatory pathways have encountered substantial safety issues that have hindered successful therapeutic implementation6.
A potential solution is to break with the traditional drug development paradigm and engineer the delivery of immunotherapeutics, focusing their action on target tissues (that is, tumours and tumour-draining lymph nodes) or cell types, to control the timing and location of immunomodulation. To achieve this goal, approaches based in nanomedicine — the formulation of drugs in carrier materials that are less than ~100 nm in size — may offer the means to increase both the safety and therapeutic efficacy of many immunotherapies. Formulation of immunotherapeutics in nanoparticles composed of lipids, polymers or other materials has been used to alter systemic exposure, promote accumulation in tumours, enhance uptake in innate immune compartments and alter signalling at the single-cell level. Such approaches introduce new complexities in drug development, but these are technical hurdles that numerous clinical stage companies have demonstrated to be readily surmountable. In this Review, we discuss the mechanisms by which nanomedicine-based treatments act on the immune system to enhance immunotherapies in preclinical models, the challenges facing these approaches and the early stages of clinical translation of these concepts. Protein engineering (such as antibody–cytokine fusions) and generation of tumour-tropic viruses represent important alternative strategies to achieve some of the same goals and have been recently reviewed7–9. We focus here on approaches complementary to strategies founded in genetic engineering and molecular biology and apply the traditional definition of nanomedicine as biological or small molecule drugs that are modified through synthetic chemistry or materials science approaches into nanoscale formulations for effective delivery, including chemical conjugation, encapsulation or physical incorporation within other materials. In the interest of limiting the scope, we also exclude discussions of the use of nanoparticles in vaccines, which has been the subject of other recent reviews and has its own large body of relevant literature10–13.
Mechanisms of nanoparticle therapeutics
Nanomedicine-based drug formulations were originally developed to alter the pharmacokinetics and toxicity profiles of chemotherapy agents, and promote their accumulation in tumours (Box 1). The ability to concentrate drugs within tumour cells and/or the tumour microenvironment (TME) is also of relevance for enhancing immunotherapy. However, nanomaterials also enable novel mechanisms of action for immunotherapy agents, including the ability to display ligands to immune cells, regulate intracellular delivery of cell-impermeable compounds, and control the timing of drug release and/or activation.
Box 1 |. An overview of nanomedicine.
The term nanomedicine was popularized in the late 1990s to describe the application of the emerging field of nanotechnology, which seeks to exploit the unique properties and behaviours of structures at the nanometer length scale, to human health135,136. The concept encompasses a broad range of approaches to disease treatment and diagnosis and the definition of nanomedicine is being refined as this discipline matures. In the drug delivery field, nanomedicines are formulations of therapeutics with polymers, lipids, metals or inorganic materials, which have at least one physical dimension of ~100 nm or less. Chemical conjugates of protein drugs (such as drug–protein and polymer–protein conjugates) are also sometimes considered nanomedicines due to similarities in their therapeutic goals and pharmacokinetic behaviour. Polymers and nanoparticles first became of interest in cancer therapy for their potential to alter drug pharmacokinetics, and later, for their capacity to accumulate in tumours through the enhanced permeation and retention (EPR) effect137,138. The EPR paradigm is based on the idea that particles of 10–100 nm in size in the bloodstream are too large to escape the vasculature and enter healthy tissues or to be cleared by the kidneys, but can escape from dysfunctional vasculature into tumours if they are not cleared too rapidly by the reticuloendothelial system. Within a tumour, defective lymphatics prevent the particles from being cleared from the tissue, leading to accumulation. Nanomedicines can also be functionalized with targeting molecules such as antibodies, which can increase internalization of drugs into target cells — in this case, drug-to-antibody ratios can be more than 10,000, greatly in excess of traditional antibody–drug conjugates45. There are currently approximately 50 nanomedicine therapies approved by the U.S. Food and Drug Administration for cancer and other diseases139. Challenges include the fact that current technologies typically only deliver a few percent of the injected dose into the tumour140 and clinical evidence suggests that the efficiency of EPR-based targeting varies significantly among patients141. However, nanomedicines aimed at enhancing immunotherapy often have a different set of delivery requirements than systems delivering chemotherapies and can act by a number of mechanisms distinct from chemotherapy. Because the immune system acts as a highly dynamic network, effective stimulation of a small number of leukocytes in tumours or lymphoid organs can lead to large changes in the microenvironment driven by a cascade of cell-to-cell communication. Challenges include the need to establish robust manufacturing strategies tailored to the objectives of immunotherapy, and the need for better preclinical models to evaluate immune-related toxicity as early as possible in the development pipeline142. Improvements in tools to monitor ongoing immune responses both in preclinical models and in patients undergoing treatment may be key to ensuring the high promise of nanomedicine for cancer immunotherapy is met in the clinic.
Promoting immunogenic tumour cell death.
Most traditional antitumour drugs aim to kill cancer cells, but not all pathways of cell death are immunologically equivalent. Tumour cell death events that promote an antitumour immune response are referred to as immunogenic cell death (ICD)14. ICD is associated with the extracellular release of damage-associated molecular patterns (DAMPs) such as ATP and high mobility group protein B1 (HMGB1), and the surface exposure of calreticulin and heat shock protein 90 (HSP90). These factors promote tumour antigen uptake by antigen-presenting cells and their subsequent activation. Traditional ablative cancer therapies, such as chemotherapy or ionizing radiation, vary in their ability to induce ICD and their immune potentiation can be offset by toxicities to responding immune cells. Nanomedicine formulations are an attractive modality to promote ICD because they can concentrate cytotoxic agents in tumour cells. In addition, nanomaterials can be designed to directly interact with external energy sources, enabling amplification of ICD induced by treatments such as radiotherapy and magnetic hyperthermia15.
Nanoparticle formulation of chemotherapeutics that promote ICD can enhance antitumour immunity by promoting more effective delivery of the drugs to tumour cells (Fig. 1a). For example, Doxil®, a clinically-approved PEGylated liposomal formulation of the anthracycline chemotherapeutic doxorubicin, synergized with checkpoint blockade in the treatment of mouse models of colon adenocarcinoma and fibrosarcoma, leading to stronger antitumour activity compared with treatment with free doxorubicin16. Doxil induced increased intratumoural CD8+ T cell infiltration, decreased regulatory T (Treg) cells, and increased CD80 expression by myeloid cells (including dendritic cells (DCs)) compared with free doxorubicin. Consistent with the importance of adaptive immunity in the effectiveness of the nanoparticle formulation of doxorubicin, efficacy was lost in athymic nude mice. Following these results, combination therapy of Doxil with checkpoint blockade is now being tested in a Phase IIb clinical trial in patients with metastatic breast cancer (Table 1). A more recent strategy to improve doxorubicin delivery and ICD relied on synthetic high-density lipoprotein nanodiscs with the drug covalently tethered to the nanoparticles through an acid-labile linkage. Delivery via nanodiscs upregulated markers of ICD in tumours and synergized with anti-PD1 relative to free drug in two mouse models of colon cancer.17
Figure 1 |. Nanomedicines enable unique modes of action in immunotherapy.
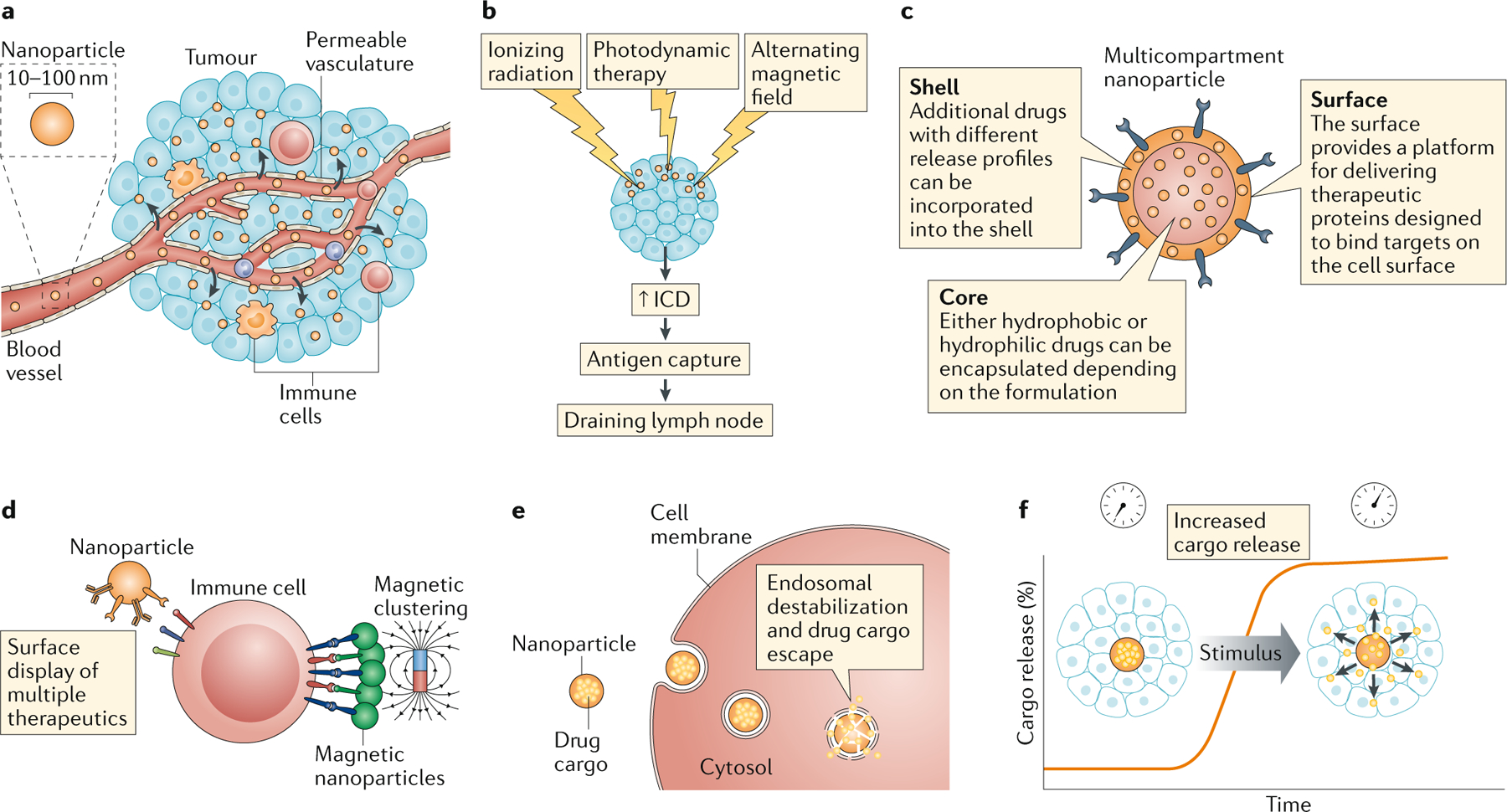
a | Nanomedicines, such as nanoparticles, accumulate within tumours via the enhanced permeation and retention effect, concentrating the drug in tumour sites. b | Nanoparticles can be designed to interact with external energy sources, such as ionizing or non-ionizing radiation or magnetic fields to enhance immunogenic cell death [Au:OK?] YES . c | Nanomedicines enable combinations of therapeutics, including drugs with very different properties, to be co-delivered to tumour sites. d | Multiple ligands can be arrayed on the surfaces of polymers and nanoparticles to enhance engagement of immunostimulatory receptors. e | Nanoparticles can be formulated to destabilize endosomal membranes and promote drug delivery into the cytosol. f | Nanoparticles enable control of the kinetics of drug release, either pre-programmed through the particle chemistry or through responsiveness to external stimuli such as light or heat.
