

SUMMARY
Mechanisms of experience-dependent plasticity have been well characterized in mouse primary visual cortex (V1), including a form of potentiation driven by repeated presentations of a familiar visual sequence (“sequence plasticity”). The prefrontal anterior cingulate cortex (ACC) responds to visual stimuli, yet little is known about if and how visual experience modifies ACC circuits. We find that mouse ACC exhibits sequence plasticity, but in contrast to V1, the plasticity expresses as a change in response timing, rather than a change in response magnitude. Sequence plasticity is absent in ACC, but not V1, in a mouse model of a neurodevelopmental disorder associated with intellectual disability and autism-like features. Our results demonstrate that simple sensory stimuli can be used to reveal how experience functionally (or dysfunctionally) modifies higher-order prefrontal circuits and suggest a divergence in how ACC and V1 encode familiarity.
Graphical Abstract
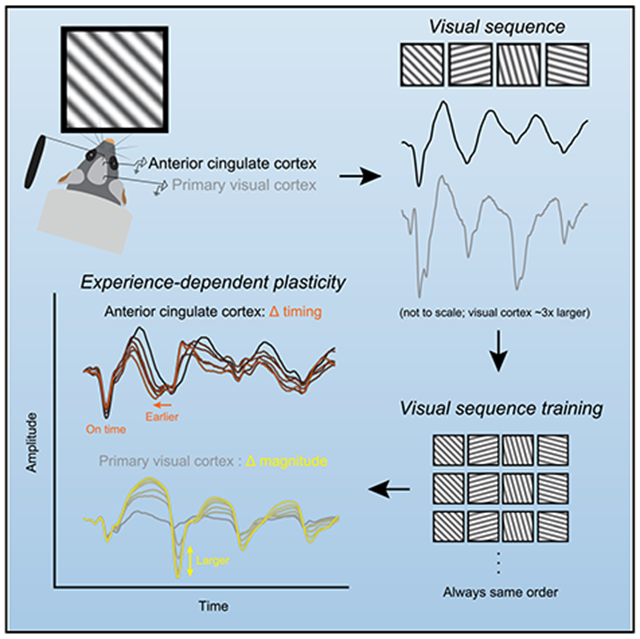
In Brief
Sidorov et al. demonstrate that patterned visual input can drive experience-dependent plasticity in the timing of neural responses in mouse anterior cingulate cortex.
INTRODUCTION
The prefrontal anterior cingulate cortex (ACC), which is associated with a range of processes including cognitive control, error detection, and pain perception (Carter et al., 1998; Hyman et al., 2017; Kerns et al., 2004; Shenhav et al., 2013; Vogt, 2005), regulates behavior through broad top-down inputs to many areas of the mouse brain (Fiser et al., 2016; Hu et al., 2019; Leinweber et al., 2017; Rajasethupathy et al., 2015; Zhang et al., 2014, 2016). The ACC is thus well positioned to modulate widely distributed networks, and plasticity within ACC is likely to have broad consequences. Rodent ACC receives direct input from visual cortex (Han et al., 2018; Oh et al., 2014; Sreenivasan et al., 2017; Wang et al., 2012; Zhang et al., 2016; Zingg et al., 2014) and is rapidly activated by simple visual stimuli (Ma et al., 2016; Mohajerani et al., 2013; Murakami et al., 2015; Sreenivasan et al., 2017). In mouse primary visual cortex (V1), controlled visual experience can drive bidirectional plasticity in the magnitude of visually evoked potentials (VEPs), with the direction of plasticity depending on the type of visual manipulation. For example, monocular deprivation can result in depression of the VEP, while repeated exposure to a familiar stimulus or familiar sequence drives potentiation of the VEP (Frenkel and Bear, 2004; Frenkel et al., 2006; Gavornik and Bear, 2014; Gordon and Stryker, 1996). Here, we tested the hypothesis that patterned visual input can also drive experience-dependent plasticity in the ACC.
First, we confirmed that ACC receives anatomical input from visual cortex, and that visual stimuli can reliably drive VEPs in the ACC in vivo. We then trained mice using visual sequences and found that the ACC encodes familiarity by modifying the timing, rather than the magnitude, of VEPs. This result suggests mechanistic divergence in how a primary sensory region (V1) and a multi-modal associational cortical region (ACC) adapt to visual experience.
Abnormal sensory processing and synaptic plasticity are common features of autism and related neurodevelopmental disorders, seen in both humans and mouse models (Bourgeron, 2015; Ebert and Greenberg, 2013; Pignatelli et al., 2013; Sidorov et al., 2013; Sinclair et al., 2017). Dysfunction in primary visual circuits has been described extensively in these models (Dölen et al., 2007; Haberl et al., 2015; Kim et al., 2016; Krishnan et al., 2015; Sato and Stryker, 2010; Wallace et al., 2012; Yashiro et al., 2009). But despite the importance of prefrontally mediated behaviors to the autism phenotype (Brumback et al., 2018; Guo et al., 2019; Lai et al., 2014; Selimbeyoglu et al., 2017; Willsey et al., 2013), substantially less is known regarding sensory processing and plasticity in prefrontal circuits. Therefore we also tested how ACC encodes and adapts to visual experience in a mouse model of Angelman syndrome, a single-gene autism-like disorder characterized by cognitive impairment, motor and speech disorders, epilepsy, and a unique “hypersocial” behavioral profile (Thibert et al., 2013). Overall, we demonstrate: (1) visual sequences can be used to drive mouse ACC, (2) patterned visual experience modifies ACC circuits, and (3) visual sequences fail to modify ACC circuits in a mouse model of a neurodevelopmental disorder.
RESULTS
ACC Receives Direct Input from Visual Cortex and Responds to Visual Stimuli
The primary objective of this study was to assess whether visual stimuli could be used to drive experience-dependent plasticity within ACC. First, we sought to confirm and expand upon prior work demonstrating that (1) ACC receives direct input from visual cortex (Gamanut et al., 2018; Han et al., 2018; Oh et al., 2014; Sreenivasan et al., 2017; Wang et al., 2012; Zhang et al., 2016; Zingg et al., 2014), and (2) ACC is visually responsive in vivo (Ma et al., 2016; Mohajerani et al., 2013; Murakami et al., 2015; Sreenivasan et al., 2017). We injected the retrograde tracer cholera toxin B (CTB594; Conte et al., 2009) into ACC Figure 1A) to visualize sources of direct, monosynaptic input. Cells in both V1 and medial secondary visual cortex V2 (V2M) were labeled with CTB (Figures 1B and S1A). V2M showed more labeling than V1 (Figure 1C; t(3) = 5.976, p = 0.0094, paired t test), confirming a largerinput projection to ACC from V2M than from V1 (Wang et al., 2012). As expected, we also detected CTB in mediodorsal (MD) and anteromedial (AM) thalamus, two well-established sources of non-visual input to ACC (Delevich et al., 2015; Divac et al., 1993; Shibata and Kato, 1993; Figure S1B). Anterograde labeling of V2M and V1 projections with AAV5-CaMKII-GFP confirmed that both regions target ACC with direct, monosynaptic projections (Figures 1D and 1E). While substantial V2M projections targeted most of ACC, the relatively sparse V1 projections appeared to be spatially restricted to more dorsal and caudal regions of ACC (Figures S1C and S1D). We confirmed the functional relevance of visual inputs to ACC by pairing optogenetic stimulation of V2M and V1 axons with whole-cell recordings from ACC neurons in ex vivo slices. Here, we focused on ventral ACC, as this was the site of most experiments in vivo (Figures 2, 3, 4, and 5). Optogenetic activation of V2M axons was sufficient to drive synaptic activity in both excitatory and inhibitory neurons in ventral ACC (Figures 1F and S1E). Optogenetic activation of V1 axons drove synaptic activity in dorsal ACC but did not effectively drive ventral ACC (Figures S1F and S1G).
Figure 1. ACC Receives Direct Input from Primary and Secondary Visual Cortex.
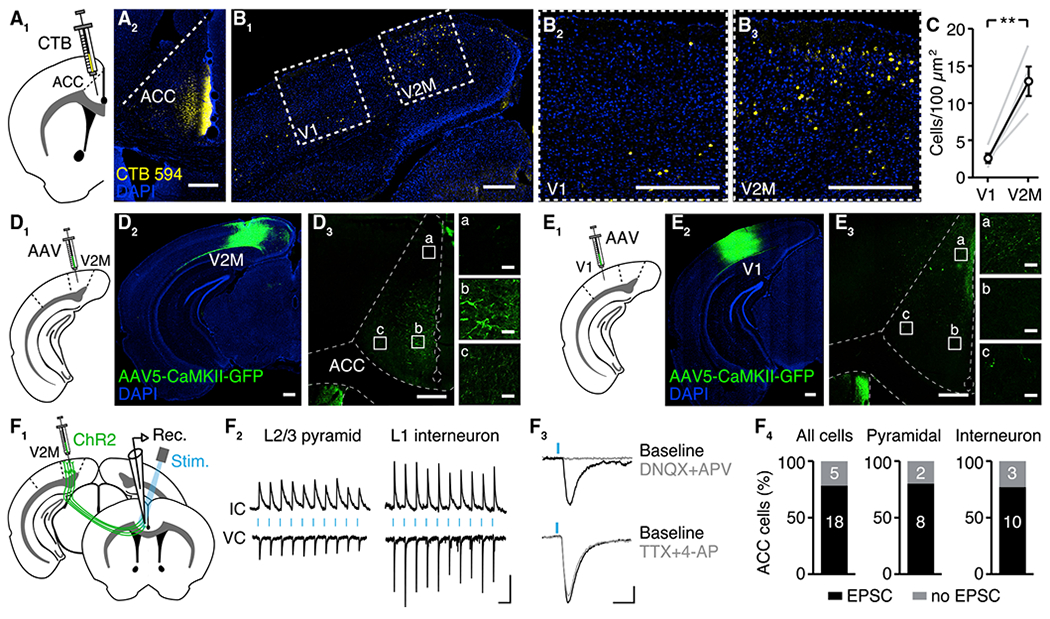
(A) Retrograde labeling of projections to ACC. (A1) Schematic. (A2) Example injection site of retrograde-label CTB594 into ACC. Scale bar: 300 μm.
(B) Visualizing input sources to ACC. (B1) Wide view of visual cortex. (B2) Closeup view of V1. (B3) Closeup view of V2M. Scale bars: 300 μm, except (B2) and (B3) insets, 30 μm.
(C) The number of cell bodies expressing CTB594 is greater in V2M than in V1 (n = 4 mice, **p < 0.01).
(D) Anterograde labeling of V2M projections to ACC. (D1) Schematic. (D2) Example of injection site of anterograde-label AAV5-CaMKII-GFP into V2M. (D3) Axonal projections to ACC (A/P +0.6 mm). Scale bars: 300 μm, except (D3) inset, 30 μm.
(E) Anterograde labeling of V1 projections to ACC. (E1) Schematic. (E2) Example of injection site of anterograde label into V1. (E3) Axonal projections to ACC (A/P +0.6 mm). Scale bars: 300 μm, except (E3) inset, 30 μm.
(F) Optogenetic activation of V2M axons drives ventral ACC neurons in slice. (F1) Schematic. (F2) Example of ACC neurons. (F3) Confirmation that opto-EPSCs are synaptic in nature. (F4) In the majority of ACC neurons, optogenetic stimulation of V2M axons drove EPSCs. Scale bars: 100 ms, 30 pA/1 mV. IC, current clamp; VC, voltage clamp.
Error bars represent SEM.
See also Figure S1.
Figure 2. Visual Sequences Drive Activity in ACC.
