

This study examines the US Food and Drug Administration’s approval process for biosimilar biologic products by evaluating the site and facility inspections, animal and human studies, and comparative efficacy trials conducted to support the applications.
Key Points
Question
Ten years after an abbreviated pathway was established for approval of biosimilar biologic products in the US, what biosimilars have been approved, and how extensively were they tested?
Findings
In this study of 23 biosimilars for 9 reference products approved by the US Food and Drug Administration (FDA), 1 novel and frequent requirement for approval was a year-long comparative efficacy trial that included not only a comparison with the reference product but also a period in which some patients were switched between products. The issues that the FDA identified in biosimilar applications most often required addressing problems in facilities or manufacturing process.
Meaning
Findings of this study suggest that comparative efficacy trials for biosimilar products are as rigorous as and often larger, longer, and more costly than many pivotal trials for initial approval of new molecular entities, in contrast to trials for most small-molecule generic drugs, which compare plasma concentrations over time in healthy volunteers.
Abstract
Importance
Biosimilar biologic products were authorized in 2010, after the US Congress established an expedited pathway for approval of clinically similar versions of approved biologic products. Unlike for most small-molecule generic drugs, approval requirements for a biosimilar included animal studies and a comparative efficacy clinical trial.
Objective
To analyze the evidence required to support a biosimilarity license application, examine the US Food and Drug Administration (FDA) evaluation process, and estimate the costs of the key clinical trial evidence.
Design
This study evaluated all biosimilar biologic products approved from January 2010 through October 2019, using the publicly available FDA review documents, disclosures from ClinicalTrials.gov, and the published peer-reviewed literature. The costs of efficacy clinical trials were estimated using licensed proprietary software.
Main Outcomes and Measures
The following elements of each approved biosimilar were evaluated: the extent of human clinical testing to establish that the biosimilar had no clinically meaningful differences with the reference product, results of comparative animal studies, and FDA-cited application deficiencies. The cited deficiencies included the following categories: (1) facility inspection, (2) manufacturing or product quality, (3) animal studies, (4) laboratory analytical studies, (5) phase 1 and/or immunogenicity studies, and (6) phase 3 comparative efficacy trials.
Results
As of October 2019, the FDA had approved 23 biosimilar biologics for 9 reference products. The 29 clinical trials that established that the efficacy of the biosimilar products was comparable to that of the reference products enrolled a median (interquartile range [IQR]) of 504 (258-612) patients, had a median (IQR) estimated cost of $20.8 ($13.8-$35.3) million, and had a median (IQR) treatment duration of 52 (28-68) weeks. Substantial deficiencies temporarily halted the review of 9 applications, and the most frequent deficits were failed facilities inspections (n = 5) and manufacturing process quality problems (n = 6). The approved biosimilar submissions included 51 animal studies on species that included mice, rats, rabbits, dogs, and cynomolgus monkeys. Negative outcomes in 2 animal studies were attributed to differences between human and test species. The FDA generally met the standard 12-month review deadlines or stopped the review clock when serious deficiencies were identified.
Conclusions and Relevance
This study found that most comparative efficacy trials supporting the FDA approval of biosimilars appeared to be as rigorous as and often larger, longer, and more costly than pivotal trials for new molecular entities. Further research is needed into whether less costly comparative efficacy trials could provide adequate evidence of biosimilarity and whether animal studies contribute useful scientific evidence.
Introduction
In 2010, the Biologics Price Competition and Innovation (BPCI) Act of 2009 was enacted in the US, creating an abbreviated pathway for the approval of biologic products shown to be biosimilar to a previously approved reference product. The pathway was tailored to address the properties of biologic products, which in contrast to small-molecule drugs, are large, complex molecules produced through living systems rather than through chemical processes. Biosimilarity was defined as having “no clinically meaningful differences between the biosimilar biologic product and the reference product in terms of safety, purity and potency of the product.”1
Although small-molecule generic drugs accounted for 90% of US prescriptions in 2018,2 net spending for all biologics totaled $125 billion, or 36% of all spending for medication in the US, a 50% increase since 2014. The framers of the BPCI Act sought to achieve cost savings through increased market competition.
Although established standards existed for approving generics for small-molecule drugs, for which the primary evidence was a modest bioequivalence study,3 the detailed testing and approval standards needed to establish biosimilarity triggered a debate.4 What kind of and how many efficacy studies are required to establish clinical equivalence? Should biosimilars, which are not structurally identical to the reference product, be allowed to have identical chemical names as do generics? Are comparative efficacy trials that could take years to complete too burdensome?
Beginning in 2012, the US Food and Drug Administration (FDA) approval requirements for biosimilars began to appear, first as draft guidance documents and thereafter as final guidance documents. Biosimilar molecules were required to be “highly similar not withstanding minor differences in chemically inactive components.”1 Both biosimilar and all new biologic products would have 4-letter suffixes (eg, trastuzumab-anns) that would distinguish reference products from biosimilars.5 Although the guidance for biosimilarity outlined 6 basic requirements (Box),6 it also signaled that the FDA intended to exercise regulatory flexibility. The guidance stated that establishing no clinically significant differences depended on the totality of evidence and proceeded with a stepwise approach.6 The guidance document contained multiple criteria and varying approaches to describe 1 or more comparative clinical studies.
Box. Typical Requirements for Biosimilar Approval in USa.