Table 1 |.
Clinical translation of cancer immunotherapy nanomedicines
| refs | |||||
|---|---|---|---|---|---|
| NanoBiotix | Metal nanoparticle radioenhancers in combination with checkpoint blockade | Various solid tumours | Phase I-III | NCT03589339 | 25–27 |
| Oslo University Hospital/Bristol-Myers Squibb | PEGylated liposomal doxorubicin in combination with checkpoint blockade | Metastatic breast cancer | Phase IIb | NCT03409198 | 16 |
| Nektar Therapeutics | Reversibly-PEGylated IL-2 in combination with checkpoint blockade | Various solid tumours | Phase I, Phase II | NCT02983045, NCT03138889, NCT03282344, NCT03635983, NCT03785925, NCT03729245, NCT03435640 | 81–83 |
| Exicure | Intratumoural administration of TLR9 agonist-functionalized nanoparticles in combination with checkpoint blockade | Various solid tumours | Phase 1b/2 | NCT03684785 | 49,50 |
| Torque Therapeutics | Nanoparticle-functionalized antigen-primed T cell therapy | Various solid tumours and lymphomas | Phase I | NCT03815682 | 113,115,120 |
| Rimo Therapeutics | Metal-organic framework nanoparticles as radioenhancers combined with IDO inhibitors and/or checkpoint blockade | Various solid tumours | Phase I | NCT03444714 | 29 |
| Coordination Pharma | Nanoscale coordination polymer-based particles | Various solid tumours | Phase I | NCT03781362, NCT03953742 | 35–37 |
| Moderna Therapeutics | Lipid nanoparticle-delivered mRNA encoding OX40L, IL-23, and IL-36γ, with or without checkpoint blockade | Relapsed or refractory solid tumour malignancies or lymphoma | Phase I | NCT03739931 | 53 |
| Moderna Therapeutics | Lipid nanoparticle-delivered mRNA encoding OX40L | Relapsed or refractory solid tumour malignancies or lymphoma | Phase I | NCT03323398 | 53 |
| OncoNano | STING-activating polymer micelles | TBD | Phase I projected 2020–2021 | N/A | 48,134 |
| Tidal Therapeutics | Nanoparticles for gene transfer to macrophages and lymphocytes | TBD | Phase I projected 2020–2021 | N/A | 127,128 |
Although capable of inducing ICD, radiotherapy is rarely effective as a monotherapy for promoting sustained antitumour immunity18. However, numerous clinical studies are currently exploring radiotherapy as a component of combination immunotherapies19. Radiation kills tumour cells by inducing non-repairable DNA damage, but it can also trigger activation of the cGAS–STING pathway and subsequent production of pro-inflammatory cytokines, promoting innate and adaptive antitumour immunity20. The dose of radiotherapy must be carefully tuned as high radiation doses lead to attenuated STING activation due to DNA exonuclease TREX1-dependent cytosolic DNA degradation21. An initial, successful pro-inflammatory radiotherapy response can also be blunted by the recruitment of immunosuppressive immune cells to the tumour22. One long-standing concern has been that even localized radiotherapy may be detrimental to antitumour immunity due to damaging or suppressing tumour-infiltrating T cells, but recent findings from preclinical studies suggest that radiotherapy can actually energize tumour-resident T cells, which are comparatively more radioresistant than circulating T cells23.
Several approaches have used nanoparticles to improve immune priming provoked by radiotherapy. For example, biodegradable polymer nanoparticles designed to bind proteins from the surrounding solution were shown to promote T cell priming following radiotherapy. Intratumoural injection of the nanoparticles following radiotherapy led to capture of protein antigens released from dying cancer cells by the particles, which subsequently trafficked through lymphatics and were internalized by phagocytic professional antigen-presenting cells in the draining lymph node24. Nanomedicines can also be used as radioenhancers that directly interact with ionizing radiation to upregulate ICD25 (Fig. 1b). Materials containing elements with a high atomic number (Z) can both absorb and scatter radiation, and thus high-Z nanoparticles that accumulate in tumours can potentiate radiotherapy25. A recent clinical trial demonstrated the ability of intratumourally-injected Hafnium oxide nanoparticles to double the frequency of pathological complete responses to radiotherapy in patients with sarcomas26, leading to initial clinical approval in Europe. One of the mechanisms by which radiotherapy promotes antitumour immunity is through the release of DNA into the cytoplasm, which activates the cGAS-STING pathway. In preclinical studies, high-Z nanoparticles have been shown to enhance irradiation-induced STING activation in human tumour cells27. In mice, intratumourally administered high-Z metal-organic nanoparticles improved the immunogenicity of low-dose radiation therapy when combined with antibodies against programmed cell death 1 ligand 1 (PD-L1) as evidenced by shrinkage of both irradiated primary tumours and secondary non-irradiated tumours28,29. This enhanced abscopal response was dependent on both CD8+ and CD4+ T cells. Motivated by such findings, nanoparticle radiosensitizers have recently entered clinical testing in combination with checkpoint blockade (Table 1).
Hyperthermia within tumours can induce ICD and promote an adaptive immune response, but precise heating of tumours without damage to surrounding tissues is often difficult to achieve. However, localized hyperthermia can be realized by using an externally applied alternating magnetic field to excite paramagnetic iron oxide nanoparticles within the TME. This approach induces tumour ICD and promotes a CD8+ T cell-mediated immune response as demonstrated in preclinical models of glioma, colon adenocarcinoma and melanoma30,31. Nanoparticle-mediated hyperthermia induced the production of a range of pro-inflammatory cytokines within treated tumours and activated DCs. Combination therapy using magnetic nanoparticle-induced hyperthermia, radiation therapy and a virus-like particle adjuvant was effective in canines with spontaneously arising oral melanoma32. Motivated by these preclinical findings, the use of magnetic nanoparticles to induce tumour hyperthermia and ICD is being tested clinically. As iron oxide nanoparticles are considered relatively nontoxic, are amenable to functionalization with targeting molecules and can be imaged using magnetic resonance imaging, they have the potential to be clinically successful in the near future if clear benefits in patient survival are demonstrated.
Nanomaterials have also been used to enhance combination therapies, either via co-encapsulation of multiple drugs in the same particle to guarantee co-delivery to target cells or by combining drugs with modes of action that synergize to produce supra-additive benefits33,34 (Fig. 1c). Combinatorial drug loading has been facilitated by the development of chemistries that enable small molecule drugs to be directly polymerized into a solid particle, and by methods to form nanoparticles with a solid core of one material surrounded by a shell of another material. For example, oxaliplatin was derivatized with metal coordinating groups that allow it to polymerize into a solid particle in the presence of metal ions (for example, zinc). When such nanoparticles were prepared with a phospholipid shell carrying a second reactive oxygen species-producing drug, dihydroartemisinin, the resulting combination therapy-loaded particles elicited pronounced ICD in tumours and synergized with anti-PD-L1 therapy in mouse models of colorectal adenocarcinoma35. Such nanoscale coordination polymer formulations have recently entered phase I clinical trials carrying undisclosed TME-modulating compounds (Table 1). Nanoparticles have also been designed that both interact with external energy sources and carry immunostimulatory drugs. Inorganic nanoparticles that are 25 nm in diameter coated with a lipid-anchored photosensitizer enhanced calreticulin exposure on the surface of tumour cells and immune cell infiltration in murine breast tumours when combined with infrared light irradiation compared with the free photosensitizer36. Encapsulation of oxaliplatin in the same nanoparticle allowed combined ICD-inducing effects of photodynamic therapy and chemotherapy, and elicited regressions of both primary irradiated tumours and non-irradiated secondary tumours in murine models of colorectal cancer37.
Ligand presentation to immune cells.
Many key immunoregulatory receptor engagements, especially costimulatory signals provided to T cells and natural killer (NK) cells, occur at cell–cell junctions. Nanomedicines offer the potential to mimic such an interface by presenting an array of ligands from the particle surface on physiologically relevant size scales (Fig. 1d). For example, agonist anti-CD137 antibodies conjugated to the surfaces of liposomes were shown to activate CD8+ T cells approximately tenfold more potently than equivalent concentrations of soluble anti-CD137 antibody38. In the setting of T cell activation, 30 nm diameter magnetic nanoparticles bearing immobilized peptide–MHC molecules together with nanoparticles bearing anti-CD28 antibody induce minimal T cell triggering because they are individually too small to induce sufficient receptor crosslinking and each nanoparticle only presents a single signal39. However, application of a magnetic field to induce nanoparticle clustering triggered robust T cell activation, proliferation and induction of effector functions. Particles can also present multiple ligands, either to co-engage multiple receptors on target immune cells or to engage multiple cell types simultaneously. Nanoparticles presenting both anti-PD1 and anti-OX40 antibodies elicited enhanced T cell activation and therapeutic efficacy in the treatment of murine melanoma and breast tumours in comparison to therapy with the same agents administered as a simple drug mixture40.