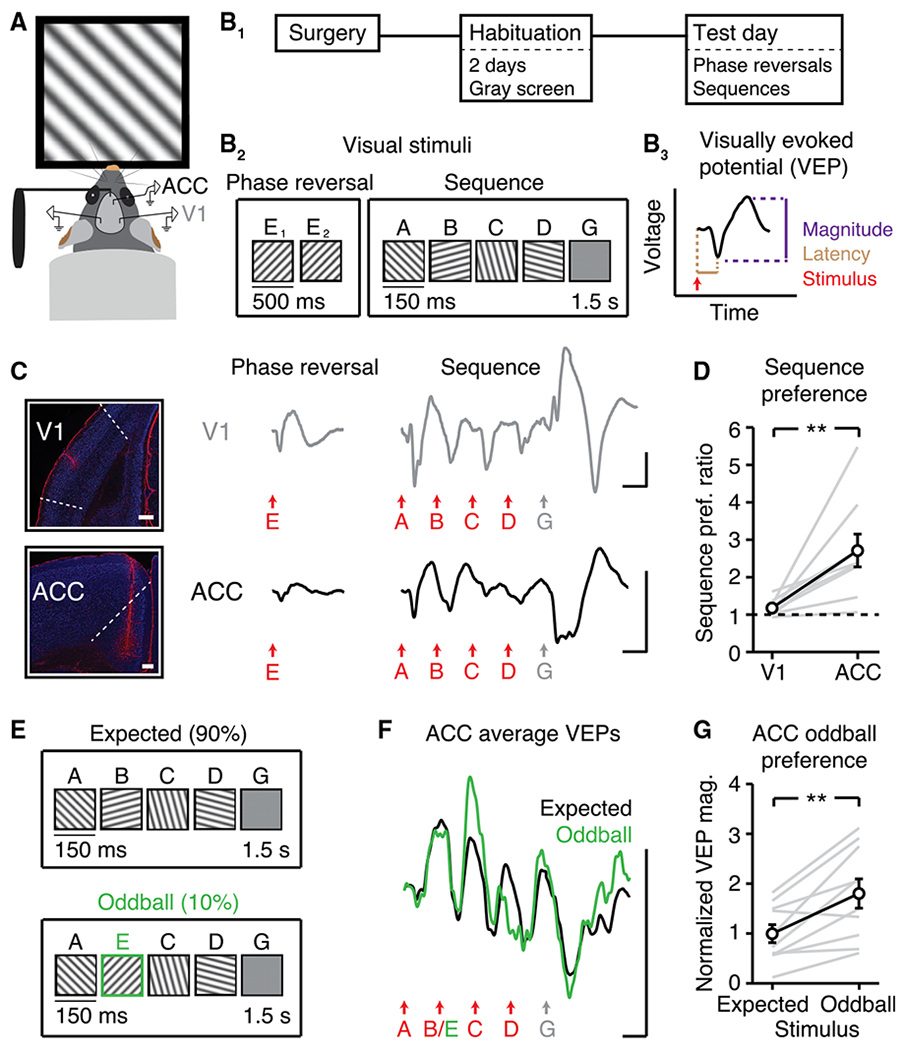
(A) Head-fixed mice viewed visual stimuli.
(B) (B1) Recordings followed two days of habituation. (B2) On test day, mice viewed phase reversals and sequences in the same recording session (n = 9). Letters “A” to “E” represent distinct orientations; “G” represents gray screen. (B3) Schematic shows how VEP magnitude and latency were quantified.
(C) V1 VEPs (top) and ACC VEPs (bottom) driven by phase reversals and sequences. Images in left panel show example electrode locations (red, GFAP; blue, NeuN; scale bars: 300 μm).
(D) Within animals, ACC showed an increased sequence preference relative to V1. Sequence preference ratio is defined, within animal, as (sequence VEP magnitude) / (phase reversal VEP magnitude). Sequence VEP magnitude is defined as (“A” VEP + “B” VEP + “C” VEP + “D” VEP) / 4.
(E) A new cohort (n = 10) viewed 500 sequences: 50 AECD (“oddball”) interleaved every 10th sequence within 450 ABCD (“expected”).
(F) ACC VEPs during expected and oddball sequences.
(G) ACC responded more strongly to oddball stimulus “E” than expected stimulus “B.”
All traces represent group average VEPs. Scale bars for all traces: 50 μV, 100 ms. **p < 0.01. Error bars represent SEM.
See also Figure S2.
Figure 3. Patterned Visual Experience Drives Sequence-Specific Plasticity in ACC.
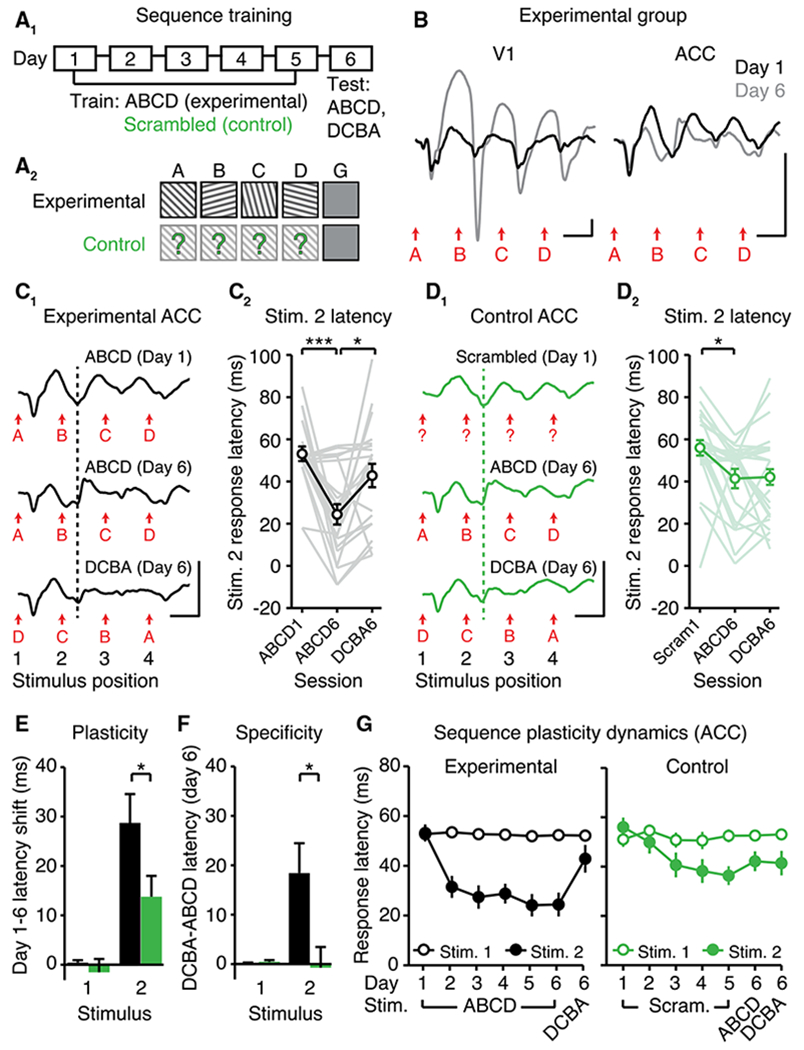
(A1) Sequence training over six days using either (A2) familiar (experimental, n = 21) or pseudorandomized sequences (control, n = 24).
(B) VEPs illustrate experience-dependent plasticity in VEP magnitude in V1 (left) and in VEP latency in ACC (right).
(C) Experience-dependent plasticity in the latency to stimulus 2 (B position) in experimental mice (black). (C1) VEPs. (C2) Quantification.
(D1 and D2) In control mice (green), residual sequence plasticity was non-specific. Dotted lines are aligned to the peak negativity driven by the second stimulus (“B” or random) on day 1.
(E) “Plasticity” refers to the within-animal latency shift between days 1 and 6 in experimental (black) and control (green) mice.
(F) “Specificity” refers to the within-animal difference in response latency (ABCD versus DCBA) on day 6.
(G) Response latencies to stimuli 1 (A position) and 2 (B position) across training.
All traces represent group average VEPs. Scale bars: 50 μV, 100 ms. *p < 0.05; **p < 0.01; ***p < 0.0001. Error bars represent SEM.
See also Figure S3.
Figure 4. Chemogenetic Inhibition of ACC during Training Does Not Affect Sequence Plasticity in V1.
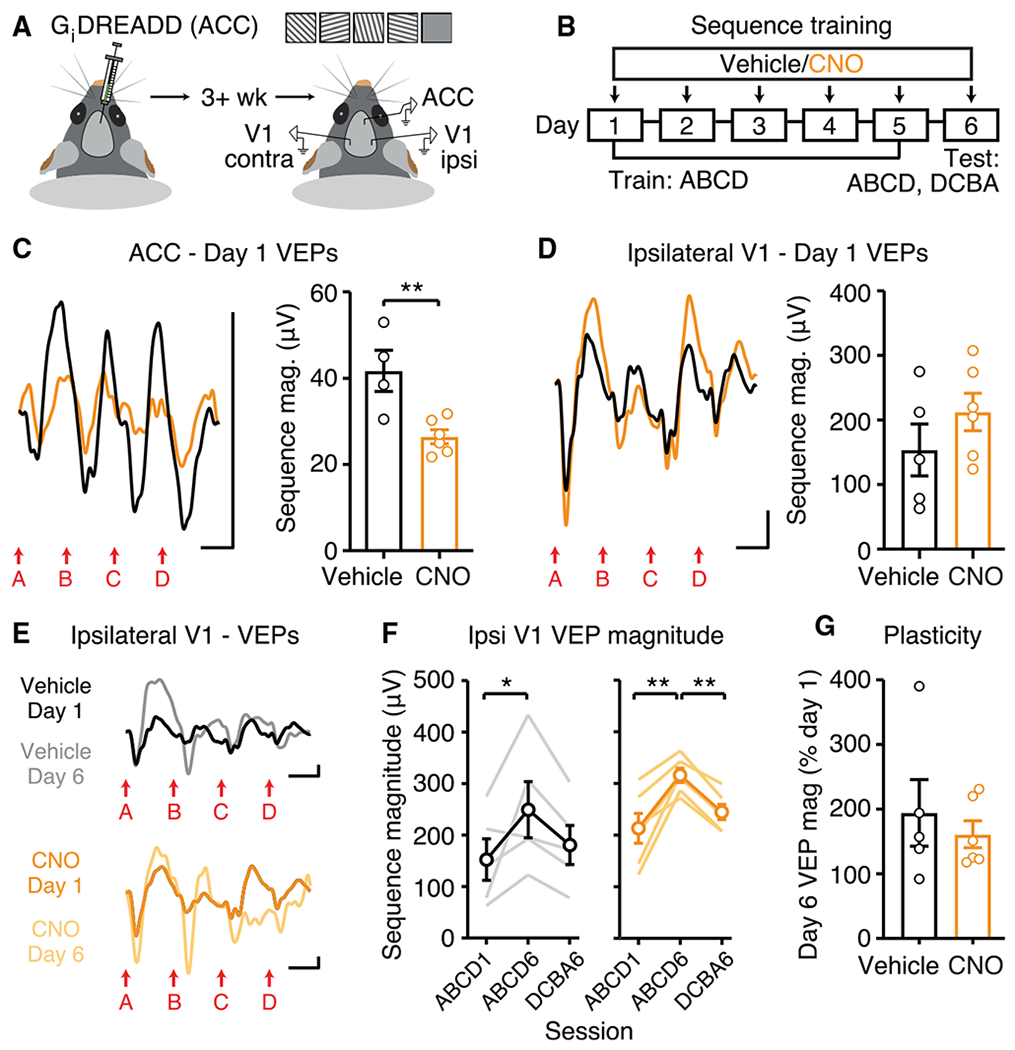
(A) Injection of inhibitory DREADD into ACC 3+ weeks prior to recording in ACC and bilateral V1.