Inspected manufacturing facility
Manufacturing process or product quality
Laboratory analytical studies of molecule
Animal studies in multiple species
Phase 1 comparative clinical trial in healthy volunteers
Phase 3 comparative efficacy trial in 1 indication
In March 2015, the FDA approved the first biosimilar biologic product for the US market, filgrastim-sndz. Development of the US approval process was slower than in the European Union. The European Medicines Agency (EMA) approved its first biosimilar product, somatotropin, in 2006.7 The Figure shows the timeline of key events in the history of biosimilar biologic products.
Figure. Timeline of Key Events in Regulation of Biosimilar Biologic Products.
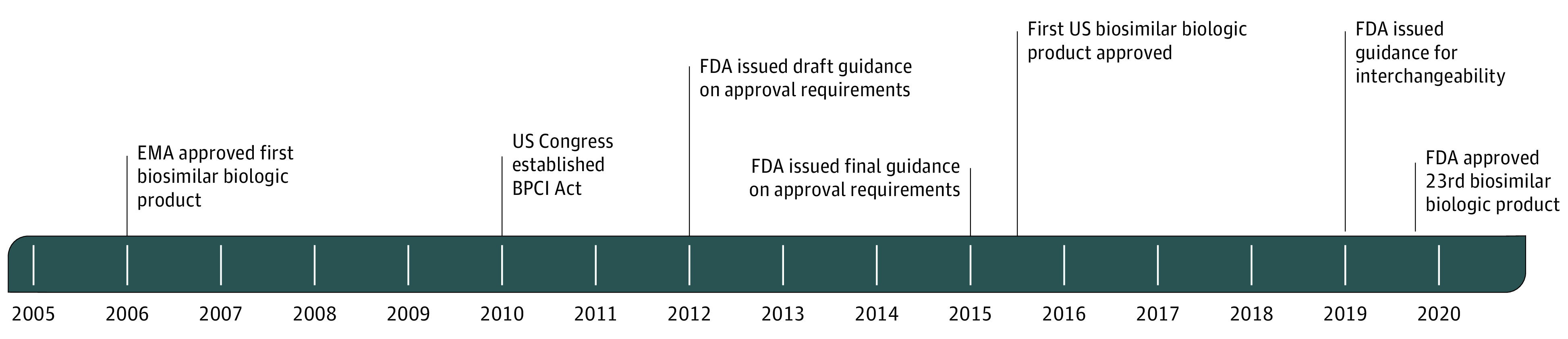
BPCI Act indicates Biologics Price Competition and Innovation Act of 2009; EMA, European Medicines Agency; and FDA, US Food and Drug Administration.
In this study, we examined the FDA approval requirements, reviews, and comparative clinical studies for all approved biosimilar biologic products in the US. The objective was to analyze the evidence required to support a biosimilarity claim, examine the FDA evaluation process, and estimate the costs of the key clinical trial evidence.
Methods
The biosimilar biologics, reference products, and approval eligibility were identified through the Purple Book,8 the official listing of licensed biologic products regulated by the FDA Center for Drug Evaluation and Research. The edition used for this study included all biosimilar approval actions through October 2019. This study did not involve personally identifiable information and thus was exempted from review by the Johns Hopkins Bloomberg School of Public Health Institutional Review Board.
We assessed FDA reviews and approvals of the Biologics License Applications (BLAs) through the Summary, Clinical, Pharmacology, or Cross Disciplinary Reviews that are publicly available on the drugs@FDA website. We obtained additional detail about the clinical studies cited in the BLAs from ClinicalTrials.gov and published peer-reviewed articles. These FDA reviews contained specific descriptors that enabled us to identify the sponsor-required entries in ClinicalTrials.gov and the peer-reviewed literature. We ascertained whether the approved products had been launched as of March 1, 2020, through the National Library of Medicine RxNorm database of drug and biologic products currently on the market.9
We evaluated the FDA approval requirements as implemented through these elements of each biosimilar application: comparative efficacy testing in humans and animals, clinical trial site inspections, and agency decisions to halt review of a BLA until substantial deficiencies were addressed. In addition, we examined the timeliness of the FDA review and approval process. These end points provided not only a perspective on the rigor of the FDA reviews but also insights into the problems that might occur in the development of a biosimilar biologic product.
Variables and other data for this study were collected by a member of the study team and verified by another member. Specifically, data on FDA review practices were collected by J.L.B., and data on trial characteristics and cost estimates were collected by M.C.M.; all data were verified by T.J.M.
Comparative Efficacy Clinical Trials
We assessed all comparative efficacy clinical trials in humans that were specifically identified in the FDA reviews, whether they were phase 1 studies of bioequivalence in healthy volunteers or head-to-head phase 3 studies for an approved indication in patients with disease. The FDA guidance also requires an assessment of comparable immunogenicity, usually measured by blood levels of antidrug antibodies. Immunogenicity assays were typically appended to 1 of the other phase 1 or phase 3 studies. However, if separate immunogenicity trials were conducted, we included them among the comparative efficacy trials.