Nanoparticles have also been used to present clusters of death-inducing ligands from the surface of immune cells to circulating tumour cells. Liposomes surface-conjugated with many copies of recombinant E-selectin and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) bound to T cells, monocytes and NK cells in blood via E-selectin ligand, and presented clustered TRAIL to circulating tumour cells, leading to their engagement with the death ligands and tumour cell killing41. TRAIL-presenting nanoparticles have been shown to inhibit metastasis and prolong survival in murine and human xenograft tumour models42,43.
Polymer nanoparticles surface-conjugated with an antibody against human epidermal growth factor (HER2) and the “eat me” signal calreticulin in an optimized 1:3 anti-HER2:calreticulin ratio slowed HER2+ tumour growth by binding to tumour cells and presenting calreticulin to DCs in the TME, promoting phagocytosis. The resulting increased tumour antigen presentation by DCs led to enhanced CD8+ T cell infiltration into tumours44. In a second example, lipid nanoparticles were used to present signal-regulatory protein α (SIRPα)-blocking antibodies, reprogramming tumour-associated macrophages from an immunosuppressive to a tumouricidal phenotype while simultaneously delivering a small-molecule inhibitor of macrophage colony-stimulating factor 1 receptor (CSF1R) signalling45. This approach significantly outperformed simultaneous dosing of both agents in the absence of the nanoparticle formulation as measured by tumour regression and survival. The improved efficacy was attributed to the fact that the antibody acts both as a targeting molecule, localizing the drug-loaded particles on macrophages, and as a direct therapeutic, inhibiting SIRPα signalling. Thus, nanomedicine formulations can alter signalling mediated by immunotherapy drugs that target cell surface receptors in several ways to enhance their net antitumour activity.
Intracellular delivery of danger signals and nucleic acids.
One of the major applications of nanomaterials in medicine is promoting cytosolic drug delivery. Hydrophilic and charged compounds such as nucleic acids are generally internalized by cells via endocytosis, but thereafter remain trapped in the endolysosomal pathway. Nanomaterials have been extensively studied as surrogates of natural viruses to promote access of nucleic acids and other drugs to the cytosol (Fig. 1e). In the setting of cancer immunotherapy, this is particularly of interest for delivering compounds to activate cytosolic danger sensor proteins or to deliver RNA or DNA that encode immunomodulatory proteins. Polymer vesicles (polymersomes) have recently been reported that encapsulate a cyclic dinucleotide agonist of STING for delivery to the cytoplasm46. Once internalized by cells, the polymersomes disrupt endosomal membranes in response to endosome acidification, releasing the STING agonist cargo into the cytoplasm. These nanomaterials increased the apparent potency of cyclic dinucleotides by more than 100-fold, and in turn exhibited significantly increased antitumour efficacy in melanoma models following intratumoural administration. Enhanced STING activation has also been reported for liposomal formulations of cyclic dinucleotides incorporating pH-responsive lipids47.
Another intriguing approach is the use of synthetic polymers that directly engage danger signal pathways. pH-responsive block copolymer micelles were recently described that disrupt endosomes and directly interact with STING, triggering its activation and downstream signalling48. Nanoparticles can also be functionalized with high densities of danger signals such as Toll-like receptor (TLR) agonists, and these immunostimulators are taken up by myeloid cells and antigen-presenting cells more efficiently than free TLR ligands, leading to enhanced immunostimulation49,50. Each of these approaches is moving into clinical testing (Table 1).
Delivery of nucleic acid cargos encoding therapeutic proteins can further provide new possibilities for manipulating immune-stimulatory or immune-suppressive pathways.51 Systemic administration of a lipid and protamine-based nanoparticle loaded with plasmid DNA encoding a PD-L1 trap protein synergized with oxaliplatin chemotherapy to delay tumour growth in models of metastatic colon cancer52. Intratumoural administration of lipid nanoparticles delivering mRNA encoding the cytokines IL-23 and IL-36γ and the T cell costimulator OX40L led to in situ vaccination and CD8+ T cell-dependent tumour regression in colon cancer and melanoma models, and this approach is currently being testing in patients53 (Table 1).
Temporal control of immunostimulation.
The dosing schedule of immunotherapy can have profound effects on treatment efficacy in preclinical models. Timing can be important both in terms of the duration that immune stimuli are present and active in tumours (or tumour-draining lymph nodes) and in the ordering of cues delivered in combination therapies54. Nanoparticles have long been used to provide sustained release of drugs into tissues or the circulation that otherwise exhibit rapid clearance, and can serve similar roles in immunotherapy55 (Fig. 1f). In addition, nanomedicine formulations can be designed to interact with external energy sources such as light or heat to permit precisely controlled timing of drug release. This approach is exemplified by studies of near-infrared light-activatable nanoparticles complexed with TLR9 agonist CpG-containing DNA56. In this approach, photolabile DNA strands complementary to CpG DNA were linked to the surface of near-infrared light-sensitive upconversion nanoparticles. Hybridization of CpG oligonucleotides with the nanoparticle-linked DNA complexed the TLR9 agonist to the particles in a “silent” state. Systemic injection of the nanoparticles to breast tumour-bearing mice led to accumulation in tumours. Subsequent near-infrared light illumination of the tumours cleaved the photolabile DNA anchor, releasing active CpG DNA and triggering localized inflammation in the TME without systemic side effects, driving immune infiltration of tumours and enhanced survival.
The preceding examples highlight how nanomedicine can enable the more precise application of existing immunotherapy strategies, control timing and location of immunostimulation, and alter signalling from immunostimulatory biologicals. In the following sections, we discuss how nanomedicine formulations alter the outcome of immunotherapies, focusing first on locally applied therapies and then discussing systemic and cell-based modalities.
Enhancing localized immunotherapy
Local injections of immunotherapy agents intratumourally or peritumourally to directly modulate the TME or tumour-draining lymph nodes is a treatment strategy in preclinical and clinical development. Localized treatment is based on the expectation that T cell priming in tumour-draining lymph nodes will lead to trafficking of lymphocytes to distal untreated tumours57–59. While such systemic abscopal responses have been anecdotally reported in patients for many years, recent efforts exploring rational combination therapies have begun to show increasing frequencies of systemic antitumour responses60–62. Relative to systemic therapy, local treatment has the potential to increase the safety of immunotherapy by limiting systemic exposure, allow more effective modulation of the TME via increased concentration or persistence of drugs in the TME, and may enable the use of combination treatments that would otherwise be too toxic for systemic administration59. Two local immunotherapies are currently approved by the FDA for cancer treatment: intravesicular injections of Bacille–Calmette Guerin (BCG) bacteria (the live-attenuated tuberculosis vaccine) for treatment of bladder cancer and intratumoural injections of an engineered herpes simplex virus type-1 (talimogene laherparepvec) for the treatment of melanoma63,64. Nanoparticle formulations may have an important role to play in this treatment setting by providing a means to promote retention of immunotherapy agents in tumours and to target therapeutics to tumour-draining lymph nodes.
Promoting immunotherapy retention in tumours.
The goal of localized immunotherapy is to confine administered drugs to the tumour and/or tumour-draining lymph nodes, maximizing stimulation in the TME while minimizing toxicities due to systemic immunostimulation. However, intratumourally injected drugs tend to rapidly disseminate into the systemic circulation, driven in part by the high interstitial fluid pressure in tumours38,65–68. Unlike small molecules or free proteins, nanoparticles can be physically trapped in the TME following injection because the interstitial space in tumours is filled by a collagen-rich extracellular matrix69 (Fig. 2). The collagen fibres form a meshwork with irregular spacings ranging from 20 nm to 130 nm that traps particles of a similar or larger size. This has been experimentally demonstrated in various tumour models, in which nanoparticles of approximately 100 nm in diameter or larger diffuse either very slowly or are effectively immobilized in the tumour interstitium70–72. Intratumoural injection of immunostimuli chemically conjugated to liposomes of 100–200 nm in diameter leads to their distribution through the local lesion and some accumulation in tumour-draining lymph nodes, but no detectable dissemination into the blood38,68. Such alterations in biodistribution have enabled the safe administration of combination treatments that were lethally toxic as free drugs, with concomitant enhancements in antitumour activity. Based on similar principles, recombinant plant virus nanoparticles with a diameter of 30 nm when injected intratumourally or administered by lung inhalation act through as-yet-undefined innate immunostimulatory pathways to promote antitumour immunity and cancer regression73. In a related approach where the tumor is surgically removed and thus does not provide a defined site for administration, a nanoparticle encapsulating anti-CD47 suspended in a fibrin gel matrix to promote retention was sprayed on a tumour resection site to provide localized and sustained release of the antibody and thus block the “don’t eat me” signal provided by residual tumour cell CD47 binding to SIRPα on macrophages74.
Figure 2 |. Nanomedicines improve tumour retention and lymph node trafficking.