(B) Daily administration of 5 mg/kg CNO or saline vehicle 1 h prior to recordings.
(C) Day 1: ACC VEPs are smaller in CNO-treated group (Vehicle: n = 4, CNO: n = 6).
(D) Ipsilateral V1 VEPs are normal in CNO-treated group (Vehicle: n = 5, CNO: n = 6).
(E) VEPs in V1 ipsilateral to chemogenetic ACC inhibition.
(F and G) Within-group comparisons (F) and across-group comparisons (G) show normal sequence plasticity in V1 ipsilateral to chemogenetic ACC inhibition.
All traces represent group average VEPs. Scale bars: 50 μV, 100 ms. *p < 0.05; **p < 0.01. Error bars represent SEM.
See also Figure S4.
Figure 5. Sequence Plasticity Is Absent in ACC, but Normal in V1, in Angelman Syndrome (AS) Model Mice.
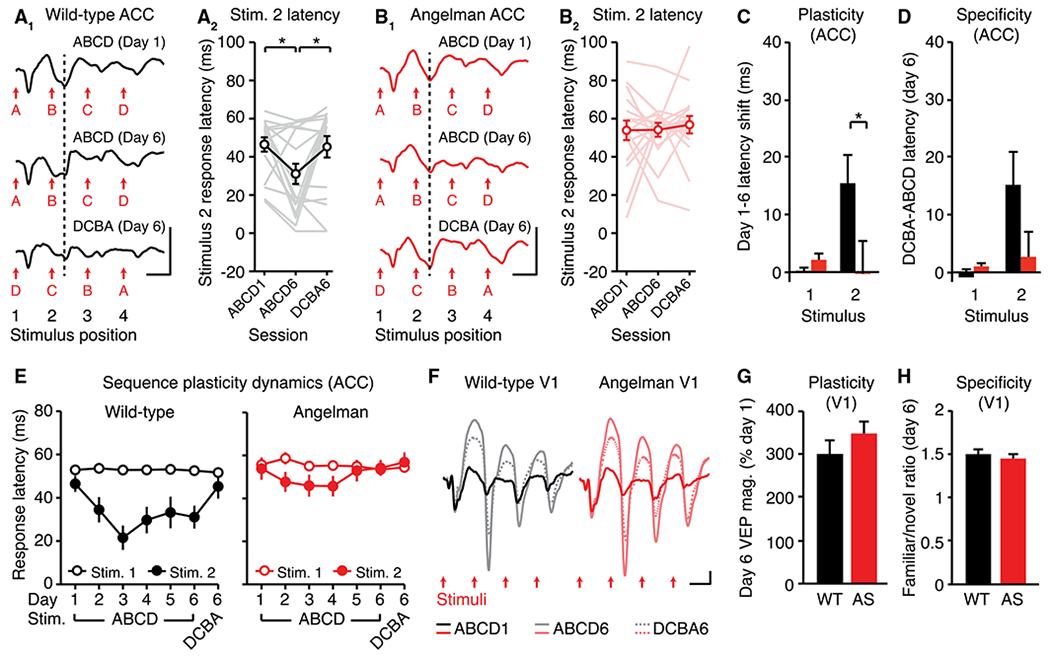
(A) Expression of sequence plasticity in wild-type (WT) ACC. (A1) VEPs. (A2) Quantification of the latency to stimulus 2 (n = 17).
(B) Sequence plasticity is absent in AS mouse ACC. (B1) VEPs. (B2) Quantification of the latency to stimulus 2 (n = 17).
(C and D) Amount of plasticity (C) and specificity of plasticity (D) to the trained sequence in ACC.
(E) Response latencies to stimuli 1 and 2 across training.
(F to H) VEPs (F) in V1 on day 1 and day 6 of sequence training illustrate normal (G) sequence plasticity and (H) specificity in V1 (WT: n = 18, AS: n = 19).
All traces represent group average VEPs. Scale bars: 50 μV, 100 ms. *p < 0.05. Error bars represent SEM.
See also Figure S5.
Next, we recorded local field potentialsin ACC and V1 as awake, head-fixed mice viewed full-field visual stimuli in an otherwise dark, quiet environment (Figure 2A). We confirmed that full-field oriented gratings and full-field changes in luminance (black to gray) were sufficient to drive activity in ACC (Figures S2A and S2B; Ma et al., 2016; Mohajerani et al., 2013; Murakami et al., 2015; Sreenivasan et al., 2017). Overall, these results confirmed that direct projections from visual cortex can activate ACC microcircuits and that simple visual stimuli can evoke neural activity locally in ACC.
ACC Encodes Visual Sequences In Vivo
We next sought to expand the scope of visual stimulation and identify visual stimuli that could be used to drive experience-dependent plasticity in ACC. Mice viewed two patterns of visual stimulation in the same recording session: (1) phase reversals, and (2) sequences of sinusoidal gratings (Figures 2B1 and 2B2). Sequences consisted of four distinct stimuli (“ABCD,” letters represent orientations), separated by a gray screen. We tested these stimulation protocols because they have both been previously used to drive plasticity in V1 when shown consecutively over days (Cooke et al., 2015; Frenkel et al., 2006; Gavornik and Bear, 2014). We quantified VEPs (Porciatti et al., 1999; Figure 2B3) by averaging field potentials locked to stimuli across 200 trials. As expected, V1 was reliably driven by both phase reversals and sequences (Figures 2C, top, and S2C1). Relative to V1, ACC responded more strongly to sequences (Figures 2C, bottom, and S2C2): within mice, the ratio of sequence VEP magnitude to phase reversal VEP magnitude (“sequence preference ratio”) was greater in ACC than in V1 (Figure 2D; t(8) = 3.608, p = 0.0069, paired t test). Visual sequences drove VEPs in V1 and ACC in all experimental mice (Figure S2C3).
The ability of ACC to respond to visual sequences was verified at the level of single neuron responses. Single unit activity within ACC could be driven by sequences (Figure S2D), confirming that the population-level VEP, which largely reflects synaptic currents, is representative of underlying activity. In addition to oriented gratings, ACC was also activated by sequences of white objects on a black background (Bussey et al., 2008; Figure S2E). However, when mice were anesthetized with ketamine/xylazine, visual sequence encoding was disrupted in ACC. Under anesthesia, ACC VEPs were no longer reliably time-locked to stimuli, despite reliable VEPs in V1 (Figure S2F).
The difference in responsiveness to sequences and phase reversals (Figure 2D) suggested that not all visual stimuli are equally effective in driving ACC. We hypothesized that this differential responsiveness is not caused by differences in basic parameters of the stimulus (e.g., grating “E” and grating “A” are identical except for their orientation), but rather by the context of when and how stimuli are presented. To directly test the importance of context, we used an oddball sequence paradigm in a new cohort. Mice viewed expected stimulus “B” on 90 percent of trials and oddball stimulus “E” on 10 percent of trials (Figure 2E). Oddball stimuli drove larger VEPs than expected stimuli in ACC (Figures 2F and 2G; t(9) = 4.121, p = 0.0026, paired t test). Overall, visual sequences evoked context-dependent activity in ACC in awake mice that was reliably resolved at the level of population-level VEPs. Visual sequences may thus be presented across multiple sessions to assess whether familiar sequences can drive plasticity in ACC circuits, as they do in V1 (Gavornik and Bear, 2014).
ACC Expresses Sequence Plasticity Using Timing
Exposure to a sequence of visual stimuli over days drives plasticity in the magnitude of V1 VEPs (Gavornik and Bear, 2014). To test if ACC is capable of undergoing sequence plasticity, we implanted electrodes into both ACC and V1. We then trained mice using either sequence ABCD (“experimental”) or pseudorandomly scrambled arrangements of stimuli A, B, C, and D that varied every trial (“control”; Figure 3A). After five days of training, on test day (day 6) we presented both sequence ABCD and sequence DCBA. Thus, on test day, ABCD was familiar and DCBA was novel to experimental mice, whereas ABCD and DCBA were functionally equivalent to control mice (because neither sequence had been displayed at a level higher than chance during training). This experimental design allowed us to answer two related questions: (1) does plasticity occur in ACC to a trained sequence, and if so, (2) is plasticity specific to the trained sequence?
In the experimental group, sequence plasticity in V1 was expressed by a potentiation of VEP magnitude, as expected (Figures 3B, S3A, and S2B). There was no change in VEP magnitude in ACC as a result of training (Figures S3C and S3D). However, sequence training drove plasticity in the timing, rather than the magnitude, of ACC VEPs (Figures 3B and 3C). We quantified timing by measuring the latency to peak VEP negativity (“response latency”) following stimuli (Figure 2B3). In the experimental group, ACC responded faster on day 6 than on day 1 to stimulus B (Figure 3C; RM ANOVA: main effect of time: F(2,40) = 11.6, p < 0.0001, post hoc ABCD1 versus ABCD6: p < 0.0001). There was no change in the response latency to stimulus A (Figure S3E). We hypothesize that the plasticity in the latency to stimulus B but not A reflects the learned expectation that stimulus B follows A. Within animals, there was not a statistically significant correlation between plasticity in ACC and in V1 (Figure S3F). Moving forward, we use the shorthand phrase “ACC sequence plasticity” to refer to the leftward shift in VEP latency to expected stimulus B, and “V1 sequence plasticity” to refer to the potentiation of VEP magnitude driven by sequence ABCD.
A key feature of V1 sequence plasticity is that it is largely specific to the trained sequence (i.e., does not generalize to novel sequence DCBA; Gavornik and Bear, 2014; Figure S3B). We tested whether ACC sequence plasticity was sequence specific using two approaches: (1) by comparing ABCD and DCBA responses on test day in the experimental group, and (2) by comparing plasticity between experimental and control groups. In the experimental group, ACC sequence plasticity was specific to trained sequence ABCD (Figure 3C; post hoc ABCD6 versus DCBA6: p = 0.0123, post hoc ABCD1 versus DCBA6: p = 0.2860). In the control group, there was a statistically significant latency shift to the second stimulus (the random element in the B position; Figure 3D; RM ANOVA: main effect of time: F(2,46) = 6.489, p = 0.0033, post hoc Scram1 versus ABCD6: p = 0.0119). This finding suggests that a component of ACC sequence plasticity can be non-specific, i.e., plasticity reflecting the expectation that a stimulus would occur after the first stimulus, without being sensitive to which stimulus occurred. To directly assess the amount of sequence-specific plasticity versus non-specific plasticity within animals, we compared (ABCD1 to ABCD6) and (ABCD1 to DCBA6) for stimulus 2 (B position) in the experimental group. The amount of sequence-specific plasticity was greater than the amount of non-specific plasticity (Figure S3G; t(20) = 3.055, p = 0.0062, paired t test).
We next directly compared sequence plasticity between experimental and control groups. We assessed both the within-animal latency shift across days (Figure 3E, “plasticity” = day-1 latency minus day-6 latency) and the degree to which this shift was sequence specific (Figure 3F, “specificity” = day-6 DCBA latency minus day-6 ABCD latency). We quantified plasticity and specificity for both the first and second stimuli (A and B positions). There was no plasticity to stimulus 1 in either group and no difference between experimental and control groups (Figure 3E; t(43) = 0.6661, p = 0.5089, Student’s t test). Plasticity to stimulus 2 was greater in the experimental group than the control group (Figure 3E; t(43) = 2.099, p = 0.0417). Likewise, there was no specificity to stimulus 1 in either group and no difference between experimental and control groups (Figure 3F; t(43) = 1.064, p = 0.2934). Specificity to stimulus 2 was greater in the experimental group than the control group (Figure 3F; t(43) = 2.652, p = 0.0112). Overall, these results suggest that a substantial proportion of sequence plasticity in ACC is due to experiencing a familiar sequence of stimuli in a specific order, despite a lesser contribution of non-specific plasticity.