To establish the characteristics and estimated costs of the comparative efficacy trials, we adapted the methods described in previous studies10,11,12 to assess the pivotal clinical efficacy trials for new molecular entities. Briefly, for each trial, we collected 52 variables that described not only the scope of the trial, such as disease condition, enrollment size, treatment duration, end points, and clinical visits, but also the characteristics associated with study costs, such as number of study sites, global site locations, and number of patients screened. To estimate costs, we licensed a proprietary pharmaceutical industry clinical trial cost-estimating tool (CostPro Mid-Level Tool; IQVIA).13 The CostPro software produces trial cost estimates derived from 2000 clinical trial contracts awarded to contract research organizations and integrates information from 200 000 clinical trial sites in 60 countries. The trial duration included not only the period to establish effectiveness but also the extensions during which patients were switched between products. For oncology product trials, which typically continued indefinitely, the trial duration was defined as the period from the date of reported trial start to the date when the FDA accepted the data for comparability or, if unavailable, the date when the sponsor filed the BLA. To estimate the time required for sites to conduct a comparative efficacy study, we calculated the difference between study start date reported on ClinicalTrials.gov and the actual completion date in that data source.
Animal Studies and Complete Response Actions
For each approved biosimilar, we examined whether animal studies were required, the number and species involved, and the FDA judgment about whether they provided evidence of unacceptable biological differences between the biosimilar and reference products or evidence of safety concerns.
We screened every biosimilar review to ascertain whether the FDA found substantial deficiencies in the application that had to be addressed before approval. A Complete Response letter declares that the agency will not approve the application in its present form14 and identifies specific deficiencies ranging from minor informational or labeling issues to questions so substantial that addressing them would require an additional clinical study. The Complete Response letter is the most severe adverse regulatory action compared with an Information Request letter for additional data or an Approvable letter, which declares that the agency can approve the application if a specific deficiency is resolved.
We also categorized the cited deficiencies into these 6 basic elements of a biosimilar application: (1) facility inspection, (2) manufacturing or product quality, (3) animal studies, (4) laboratory analytical studies, (5) phase 1 and/or immunogenicity studies, and (6) phase 3 comparative efficacy trials. We hypothesized that these actions would not only provide qualitative information about the rigor of the FDA review process but also indicate the kind of problems that might arise in the process of developing an approvable biosimilar product.
Site Inspections and Review Timeliness
We assessed whether the FDA evaluation of clinical trial evidence also included independent clinical trial site inspections similar to those used for evaluating new molecular entities. Each BLA was examined for the number of sites inspected and the number of countries visited. We used the timeliness of each FDA review to assess whether the agency had resources and policy priority to handle the evaluation of this new category of biologic product. The duration of the FDA review was measured from the date that the BLA was filed until the first formal regulatory action (either approval or a Complete Response letter).
Statistical Analysis
We used the median (interquartile range [IQR]) to report the overall characteristics (such as patient enrollment) and estimated costs of comparative efficacy trials. However, for each individual trial, we calculated the mean and 95% CI for 6 CostPro outputs. The data for this study were collected in databases (Microsoft Access and Excel; Microsoft Corp), and the cost estimates were generated with a software tool (CostPro; IQVIA). Results were analyzed with Stata, Release 16 (StataCorp LLC). Data were analyzed from September 2019 to March 2020.
Interchangeability
We were unable to evaluate a separate but related expedited pathway for approval of biosimilars, a designation called interchangeability. Under the BPCI Act, patients may be switched between reference products and interchangeable biosimilars without the explicit permission of prescribing physicians, unless prescribed as dispense as written (identical to the small-molecule drugs). The primary additional requirement was a clinical trial designed to assess whether risks, benefits, and immunogenicity remained comparable when patients alternated between the biosimilar and reference product. However, the guidance for interchangeable products was not finalized until 201915; no equivalent category existed in the EMA, and, as of October 2019, no interchangeable products had been approved.
Results
The FDA had approved 23 biosimilars for 9 reference products as of October 2019. Table 1 provides an overview of the status of approved and eligible biosimilar products under the BPCI Act. Table 2 lists the approved biosimilars by reference product and drug class.16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44 The 12-year market exclusivity period had expired for an additional 56 products, and an additional 111 products are eligible to begin clinical testing. FDA marketing approval did not always result in a prompt launch of the biosimilar product. Eleven licensed biosimilars had not been launched as of March 2020 because of secondary patent litigation or for other commercial reasons (Table 2).16,17,18,19,20,21,22,25,35,39,41,44
Table 1. Status of Biosimilar Biologic Products in the US, October 2019.
| Status | No. | |
|---|---|---|
| Reference products | Biosimilar products | |
| Launched in US | 7 | 12 |
| FDA approved | 9 | 23 |
| Eligible for FDA approvala | 56 | NA |
| Eligible for developmentb | 111 | NA |
Abbreviations: FDA, US Food and Drug Administration; NA, not applicable.
12 years after reference product approval.
8 years after reference product approval.