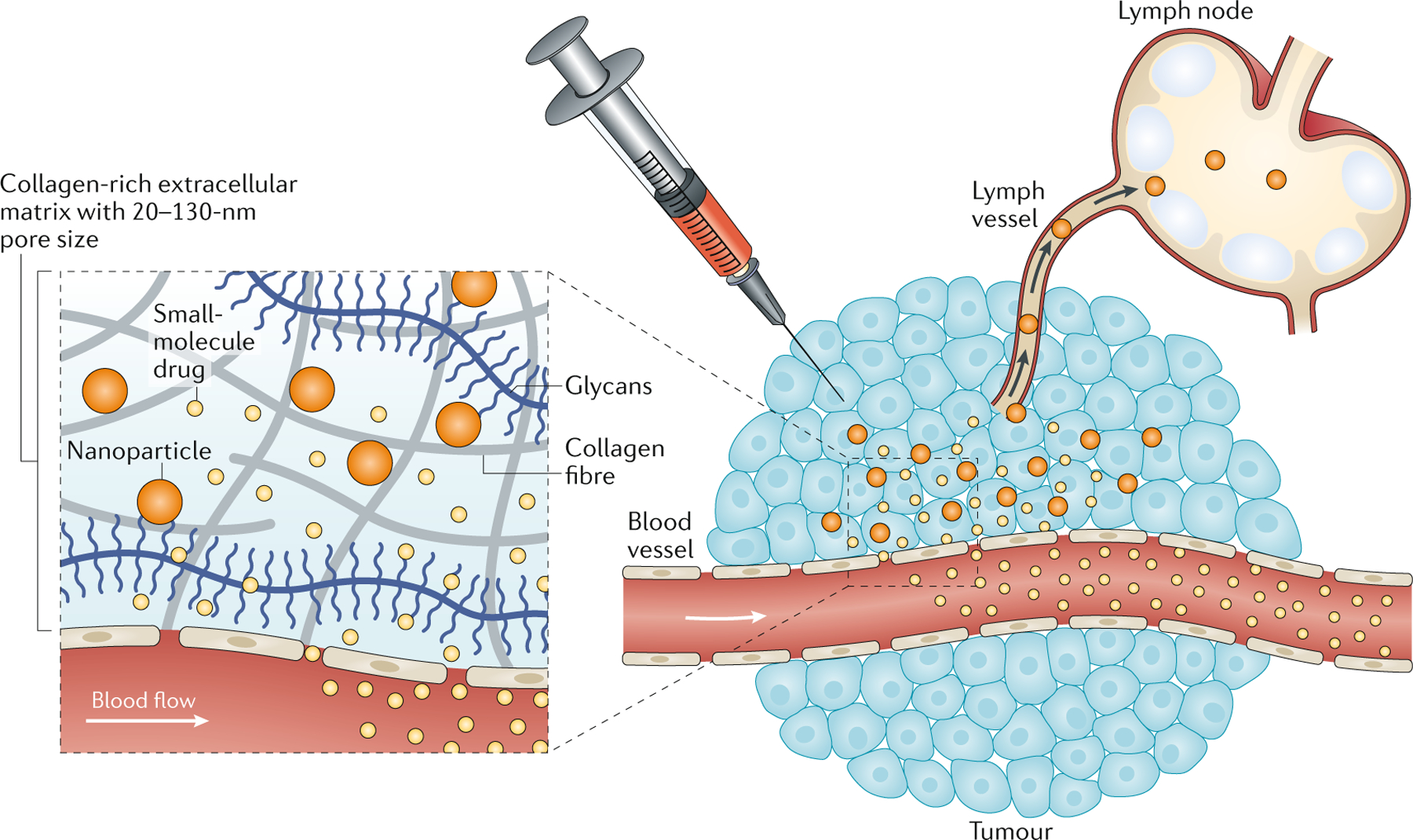
When administered locally, the tumour’s dense extracellular matrix, composed of a collagen-rich hydrogel with a 20–130 nm pore size, preferentially retains nanomedicines and promotes their trafficking to the lymph node, whereas small molecule drugs are rapidly cleared into the systemic circulation due to their small size and the high interstitial fluid pressure in tumours.
Targeting lymphoid tissues.
Lymphoid organs can also be sites of interest for immunomodulation in cancer. Tumour-draining lymph nodes accumulate tumour antigens, and tumour antigen-specific T cells accumulate in these sites75. However, tumours release cytokines and other factors to suppress the draining lymph nodes76, Treg cells preferentially migrate to these sites77, and tumour-draining lymph nodes are a preferential site for tumour cell metastasis — all factors that promote tolerance rather than immunity. Thus, delivery of immunostimulatory compounds to tumour-draining lymph nodes has been pursued to reverse this immunosuppression and exploit the presence of tumour antigen and pre-existing antigen-specific T cells at these sites. Particles with sizes in the range of 5–50 nm injected into tissues are too large to efficiently enter the blood vasculature, and instead preferentially enter lymphatic vessels, and are thus targeted to downstream lymph nodes78. Based on this principle, TLR agonists conjugated to 30 nm diameter polymer nanoparticles injected intradermally near melanomas led to an accumulation of the TLR agonists in tumour-draining lymph nodes, activation of DCs in this site and induction of T cell priming that slowed tumour progression and enhanced survival75,79. Nanoparticles can also be targeted to DCs and myeloid cells in tumours and spleen, as discussed in the following section.
Localized immunotherapy is attractive for its potential to enable very potent combinations of immunostimulatory agents to be safely used to dramatically reprogramme the TME. The effectiveness of this strategy in the clinic hinges on the capacity of systemic immunity being produced from such local stimulation, and will likely require support from systemic checkpoint blockade to be fully effective, to support T cells infiltrating distal untreated tumour sites. The alternative is to pursue approaches to systemically concentrate immunotherapies in disseminated tumours, as discussed in the next section.
Systemic targeting of tumours
Altering pharmacokinetics of immunotherapy agents for enhanced safety.
One challenge facing immunotherapy agents, especially immune agonists such as drugs that engage costimulatory receptors, is the high level of toxicity observed when such drugs are administered systemically and stimulate circulating lymphocytes. Nanomedicine formulations are being developed to promote greater immune activation within tumours while limiting extratumoural stimulation (Fig. 3a). For example, lethal side effects from systemic administration of an anti-CD137 agonist antibody and IL-2 were eliminated by conjugating these proteins to the surfaces of liposomal carriers80. Liposome conjugation substantially reduced the half-life of the drugs in the bloodstream, while maintaining their accumulation to equivalent levels in tumours via the enhanced permeation and retention (EPR) effect (Box 1), enabling robust stimulation of antitumour T cell responses while ablating the major toxicity observed with the free drugs. A second strategy that is now in clinical development manipulates the bioactivity of immunostimulatory cytokines by administering them as an inactive prodrug that is masked with polyethylene glycol (PEG) chains. NKTR-214 is a recombinant human IL-2 cytokine that is engineered to include on average six cleavable PEG chains and only becomes active when the majority of the PEG chains are hydrolysed81,82. Interestingly, this PEGylation approach provides not only sustained dosing but also selectively decreases the affinity of IL-2 for the heterotrimeric IL-2 receptor complex (IL-2Rαβγ) that is abundantly expressed by Treg cells, relative to the heterodimeric IL-2 receptor complex (IL-2Rβγ), which is preferentially expressed on CD8+ T cells, and thus favours the activation of CD8+ T cells over Treg cells81. Phase 1 and 2 clinical trial results of NKTR-214 have been promising in terms of toxicity and signs of efficacy83. NKTR-214 blurs the distinction between biological drug and nanoparticle, as at the core of each “particle” is a single protein. However, it is useful to examine NKTR-214 through the lens of nanomedicine because the synthetic PEG polymer comprises approximately 88% of the mass of the conjugate, and this labile PEG coating profoundly modulates the pharmacokinetics, pharmacodynamics and receptor selectivity of the drug.
Figure 3 |. Systemic targeting of tumours by intravenously administered nanomedicines.
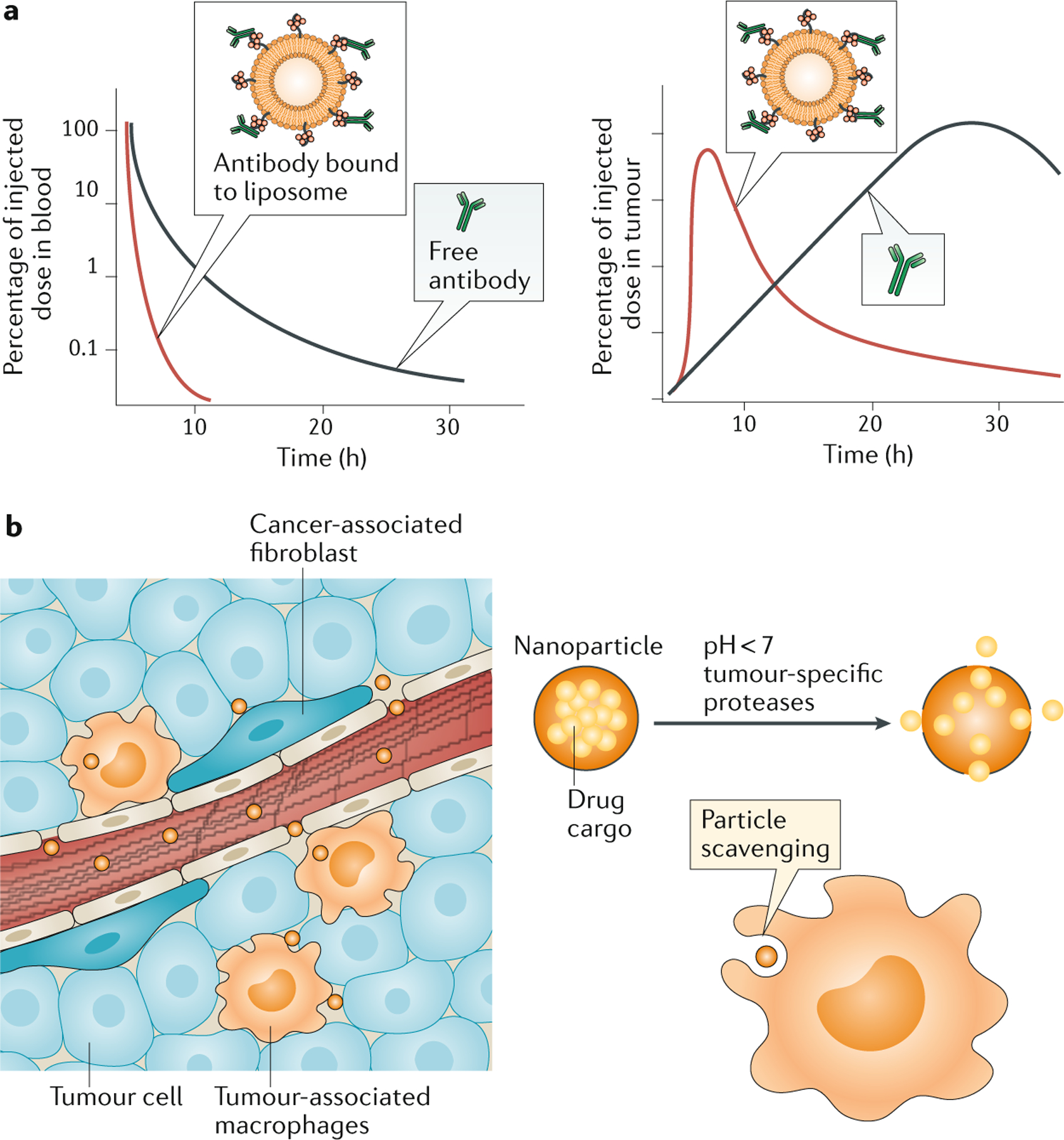
a |, Nanoparticles alter the pharmacokinetics of immunomodulatory drugs in a manner that can increase safety and efficacy. Illustrated is the case of an antibody therapeutic, which as a free drug will exhibit a long half-life in the blood and slowly accumulate in tumours. The same drug conjugated to a nanoparticle scaffold can be delivered to similar levels into the tumour in a shorter time window, accompanied by more rapid clearance from the systemic circulation, providing similar stimulation in the tumour microenvironment but substantially lowering systemic exposure. b | In addition to permeable blood vessels to promote nanoparticle localization via the enhanced permeation and retention effect, the tumour microenvironment has unique attributes that can be leveraged to effect drug targeting. For example, phagocytic myeloid cells, such as tumour-associated macrophages, readily scavenge nanoparticles and they can act as depots for drugs that act directly on tumours, such as chemotherapeutics, or can themselves be the targets for immunomodulatory drugs designed to decrease immunosuppression. In addition, tumour-specific properties, such as a slightly acidic pH or the presence of higher than normal levels of certain enzymes, can act as triggers for drug release from nanoparticles, and such systems have been used to both directly target cancer cells and also modulate stromal cells, such as cancer-associated fibroblasts or immunosuppressive immune cells.