ACC sequence plasticity was evident as soon as day 2 of training in the experimental group (Figure 3G). Therefore, we asked whether ACC sequence plasticity occurs within a single session by comparing VEPs across four 50-trial blocks. Sequence plasticity in ACC emerged between sessions, but not within sessions (Figure S3H), suggesting that time is required for consolidation to occur. Expression of ACC sequence plasticity was maintained when a subset of experimental animals was re-tested one week following training (Figures S3I1 and S3I2). However, ketamine/xylazine anesthesia impaired the expression of plasticity at re-test, as proper encoding of stimuli was lost in ACC (Figure S3I3). In V1, sequence plasticity was also maintained at re-test and expression of plasticity was also impaired at re-test under ketamine/xylazine anesthesia (Figures S3I4 and S3I5).
Anatomical tracing (Figure 1) suggested that V2M may send denser projections to ventral ACC and V1 may send denser projections to dorsal ACC. We therefore asked whether dorsal ACC could (1) be driven by visual sequences, and (2) express sequence plasticity. VEPs in dorsal ACC were of a similar magnitude to ventral ACC and sequence plasticity was expressed in dorsal ACC (Figure S3J). These results suggest that potential variations in anatomical input do not grossly affect the expression of sequence plasticity locally within ACC. As arousal and locomotion can modulate visual processing and affect visual plasticity in V1 (Fu et al., 2014; Kaneko and Stryker, 2014; Niell and Stryker, 2010; Vinck et al., 2015), we also tested whether sequence plasticity could be expressed when mice are head fixed on a running wheel, rather than enclosed in a tube. Sequence plasticity was expressed normally in ACC under these freely moving conditions (Figure S3K).
Chemogenetic Inhibition of ACC Does Not Impair Sequence Plasticity in V1
In addition to receiving inputs from visual cortex, ACC sends anatomical projections to V1. These top-down projections can modulate visual encoding and visually guided behavior (Leinweber et al., 2017; Zhang et al., 2014). Thus, we asked whether inhibition of ACC during sequence presentations would affect plasticity in downstream V1 (Figures 4A and 4B). Inhibition of ACC with Gi DREADD + clozapine N-oxide (CNO) resulted in a decrease in day-1 VEP magnitude in ACC, relative to Gi DREADD + vehicle (Figure 4C; t(8) = 3.565, p = 0.0073). This result confirms the effectiveness of a chemogenetic approach to inhibit ACC in vivo. Inhibition of ACC also appears to impair sequence plasticity locally within ACC (Figures S4A–S4D), although quantification was difficult due to the decreased baseline VEP. However, inhibition of ACC did not affect day-1 VEPs (Figure 4D; t(9) = 1.216, p = 0.2550) or sequence plasticity (Figures 4E–4G) in V1 ipsilateral to the DREADD injection site. In ipsilateral V1, sequence plasticity was expressed in both vehicle (Figure 4F; RM ANOVA: main effect of time: F(2,8) = 4.801, p = 0.0427, post hoc ABCD1 versus ABCD6: p = 0.0399) and CNO-treated (Figure 4F; main effect of time: F(2,10) = 17.18, p = 0.0006, post hoc ABCD1 versus ABCD6: p = 0.0005, post hoc ABCD6 versus DCBA6: p = 0.0068) groups, and there was no difference in the amount of plasticity between groups (Figure 4G; t(9) = 1.216, p = 0.2550). As expected, inhibition of ACC did not affect sequence plasticity in V1 contralateral to the injection site (Figures S4E–S4G). There was no correlation between the magnitude of day-1 VEPs in ACC and subsequent plasticity in ipsilateral V1 in the Gi DREADD + CNO group (Figure S4H). Overall, these results suggest that sequence plasticity can persist in V1 despite partial inhibition of ACC feedback.
Impaired ACC Sequence Plasticity and Normal V1 Sequence Plasticity in a Mouse Model of Neurodevelopmental Disease
Impaired experience-dependent plasticity has been described in primary sensory circuits in multiple mouse models of neurodevelopmental disorders, such as Fragile X, Rett, and Angelman syndromes (Dölen et al., 2007; Harlow et al., 2010; Kim et al., 2016; Krishnan et al., 2015; Noutel et al., 2011; Yashiro et al., 2009). Sequence plasticity represents an experimental paradigm by which experience-dependent adaptations can be measured in parallel in primary sensory and prefrontal circuits. We assessed sequence plasticity in ACC and V1 in a mouse that genetically models Angelman syndrome (Ube3am−/p+; Jiang et al., 1998). ACC sequence plasticity, defined as the latency shift to expected stimulus B, was observed in wild-type (WT) littermate controls (Figure 5A, RM ANOVA, main effect of time: F(2,32) = 5.028, p = 0.0126, post hoc ABCD1 versus ABCD6: p = 0.0226, post hoc ABCD6 versus DCBA6: p = 0.0396), but not in Angelman syndrome model mice (Figure 5B, no main effect of time: F(2,32) = 0.1798, p = 0.8363). A direct comparison of plasticity between groups confirmed impaired plasticity to stimulus B in Angelman mice (Figure 5C; t(32) = 2.061, p = 0.0475). A direct comparison of specificity did not reveal a statistically significant difference between WT and Angelman mice (Figure 5D; t(32) = 1.620, p = 0.1150). Sequence plasticity in ACC followed the expected time course in WT mice but not Angelman-model mice (Figure 5E). Long-range projections from visual cortex to ACC did not appear to be grossly different between groups (Figures S5A–S5D).
In the same experimental cohort, sequence plasticity was normal in V1 of Angelman-model mice (Figures 5F and S5E). Both the amount of plasticity from day 1 to day 6 (Figure 5G, t(35) = 0.9142, p = 0.3668) and the specificity of plasticity to the trained sequence on day 6 (Figure 5H; t(35) = 0.782, p = 0.4395) were not different between groups. In addition, stimulus-specific response potentiation, driven by phase reversals of a single familiar orientation (Frenkel et al., 2006), was normal in V1 of Angelman-model mice (Figures S5F–S5J). There was no group difference in day-1 sequence VEP magnitude, either in V1 (Figure S5K) or in ACC (Figure S5L), suggesting that sequence encoding is normal in Angelman-model mice. ACC oddball responses in Angelman-model mice were similar to those in WT controls (Figure S5M–S5O). Overall, we conclude that sequence plasticity is normal in V1, but impaired in ACC, in Angelman-model mice. Impairments are likely specific to plasticity, as sequence encoding and oddball responses are normal in ACC of Angelman-model mice.
DISCUSSION
Rodent ACC receives input from the visual cortex (Gamanut et al., 2018; Han et al., 2018; Oh et al., 2014; Sreenivasan et al., 2017; Wang et al., 2012; Zhang et al., 2016; Zingg et al., 2014; Figure 1), and ACC is visually responsive to simple stimuli such as light flashes (Ma et al., 2016; Mohajerani et al., 2013; Sreenivasan et al., 2017) and drifting gratings (Murakami et al., 2015; Figure S2). It is not yet known whether visual experience modifies ACC circuits, and if so, whether mechanisms of experience-dependent plasticity are conserved between ACC and V1. This study shows that visual sequences drive activity in ACC, and that repeated exposure to a familiar sequence drives plasticity in the timing of ACC responses.
Sequences of visual stimuli were sufficient to drive activity in ACC that was detectable at the population level using VEPs in awake animals. ACC showed preferential tuning to sequences over phase reversals (Figure 2D), and responded more strongly to unexpected visual oddballs than to expected stimuli within a sequence (Figure 2G). Together, these data suggest that ACC is sensitive to the context in which a stimulus is presented. The same stimulus may drive a greater response when it is not expected (i.e., oddball) or when it follows a stimulus of a different orientation (i.e., sequences). The increased response to oddball stimuli aligns with evidence that rodent ACC may encode error signals and is conceptually similar to the error-related negativity observed in human ACC (Alexander and Brown, 2019; Gehring et al., 1993; Gehring et al., 1995; Hyman et al., 2017).
The ability of ACC to encode visual sequences enabled us to assess whether repeated exposure to a familiar sequence drives plasticity within ACC, and if so, whether mechanisms of sequence plasticity are similar between ACC and V1. We found that in ACC, experience drives plasticity in VEP timing (Figure 3) but not VEP magnitude, as in V1 (Gavornik and Bear, 2014; Figure S3). Other aspects of plasticity were similar between ACC and V1, such as a requirement for consolidation between sessions and the degree of specificity to the trained sequence (Frenkel et al., 2006). Experience-dependent changes in ACC VEP timing were not uniform across stimuli. Plasticity occurred in the response latency to the second stimulus in a sequence, which always follows the first stimulus, but not in the latency to the first stimulus, which always follows a long-duration gray screen. This result suggests that ACC sequence plasticity may reflect a prediction, or learned expectation, that the second stimulus follows the first. Plasticity to the third and fourth responses in a sequence was more difficult to assess because these VEPs were less consistent and were not distinguishable from noise in some mice (Figure S2C). ACC VEPs were unreliable under anesthesia, but ACC sequence plasticity occurred in awake animals regardless of whether animals were head-fixed in a tube or on a running wheel. Together, these results suggest that some level of arousal/wakefulness is required for proper sequence encoding, but that this type of plasticity does not appear to be further enhanced by motor activity.
ACC provides top-down input to many brain areas, including the hippocampus and V1, and direct modulation of these projections affects sensory processing and behavior in vivo (Fiser et al., 2016; Hu et al., 2019; Rajasethupathy et al., 2015; Zhang et al., 2014, 2016). For example, a recent study by Fiser et al. (2016) found that as mice are trained to expect ordered stimuli in a virtual maze, a subset of projections from ACC to V1 increase their activity in anticipation of an expected stimulus. Follow-up work confirmed that top-down projections to V1 from ACC and adjacent M2 provide predictive information regarding visual flow (Leinweber et al., 2017). Overall, this body of work suggests that ACC provides V1 with predictive information. We hypothesize that experience-dependent plasticity within ACC may be required for ACC to export predictive feedback information to V1. However, local chemogenetic inhibition of ACC during sequence training did not affect sequence plasticity downstream in V1 (Figure 4). This result, plus the lack of correlation between plasticity in ACC and plasticity in V1 within animals (Figure S3), suggests that proposed predictive feedback from ACC may not be necessary for V1 to encode stimulus-specific potentiation. However, it is possible that the lack of effect on V1 plasticity may be due in part to incomplete inhibition of ACC. The use of inhibitory DREADDs consistently achieved an ~40% reduction in the magnitude of ACC VEPs (Figure 4C) in the area with viral expression. However, it is likely that we targeted only a subset of total projections from ACC to V1. Thus, less than a 40% effect might be expected in downstream V1. But even within animals, the amount of ACC inhibition achieved chemogenetically was not correlated with the amount of sequence plasticity in ipsilateral V1 (Figure S4). Overall, we conclude that partial inhibition of ACC does not have a measurable effect on sequence plasticity in downstream V1.