Table 2. US Food and Drug Administration Approvals of Biosimilar Biologics and Their Reference Products.
| Therapeutic category/brand namea | Chemical name | Reference product | Approval date | Launched?b |
|---|---|---|---|---|
| Anti-TNF | ||||
| Amjevita16,17 | adalimumab-atto | Humira | 9/23/2016 | No |
| Cyltezo18 | adalimumab-adbm | Humira | 8/25/2017 | No |
| Hadlima19 | adalimumab-bwwd | Humira | 7/23/2019 | No |
| Hyrimoz20 | adalimumab-adaz | Humira | 10/20/2018 | No |
| Erelzi21 | etanercept-szzs | Enbrel | 8/30/2016 | No |
| Eticovo22 | etanercept-ykro | Enbrel | 4/25/2019 | No |
| Renflexis23 | infliximab-abda | Remicade | 4/21/2017 | Yes |
| Inflectra24 | infliximab-dyyb | Remicade | 4/5/2016 | Yes |
| Ixifi25 | infliximab-qbtx | Remicade | 12/13/2017 | No |
| Hematopoiesis | ||||
| Nivestym26,27,28 | filgrastim-aafi | Neupogen | 7/20/2018 | Yes |
| Zarxio29 | filgrastim-sndz | Neupogen | 3/6/2015 | Yes |
| Udenyca30,31 | pegfilgrastim-cbqv | Neulasta | 11/2/2018 | Yes |
| Fulphila32 | pegfilgrastim-jmdb | Neulasta | 6/4/2018 | Yes |
| Retacrit33,34 | epoetin alfa-epbx | Epogen | 5/15/2018 | Yes |
| Oncology | ||||
| Ontruzant35 | trastuzumab-dttb | Herceptin | 1/18/2019 | No |
| Kanjinti36 | trastuzumab-anns | Herceptin | 6/13/2019 | Yes |
| Ogivri37 | trastuzumab-dkst | Herceptin | 12/1/2017 | Yes |
| Herzuma38 | trastuzumab-pkrb | Herceptin | 12/14/2018 | Yes |
| Trazimera39 | trastuzumab-qyyp | Herceptin | 3/11/2019 | No |
| Mvasi40 | bevacizumab-awwb | Avastin | 9/14/2017 | Yes |
| Zirabev41 | bevacizumab-bvzr | Avastin | 6/27/2019 | No |
| Truxima42,43 | rituximab-abbs | Rituxan | 11/28/2018 | Yes |
| Ruxience44 | rituximab-pvvr | Rituxan | 7/23/2019 | No |
Abbreviation: TNF, tumor necrosis factor.
Nivestym and Udenyca approved with phase 1 trials only.
As of March 1, 2020.
Comparative Efficacy Trials
The statutory mandate to demonstrate no clinically meaningful differences was implemented in a requirement for at least 1 clinical trial that compared the efficacy of the biosimilar with that of the reference product. Costs and characteristics of the comparative efficacy trials are shown in Table 3. Overall, the 29 clinical trials16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44 that established that the efficacy of the biosimilar products was comparable to that of the reference products enrolled a median (IQR) number of 504 (258-612) patients, had a median (IQR) estimated cost of $20.8 ($13.8-$35.3) million, and had a median (IQR) treatment duration of 52 (28-68) weeks.
Table 3. Characteristics and Costs of Biosimilar Comparative Efficacy Trials.
| Characteristic | No. | Median (IQR) | |||
|---|---|---|---|---|---|
| Trials | Biosimilar products | Patients enrolled, No. | Treatment duration, wk | Estimated costa | |
| Overall | 29 | 23 | 504 (258-612) | 52 (28-68) | 20.8 (13.8-35.3) |
| By trial phase | |||||
| Phase 1 | 5 | 2 | 122 (60-256) | 15 (14-15) | 1.9 (1.6-1.9) |
| Phase 3 | 24 | 21 | 538 (372-644) | 55 (46-78) | 27.6 (18.0-36.7) |
| By therapeutic category | |||||
| Anti-TNF | 10 | 9 | 564 (526-606) | 61 (52-78) | 19.0 (14.0-32.8) |
| Oncology | 10 | 9 | 573 (394-719) | 60 (52-156) | 36.7 (34.5-71.3) |
| Hematopoietic | 9 | 5 | 218 (122-303) | 25 (15-28) | 2.1 (1.9-15.6) |
Abbreviations: IQR, interquartile range; TNF, tumor necrosis factor.
In millions of US dollars.
For 21 of the 23 (91%) approved biosimilars, comparative efficacy was established in 1 or more phase 3 trials in diseased patients for an approved reference product indication and included both an initial comparative efficacy period and additional follow-up, during which a subset of patients was switched between the biosimilar and reference products. These 24 studies enrolled a median (IQR) number of 538 (372-644) patients, had a median (IQR) treatment duration of 55 (46-78) weeks, and had a median (IQR) estimated cost of $27.6 ($18.0- $36.7) million (Table 3).16,17,18,19,20,21,22,23,24,25,29,32,33,34,35,36,37,38,39,40,41,42,43,44
For 2 hematopoietic reference products, the FDA accepted phase 1 trials in healthy volunteers: 1 of 2 biosimilars for a filgrastim reference product, and 1 of 2 biosimilars for a pegfilgrastim reference product. Clinical equivalence for pegfilgrastim-cbqv was based on 2 phase 1 studies with a pharmacodynamic end point of absolute neutrophil count.30,31 The FDA concluded that this pharmacodynamic biomarker was the same in patients who were healthy as in those with cancer and was an adequate measure of clinical effects. For filgrastim-aafi, equivalence was established in 3 open-label crossover studies in healthy volunteers with a pharmacodynamic biomarker of CD34+ cell counts.26,27,28 The characteristics and costs for these phase 1 studies were markedly lower, enrolling a median (IQR) number of 122 (60-256) patients, for a median (IQR) treatment duration of 15 (14-15) weeks, and with a median (IQR) estimated cost of $1.9 ($1.6-$1.9) million.