Nanomedicine formulations are also being pursued as a means to enable safe systemic administration of innate immune stimulators such as TLR agonists, STING agonists and ligands for other danger sensors. STING agonists have been formulated within nanoparticles designed to both target tumours via the EPR effect and improve drug potency by promoting endosomal escape into the cytosol where the STING receptor resides. Whereas STING agonists are generally ineffective when given systemically, this approach has allowed intravenous administration to potentiate immunotherapy in mouse models of melanoma46,84 and breast cancer47. An important issue that remains to be studied more deeply is whether such approaches are safe when treating with the doses of STING agonist that are needed for treatment of large tumours, as nanoparticle clearance by macrophages and DCs of the spleen and liver or other tissue sites may be a source of off-tumour, on-target toxicity.
Targeting myeloid cells.
Systemic targeting of myeloid cells using nanomedicine is a clinically attractive approach because myeloid cells often have a suppressive role as myeloid-derived suppressor cells (MDSCs) or tumour-associated macrophages (TAMs), and because they are logical targets due to their propensity to phagocytose nanoparticles and microparticles85 (Fig. 3b). In murine tumour models, intravenously administered nanoparticles readily accumulate in TAMs86,87, which can serve as cellular reservoirs for antitumour therapeutics88. Radiation therapy can increase TAM numbers, induce macrophage-mediated changes in vascular leakage, and thereby further enhance nanoparticle accumulation in TAMs89. Notably, myeloid cells have been reported to take up tenfold more nanoparticles than tumour cells even when the particles are conjugated with a ligand intended to promote tumour cell uptake90. Recent preclinical studies have sought to exploit this effect to deplete suppressive myeloid populations in cancer. Polymeric nanoparticles or micelles 20–30 nm in diameter administered intradermally rapidly reach the systemic circulation by passing through lymphatics to the thoracic duct, and subsequently accumulate in macrophages and myeloid cells in the spleen and tumours91,92. Formulation of the chemotherapy agent 6-thioguanine in these particles led to depletion of MDSCs in both sites, enhancing adoptive T cell therapy91, a result that has also been reported for subcutaneously administered lipid nanoparticles carrying a hydrophobic gemcitabine derivative93.
An alternative to depleting suppressive myeloid cells is to reprogramme them to a phenotype that promotes antitumour immunity. Innate immunostimulants that mimic danger signals produced by pathogens induce pro-inflammatory cytokine production and polarize macrophages from an immunosuppressive state towards a tumoricidal phenotype94. Repetitive systemic administration of cyclodextrin nanoparticles loaded with the TLR7 and TLR8 agonist R848 led to preferential nanoparticle accumulation in TAMs within mouse tumours and promoted both macrophage re-education and a CD8+ T cell-dependent immune response, with a subset of mice completely rejecting the initial tumour and subsequent rechallenge95. Several recent studies have also used nanoparticles to target small interfering RNAs or micro RNAs to tumour-associated myeloid cells to promote reprogramming of the TME and antitumour immunity, exploiting the capacity of nanoparticles to both target myeloid cells and promote transfection96–98. Interestingly, the FDA-approved iron supplement ferumoxytol, which is composed of dextran-coated iron oxide nanoparticles, was recently found to reprogramme macrophages towards an antitumour phenotype. When administered systemically before intravenous administration of tumour cells, ferumoxytol prevented the seeding of liver tumours99. Understanding the specific attributes of these iron-oxide nanoparticles that lead to alterations in macrophage function may suggest new approaches to myeloid cell reprogramming.
An intriguing alternative to relying on spontaneous phagocytic uptake of particles by myeloid cells is to preferentially attract these cells to engulf the nanoparticle by using a chemokine-releasing formulation. Nanocapsules that slowly release the monocyte-macrophage attractant CC-chemokine ligand 2 were shown to promote chemoattraction of myeloid cells in vitro100. Once internalized, the same nanocapsule then released co-encapsulated small interfering RNA targeting Cebpb, which encodes a transcription factor driving immunosuppression. Cebpb knockdown by these particles was demonstrated in both the spleen and tumours following systemic administration in tumour-bearing mice.
Targeting non-immune stromal cells.
Stromal cells within the TME have important roles in suppressing immune responses and supporting tumour growth101, and have thus also been investigated as nanomedicine drug targets to potentiate immunotherapy. The proliferation and extracellular matrix production of cancer-associated fibroblasts (CAFs) impede the immune response by compressing tumour blood vessels and inducing hypoxia, but the use of small molecule drugs, such as angiotensin receptor blockers, to alter CAF activity is challenging due to systemic side-effects. Polymer nanoparticles designed to dissolve rapidly under the slightly acidic conditions found in tumours enabled selective delivery of angiotensin receptor blockers to CAFs within the TME as measured by the levels of active drug in the tumour in comparison to treatment with the free drug102. This nanomedicine therapy, when combined with checkpoint blockade, improved survival and reduced metastasis following primary tumour resection in multiple breast tumour models. A second strategy is to use nanoparticles to concentrate chemotherapy agents in CAFs for selective depletion of these suppressive cells. Acetylated carboxymethylcellulose nanoparticles were discovered to preferentially accumulate in α-smooth muscle actin+ CAFs in breast and pancreatic tumours, mediated in part by opsonization of the particles with albumin and subsequent interaction with the albumin receptor SPARC (secreted protein acidic and rich in cysteine) expressed by these cells103. Delivery of docetaxel to CAFs using these particles led to enhanced survival in models of breast and pancreatic cancer104,105.
In summary, nanoparticles have the potential to substantially alter the pharmacokinetics as well as tissue-level and cell-level biodistribution of therapeutics. These properties can be exploited to increase the safety of drugs that would otherwise be too toxic due to nonspecific stimulation of cells in circulation or healthy tissues, and also enhance efficacy due to concentrated uptake in key cellular targets such as tumour-associated myeloid cells.
Enhancing cellular immunotherapy
Alongside checkpoint blockade therapeutics, adoptive cell therapy (ACT) of cancer using CAR T cells is the second major class of immunotherapy to have made a major clinical impact to date. CAR T cells generated by virally transducing autologous patient T cells with a synthetic antigen receptor targeting CD19 have been approved for the treatment of acute lymphoblastic leukaemia and refractory large B cell lymphoma106. Recent promising data from a phase 1 clinical trial have also indicated promise for CAR T cells in treating multiple myeloma107. In addition, numerous small trials have reported positive responses in the treatment of patients with T cells transduced with tumour-specific T cell receptors (TCRs) or using natural T cell products prepared from tumour-infiltrating lymphocytes or sorted from peripheral blood108. These promising results have led to a major translational effort including the launch of numerous cell therapy companies and both academic and company-sponsored clinical trials. However, ACT treatments have had limited success in treating common epithelial cancers, motivating efforts to enhance the ability of T cells to overcome the immunosuppressive microenvironment of solid tumours. Strategies to genetically engineer new functions into immune cells will play an important role in the future of ACT106. However, a number of nanomedicine-based approaches are also being explored to enhance ACT, which provide complementary avenues to enhance the longevity, phenotype and function of cell therapy products.
Linking therapeutics to immune cells.
Many signalling pathways in T cells have been identified that can regulate lymphocyte function, metabolism and/or differentiation in ways that would be beneficial to antitumour immunity. However, targeting these pathways with drugs remains a challenge. Genetically engineering T cells to express supporting factors (such as cytokines) can be challenging because it requires avoiding therapeutic levels that are too low, too high or too prolonged109–111, and systemic administration of drugs that modulate T cell function often faces dose-limiting toxicities. An alternative is to link supporting drugs to adoptively transferred cells112. In this approach, supporting drugs are encapsulated or otherwise formulated into nanoparticles that are chemically attached to the plasma membrane of the donor cells (Fig. 4a). These nanoparticle “backpacks” are designed to release the drug at a prescribed rate or under selected microenvironmental conditions, such that the donor cell is stimulated in an engineered autocrine manner, limiting bystander stimulation of other cells and healthy tissues113. In preclinical models, this approach has enabled large doses of supporting cytokines such as IL-2 or IL-15 that are lethal when administered as free systemic drugs to be used without toxicity, substantially widening the therapeutic window for supporting cytokine therapy of ACT113–115.
Figure 4 |. Enhancing cellular immunity of cancer.