We measured experience-dependent plasticity locally within ACC, but it is possible that such plasticity (as well as enhanced oddball responses) could be inherited from visual cortex. Using a similar oddball paradigm, Homann et al. (2017) found that novel elements in a visual sequence selectively drive activity in V1. This and other work suggest that V1 itself is capable of detecting familiarity (Cooke and Bear, 2015; Cooke et al., 2015; Frenkel et al., 2006; Gavornik and Bear, 2014) and detecting when visual input violates expectations (Keller et al., 2012; Zmarz and Keller, 2016). However, we hypothesize that sequence plasticity is likely local to ACC, and not inherited, forthe following reasons: (1) chemogenetic inhibition of ACC during induction impairs VEPs and plasticity locally (Figure 4); (2) mechanisms of plasticity differ between V1 and ACC (Figures 3 and S3); (3) within animals, sequence plasticity in V1 and in ACC are not correlated (Figure S3); and (4) evidence from Angelman-model mice suggests that ACC plasticity deficits can emerge despite normal V1 plasticity (Figure 5). However, we have not tested and do not rule out the possibility that plasticity to sequences may also occur locally within V2M.
Retrograde and anterograde labeling confirmed prior evidence that visual cortex projects directly to ACC (Figure 1). We found that both V1 and V2M project to ACC, but that these regions may project to different anatomical targets within ACC (Figure S1). However, these potential differences are unlikely to have direct consequences for sequence plasticity within ACC, as both dorsal and ventral ACC expressed sequence plasticity (Figures 3 and S3). In humans and in rodents, the ACC is not uniform and exhibits anatomical variability in protein expression, projection targets, and behavioral function (Ash et al., 2014; Devinsky et al., 1995; Milad et al., 2007; Vogt, 2005). Therefore, it is possible that V1 and V2M projections—through variability in the microcircuits they innervate—may have different roles in encoding, plasticity, and downstream behavior. Further work will be needed to dissect the relative contribution of these long-range projections to how ACC microcircuits process visual input. It is also possible that visual information is relayed to ACC in part through alternative pathways, perhaps via non-laternal geniculate thalamic nuclei. And beyond sequence plasticity, future work will be required to better understand: (1) if and how visual cortical inputs are spatially segregated within ACC, and (2) whether visual inputs activate ACC microcircuits in a similar way that thalamic inputs activate these same circuits (Cruikshank et al., 2012; Delevich et al., 2015).
In Angelman syndrome model mice, we report normal sequence encoding in both V1 and ACC, normal oddball responses in ACC, and normal sequence plasticity in V1. However, sequence plasticity was absent in ACC in Angelman mice (Figure 5). Broadly, this suggests a dissociation between normal plasticity in “low-level” circuits and impaired plasticity in “high-level” circuits in a mouse model of an autism-like neurodevelopmental disorder. Dysregulated plasticity in primary sensory circuits is a common feature of mouse models of neurodevelopmental disorders, but disruption of primary sensory circuits alone is unlikely to drive many of the core features of these disorders. Recent studies have identified other prefrontal impairments at the circuit and behavioral levels in mouse models of Angelman syndrome and other phenotypically similar single-gene neurodevelopmental disorders (Guo et al., 2019; Kalmbach et al., 2015; Kramvis et al., 2013; Krueger et al., 2011; Rotaru et al., 2018; Sceniak et al., 2016; Sidorov et al., 2018; Siegel et al., 2017). Future work will be needed to link sequence plasticity to specific behavioral impairments in the Angelman mouse model, and to determine whether impaired plasticity generalizes to other disorders. We hypothesize that our experimental paradigm represents a timescale that would have relevance for behavior: the use of sequential 150-ms stimuli in a head-fixed mouse would be equivalent to a running mouse (~6 mph) encountering a new stimulus every 1.3 feet.
Impairments in ACC sequence plasticity have a direct correlate in a psychophysical/EEG study of individuals with autism, who showed impaired adaptation of fronto-central event-related potentials (ERPs) during a visual sequence recognition task (Thillay et al., 2016). In this task, humans were trained to expect a target stimulus following a consistent sequence of three predictive stimuli. In a control group, the latency to the N2 component of the ERP shifted earlier to reflect expectation. However, this latency shift was absent in individuals with a diagnosis of autism spectrum disorder. Imaging and functional connectivity studies have also found group differences within ACC, and between ACC and other areas, in individuals with autism (Assaf et al., 2010; Jann et al., 2015; Kennedy and Courchesne, 2008; Minshew and Keller, 2010; Zhou et al., 2016). It is clear that the importance of prefrontal circuits in higher-order cognitive processes makes study of plasticity in these regions critical. Toward this goal we provide a straightforward sequence paradigm to rapidly assess encoding and plasticity in V1 and ACC.
A limit of this study is the use of VEPs to quantify neural responses. VEPs are, by definition, a population-level response of synaptic currents derived from the local field potential in response to a visual stimulus. VEPs are a commonly used and powerful tool for assessing visual responsiveness in cortex (Porciatti et al., 1999), and VEP results have typically aligned with unit recordings in measuring experience-dependent plasticity in V1 (Aton et al., 2014; Cooke et al., 2015; Gavornik and Bear, 2014). However, VEPs do not provide information about how individual neurons encode visual information or adapt to experience. Future studies with unit recordings and/or large-scale calcium imaging would provide a clearer picture of how individual neurons in ACC respond to visual sequence training. These approaches will also enable future categorization of neurons by location, cell type, and firing properties. However, one of the advantages of VEPs and unit recordings, relative to calcium imaging, is that they have faster kinetics and are better suited to measure small changes in timing (Chen et al., 2013; Kim et al., 2020).
Overall, our work demonstrates that visual sequences may be used to drive experience-dependent plasticity in vivo in ACC circuits. Sequence plasticity is expressed in ACC by a change in timing of visually evoked responses, which may reflect prediction or expectation. A mouse model of Angelman syndrome lacks sequence plasticity in ACC despite normal sequence plasticity in V1.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Sidorov (msidorov@childrensnational.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The code used to analyze visually evoked potentials are available on Github (https://github.com/jeffgavornik/VEPAnalysisSuite).
The datasets generated during this study have not been deposited in a public repository but are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experiments used littermate controls with experimenters blind to genotype. Littermates were randomly assigned to experimental groups. Mice (congenic C57BL6/J) were group-housed on a 12:12 light/dark cycle with ad libitum access to food and water. We generated experimental AS model mice (Ube3am−/p+) and wild-type littermates (Ube3am+/p+) by crossing female Ube3am+/p− and male Ube3am+/p+ breeders. Jackson Labs (Bar Harbor, ME) initially provided the AS mouse line (#016590, RRID:MGI:3694359). We used adult (~P80-P120) male and female mice in equal ratios for all experiments. All protocols were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
METHOD DETAILS
In vivo local field potential recordings
During surgeries, we anesthetized mice using i.p. injections of ketamine (40 mg/kg) and xylazine (10 mg/kg), and administered the local analgesic bupivicaine (0.25%) under the scalp. We implanted tungsten microelectrodes (FHC) in right ACC (from bregma, in mm: A/P +1.0-1.2, M/L +0.5, D/V −1.5), bilaterally in V1 (from lambda, in mm: A/P 0, M/L +3.1-3.2, D/V −0.47), and a silver ground wire in cerebellum. Dorsal ACC electrodes (Figure S3J) were implanted at a depth of −0.7 mm D/V. We attached a steel headpost anterior to the ACC electrode to allow head fixation, and used dental cement to secure a protective head cap.
After surgeries, we allowed mice to recover for at least two days. Prior to experiments, we habituated mice to the recording apparatus for two consecutive days for 15 minutes per day (Figure 2B1). During habituation and most experiments, mice were awake and head-fixed in a dark, quiet environment with their body in a tube while facing a computer monitor (Figure 2A). Where noted (Figures S2F and S3I), animals were anesthetized with 40 mg/kg ketamine and 10 mg/kg xylazine (ketamine/xylazine) during recordings. Where noted (Figure S3K), animals were head-fixed and awake on a running wheel (design adapted from Heiney et al., 2014). We recorded data using two acquisition systems, always using the same system for an entire experimental cohort. System 1 amplified LFP recordings 1,000x using single-channel amplifiers (Grass Technologies) with 0.1 Hz high-pass and 100 Hz low-pass analog filtration preceding acquisition and digitization at 4 kHz using Spike2 software (CED). System 2 amplified and digitized data using the OmniPlex D Neural Data Acquisition System (Plexon), with an acquisition rate of 40 kHz (downsampled to 1 kHz prior to analysis) and with 0.1 Hz high-pass and 200 Hz low-pass filtration.
Visual stimuli
For experiments in Figure 2A–2D, we used stimulus protocols previously shown to drive strong, reliable V1 VEPs to both phase reversals (Cooke et al., 2015) and sequences (Gavornik and Bear, 2014). For phase reversals, mice viewed 200 cycles of a single orientation (E: 135°), phase reversing (E-1, E2) at 2 Hz (400 total phase reversals; Figure 2B2). For sequences, each element of a visual sequence (A: 45°, B: 105°, C: 15°, D: 75°) was presented for 150 ms, with a 1.5 s gray screen following each trial (Figure 2B2). Mice viewed 200 sequence trials. Cardinal orientations were not used to avoid the potential confound of larger VEPs driven by cardinal orientations relative to oblique orientations (Frenkel et al., 2006). For oddball experiments (Figures 2E–2G and S5M–S5O), mice viewed 500 sequence trials of either expected sequence ABCD (90%) or oddball sequence AECD (10%). AECD was presented every 10th trial (Homann et al., 2017). 500 trials were used because 20 (200 / 10) trials would not be sufficient for averaging to reliably quantify oddball VEPs. Similar oddball protocols have been used in the mouse auditory system (Featherstone et al., 2015). In a subset of animals, we compared the “every 10th trial” oddball protocol with a randomized oddball presentation on 10% of trials, and found these methods to be equally effective at driving oddball responses (data not shown). Object sequences (Figure S2E) consisted of stimuli initially used by Bussey et al. (2008). For all experiments, mice viewed stimuli (phase reversals and sequences) in blocks of 50 trials, each separated by a 30 s gray screen. For experiments in Figure 3, WT littermates were randomly assigned into experimental and control groups. Control stimuli were pseudorandom arrangements of A, B, C, D every trial. For experiments in Figures 4 and S4, WT littermates were randomly assigned to Gi DREADD + CNO and Gi DREADD + vehicle groups.
VEP analysis
The VEP represents an average of all trials within a single session (Frenkel et al., 2006; Gavornik and Bear, 2014; Porciatti et al., 1999). We analyzed session average VEPs using custom MATLAB software (https://github.com/jeffgavornik/VEPAnalysisSuite) (Gavornik and Bear, 2014). VEP magnitude refers to the voltage difference between the peak negativity and peak positivity following a stimulus. Response latency refers to the time at which the maximal negative-going peak following the stimulus occurs (Figure 2B3). ACC oddball preference (Figures 2G and S5O) is defined as (oddball “E” ACC VEP/expected “B” ACC VEP) within animals, and is plotted normalized to the group average in expected “B” VEP magnitude. For running wheel experiments (Figure S3K), we calculated the VEP for all trials throughout the entire session. Prior work using this wheel resulted in mouse locomotion for roughly ~25% of a session (Siegel et al., 2017).
VEPs under anesthesia
Experiments in Figure S2F were conducted in the same mice as in Figure 2B–2D, on test day. Experiments in Figure S3I were conducted in the same mice as in Figure 3A–3G, seven days after test day 6. In both cases, mice were anesthetized with ketamine (40 mg/kg) and xylazine (10 mg/kg) 30-60 minutes after awake recordings and tested immediately. For experiments in Figure S3I, only a subset of animals (n = 7 / 21 in ACC; n = 7 / 22 in V1) were re-tested at Day 13. We confirmed that ketamine/xylazine anesthesia increased spectral power in the V1 local field potential in the 2-4 Hz range (data not shown) (Michelson and Kozai, 2018).