The 29 comparative efficacy trials not only had a median (IQR) treatment duration of 52 (28-68) weeks but also took additional time because multiple global sites had to be established and patients had to be recruited. The phase 3 trials16,17,18,19,20,21,22,23,24,25,29,32,33,34,35,36,37,38,39,40,41,42,43,44 took a median (IQR) period of 26 (21-35) months from the reported study start date to actual completion date. The phase 1 studies26,27,28,30,31 were completed more quickly, with a median (IQR) period of 3 (3-4) months.
When clinical equivalence was established in a clinical trial for 1 indication for the reference product and the biosimilars, the FDA biosimilar guidance permitted efficacy to be inferred “through extrapolation of clinical data across indications,”6(p21) although the agency said it would judge these extrapolations on a case-by-case basis. Thus, a single phase 3 trial for 1 indication for the reference product could be accepted as evidence of overall biosimilarity even if the reference product had multiple approved indications. Biosimilar BLAs for rituximab-abbs and adalimumab-atto included 2 comparative efficacy trials for different indications already approved for the reference product.16,17,42,43 Some reference product indications with unexpired market or orphan drug exclusivity were excluded from the biosimilar prescribing information.
Animal Studies
The approved biosimilar submissions included 51 animal studies, with 1 to 7 experiments conducted for each of the 23 approved biosimilars. Species included mice, rats, rabbits, dogs, and cynomolgus monkeys. The most frequent study design was a comparative toxicological assessment in cynomolgus or unspecified species of monkeys (n = 14). For 21 of 23 biosimilars, the FDA found various evidence of similarity, identified no safety concerns, or reported no biologically significant differences. However, for pegfilgrastim-cbqv, the FDA found residual uncertainty from a 4-week comparative study in cynomolgus monkeys because at 4 weeks the systematic exposure was lower in the biosimilar-treated monkeys compared with exposure to the reference product. For epoetin alfa-epbx, the agency noted differences in immunogenicity between the biosimilar and reference products in a rat study. However, in both cases, the animal study differences were considered to be better addressed in clinical trials and did not otherwise change approval or labeling.
FDA Reviews, Site Inspections, and Application Quality
The FDA reviews of biosimilar BLAs were conducted by the Center for Drug Evaluation and Research with user fee charges and timely review completion goals similar to those for the new molecular entities. Biosimilars did not qualify for any of the expedited review programs45 and received a standard 12-month review designation. For the 14 applications that were completed without delays directly associated with application deficiencies, the agency completed the reviews in a mean (SD) time of 350 (49.6) days.
In addition to examining the data in a BLA, the FDA conducted direct inspections of manufacturing facilities and selected clinical trial sites. For 9 of 23 biosimilars (39%), the agency halted review of the BLA, issuing a Complete Response letter that identified specific deficiencies serious enough that the agency would not further consider the application until or unless the deficiencies had been satisfactorily addressed. The application sponsors then took from 59 to 433 days to address the deficiencies and resubmit the BLA. The most frequently cited deficits were problems in product quality and manufacturing process (n = 6) and failed facility inspections (n = 5). Additional detail for types of deficiencies identified are shown in Table 4. In addition, all but 1 Complete Response letters cited deficiencies in 2 to 4 different categories.
Table 4. Major Deficiencies Cited in Biosimilar Applicationsa.
| Approval requirement problems | Products affected, No. |
|---|---|
| Facility inspection | 5 |
| Manufacturing quality | 6 |
| Animal studies | 0 |
| Laboratory analytical | 3 |
| Phase 1 bioequivalence | 1 |
| Phase 3 efficacy | 2 |
Complete Response halted US Food and Drug Administration review until deficiencies were resolved.
Another challenge to timely FDA reviews was the need to inspect individual clinical trial sites in a global enterprise; the trials were conducted in a mean (SD) number of 12 (9) countries. The agency inspected 1 to 7 clinical sites for the trials for each approved biosimilar, finding no violations at the sites associated with 15 biosimilar clinical trials and voluntary corrective actions needed at 8 sites. However, other more serious deficiencies in phase 1 or phase 3 overall clinical trial conduct were noted in 3 Complete Response letters.
Discussion
In this study, we examined the 23 biosimilar biologic products that were FDA approved under the abbreviated pathway established in the BPCI Act (enacted in 2010) for this new category of therapeutic agents. The required evidence of no clinically meaningful differences between a biosimilar and a reference product was supported by comparative efficacy trials involving a median of 504 patients who were observed for a median of 52 weeks. Establishing evidence of biosimilarity also included inspection of facilities, evaluation of the manufacturing process, analysis of molecular structure and function, animal studies, and phase 1 studies in healthy volunteers.
The FDA performance and actions in reviewing biosimilar applications were timely and had analytical and inspectional depths that were similar to those seen in previous reviews of new molecular entities.10,11,12 With the imposition of user fees and compliance with the 12-month review deadlines, the FDA appeared to have avoided the application backlogs and equivalence controversies reported in the past concerning both generic small-molecule drugs and medical devices.