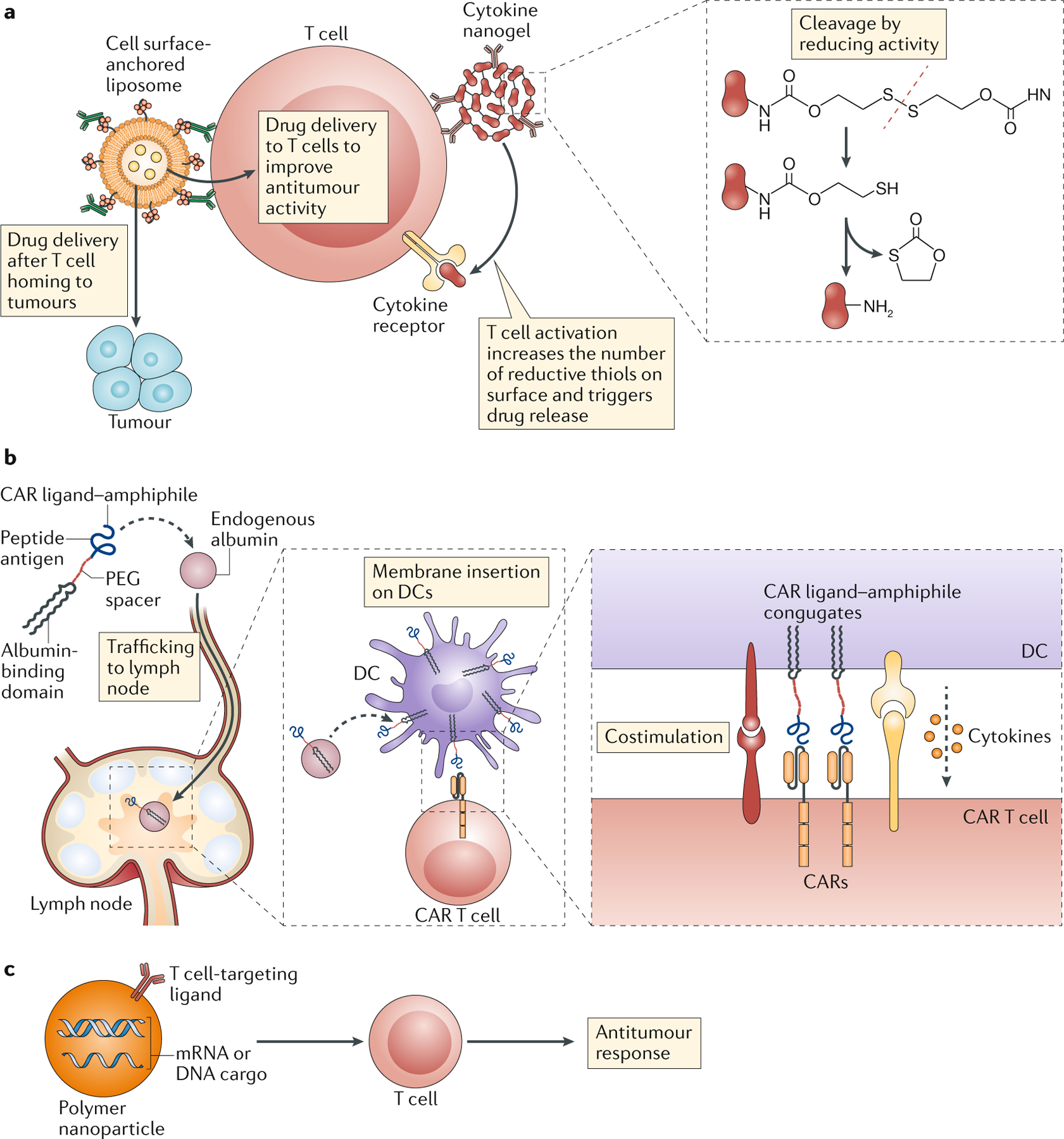
a | Conjugation of drug-releasing nanoparticles to the plasma membrane of T cells has been used to deliver drugs to tumours or tumour-infiltrating immune cells or to provide a continuous, autocrine supply of cytokines (such as IL-15 and IL-2) to the carrier cell, promoting T cell expansion and effector functions. b | Polymer amphiphiles conjugated to a chimeric antigen receptor (CAR) ligand bind endogenous albumin and traffic to lymph nodes where they are displayed on dendritic cells and provide stimulation to antitumour CAR T cells, effectively acting as a boosting vaccination. c | Polymer nanoparticles carrying either mRNA or plasmid DNA and displaying a T cell-targeting ligand genetically reprogramme endogenous lymphocytes to promote antitumour immune responses.
Cell-conjugated nanoparticles have also been used to backpack lymphocytes with small molecule supporting drugs116–118. Notably, conjugation of nanocarriers to cell surface proteins causes the nanoparticles to follow the cell surface localization of their protein anchors: particles preferentially conjugated to CD45, integrin αL (also known as LFA1) and CD2 were found to traffic into the immune synapse during the early stages of T cell activation118, mirroring the transport of these receptors during T cell priming119. The timing of cell stimulation can be controlled by linking release of the encapsulated drug to local changes in the environment. For example, nanoparticles were designed to release IL-15 in response to the upregulation of cell surface reducing conditions as T cells become triggered through the TCR, amplifying T cell function in tumours and tumour-draining lymph nodes while minimizing systemic stimulation115. This approach of backpacking T cells with cytokines has recently entered clinical trials for a variety of solid tumour types (Table 1).
Attaching nanoparticle formulations to immune cells substantially alters the biodistribution of the nanomedicine — for example, providing greatly enhanced accumulation of particles in tumours when attached to tumour-reactive T cells113. Thus, this approach can also be used to enhance the delivery of drugs that are meant for other cells in the TME. For example, attachment of lipid nanoparticles carrying a potent but poorly soluble camptothecin chemotherapy agent to T cells led to pronounced enhancements of drug accumulation in tumours in a lymphoma model, allowing enhancements in survival, whereas the systemic administration of free drug had no impact on tumour progression120.
Targeting drugs to circulating lymphocytes.
Using nanomaterials as a strategy to associate drug reservoirs with donor cells in ACT provides the opportunity for exquisite control over cell stimulation but is a single-timepoint intervention; the temporal window for therapeutic intervention is limited to the timing of cargo release from the drug carrier. An alternative is to use nanomaterials to directly target drugs to lymphocytes in vivo, through chemically conjugated antibodies or other targeting moieties that will bind to target cell surface receptors. This strategy can be used to target either endogenous immune cells or adoptively transferred cells, but it may be particularly efficient in the case of ACT, in which the donor cells serve as an enriched tumour-specific target population. For example, liposomes targeted to T cells using surface-conjugated IL-2 or antibody fragments against a congenic cell surface receptor allowed more than 95% of circulating T cells delivered by ACT to be labeled following a single injection, leading to significant enhancements in expansion of transferred T cells in a melanoma model117,121. By comparison, targeting of endogenous T cells using polymeric nanoparticles conjugated with anti-PD1 antibody allowed approximately 5% of circulating PD1+ T cells to take up nanoparticles within 1 hr of injection122. Despite this modest particle uptake, administration of anti-PD1-targeted particles carrying small molecule inhibitors of transforming growth factor-β or a TLR7 agonist led to pronounced therapeutic activity that was absent from particle formulations lacking the targeting moiety or equivalent doses of free drug.
An alternative to delivering nanomaterials to cells in the blood is to target compounds to lymph nodes, with the goal of engaging lymphocytes as they recirculate through these secondary lymphoid organs. As described above, nanoparticles can efficiently traffic from parenteral injection sites into lymphatic vessels, and hence to lymph nodes. Lymphatic targeting can also be achieved by using endogenous lymph-transported proteins as chaperones: this has been demonstrated using polymer conjugates that bind to albumin123,124. Recently, an approach to target stimulation to CAR T cells was demonstrated, whereby an albumin-binding lipid tail was linked via a PEG spacer to a small molecule or peptide ligand for a CAR. This lipid–PEG–CAR ligand construct was efficiently shuttled to lymph nodes following parenteral injection, and decorated the surfaces of macrophages and DCs through insertion of the lipid tail into antigen-presenting cell plasma membranes (Fig. 4b). Boosting of CAR T cells by DCs coated with the CAR ligand led to enhanced CAR T cell expansion, increased effector functions and enhanced tumour rejection in several syngeneic mouse tumour models125.
[H2] Gene delivery to lymphocytes in vivo.
Ex vivo genetic manipulation or expansion in ACT is laborious and costly126. Nanoparticle formulations designed to target specific lymphocyte subpopulations in vitro or in vivo and promote their transfection with encapsulated nucleic acids thus represent a third major application of nanomedicine to cellular immunotherapy. Viral vectors are the primary means of producing clinical cell products transduced with CARs or TCRs, but viral production that adheres to good manufacturing practice is a significant financial cost, viral vectors can be limited by the size of transgene that can be introduced, and often do not provide control over the cell types that are transduced. By contrast, synthetic nanoparticle formulations can be designed to specifically transduce target cell subpopulations through antibody-directed or ligand-directed cell binding, carry larger gene cargos, and can be produced at scale by simple chemical synthesis. To this end, it was recently demonstrated that polymeric nanoparticles can be used to carry out targeted gene expression or gene editing in primary T cells through in vitro addition of antibody-targeted particles carrying mRNA127.
Going a step further, strategies to generate CAR/TCR-transduced T cells directly in vivo would represent a major breakthrough to enable the treatment of a larger population of patients more rapidly at lower cost. Synthetic nanoparticles are not prone to the same issues of pre-existing or treatment-induced anti-vector immunity or off-target toxicities that can be of concern with viral vectors, but they suffer from lower efficiencies in cellular transduction compared with their biological counterparts. However, this latter drawback may be less of a concern in the setting of in vivo CAR or TCR delivery, because transduced T cells are intrinsically self-amplifying as they encounter tumour antigen. This feature was observed in preclinical studies using a polymeric nanoparticle formulation to target transposon-based DNA carrying a CAR to circulating T cells in a lymphoma model, where transduction of a small founder population of T cells led to substantial T cell expansion over the course of 1–2 weeks and substantial antitumour activity128 (Fig. 4c). This nanomedicine-based approach to deliver CARs in vivo is now moving to clinical testing (Table 1). Lipid nanoparticles capable of delivering mRNA into DCs have also recently been reported129, and moved into early clinical testing for vaccines, suggesting multiple avenues for systemically targeting immune cell populations for gene delivery.
Altogether, nanomedicine approaches appear promising both as agents for ex vivo engineering of therapeutic cells and as a platform for directly modifying cells in vivo. The potential of synthetic formulations to achieve delivery of nucleic acids to target cell in vivo without induction of strong anti-vector immunity could open new opportunities for cellular immunotherapies, while also removing many of the challenging logistics associated with patient-specific cell manufacturing.