VEPs in awake animals on running wheel
For experiments in Figure S3K, mice were briefly anesthetized with isoflurane just prior to loading onto the running wheel to reduce the chances of headcaps coming off due to running-induced torsion. We allowed animals to fully recover from light isoflurane while habituating to the gray screen for 15 minutes prior to recordings. In this cohort (Figure S3K), the “DCBA” stimulus on Day 6 was not run correctly (animals instead saw a second session of “ABCD”). This error was discovered on Day 7, after 3/9 animals had already been sacrificed. We tested “DCBA” in the remaining six animals on Day 7. This represents a deviation from the standard protocol and is indicated with a † symbol in the figure.
In vivo tetrode recordings, unit isolation, and unit analysis
Mice received chronic tetrode implants for single-unit recordings (Figure S2D). For these mice, a 1.0 × 1.5 mm craniotomy was made above the right ACC (in mm, from bregma: A/P +0.5 – 2.0, M/L 0.0 – 1.0). A durotomy was performed and a microdrive housing a bundle of 4–8 tetrodes was positioned within the craniotomy with tips lowered to approximately 300 μm below brain surface. A stainless steel housing was placed around the bundle and sealed with gel cyanoacrylate. The housing, microdrive legs, and headbar were secured to the skull with dental cement as described above.
Tetrodes were constructed from platinum-iridium polyimide-coated wire (18 μm diameter, post-implantation impedance range = 0.5–1.2 MΩ at 1 kHz, California Fine Wire). Tetrode signals were amplified and digitized at 40 kHz using the OmniPlex D system (Plexon), and band-pass filtered between 0.3 and 6 kHz. Single units were isolated offline using interactive cluster cutting software (Plexon). Isolated clusters of supra-threshold neural events with common waveform parameters (such as peak amplitude and energy) were identified as single-units (Siegel et al., 2012; Siegel, 2014, 2016). Only single-units with clustered points that showed little or no overlap (< 10%) with another cluster and with un-isolated background activity were included. Neural data were synchronized with the presentation of training stimuli by triggering the Plexon I/O port with the same TTL pulses used to trigger visual stimuli.
Trial x Time raster plots were created for each isolated single unit. For analysis, detected spikes were binned (50 ms) for each trial, and each post-stimulus trial time bin was compared to the average number of spikes/bin observed during baseline (600 ms pre stimulus) using a two-tailed paired t test (with Bonferroni correction for the number of trial time bins compared, alpha = 0.05/12 time bins, p < 0.004 for each trial bin) (Siegel et al., 2012; Siegel, 2014, 2016). A 600 ms baseline was used to match the total duration of the visual stimuli (150 ms x 4), and this baseline period was determined a priori. Significant trial bins indicated reliable increases or decreases in spiking in response to visual stimuli by a given single unit during a session.
Anatomical circuit mapping with viral vectors and CTB
Adeno-associated virus (AAV) vectors (~1012 viral molecules per mL), AAV5-CaMKII-GFP, and AAV5-CaMKII-hChR2-EYFP, were produced by the University of North Carolina Vector Core Facilities (Chapel Hill, NC) and have been described previously (Li et al., 2013; Miyamichi et al., 2011; Zhang et al., 2007). All viral vectors were stored in aliquots at −80°C until use.
Mice were anesthetized with isoflurane (10% induction and 2% maintenance) and placed on a stereotaxic frame. A craniotomy (~0.5 mm diameter) was made above the injection site. The following coordinates were used for unilateral injections (relative to bregma, in mm): V1: M/L +2.5 mm, A/P −3.28mm, D/V −0.48, V2M: M/L +1.5, A/P −2.7, D/V-0.48 mm, ACC: M/L +0.24, A/P +0.86, D/V −1.13. The injection at V2M was located close to the posteromedial secondary visual area (Wang et al., 2012). The final volumes of AAV and cholera toxin subunit B (CTB, Alexa Fluor 594 Conjugate, Invitrogen) were 0.5 μL and 0.2 μl, respectively. To ensure minimal leak into surrounding brain regions, injection pipettes remained in the injection site for ~10 min after injection before being slowly withdrawn. We waited 4-7 weeks for viral expression and 2 weeks for CTB expression. To compare CTB expression in V1 and V2M, we adapted the method from Wang et al. (2012), which demonstrates expression of type 2 muscarinic acetylcholine receptor (M2 AChR) in V1, but not in adjacent V2M. The CTB labeling cells in V1 and V2M were quantified in the area with and without M2 AChR expression, respectively (Figure S1A).
Images for quantification were captured from consistent coronal section planes across different mouse brains. The CTB-labeling image (100 × 100 μm) was converted to 8-bit in black and white, and its threshold was adjusted using Huang method in Fiji software. This Huang threshold method was chosen to reveal high intensities of CTB labeling and was applied to all sections analyzed. The number of CTB-expressing cells was quantified using Analyze Particles function in Fuji software (Figures 1C and S5D).
Immunohistochemistry
Mice were anesthetized with sodium pentobarbital (40 mg/kg) before transcardial perfusion with PBS, immediately followed by 4% paraformaldehyde (PFA) in PBS, pH 7.4. Brains were post fixed overnight at 4°C before sequential 24 h incubations in PBS with 10%, 20%, and 30% sucrose. Brains were sectioned coronally at 40 μm using a freezing sliding microtome (Thermo Scientific). Sections were stored at 4°C in a cryopreservative solution (45% PBS, 30% ethylene glycol, 25% glycerol, by volume). For immunostaining, brain sections were rinsed several times with PBS before blocking with 5% normal goat serum and 0.02% Triton X-100 in PBS for 1 h at room temperature. Sections were then incubated with primary antibodies, including rat anti-M2 AChR antibody (1:500, MAB367, Millipore) and rabbit GFP antibody (1:500, NB600-308, Novus), diluted in normal goat serum at 4°C overnight, and then rinsed several times with 0.02% Triton X-100 in PBS. Tissues were then incubated with secondary antibodies, including Alexa Fluor-488 goat anti-rabbit IgG (1:500, #A11008, Invitrogen) and Alexa Fluor-568 goat anti-rat IgG (1:500, #A11077, Invitrogen) for 1 hour. 4’,6-diamidino-2-phenylindole (DAPI, 7mg/mL, D1306, Invitrogen) was added during the secondary antibody incubation for nuclear counterstaining. Images were acquired with a Zeiss LSM 780 or 710 confocal microscope.
Electrode visualization
The glial marker glial fibrillary acidid protein (GFAP) was used to visualize electrode placement (Figures 2C and S3J1). After experiments, mice were killed and whole brains were extracted and embedded in O.C.T. compound (Optimal Cutting Temperature, Tissu-Tek) and flash frozen on dry ice and stored at −80°C. Brains were then sectioned using a cryostat (CryoStar NX50, Thermo Scientific) to a thickness of 15-20 μm. Sections were fixed in 4% PFA, rinsed, and blocked with 5% normal goat serum as above. Primary antibodies included mouse IgG1 anti-NeuN (1:500; MAB377, Millipore) and rabbit IgG anti-GFAP (1:800; Z0334, Dako), and secondary antibodies included AlexaFluor 488-conjugated anti-mouse IgG1-gamma 1 (1:500; A21121, Invitrogen) and AlexaFluor 568-conjugated anti-rabbit IgG (1:500; A11011, Invitrogen). Images were acquired using fluorescent microscopy (Nikon Eclipse 80i or Nikon Eclipse Ti2).
Slice preparation for whole-cell electrophysiology
Mice (P70-90) were anesthetized with pentobarbital (40 mg/kg) and intracardially perfused with ice-cold dissection buffer (in mM: 87 NaCl, 2.5 KCl, 1.25 NaH2PO4*H2O, 25 NAHCO3, 10 D-glucose, 75 sucrose, 1.3 ascorbic acid, 7 MgCl2 and 0.5 CaCl2) bubbled with 95% O2–5% CO2 after disappearance of corneal reflexes. Brains were then rapidly removed and immersed in ice-cold dissection buffer. ACC sections ipsilateral to the virus injections were dissected, and 350-μm thick coronal slices were prepared using a vibrating microtome (Leica VT1200S). Slices recovered for 20 min in a 35°C submersion chamber filled with oxygenated artificial cerebrospinal fluid (ACSF; in mM: 124 NaCl, 3 KCl, 1.25 KH2PO4*H2O, 26 NaHCO3, 20 D-Glucose, 1 MgCl2, 2 CaCl2) and then kept at room temperature for > 40 min until use.
Whole-cell recordings
Cells were visualized using a Zeiss Examiner microscope equipped with infrared differential interference contrast optics. Patch pipettes were pulled from thick-walled borosilicate glass (P2000, Sutter Instruments Novato, CA). Open-tip resistances were between 2.5–4 ΩU, and pipettes were backfilled with the internal solution containing (in mM): 112 K-gluconate, 8 KCl, 10 HEPES, 0.2 EGTA, 4 ATP-Mg, 0.3 GTP-Na, and 10 Na2 phosphocreatine, and 0.015 Alexa-594 (Life Technologies) with pH adjusted to 7.25 and osmolarity adjusted to ~290 mOSM with sucrose. Voltage and current clamp recordings were performed in the whole-cell configuration using a patch-clamp amplifier (Multiclamp 700B, Molecular Devices), and data were acquired using pClamp 10 software (Molecular Devices). Pipette seal resistances were > 1GΩ, and pipette capacitive transients were minimized before breakthrough. Voltages were not corrected for a liquid junction potential. Changes in series and input resistance were monitored throughout the experiment by giving a test pulse every 30 s and measuring the amplitude of the capacitive current. Cells were discarded if series resistance rose above 30 MΩ. We categorized neurons as pyramidal cells or interneurons based on morphology, location, and firing rate. In voltage-clamp experiments, evoked postsynaptic currents (EPSCs) were recorded at −73 mV (chloride reversal potential under our recording conditions) in the presence of picrotoxin (50 μM, Sigma) and APV (50 μM, Tocris). All drugs (DNQX, 20 μM, Abcam; TTX, 1 μM, Abcam; 4-AP, 1mM, Abcam) were applied through the bathing solution. In current-clamp experiments, the membrane potentials (Vrest) of pyramidal neurons and interneurons were measured within 3 min of break-in. Intrinsic membrane and synaptic properties of the neurons were characterized by injecting suprathreshold positive currents in steps of 20 pA for 350 ms.
Optogenetic stimulation
Widefield ChR2-mediated photostimulation was provided through a X20/0.8NA objective using single-photon excitation through a 470 nm l-filter. Light power was provided by a Lambda DG-4 300W Xenon bulb (Sutter Instruments). This light was coupled to a Mosaic microelectro-mechanical-system digital micromirror device (Andor Technology) and was shuttered via pClamp-delivered TTL pulse to the Lambda DG-4 as previously described (Larsen et al., 2014; Sparta et al., 2011). Synaptic responses to photostimulation of the ChR2-expressing axons were measured from postsynaptic cortical neurons (which were ChR2 negative) recorded in whole-cell current and voltage clamp. The laser was directly delivered to the cortical column of the recorded cell with the square illumination patterns (227x227 μm) over YFP-positive L1 and L2/3 somata using brief light pulses (3 ms duration, 10 Hz train, 13.6 mW/mm2 power).