Trial Comparisons With New Molecular Entities
When phase 3 efficacy trials were required (21 of 23 approved biosimilars), the median patient enrollment, treatment duration, and estimated costs often exceeded those reported in published studies of FDA pivotal trials required for new molecular entities.10,46 Although the comparisons are not exact, the median (IQR) cost was $19 ($12-$33) million for 138 pivotal trials supporting the approval of 59 new molecular entities in 2015 to 201611 compared with the $27.6 ($18.0-$36.7) million for the 24 trials in the current study. A study of FDA approvals from 2005 to 2012 reported a median (IQR) patient enrollment of 446 (205-678) patients46 compared with a median (IQR) number of 538 (372-644) patients in the phase 3 studies and a median (IQR) number of 122 (60-256) patients in the phase 1 trials that provided evidence of clinical equivalence in the current study. For the phase 3 biosimilar efficacy trials, 100% had a trial treatment duration that exceeded 26 weeks compared with 35% of the pivotal trials supporting the approvals in 2015 to 2016 and 25% of pivotal trials in the 2005 to 2012 approvals. These comparisons provide a useful context, but they are not exact comparisons given that the previous findings included biologic products and small-molecule drugs and that phase 3 trials were usually but not always required for marketing approval.
Data from the current and previous research on pivotal trials10,11,12 explain why comparative efficacy trials are so large, costly, and long. Patient enrollment requirements increase when more statistical power is needed to document small differences with a comparator. Because the hypothesis is that no differences exist between biosimilars and their comparators, the FDA required a noninferiority design, with a 90% to 110% CI.47 A previous examination of pivotal trials10,11,12 found that the costliest trials were those that required evidence of noninferiority to an existing treatment. In addition, trial duration was nearly doubled because, after the initial efficacy comparison period, some patients had to be switched to the comparator to assess any differences from a product switch.
Given that pivotal trials for new molecular entities provide the primary and essential new scientific evidence that will guide the treatment of thousands to millions of future patients, an ethical and policy question arises about whether numerous, larger and more costly trials should be conducted to demonstrate that no meaningful differences exist from a proven treatment. However, additional research is needed to assess whether less-costly and time-consuming studies of bioequivalence, use of real-world evidence, or use of other alternatives could provide adequate scientific evidence of biosimilarity. Currently, the FDA and EMA requirements for comparative efficacy trials are similar.
Animal studies, although required by the BPCI Act, did not appear to provide useful scientific information given that human data were available both for the reference product and in clinical trials for the biosimilar product. These studies are not required for EMA approval.48
Furthermore, adequately supported proposals for re-evaluating the approval requirements for biosimilar biologic products need to include the examination of additional information that the FDA does not publicly disclose, in contrast to the EMA. That information is related to BLAs that were withdrawn, rejected, or subject to lengthy suspensions that extended beyond the review period for this study. For example, we could not identify even 1 phase 3 comparative efficacy trial that did not achieve equivalency end points. However, if several such trials led to an application withdrawal or rejection, they would serve as important evidence for a reconsideration of biosimilar legal requirements.
The Interchangeability category, which allows substitution without the participation of the prescribing physician, also requires additional examination to ascertain whether it should be retained, amended, or eliminated. No final guidance was published until 19 years after the BPCI Act established the Interchangeability category, and none had been approved as of March 2020. Interchangeability is also an approval category that is not included in EMA regulations.48
Limitations
This study has several limitations. First, we used only those comparative efficacy studies that were publicly cited by the FDA as pivotal in establishing no clinically meaningful differences between the biosimilars and their comparators. Phase 1 studies for the same drugs approved with phase 3 trials were not included. Second, although this study included every biosimilar approved through October 2019, we were unable to assess any BLAs that were rejected or withdrawn, given that unlike the EMA, the FDA does not disclose this information. Third, the estimated costs for the trials are subject to a series of limitations, which have been previously described.10 Briefly, these estimated costs for conducting a trial used an industry software tool that was designed for this purpose. Actual costs could be higher or lower than the estimates that we used. In addition, these costs did not include the sponsor costs for trial design and additional monitoring or the cost of the medication. Although the trial cost estimate data are an important component of demonstrating comparable efficacy, they do not reflect the overall costs of developing, testing, and obtaining regulatory approval for a biosimilar biologic product. The overall development costs include many additional and costly components, including establishing a manufacturing facility and conducting analytical, animal, and preliminary human studies. Fourth, although we examined every biosimilar in the US approved under the BPCI Act, other routes to approval exist for similar biologic products. For example, insulins were approved under a different statute until 2020, and some similar biologics were approved under BLAs without comparison to a reference product (eg, somatropin products). In these cases, the biologics were approved as new molecular entities without reference to or depending on data from a reference product.
Conclusions
In the 10 years since the BPCI Act was enacted, the FDA has approved 23 biosimilars for 9 reference products. Comparable efficacy was usually established in a single indication but often required clinical studies that were often larger, longer, and more costly than those required for new molecular entities. Further research and discussion are needed on whether trials with less costly designs could provide adequate evidence of biosimilarity and whether animal studies contribute useful scientific information.
Footnotes
Specific requirements may be waived or increased. Immunogenicity assay also required but usually as part of phase 1 or phase 3.
References
- 1.Biologics Price Competition and Innovation Act: Approval Pathway for Biosimilar Biologic Products. 42 USC §262(k)(2)(A) (2010).