Conclusion and future perspectives
As demonstrated by the many examples discussed above, nanomedicine has the potential to enhance cancer immunotherapies in diverse ways, and preclinical evidence provides clear motivation for clinical testing of many concepts. Initial human trials are now underway for several of these approaches and the next few years should provide substantial insight into the clinical potential of these technologies.
The field faces several immediate challenges, where solutions could have significant impact on clinical translation of immunotherapy. Safe methods to systemically target potent innate immune stimulators such as STING or TLR agonists to tumours remain to be developed. Although these agents demonstrate potent antitumour activity when administered intratumourally in preclinical models and are currently being evaluated for intratumoural administration in early clinical trials, intratumoural administration does not guarantee an avoidance of systemic distribution and subsequent toxicity from such small molecule compounds, and precludes direct action on disseminated disease. The tropism of nanoparticles for myeloid cells in the circulation, spleen and liver makes it unclear if nanomedicine-formulated innate stimulators will be a safe solution. A second major challenge is the robust delivery of genetic material to lymphocytes in vivo, such as CARs, TCRs or other supporting genes. As noted above, the first attempts to use polymeric materials to target RNA or DNA into T cells have recently been reported, but the efficiency of transfection in vivo remains very low and it is unclear if short-lived mRNAs or randomly-integrating transposon DNAs are the right solution. However, the potential impact of a safe and effective means for gene delivery to lymphocytes or other immune cells in vivo cannot be underestimated.
Emerging areas of immunology and immunotherapy also present new opportunities for the future. Recently, the phenomenon of long-lived reprogramming of myeloid cells following exposure to certain innate immune stimuli has begun to be understood at the molecular and genetic level. This process known as trained immunity is characterized by hyperresponsiveness of myeloid cells to innate stimuli such as TLR agonists, is induced through epigenetic and metabolic reprogramming of myeloid cells and/or their precursors by stimuli such as β-glucan or systemic BCG vaccination130. Such myeloid training has been shown to mediate heterotypic protection against bacterial infections131,132. Therapeutically inducing trained immunity has been proposed as a strategy that may be relevant in cancer, and the ability of nanoparticles to efficiently target the bone marrow and myeloid cells makes nanomedicine-based approaches to this task appear attractive133. Such approaches that specifically amplify innate immune attack against tumours could be an important complement to current therapies that predominantly focus on adaptive immunity.
Acknowledgements
This work was supported in part by the US National Institutes of Health (awards CA235375, EB022433 and CA206218), the Mayo Clinic-Koch Institute Cancer Solutions Team Grant funding, the Marble Center for Nanomedicine, and the Ragon Institute of MGH, MIT and Harvard. D.J.I. is an investigator of the Howard Hughes Medical Institute.
GLOSSARY
- cGAS–STING pathway
An intracellular signaling pathway that responds to cytosolic double-stranded DNA through the sensor enzyme cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase (cGAS) to produce the second messenger cyclic GMP-AMP, which subsequently activates STING and can stimulate cells to produce type-I interferon and other cytokines.
- Abscopal effect
Immunological response to radiation or other localized therapies whereby the treatment of a malignant lesion results in the regression or stabilization of distant, non-treated lesions.
Footnotes
Competing interests
D.J.I. and E.L.D. are co-inventors on patents related to nanoparticle delivery of innate immune stimulators assigned to Massachusetts Institute of Technology (MIT). D.J.I. is an inventor on patents related to nanomedicine-based immunotherapy assigned to MIT that have been licensed to Torque Therapeutics, Elicio Therapeutics and Strand Therapeutics, of which D.J.I. is a co-founder.
Peer review information
Nature Reviews Immunology thanks B. Kim and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gettinger SN et al. Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J. Clin. Oncol 33, 2004–2012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lebbe C et al. Survival follow-up and ipilimumab retreatment of patients with advanced melanoma who received ipilimumab in prior phase II studies. Ann. Oncol 25, 2277–2284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larkin J et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Postow MA, Sidlow R & Hellmann MD Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med 378, 158–168 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Gangadhar TC & Vonderheide RH Mitigating the toxic effects of anticancer immunotherapy. Nat. Rev. Clin. Oncol 11, 91–99 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Bommareddy PK, Shettigar M & Kaufman HL Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol 18, 498–513 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Neri D Antibody–cytokine fusions: versatile products for the modulation of anticancer immunity. Cancer Immunol. Res 7, 348–354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kureshi R, Bahri M & Spangler JB Reprogramming immune proteins as therapeutics using molecular engineering. Curr. Opin. Chem. Eng 19, 27–34 (2018). [Google Scholar]
- 10.Lebre F, Hearnden CH & Lavelle EC Modulation of immune responses by particulate materials. Adv. Mater 28, 5525–5541 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Smith JD, Morton LD & Ulery BD Nanoparticles as synthetic vaccines. Curr. Opin. Biotechnol 34, 217–224 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Kelly HG, Kent SJ & Wheatley AK Immunological basis for enhanced immunity of nanoparticle vaccines. Expert Rev. Vaccines 18, 269–280 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Bachmann MF & Jennings GT Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol 10, 787–796 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Krysko DV et al. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Duan X, Chan C & Lin W Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew. Chem. Int. Ed. Engl 58, 670–680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rios-Doria J et al. Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia 17, 661–670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuai R et al. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv 4, eaao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walle T et al. Radiation effects on antitumor immune responses: current perspectives and challenges. Ther. Adv. Med. Oncol 10, 1–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vacchelli E et al. Trial watch: immunotherapy plus radiation therapy for oncological indications. Oncoimmunology 5, e1214790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li T & Chen ZJ The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 215, 1287–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanpouille-Box C et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun 8, 15618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang H et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun 8, 1736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arina A et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun 10, 3959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min Y et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol 12, 877–882 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated a new mechanism of action for nanoparticles in augmenting in situ vaccination by capturing antigens released from tumour cells during radiotherapy and promoting uptake by antigen-presenting cells.
- 25.Rancoule C et al. Nanoparticles in radiation oncology: From bench-side to bedside. Cancer Lett. 375, 256–262 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Bonvalot S et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): a multicentre, phase 2–3, randomised, controlled trial. Lancet Oncol. 20, 1148–1159 (2019). [DOI] [PubMed] [Google Scholar]; Randomized clinical trial demonstrating the use of inorganic nanoparticles in potentiating radiotherapy in sarcoma patients.
- 27.Marill J, Mohamed Anesary N & Paris S DNA damage enhancement by radiotherapy-activated hafnium oxide nanoparticles improves cGAS-STING pathway activation in human colorectal cancer cells. Radiother. Oncol 141, 262–266 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Ni K et al. Nanoscale metal-organic frameworks enhance radiotherapy to potentiate checkpoint blockade immunotherapy. Nat. Commun 9, 2351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu K et al. Low-dose X-ray radiotherapy-radiodynamic therapy via nanoscale metal-organic frameworks enhances checkpoint blockade immunotherapy. Nat. Biomed. Eng 2, 600–610 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Toraya-Brown S et al. Local hyperthermia treatment of tumors induces CD8+ T cell-mediated resistance against distal and secondary tumors. Nanomedicine: N.B.M 10, 1273–1285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanase M et al. Antitumor immunity induction by intracellular hyperthermia using magnetite cationic liposomes. Jpn. J. Cancer Res 89, 775–782 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoopes PJ et al. Treatment of canine oral melanoma with nanotechnology-based immunotherapy and radiation. Mol. Pharm 15, 3717–3722 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y et al. An immunostimulatory dual-functional nanocarrier that improves cancer immunochemotherapy. Nat. Commun 7, 13443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park J et al. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater 11, 895–905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrated an elegant strategy to co-deliver small molecule drugs (inhibitor of TGFβ) and protein drugs (IL-2) to tumours using nanoparticles.
- 35.Duan X et al. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat. Commun 10, 1899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan X et al. Photodynamic therapy mediated by nontoxic core–shell nanoparticles synergizes with immune checkpoint blockade to elicit antitumor immunity and antimetastatic effect on breast cancer. J. Am. Chem. Soc 138, 16686–16695 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He C et al. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun 7, 12499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwong B, Gai SA, Elkhader J, Wittrup KD & Irvine DJ Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 73, 1547–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kosmides AK, Necochea K, Hickey JW & Schneck JP Separating T Cell targeting components onto magnetically clustered nanoparticles boosts activation. Nano Lett. 18, 1916–1924 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mi Y et al. A dual immunotherapy nanoparticle improves T-cell activation and cancer immunotherapy. Adv. Mater 30, 1706098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell MJ, Wayne E, Rana K, Schaffer CB & King MR TRAIL-coated leukocytes that kill cancer cells in the circulation. Proc. Natl. Acad. Sci. USA 111, 930–935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates the use of liposomal particles to “present” TRAIL from the surfaces of circulating leukocytes, promoting killing of circulating tumour cells.
- 42.Jyotsana N, Zhang Z, Himmel LE, Yu F & King MR Minimal dosing of leukocyte targeting TRAIL decreases triple-negative breast cancer metastasis following tumor resection. Sci. Adv 5, eaaw4197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nair PM et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display. Proc. Natl. Acad. Sci. USA 112, 5679–5684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan H et al. Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat. Nanotechnol 12, 763 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Kulkarni A et al. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat. Biomed. Eng 2, 589–599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shae D et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol 14, 269–278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng N et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1–insensitive models of triple-negative breast cancer. JCI Insight 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo M et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first demonstration of a synthetic polymer particle that appears to directly interact with STING to promote interferon production and T cell priming.
- 49.Guan C et al. RNA-based immunostimulatory liposomal spherical nucleic acids as potent TLR7/8 modulators. Small 14, e1803284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radovic-Moreno AF et al. Immunomodulatory spherical nucleic acids. Proc. Natl Acad. Sci. USA 112, 3892–3897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pastor F et al. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov 17, 751–767 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Song W et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun 9, 2237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hewitt SL et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36gamma, and OX40L mRNAs. Sci. Transl. Med 11, eaat9143 (2019). [DOI] [PubMed] [Google Scholar]; This work demonstrated the use of in vitro screening to define candidate immunotherapy cues that would exert synergy in priming antitumour immunity, and then delivery of these cues to tumours using lipid nanoparticles carrying mRNA encoding the target genes.