Chemogenetic inhibition of ACC in vivo
Mice were anesthetized with isoflurane (10% induction and 2% maintenance) and placed on a stereotaxic frame. A craniotomy (~0.5 mm diameter) was made above the injection site. The inhibitory DREADD hM4Di (“Gi DREADD,” rAAV8/hSyn-HM4Di-mCherry (7.4 × 1012 vg/ml), 500 nl) (Armbruster et al., 2007) was injected into right ACC (M/L +0.3, A/P +0.5, D/V −0.9). LFP electrodes were implanted during the same surgery both into right ACC (from bregma, in mm: M/L +0.3, A/P +0.5, D/V −1.0) and bilaterally into V1 (from lambda, in mm: A/P 0, M/L +3.1-3.2, D/V −0.47). Mice were given 3+ weeks for recovery and viral expression before two days of habituation to gray screen and 6 days of recording (Figure 4A and 4B). Prior to Day 1 of recording, mice were randomly assigned into CNO and vehicle groups. Mice were injected i.p. with 5 mg/kg CNO (Enzo Life Sciences) in saline or saline vehicle. CNO/vehicle injections occurred one hour prior to recordings on all six days of recordings.
QUANTIFICATION AND STATISTICAL ANALYSIS
All VEP traces shown in figures represent group averages. All data are represented as mean ± SEM. For main figures, statistical details can be found in Results text and “n” can be found in figure legends. For supplemental figures, statistical details and “n” can be found in supplementary figure legends. We used Prism 8 for statistics. Statistical comparisons in Figures 1C, 2D, 2G, 3E, 3F, 4C, 4D, 4G, 5C, 5D, 5G, and 5H, S3G, S4D, S4G, S5I, S5J, S5K, S5L, S5N, and S5O used two-tailed Student’s t tests. Statistical comparisons in Figures 3C, 3D, 4F, 5A, and 5B, S3B, S3D, S3E, S3J, S3K, S4C, and S4F, used a repeated-measures (RM) ANOVA and a post hoc Bonferroni test when applicable. Statistical comparisons in Figures S3F and S4H used a linear regression analysis to assess correlations. Statistical comparisons in Figure S3I and S3K used a mixed-effects analysis instead of a RM ANOVA because of missing values. For Figure S3I, values were missing because only a subset of animals were re-rested on Day 13 (see VEPs under anesthesia). For Figure S3K, values were missing because three animals were sacrificed between Day 6 and Day 7 (see VEPs in awake animals on running wheel).
In rare cases, VEPs polluted by electrical noise or a poor connection were excluded from analysis while blind to treatment and/or genotype. For example, Figure 3 reports n = 21 experimental mice for ACC but Figure S3 reports n = 22 experimental mice for V1. In this group, there were a total of 23 mice. 2/23 ACC VEPs were excluded and 1/23 V1 VEP was excluded. For correlational analyses (Figure S3F), only mice with both ACC and V1 VEPs were included (n = 20). For in vivo chemogenetic studies (Figures 4 and S4), there were a total of n = 5 mice in the Vehicle group and n = 6 mice in the CNO group. In the Vehicle group, 1/5 ACC VEPs were excluded, 0/5 ipsilateral V1 VEPs were excluded, and 0/5 contralateral V1 VEPs were excluded. In the CNO group, 0/6 ACC VEPs were excluded, 0/6 ipsilateral V1 VEPs were excluded, and 2/6 contralateral V1 VEPs were excluded. For Figure 5, there were a total of n = 18 WT mice (1/18 ACC VEP excluded, 0/18 V1 VEP excluded) and n = 21 AS mice (4/21 ACC VEP excluded, 2/21 V1 VEP excluded).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-M2 AChR antibody | Millipore | Cat#MAB367; RRID: AB_94952 |
| Rabbit GFP antibody | Novus | Cat#NB600-308; RRID: AB_10003058 |
| Alexa Fluor 488 goat anti-rabbit IgG | Invitrogen | Cat#A11008; RRID: AB_143165 |
| Alexa Fluor 568 goat anti-rat IgG | Invitrogen | Cat#A11077; RRID: AB_141874 |
| IgG1 anti-NeuN | Millipore | Cat#MAB377; RRID: AB_2298772 |
| Rabbit IgG anti-GFAP | Dako | Cat#Z0334; RRID: AB_10013382 |
| Alexa Fluor 488 goat anti-mouse IgG1 | Invitrogen | Cat#A21121; RRID: AB_2535764 |
| Alexa Fluor 568 goat anti-rabbit IgG | Invitrogen | Cat#A11011; RRID: AB_143157 |
| Bacterial and Virus Strains | ||
| AAV5-CaMKII-GFP | UNC Vector Core | N/A |
| AAV5-CaMKII-hChR2-EYFP | UNC Vector Core | N/A |
| rAAV8/hSyn-HM4Di-mCherry | UNC Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CTB594 | Invitrogen | Cat#C34777 |
| Clozapine-N-oxide | Enzo Life Sciences | Cat#BML-NS105-0005 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Labs | Cat#000664 |
| Mouse: Ube3am−/p+: Ube3atm1Alb/J | Jackson Labs | Cat#016590; RRID:MGI:3694359 |
| Software and Algorithms | ||
| MATLAB | MathWorks | N/A |
| Prism | GraphPad | N/A |
| Other | ||
| Tungsten microelectrodes | FHC | Cat#30070 |
| Tetrode wire | California Fine Wire | Cat#100167 |
Highlights.
Visual sequences can be used to drive mouse anterior cingulate cortex (ACC)
Training to an expected sequence drives plasticity in the timing of ACC responses
Sequence plasticity is absent in ACC, but not V1, in Angelman syndrome mice
ACKNOWLEDGMENTS
We thank Eric Gao for assisting in perfusing mice and sectioning tissue. This work was supported by NIH grants K99EY028964 (to M.S.S.) and R01HD093771 (to B.D.P.), and NIH training grant T32HD040127 (to M.S.S. and B.W.). Microscopy was performed at the Neuroscience Microscopy Core Facility, supported, in part, by funding from the NIH-NINDS Neuroscience Center Support Grant P30–NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant U54–HD079124.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108152.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alexander WH, and Brown JW (2019).The role of the anterior cingulate cortex in prediction error and signaling surprise. Top. Cogn. Sci 11, 119–135. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash ES, Heal DJ, and Clare Stanford S (2014). Contrasting changes in extracellular dopamine and glutamate along the rostrocaudal axis of the anterior cingulate cortex of the rat following an acute d-amphetamine or dopamine challenge. Neuropharmacology 87, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaf M, Jagannathan K, Calhoun VD, Miller L, Stevens MC, Sahl R, O’Boyle JG, Schultz RT, and Pearlson GD (2010). Abnormal functional connectivity of default mode sub-networks in autism spectrum disorder patients. Neuroimage 53, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aton SJ, Suresh A, Broussard C, and Frank MG (2014). Sleep promotes cortical response potentiation following visual experience. Sleep (Basel) 37, 1163–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci 16, 551–563. [DOI] [PubMed] [Google Scholar]
- Brumback AC, Ellwood IT, Kjaerby C, lafrati J, Robinson S, Lee AT, Patel T, Nagaraj S, Davatolhagh F, and Sohal VS (2018). Identifying specific prefrontal neurons that contribute to autism-associated abnormalities in physiology and social behavior. Mol. Psychiatry 23, 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey TJ, Padain TL, Skillings EA, Winters BD, Morton AJ, and Saksida LM (2008). The touchscreen cognitive testing method for rodents: how to get the best out of your rat. Learn. Mem 15, 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Braver TS, Barch DM, Botvinick MM, Noll D, and Cohen JD (1998). Anterior cingulate cortex, error detection, and the online monitoring of performance. Science 280, 747–749. [DOI] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte WL, Kamishina H, and Reep RL (2009). Multiple neuroanatomical tract-tracing using fluorescent Alexa Fluorconjugates of cholera toxin subunit B in rats. Nat. Protoc 4, 1157–1166. [DOI] [PubMed] [Google Scholar]
- Cooke SF, and Bear MF (2015). Visual recognition memory: a view from V1. Curr. Opin. Neurobiol 35, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Komorowski RW, Kaplan ES, Gavornik JP, and Bear MF (2015). Visual recognition memory, manifested as long-term habituation, requires synaptic plasticity in V1. Nat. Neurosci 18, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Ahmed OJ, Stevens TR, Patrick SL, Gonzalez AN, Elmaleh M, and Connors BW (2012). Thalamic control of layer 1 circuits in prefrontal cortex. J. Neurosci 32, 17813–17823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delevich K, Tucciarone J, Huang ZJ, and Li B (2015). The mediodorsal thalamus drives feedforward inhibition in the anterior cingulate cortex via parvalbumin interneurons. J. Neurosci 35, 5743–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Morrell MJ, and Vogt BA (1995). Contributions of anterior cingulate cortex to behaviour. Brain 118, 279–306. [DOI] [PubMed] [Google Scholar]
- Divac I, Mogensen J, Petrovic-Minic B, Zilles K, and Regidor J (1993). Cortical projections of the thalamic mediodorsal nucleus in the rat. Definition of the prefrontal cortex. Acta Neurobiol. Exp. (Warsz.) 53, 425–429. [PubMed] [Google Scholar]
- Dölen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of Fragile X syndrome in mice. Neuron 56, 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone RE, Shin R, Kogan JH, Liang Y, Matsumoto M, and Siegel SJ (2015). Mice with subtle reduction of NMDA NR1 receptor subunit expression have a selective decrease in mismatch negativity: Implications for schizophrenia prodromal population. Neurobiol. Dis 73, 289–295. [DOI] [PubMed] [Google Scholar]
- Fiser A, Mahringer D, Oyibo HK, Petersen AV, Leinweber M, and Keller GB (2016). Experience-dependent spatial expectations in mouse visual cortex. Nat. Neurosci 19, 1658–1664. [DOI] [PubMed] [Google Scholar]
- Frenkel MY, and Bear MF (2004). How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 44, 917–923. [DOI] [PubMed] [Google Scholar]
- Frenkel MY, Sawtell NB, Diogo AC, Yoon B, Neve RL, and Bear MF (2006). Instructive effect of visual experience in mouse visual cortex. Neuron 51, 339–349. [DOI] [PubMed] [Google Scholar]
- Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Darcy DP, Nicoll RA, Huang ZJ, and Stryker MP (2014). A cortical circuit for gain control by behavioral state. Cell 156, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamanut R, Kennedy H, Toroczkai Z, Ercsey-Ravasz M, Van Essen DC, Knoblauch K, and Burkhalter A (2018). The mouse cortical connectome, characterized byan ultra-dense cortical graph, maintains specificity by distinct connectivity profiles. Neuron 97, 698–715 e610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavornik JP, and Bear MF (2014). Learned spatiotemporal sequence recognition and prediction in primary visual cortex. Nat. Neurosci 17, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring WJG, Coles MG, Meyer DE, and Donchin E (1993). A neural system for error detection and compensation. Psychol. Sci 4, 385–390. [Google Scholar]
- Gehring WJ, Goss B, Coles MG, Meyer DE, and Donchin E (1995). A brain potential manifestation of error-related processing. Electroencephalogr. Clin. Neurophysiol. Suppl 44, 261–272. [PubMed] [Google Scholar]
- Gordon JA, and Stryker MP (1996). Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J. Neurosci 16, 3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Chen J, Chen Q, Ren K, Feng D, Mao H, Yao H, Yang J, Liu H, Liu Y, et al. (2019). Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat. Neurosci 22, 1223–1234. [DOI] [PubMed] [Google Scholar]
- Haberl MG, Zerbi V, Veltien A, Ginger M, Heerschap A, and Frick A (2015). Structural-functional connectivity deficits of neocortical circuits in the Fmr1 (-/y) mouse model of autism. Sci. Adv 1, e1500775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kebschull JM, Campbell RAA, Cowan D, Imhof F, Zador AM, and Mrsic-Flogel TD (2018). The logic of single-cell projections from visual cortex. Nature 556, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, and Contractor A (2010). Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiney SA, Wohl MP, Chettih SN, Ruffolo LI, and Medina JF (2014). Cerebellar-dependent expression of motor learning during eyeblink conditioning in head-fixed mice. J. Neurosci 34, 14845–14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann J, Koay SA, Glidden AM, Tank DW, and Berry MJ II. (2017). Predictive coding of novel versus familiar stimuli in the primary visual cortex. bioRxiv, 197608. [Google Scholar]
- Hu F, Kamigaki T, Zhang Z, Zhang S, Dan U, and Dan Y (2019). Prefrontal corticotectal neurons enhance visual processing through the superior colliculus and pulvinar thalamus. Neuron 104, 1141–1152 e1144. [DOI] [PubMed] [Google Scholar]
- Hyman JM, Holroyd CB, and Seamans JK (2017). A novel neural prediction error found in anterior cingulate cortex ensembles. Neuron 95, 447–456 e443. [DOI] [PubMed] [Google Scholar]
- Jann K, Hernandez LM, Beck-Pancer D, McCarron R, Smith RX, Dapretto M, and Wang DJ (2015). Altered resting perfusion and functional connectivity of default mode network in youth with autism spectrum disorder. Brain Behav. 5, e00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, and Beaudet AL (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811. [DOI] [PubMed] [Google Scholar]
- Kalmbach BE, Johnston D, and Brager DH (2015). Cell-type specific channelopathies in the prefrontal cortex of thefmr1-/y mouse model of Fragile X syndrome. eNeuro 2, ENEURO.0114-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, and Stryker MP (2014). Sensory experience during locomotion promotes recovery of function in adult visual cortex. eLife 3, e02798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller GB, Bonhoeffer T, and Hübener M (2012). Sensorimotor mismatch signals in primary visual cortex of the behaving mouse. Neuron 74, 809–815. [DOI] [PubMed] [Google Scholar]
- Kennedy DP, and Courchesne E (2008). Functional abnormalities of the default network during self- and other-reflection in autism. Soc. Cogn. Affect. Neurosci 3, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns JG, Cohen JD, MacDonald AW 3rd, Cho RY, Stenger VA, and Carter CS (2004). Anterior cingulate conflict monitoring and adjustments in control. Science 303, 1023–1026. [DOI] [PubMed] [Google Scholar]
- Kim H, Kunz PA, Mooney R, Philpot BD, and Smith SL (2016). Maternal loss of Ube3a impairs experience-driven dendritic spine maintenance in the developing visual cortex. J. Neurosci 36, 4888–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Chaloner FA, Cooke SF, Harnett MT, and Bear MF (2020). Opposing somatic and dendritic expression of stimulus-selective response plasticity in mouse primary visual cortex. Front. Cell. Neurosci 13, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramvis I, Mansvelder HD, Loos M, and Meredith R (2013). Hyperactivity, perseveration and increased responding during attentional rule acquisition in the Fragile X mouse model. Front. Behav. Neurosci 7, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K,Wang BS, Lu J, Wang L, Maffei A, Cang J, and Huang ZJ (2015). MeCP2 regulates the timing of critical period plasticity that shapes functional connectivity in primary visual cortex. Proc. Natl. Acad. Sci. USA 112, E4782–E4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DD, Osterweil EK, Chen SP, Tye LD, and Bear MF (2011). Cognitive dysfunction and prefrontal synaptic abnormalities in a mouse model of Fragile X syndrome. Proc. Natl. Acad. Sci. USA 108, 2587–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Lombardo MV, and Baron-Cohen S (2014). Autism. Lancet 383, 896–910. [DOI] [PubMed] [Google Scholar]
- Larsen RS, Smith IT, Miriyala J, Han JE, Corlew RJ, Smith SL, and Philpot BD (2014). Synapse-specific control of experience-dependent plasticity by presynaptic NMDA receptors. Neuron 83, 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinweber M, Ward DR, Sobczak JM, Attinger A, and Keller GB (2017). A sensorimotor circuit in mouse cortex for visual flow predictions. Neuron 96, 1204. [DOI] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, and Li B (2013). Experience-dependent modification of a central amygdala fear circuit. Nat. Neurosci 16, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LQ, Ning L, Wang Z, and Wang YW (2016). Visual and noxious electrical stimulus-evoked membrane-potential responses in anterior cingulate cortical neurons. Mol. Brain 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson NJ, and Kozai TDY (2018). Isoflurane and ketamine differentially influence spontaneous and evoked laminar electrophysiology in mouse V1.J. Neurophysiol 120, 2232–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, and Rauch SL (2007). A role for the human dorsal anterior cingulate cortex in fear expression. Biol. Psychiatry 62, 1191–1194. [DOI] [PubMed] [Google Scholar]
- Minshew NJ, and Keller TA (2010). The nature of brain dysfunction in autism: functional brain imaging studies. Curr. Opin. Neurol 23, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamichi K, Amat F, Moussavi F, Wang C, Wickersham I, Wall NR, Taniguchi H, Tasic B, Huang ZJ, He Z, et al. (2011). Cortical representations of olfactory input by trans-synaptic tracing. Nature 472, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, Chan AW, Mohsenvand M, LeDue J, Liu R, McVea DA, Boyd JD, Wang YT, Reimers M, and Murphy TH (2013). Spontaneous cortical activity alternates between motifs defined by regional axonal projections. Nat. Neurosci 16, 1426–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Yoshida T, Matsui T, and Ohki K (2015). Wide-field Ca2+ imaging reveals visually evoked activity in the retrosplenial area. Front. Mol. Neurosci 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, and Stryker MP (2010). Modulation of visual responses by behavioral state in mouse visual cortex. Neuron 65, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, and Chen C (2011). Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron 70, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, Lau C, Kuan L, Henry AM, et al. (2014). A mesoscale connectome of the mouse brain. Nature 508, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignatelli M, Feligioni M, Piccinin S, Molinaro G, Nicoletti F, and Nisticò R (2013). Synaptic plasticity as atherapeutic target in the treatment of autism-related single-gene disorders. Curr. Pharm. Des 19, 6480–6490. [DOI] [PubMed] [Google Scholar]
- Porciatti V, Pizzorusso T, and Maffei L (1999). The visual physiology of the wild type mouse determined with pattern VEPs. Vision Res. 39, 3071–3081. [DOI] [PubMed] [Google Scholar]
- Rajasethupathy P, Sankaran S, Marshel JH, Kim CK, Ferenczi E, Lee SY, Berndt A, Ramakrishnan C, Jaffe A, Lo M, et al. (2015). Projections from neocortex mediate top-down control of memory retrieval. Nature 526, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru DC, van Woerden GM, Wallaard I, and Elgersma Y (2018). Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex Layer 5 neurons in a mouse model of Angelman syndrome. J. Neurosci 38, 8011–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, and Stryker MP (2010). Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. USA 107, 5611–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceniak MP, Lang M, Enomoto AC, James Howell C, Hermes DJ, and Katz DM (2016). Mechanisms of functional hypoconnectivity in the medial prefrontal cortex of Mecp2 null mice. Cereb. Cortex 26, 1938–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, Wright M, and Deisseroth K (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CN7NAP2-deficient mice. Sci. Transl. Med 9, eaah6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenhav A, Botvinick MM, and Cohen JD (2013). The expected value of control: an integrative theory of anterior cingulate cortex function. Neuron 79, 217–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, and Kato A (1993). Topographic relationship between anteromedial thalamic nucleus neurons and theircortical terminal fields in the rat. Neurosci. Res 17, 63–69. [DOI] [PubMed] [Google Scholar]
- Sidorov MS, Auerbach BD, and Bear MF (2013). Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorov MS, Judson MC, Kim H, Rougie M, Ferrer AI, Nikolova VD, Riddick NV, Moy SS, and Philpot BD (2018). Enhanced operant extinction and prefrontal excitability in a mouse model of Angelman syndrome. J. Neurosci 38, 2671–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ (2014). Modification of persistent responses in medial prefrontal cortex during learning in trace eyeblink conditioning. J. Neurophysiol 112, 2123–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ (2016). Prefrontal single-neuron responses after changes in task contingencies during trace eyeblink conditioning in rabbits. eNeuro 3, ENEURO.0057-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ, Kalmbach B, Chitwood RA, and Mauk MD (2012). Persistent activity in a cortical-to-subcortical circuit: bridging the temporal gap in trace eyelid conditioning. J. Neurophysiol 107, 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ, Chitwood RA, Ding JM, Payne C, Taylor W, Gray R, Zemelman BV, and Johnston D (2017). Prefrontal cortex dysfunction in Fragile X mice depends on the continued absence of Fragile X mental retardation protein in the adult brain. J. Neurosci 37, 7305–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair D, Oranje B, Razak KA, Siegel SJ, and Schmid S (2017). Sensory processing in autism spectrum disorders and Fragile Xsyndrome—from the clinic to animal models. Neurosci. Biobehav. Rev 76 (Pt B), 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparta DR, Stamatakis AM, Phillips JL, Hovelsø N, van Zessen R, and Stuber GD (2011). Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat. Protoc 7, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasan V, Kyriakatos A, Mateo C, Jaeger D, and Petersen CC (2017). Parallel pathways from whisker and visual sensory cortices to distinct frontal regions of mouse neocortex. Neurophotonics 4, 031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibert RL, Larson AM, Hsieh DT, Raby AR, and Thiele EA (2013). Neurologic manifestations of Angelman syndrome. Pediatr. Neurol 48, 271–279. [DOI] [PubMed] [Google Scholar]
- Thillay A, Lemaire M, Roux S, Houy-Durand E, Barthélémy C, Knight RT, Bidet-Caulet A, and Bonnet-Brilhault F (2016). Atypical brain mechanisms of prediction according to uncertainty in autism. Front. Neurosci 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinck M, Batista-Brito R, Knoblich U, and Cardin JA (2015). Arousal and locomotion make distinct contributions to cortical activity patterns and visual encoding. Neuron 86, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA (2005). Pain and emotion interactions in subregions of the cingulate gyrus. Nat. Rev. Neurosci 6, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ML, Burette AC, Weinberg RJ, and Philpot BD (2012). Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Sporns O, and Burkhalter A (2012). Network analysis of corticocortical connections reveals ventral and dorsal processing streams in mouse visual cortex. J. Neurosci 32, 4386–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, Weinberg RJ, Ehlers MD, and Philpot BD (2009). Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci 12, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, and Deisseroth K (2007). Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–639. [DOI] [PubMed] [Google Scholar]
- Zhang S, Xu M, Kamigaki T, Hoang Do JP, Chang WC, Jenvay S, Miyamichi K, Luo L, and Dan Y (2014). Selective attention. Long-range and local circuits for top-down modulation of visual cortex processing. Science 345, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xu M, Chang WC, Ma C, Hoang Do JP, Jeong D, Lei T, Fan JL, and Dan Y (2016). Organization of long-range inputs and outputs of frontal cortex for top-down control. Nat. Neurosci 19, 1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Shi L, Cui X, Wang S, and Luo X (2016). Functional connectivity of the caudal anterior cingulate cortex is decreased in autism. PLoS ONE 11, e0151879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg B, Hintiryan H, Gou L, Song MY, Bay M, Bienkowski MS, Foster NN, Yamashita S, Bowman I, Toga AW, and Dong HW (2014). Neural networks of the mouse neocortex. Cell 156, 1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmarz P, and Keller GB (2016). Mismatch receptive fields in mouse visual cortex. Neuron 92, 766–772. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used to analyze visually evoked potentials are available on Github (https://github.com/jeffgavornik/VEPAnalysisSuite).
The datasets generated during this study have not been deposited in a public repository but are available from the corresponding author on request.