- 2.IQVIA Medicine Use and Spending in the U.S.: A Review of 2018 and Outlook to 2023. IQVIA Institute; 2019. [Google Scholar]
- 3.US Food and Drug Administration Bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA:guidance for industry. Published December 2013. Accessed March 9, 2020. https://www.fda.gov/media/87219/download
- 4.US Food and Drug Administration Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. Published April 30, 2014. Accessed April 17, 2020. https://www.regulations.gov/docket?D=FDA-2011-D-0605
- 5.US Food and Drug Administration Nonproprietary naming of biological products. Guidance for Industry. Published January 2017. Accessed January 19, 2019. https://beta.regulations.gov/document/FDA-2011-D-0605-0055
- 6.US Food and Drug Administration Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for Industry. Published April 2015. Accessed January 31, 2019. https://www.fda.gov/media/82647/download
- 7.European Medicines Agency ; European Commission. Biosimilars in the EU: information guide for healthcare professionals. Accessed March 30, 2020. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
- 8.US Food and Drug Administration, Center for Drug Evaluation and Research The Purple Book: list of licensed biological products with (1) reference product exclusivity and (2) biosimilarity or interchangeability evaluations to date. Published July 23, 2019. Accessed August 16, 2019. https://www.fda.gov/media/89589/download
- 9.National Library of Medicine. Unified Medical Language System RxNorm. Published March 12, 2014. Accessed April 23, 2014. https://www.nlm.nih.gov/research/umls/rxnorm/
- 10.Moore TJ, Zhang H, Anderson G, Alexander GC. Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and Drug Administration, 2015-2016. JAMA Intern Med. 2018;178(11):1451-1457. doi: 10.1001/jamainternmed.2018.3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsiue EHC, Moore TJ, Alexander GC. Estimated costs of pivotal trials for U.S. Food and Drug Administration-approved cancer drugs, 2015-2017. Clin Trials. 2020;17(2):119-125. doi: 10.1177/1740774520907609 [DOI] [PubMed] [Google Scholar]
- 12.Qiao Y, Alexander GC, Moore TJ. Globalization of clinical trials: variation in estimated regional costs of pivotal trials, 2015-2016. Clin Trials. 2019;16(3):329-333. doi: 10.1177/1740774519839391 [DOI] [PubMed] [Google Scholar]
- 13.IQVIA. CRO CostPro : Training Manual User Guide. IQVIA; 2019. [Google Scholar]
- 14.US Food and Drug Administration, Department of Health and Human Services . Applications for approval to market a new drug; complete response letter; amendments to unapproved applications: final rule. Fed Regist. 2008;73(133):39588-39611. [PubMed] [Google Scholar]
- 15.US Food and Drug Administration Guidance for Industry: Considerations in Demonstrating Interchangeability With a Reference Product. Center for Drug Evaluation and Research, Food and Drug Administration; 2019:66. [Google Scholar]
- 16.Efficacy and safety study of ABP 501 compared to adalimumab in subjects with moderate to severe rheumatoid arthritis. ClinicalTrials.gov identifier: NCT01970475. Updated December 13, 2016. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01970475
- 17.Study to compare efficacy and safety of ABP 501 and adalimumab (Humira) in adults with moderate to severe plaque psoriasis. ClinicalTrials.gov identifier: NCT01970488. Updated April 3, 2019. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01970488
- 18.BI 695501 compared to adalimumab in patients with active rheumatoid arthritis. ClinicalTrials.gov identifier: NCT02137226. Updated January 19, 2018. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02137226
- 19.A study comparing SB5 to Humira in subjects with moderate to severe rheumatoid arthritis despite methotrexate therapy. ClinicalTrials.gov identifier: NCT02167139. Updated August 17, 2017. Accessed September 18, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02167139
- 20.Study to demonstrate equivalent efficacy and to compare safety of biosimilar adalimumab (GP2017) and Humira (ADACCESS). ClinicalTrials.gov identifier: NCT02016105. Updated May 30, 2017. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02016105
- 21.Study to demonstrate equivalent efficacy and to compare safety of biosimilar etanercept (GP2015) and Enbrel (EGALITY). ClinicalTrials.gov identifier: NCT01891864. Updated March 27, 2017. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01891864
- 22.A study comparing sb4 to Enbrel in subjects with moderate to severe rheumatoid arthritis despite methotrexate therapy. ClinicalTrials.gov identifier: NCT01895309. Updated August 17, 2017. Accessed July 2, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01895309
- 23.A study comparing SB2 to Remicade in subjects with moderate to severe rheumatoid arthritis despite methotrexate therapy. ClinicalTrials.gov identifier: NCT01936181. Updated September 14, 2017. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01936181
- 24.Program evaluating the autoimmune disease investigational drug cT-p13 in RA patients (PLANETRA). ClinicalTrials.gov identifier: NCT01217086. Updated March 12, 2013. Accessed August 22, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01217086
- 25.A study of PF-06438179 (Infliximab-Pfizer) and infliximab in combination with methotrexate in subjects with active rheumatoid arthritis (REFLECTIONS B537-02). ClinicalTrials.gov identifier: NCT02222493. Updated May 30, 2018. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02222493
- 26.A research study to test how filgrastim hospira works in the body of healthy study subjects when given by subcutaneous injection (shot) compared to an already US-approved drug Neupogen (Amgen). ClinicalTrials.gov identifier: NCT02766647. Updated May 10, 2016. Accessed December 11, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02766647