- 54.Rothschilds AM & Wittrup KD What, why, where, and when: bringing tming to immuno-oncology. Trends Immunol. 40, 12–21 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Shimizu T et al. Nanogel DDS enables sustained release of IL-12 for tumor immunotherapy. Biochem. Biophys. Res. Commun 367, 330–335 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Chu H, Zhao J, Mi Y, Di Z & Li L NIR-light-mediated spatially selective triggering of anti-tumor immunity via upconversion nanoparticle-based immunodevices. Nat. Commun 10, 2839 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Aznar MA et al. Intratumoral delivery of immunotherapy-act locally, think globally. J. Immunol 198, 31–39 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Marabelle A, Kohrt H, Caux C & Levy R Intratumoral immunization: a new paradigm for cancer therapy. Clin. Cancer Res 20, 1747–1756 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marabelle A et al. Starting the fight in the tumor: expert recommendations for the development of human intratumoral immunotherapy (HIT-IT). Ann. Oncol 29, 2163–2174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hammerich L et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med 25, 814–824 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Formenti SC et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med 24, 1845–1851 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ray A et al. A phase I study of intratumoral ipilimumab and interleukin-2 in patients with advanced melanoma. Oncotarget 7, 64390–64399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC & Nelson BH Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 18, 419–432 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Biot C et al. Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Sci. Transl. Med 4, 137ra172 (2012). [DOI] [PubMed] [Google Scholar]
- 65.van Herpen CML et al. Intratumoral rhIL-12 administration in head and neck squamous cell carcinoma patients induces B cell activation. Int. J. Cancer 123, 2354–2361 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Hanes J et al. Controlled local delivery of interleukin-2 by biodegradable polymers protects animals from experimental brain tumors and liver tumors. Pharm. Res 18, 899–906 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Hori Y, Stern PJ, Hynes RO & Irvine DJ Engulfing tumors with synthetic extracellular matrices for cancer immunotherapy. Biomaterials 30, 6757–6767 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kwong B, Liu H & Irvine DJ Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 32, 5134–5147 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pluen A et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 98, 4628–4633 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goodman TT, Olive PL & Pun SH Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomedicine 2, 265–274 (2007). [PMC free article] [PubMed] [Google Scholar]
- 71.Perrault SD, Walkey C, Jennings T, Fischer HC & Chan WC Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 9, 1909–1915 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Popovic Z et al. A nanoparticle size series for in vivo fluorescence imaging. Angew. Chem. Int. Ed. Engl 49, 8649–8652 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lizotte PH et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol 11, 295–303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Q et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol 14, 89–97 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Jeanbart L et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer Immunol. Res 2, 436–447 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Munn DH & Mellor AL The tumor-draining lymph node as an immune-privileged site. Immunol. Rev 213, 146–158 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Zhou X et al. Precise spatiotemporal interruption of regulatory T-cell-mediated CD8(+) T-cell suppression leads to tumor immunity. Cancer Res. 79, 585–597 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Schudel A, Francis DM & Thomas SN Material design for lymph node drug delivery. Nat. Rev. Mater 4, 415–428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomas SN, Vokali E, Lund AW, Hubbell JA & Swartz MA Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 35, 814–824 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y, Li N, Suh H & Irvine DJ Nanoparticle anchoring targets immune agonists to tumors enabling anti-cancer immunity without systemic toxicity. Nat. Commun 9, 6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Charych DH et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res 22, 680–690 (2016). [DOI] [PubMed] [Google Scholar]; Preclinical studies demonstrating the efficacy of an IL-2-polymer prodrug that alters binding of the cytokine to target receptors and promotes selective stimulation in tumours.
- 82.Charych D et al. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PLOS ONE 12, e0179431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bentebibel S-E et al. A first-in-human study and biomarker analysis of NKTR-214, a novel IL2Rβγ-biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov. 9, 711–721 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Nakamura T et al. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control. Release 216, 149–157 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Torres Andón F & Alonso MJ Nanomedicine and cancer immunotherapy – targeting immunosuppressive cells. J. Drug Target 23, 656–671 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Zhang F et al. Uniform brain tumor distribution and tumor associated macrophage targeting of systemically administered dendrimers. Biomaterials 52, 507–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim HY et al. Quantitative imaging of tumor-associated macrophages and their response to therapy using (64)Cu-labeled macrin. ACS Nano 12, 12015–12029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miller MA et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat. Commun 6, 8692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller MA et al. Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci. Transl. Med 9, eaal0225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Turk MJ, Waters DJ & Low PS Folate-conjugated liposomes preferentially target macrophages associated with ovarian carcinoma. Cancer Lett. 213, 165–172 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Jeanbart L, Kourtis IC, van der Vlies AJ, Swartz MA & Hubbell JA 6-Thioguanine-loaded polymeric micelles deplete myeloid-derived suppressor cells and enhance the efficacy of T cell immunotherapy in tumor-bearing mice. Cancer Immunol. Immun 64, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kourtis IC et al. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLOS ONE 8, e61646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sasso MS et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials 96, 47–62 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Jahchan NS et al. Tuning the tumor myeloid microenvironment to fight cancer. Front. Immunol 10, 1611 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rodell CB et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng 2, 578–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated how preferential nanoparticle accumulation in TAMs can be exploited to reprogramme this immunosuppressive cell population in vivo.
- 96.Zilio S et al. 4PD Functionalized dendrimers: a flexible tool for in vivo gene silencing of tumor-educated myeloid cells. J. Immunol 198, 4166–4177 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Qian Y et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano 11, 9536–9549 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Parayath NN, Parikh A & Amiji MM Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating microRNA-125b. Nano Lett. 18, 3571–3579 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Zanganeh S et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol 11, 986–994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ledo AM et al. Co-delivery of RNAi and chemokine by polyarginine nanocapsules enables the modulation of myeloid-derived suppressor cells. J. Control. Release 295, 60–73 (2019). [DOI] [PubMed] [Google Scholar]
- 101.Turley SJ, Cremasco V & Astarita JL Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol 15, 669–682 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Chauhan VP et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc. Natl. Acad. Sci. USA 116, 10674–10680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoang B et al. Docetaxel-carboxymethylcellulose nanoparticles target cells via a SPARC and albumin dependent mechanism. Biomaterials 59, 66–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ernsting MJ et al. Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle. J. Control. Release 206, 122–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Murakami M et al. Docetaxel conjugate nanoparticles that target alpha-smooth muscle actin-expressing stromal cells suppress breast cancer metastasis. Cancer Res. 73, 4862–4871 (2013). [DOI] [PubMed] [Google Scholar]
- 106.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Raje N et al. Anti-BCMA CAR T-Cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med 380, 1726–1737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yee C Adoptive T cell therapy: points to consider. Curr. Opin. Immunol 51, 197–203 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Hsu C et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood 109, 5168–5177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kerkar SP et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 70, 6725–6734 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang L et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res 21, 2278–2288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stephan MT & Irvine DJ Enhancing cell therapies from the outside in: cell surface engineering using synthetic nanomaterials. Nano Today 6, 309–325 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stephan MT, Moon JJ, Um SH, Bershteyn A & Irvine DJ Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med 16, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie YQ et al. Redox-responsive interleukin-2 nanogel specifically and safely promotes the proliferation and memory precursor differentiation of tumor-reactive T-cells. Biomater. Sci 7, 1345–1357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tang L et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol 36, 707–716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows how TCR signalling-mediated changes in cell surface biochemistry can be used as a trigger to link drug delivery to T cell activation in adoptive cell therapy.
- 116.Siriwon N et al. CAR-T Cells surface-engineered with drug-encapsulated nanoparticles can ameliorate intratumoral T-cell hypofunction. Cancer Immunol. Res 6, 812–824 (2018). [DOI] [PubMed] [Google Scholar]
- 117.Zheng Y, Tang L, Mabardi L, Kumari S & Irvine DJ Enhancing adoptive cell therapy of cancer through targeted delivery of small-molecule immunomodulators to internalizing or noninternalizing receptors. ACS Nano 11, 3089–3100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stephan MT, Stephan SB, Bak P, Chen J & Irvine DJ Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials 33, 5776–5787 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huppa JB & Davis MM T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol 3, 973–983 (2003). [DOI] [PubMed] [Google Scholar]
- 120.Huang B et al. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med 7, 291ra294 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zheng Y et al. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J. Control. Release (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmid D et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun 8, 1747 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu H et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhu G et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat. Commun 8, 1954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ma L et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 365, 162–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstration of an approach to deliver CAR T cell ligands to the surface of APCs in lymph nodes, enabling CAR T cell boosting in the native lymph node microenvironment.
- 126.June CH, O’Connor RS, Kawalekar OU, Ghassemi S & Milone MC CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). [DOI] [PubMed] [Google Scholar]
- 127.Moffett HF et al. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun 8, 389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Smith TT et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol 12, 813–820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first demonstration of the use of nanoparticles for direct gene delivery to generate CAR T cells in vivo in a preclinical mouse model.
- 129.Kranz LM et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016). [DOI] [PubMed] [Google Scholar]
- 130.Netea MG et al. Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kleinnijenhuis J et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 109, 17537–17542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Quintin J et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell host & microbe 12, 223–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mulder WJM, Ochando J, Joosten LAB, Fayad ZA & Netea MG Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov 18, 553–566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang Y et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat. Mater 13, 204–212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Freitas RA Nanomedicine, Volume I: Basic Capabilities. Vol. 1 (Landes Bioscience, 1999). [Google Scholar]
- 136.Shi J, Kantoff PW, Wooster R & Farokhzad OC Cancer nanomedicine: progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maeda H, Nakamura H & Fang J The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev 65, 71–79 (2013). [DOI] [PubMed] [Google Scholar]
- 138.Chauhan VP & Jain RK Strategies for advancing cancer nanomedicine. Nat. Mater 12, 958 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bobo D, Robinson KJ, Islam J, Thurecht KJ & Corrie SR Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res 33, 2373–2387 (2016). [DOI] [PubMed] [Google Scholar]
- 140.Wilhelm S et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 1, 16014 (2016). [Google Scholar]
- 141.Golombek SK et al. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev 130, 17–38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu J et al. Assessing immune-related adverse events of efficacious combination immunotherapies in preclinical models of cancer. Cancer Res. 76, 5288–5301 (2016). [DOI] [PubMed] [Google Scholar]