- 27.A research study to test how multiple doses of filgrastim hospira works in the body of healthy study subjects when given by subcutaneous injection (sc) (shot) compared to an already US-approved drug Neupogen (Amgen). ClinicalTrials.gov identifier: NCT02766634. Updated July 21, 2016. Accessed December 11, 2019. https://clinicaltrials.gov/ct2/show/NCT02766634
- 28.A study in healthy volunteers to assess immune response to multiple injections of filgrastim hospira or Neupogen reference product. ClinicalTrials.gov identifier: NCT02923791. Updated February 23, 2017. Accessed December 11, 2019. https://clinicaltrials.gov/ct2/show/NCT02923791
- 29.Phase III study comparing the efficacy and safety of ep2006 and filgrastim (PIONEER). ClinicalTrials.gov identifier: NCT01519700. Updated May 6, 2015. Accessed April 26, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01519700
- 30.Study to assess the pharmacokinetic and pharmacodynamic bioequivalence of CHS-1701 with Neulasta. ClinicalTrials.gov identifier: NCT02650973. Updated August 1, 2016. Accessed December 11, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02650973
- 31.Assessing the immunogenicity of 2 subcutaneous doses of CHS-1701 (coherus pegfilgrastim) with 2 subcutaneous doses Neulasta. ClinicalTrials.gov identifier: NCT02418104. Updated January 7, 2016. Accessed December 11, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02418104
- 32.Efficacy and safety study with MYL-1401H and Neulasta. ClinicalTrials.gov identifier: NCT02467868. Updated March 16, 2016. Accessed May 2, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02467868
- 33.A phase 3 study comparing the effects of subcutaneous epoetin hospira and epoetin alfa [Epogen] (Amgen) in patients with chronic renal failure requiring hemodialysis and receiving epoetin maintenance treatment. AiME—Anemia Management With Epoetin (AiME-13). ClinicalTrials.gov identifier: NCT01473420. Updated August 9, 2018. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01473420
- 34.A phase 3 study comparing the effects of intravenous epoetin hospira and epoetin alfa [Epogen] (Amgen) in patients with chronic renal failure requiring hemodialysis and receiving epoetin maintenance treatment. AiME-Anemia Management With Epoetin (AiME-01). ClinicalTrials.gov identifier: NCT01473407. Updated September 10, 2018. Accessed April 25, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01473407
- 35.A study to compare the effect of SB3 and Herceptin in women with HER2 positive breast cancer. ClinicalTrials.gov identifier: NCT02149524. Updated October 24, 2018. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02149524
- 36.Efficacy and safety study of ABP 980 compared with trastuzumab in women with HER2-positive early breast cancer (Lilac). ClinicalTrials.gov identifier: NCT01901146. Updated August 7, 2019. Accessed August 30, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01901146
- 37.Study of efficacy and safety of myl1401o + taxane vs Herceptin + taxane for 1st line, met. br. ca. (HERiTAge). ClinicalTrials.gov identifier: NCT02472964. Updated October 30, 2018. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02472964
- 38.Efficacy and safety evaluating study of CT-P6 in HER2 positive early breast cancer. ClinicalTrials.gov identifier: NCT02162667. Updated November 1, 2017. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02162667
- 39.A study of PF-05280014 [trastuzumab-Pfizer] or Herceptin [trastuzumab-EU] plus paclitaxel in HER2 positive first line metastatic breast cancer treatment (REFLECTIONS B327-02). ClinicalTrials.gov identifier: NCT01989676. Updated August 13, 2019. Accessed August 16, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01989676
- 40.Efficacy and safety study of ABP 215 compared with bevacizumab in patients with advanced non-small cell lung cancer. ClinicalTrials.gov identifier: NCT01966003. Updated October 19, 2017. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT01966003
- 41.A comparative study of PF-06439535 plus paclitaxel-carboplatin and bevacizumab plus paclitaxel-carboplatin patients with advanced non-squamous NSCLC. ClinicalTrials.gov identifier: NCT02364999. Updated February 7, 2019. Accessed August 30, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02364999
- 42.To demonstrate equivalence of pharmacokinetics and noninferiority of efficacy for CT-P10 in comparison with Rituxan (rituximab). ClinicalTrials.gov identifier: NCT02162771. Updated February 18, 2019. Accessed August 28, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02162771
- 43.To compare efficacy and safety between CT-P10 and Rituxan in patients with low tumour burden follicular lymphoma. ClinicalTrials.gov identifier: NCT02260804. Updated February 15, 2019. Accessed May 1, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02260804
- 44.A study of PF-05280586 (rituximab-Pfizer) or Mabthera (rituximab-EU) for the first-line treatment of patients with CD20-positive, low tumor burden, follicular lymphoma (REFLECTIONS B328-06). ClinicalTrials.gov identifier: NCT02213263. Updated June 20, 2019. Accessed August 22, 2019. https://www.clinicaltrials.gov/ct2/show/NCT02213263
- 45.US Food and Drug Administration Expedited programs for serious conditions–drugs and biologics: guidance for industry. Published September 1, 2017. Accessed June 22, 2019. https://www.fda.gov/downloads/Drugs/Guidances/UCM358301.pdf
- 46.Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005-2012. JAMA. 2014;311(4):368-377. doi: 10.1001/jama.2013.282034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.US Food and Drug Administration Non-inferiority clinical trials to establish effectiveness. Guidance for Industry. Published March 2010. Accessed March 1, 2020. https://www.fda.gov/media/78504/download
- 48.European Medicines Agency Guideline on similar biological medicinal products containing monoclonal antibodies–non-clinical and clinical issues. Accessed May 15, 2020. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf