

 The cGMP-dependent protein kinase in Plasmodium falciparum (PfPKG) plays multiple roles in the life cycle of the parasite.
The cGMP-dependent protein kinase in Plasmodium falciparum (PfPKG) plays multiple roles in the life cycle of the parasite.
Abstract
The cGMP-dependent protein kinase in Plasmodium falciparum (PfPKG) plays multiple roles in the life cycle of the parasite. As a result, this enzyme is a potential target for new antimalarial agents. Existing inhbitors, while potent and active in malaria models are not optimal. This communication describes initial optimization of a structurally distinct class of PfPKG inhibitors.
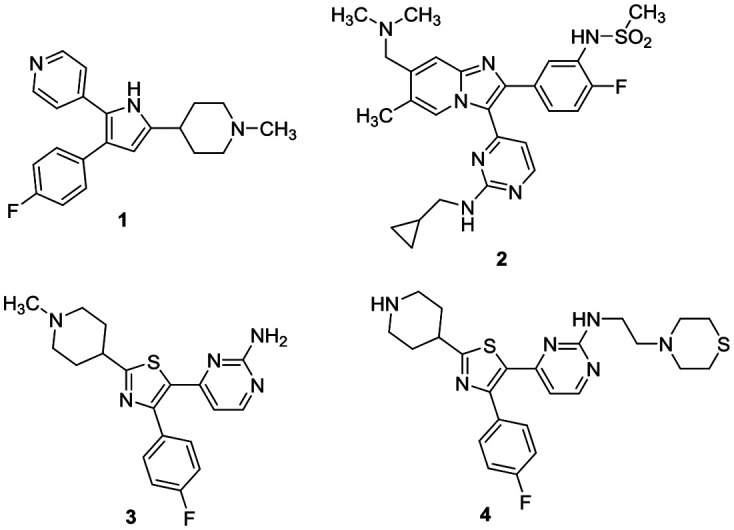
The serine/threonine cGMP-dependent protein kinase found in Plasmodium falciparum (PfPKG) is distinct from human orthologs. A key difference between the parasitic and human enzyme is a smaller threonine gatekeeper residue in the parasitic enzyme, compared to the human ortholog.1 This kinase plays a key role in the life cycle of the parasite, regulating e.g. egress of merozoites from the liver and subsequent invasion of erythrocytes.1,2 In view of the rising incidence of resistance to current agents, the potential to inhibit multiple points in the parasite life cycle stimulated interest in the kinase for the discovery of novel antimalarial agents. For example, the trisubstituted pyrrole 1, originally discovered as an inhibitor of a similar kinase in Eimeria,3 and imidazopyridine 2 (ref. 4) both were shown to be potent PfPKG inhibitors (Fig. 1). More recently, trisubstituted thiazoles exemplified by 3 and 4 have been identified.5,6 Baker and co-workers recently showed that 2 is highly selective against the human PKG ortholog and was orally active in a mouse model of P. falciparum infection.4 These highly potent PfPKG inhibitors are useful tools to help validate the target, however both molecules have pharmacokinetic issues and selected examples in the imidazopyridine series are reported to be Ames positive that limit their progression as drug candidates.7 Thus there is a clear need for identification of distinct chemotypes to continue to explore this interesting target.
Fig. 1. PfPKG inhibitors.
Our interest in exploring anti-parasitic kinase inhibitors led us to investigate this target to search for structurally different starting points as PfPKG inhibitors. A focused in house library of parasitic kinase inhibitors was screened, resulting in the identification of three related isoxazole-based hits 5a–c with activity against the parasitic kinase and excellent selectivity against the human ortholog (Fig. 2).
Fig. 2. Isoxazole based PfPKG leads.
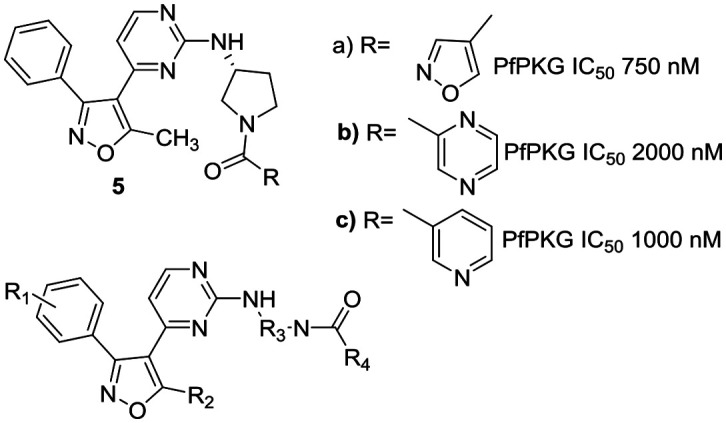
This communication describes an initial investigation of structure–activity relationships at four points in this lead scaffold: substituents on the benzene ring (R1), the methyl group on the isoxazole ring (R2), the diamine (R3) and amide (R4) as illustrated in Fig. 2. The results reported here illustrate a promising path forward in the search for candidates with suitable drug-like characteristics.
We elected to simultaneously evaluate aromatic substituents with R2 as methyl and modifications at this methyl position that incorporated a preferred phenyl substituent. The synthesis of these sets of target molecules is outlined in Schemes 1 and 2.
Scheme 1. Synthesis of initial isoxazole PfKG inhibitors.

Scheme 2. Synthesis of isoxazole amine and chlorophenyl PfPKG inhibitors.

Condensation of thiomethyl pyrimidine 6 with ethyl glyoxalate, afforded an α-ketoester that underwent cycloaddition with phenyl hydroxyliminochloride to provide pyrimidinyl isoxazole sulfide ester 7 in good yield.8 Oxidation of the methyl sulfide to the corresponding sulfone was followed by displacement in warm DMSO with (R)-3-amino-Boc-pyrrolidine. TFA-mediated deprotection and standard amide coupling furnished 8. Ester 7 was reduced using sodium borohydride at low temperature to the corresponding alcohol.5 Processing as above using the respective (R)- and (S)-enantiomers of 3-aminopyrrolidine furnished 9a and 9b. Three amine-substituted analogs (11a–c) were prepared from sulfide–alcohol 10. Bromination of 10 using CBr4/triphenylphosphine, followed by sulfide oxidation and bromine displacement at room temperature with morpholine, pyrrolidine or diethylamine provided the appropriate amine intermediates. Sulfone displacement as above was followed by TFA mediated deprotection and amide coupling. In a similar manner, 6 was deprotonated using LDA, then reacted with the Weinreb amide shown to furnish methyl ketone 12.9 Subsequent transformations as outlined above provided chloro-substituted methyl analogs 13a–c.
Analysis of these derivatives (Table 1-IC50 values based on at least 2 independent experiments) as PfPKG inhibitors using an IMAP-based kinase assay10 revealed that the enzyme has a strong preference for the (R)-enantiomer of 3-aminopyrrolidine (cf.9avs.9b), that potency can be improved by replacing a methyl group with a diethylamino moiety (11c) and that among a limited group of analogs, 3-chloro substitution was preferred (13b). An ester substituent (8) on the isoxazole ring was weakly active, and improved upon reduction to an alcohol (9a) which was slightly less active compared to methyl (3, Fig. 1). Compounds 11c and 13b were inactive (up to 10 μM) when assayed using human PKG. Selectivity against the human ortholog is an essential prerequisite for this class of compounds. More extended kinase selectivity screening has not been carried out with this series of compounds.
Table 1. Initial PfPKG SAR.
| Cpd | % Inh@10 μM | IC50 (nM) |
| 8 | 23 | ND |
| 9a | 1000 | |
| 9b | 60 | ND |
| 11a | 1000 | |
| 11b | 1400 | |
| 11c | 850 | |
| 13a | 2100 | |
| 13b | 210 | |
| 13c | 2800 |
In view of these results, we explored additional aromatic substitutions, diamine linkers and derivatization of the pyrrolidine nitrogen in the next round of optimization while holding the diethylamino substituent constant. We chose to simultaneously explore amine linker and pyrrolidine nitrogen derivatives as illustrated in Schemes 3–5. Diamines were chosen to more fully understand orientation and geometry of functional groups in the binding pocket this fragment occupies. Heterocyclic amides 16a–m were prepared from intermediate 15 as illustrated in Scheme 3. We chose to focus on heterocycles because the focused screening revealed that benzoyl- or saturated linear or carbocycle- amides were inactive as PfPKG inhibitors.
Scheme 3. Synthesis of pyrrolidine amide PfPKG inhibitors.

Scheme 4. Synthesis of tertiary amine and sulphonamide PfPKG inhibitors.

Scheme 5. Diamine linker variations.

Using intermediate 15d and selected heterocyclic aldehydes, reductive amination under standard conditions (Scheme 4) provided tertiary amine products 17a and b to evaluate the potential role of an amide carbonyl. A sulphonamide 17c was prepared to compare this functional group to a corresponding amide. A series of alternatives to (R)-3-aminopyrrolidine were investigated to explore stereochemistry, sterics and ring size in the diamine linker using a 3-isothiazolyl amide. These derivatives were obtained from 3-chlorophenyl sulfone intermediate 16 as shown in Scheme 5, using chemistry analogous to that in Scheme 1.
Table 2 provides in vitro PfPKG data for these sets of compounds (IC50 values based on at least 2 independent experiments). As with 13a–c, a 3-chloro substituent was superior to fluorine at any position on the phenyl ring (13bvs.16b). This set of fluoro derivatives displays similar SAR compared to the corresponding chlorophenyl compounds. Among the diamine linker analogs 19a–d, there is a strong preference for (R)-3-amino pyrrolidine, with only the azetidine derivative 19a showing modest inhibition of the plasmodial enzyme. This information suggests that the linker interacts with a fairly well defined site on the enzyme. Tertiary amines 17a and b were essentially inactive, while sulphonamide 17c was weakly active, suggesting that a neutral, amide nitrogen is required (cf.17a/17b/17cvs.16l/16m). This result suggests that the relatively more planar amide functional group is preferred to the more tetrahedral sulphonamide in this matched pair.
Table 2. PfPKG inhibition-pyrrolidine derivatives.
| Cpd | % Inh@10 μM | IC50 (nM) | Cpd | % Inh@10 μM | IC50 (nM) |
| 16a | 900 | 16k | 38 | ||
| 16b | 360 | 16l | 10 | ||
| 16c | 660 | 16m | 620 | ||
| 16d | 13 | 17a | 5 | ND | |
| 16e | 15 | 17b | 5 | ND | |
| 16f | 74 | 17c | 40 | ND | |
| 16g | 25 | 19a | 2600 | ||
| 16h | 120 | 19b | 60 | ND | |
| 16i | 97 | 19c | 46 | ND | |
| 16j | 47 | 19d | 75 | ND |
Among the 3-chloro heterocyclic amides 16d–m, the 3-methyl pyrazole 16l, 4-isoxazolyl 16d and 3-isothiazolyl 16e were most potent with IC50 values less than 20 nM. By comparison, in our hands positive control 1 routinely provided PfPKG IC50 values in the 30–40 nM range. This value is an order of magnitude greater than the original report describing the discovery of 1.3 The two N-methyl pyrazoles 16l and 16m were measurably more potent than the corresponding unsubstituted pyrazoles 16i and 16j, suggesting that PfPKG favors lower polarity in this region of the inhibitor. Interestingly, among the various regioisomeric pairs of compounds, the isothiazoles 16e and 16f and isoxazoles 16d and 16g show evidence of measurably different potency versus PfPKG, while pyrazole derivatives (16h, 16i, 16k, 16l) do not. Addition of another heteroatom as in 16h results in an approximate 10-fold drop in potency compared to 16d/16e/16l.
This data shows there is measurable structure–activity in the amide and that the nature and location of heteroatoms in the structure play an important role interacting with PfPKG. More details on this aspect of SAR are under active investigation.
Conclusions
This research identified three points in a lead structure (Fig. 2) of a novel chemotype that resulted in multiple PfPKG inhibitors with improved potency compared to an established PfPKG inhibitor 1. In the phenyl ring, a 3-chloro substituent is preferred over hydrogen and fluorine. The methyl group in initial leads was replaced by a potency enhancing diethylamino moiety. To date, the preferred linker (R3, Fig. 2) is an (R)-3-amino pyrrolidine in which the ring nitrogen must be a heterocyclic amide. These more potent compounds, like 1, are highly selective versus human PKG and unlike certain other known PfPKG inhbitors, have no structural alerts based on a variety of in silico models.11–13 Structure–activity is expanding to investigate other aromatic substituents and potential replacements for the phenyl ring, as well as alternatives to the diethylaminomethyl moiety on the isoxazole scaffold, additional heterocycles and variations in the amino-pyrrolidine linker. Examination of the pharmaceutical properties (e.g. water solubility, in vitro metabolic and plasma stability, CYP inhibition) of these compounds in underway, with the aim of identifying a candidate for oral bioavailability and animal efficacy studies. Ongoing work is underway in cell biology studies to evaluate selected potent PfPKG inhibitors compared to 1. This work is in progress and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This research was supported by the Margaret and Herman Sokol Endowment to JJS and DPR and by NIH RO1-AI-133633-01.
Footnotes
†Electronic supplementary information (ESI) available: General procedures and compound characterization is available. NMR and mass spectral data on all tested compounds as well as general synthetic procedures and biological evaluation. See DOI: 10.1039/c9md00511k
References
- Taylor H. M., McRobert L., Grainger M., Sicard A., Dluzewski A. R., Hopp C. S., Holder A. A., Baker D. A. Eukaryotic Cell. 2010;9:37. doi: 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley C. H. BMC Med. 2015;13:67. doi: 10.1186/s12916-015-0316-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biftu T., Feng D., Ponpipom M., Girotra N., Liang G.-B., Qian X., Bugianesi R., Simeone J., Chang L., Gurnett A., Libertore P., Dulski P., Leavitt P. S., Crumley T., Misura A., Murphy T., Rattray S., Samaras S., Tamas T., Mathew J., Brown C., Thompson D., Schmatz D., Fisher M., Wyvratt M. Bioorg. Med. Chem. Lett. 2005;15:3296. doi: 10.1016/j.bmcl.2005.04.060. [DOI] [PubMed] [Google Scholar]
- Baker D. A., Stewart L. B., Large J. M., Boyer P. W., Ansell K. H., Jiménez-Diaz M. B., El Bakkouri M., Birchall K., Dechering K. J., Bouloc N. S., Coombs P. J., Whalley D., Harding D. J., Smiljanic-Hurley E., Weldon M. C., Walker E. M., Dessens J. T., Lafuente M. J., Sanz L. M., Gamo F.-J., Ferrer S. B., Hui R., Bousema T., Angulo-Barturén I., Merritt A. T., Croft S. L., Gutteridge W. E., Kettleborough C. A., Baker D. A. Nat. Commun. 2017;8:430. doi: 10.1038/s41467-017-00572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagris D. J., Birchall K., Bouloc N., Large J. M., Merritt A., Smiljanic-Hurley E., Weldon M., Ansell K. H., Kettleborough C., Whalley D., Stewart L. B., Boyer P. W., Baker D. A., Osborne S. A. Bioorg. Med. Chem. Lett. 2018;28:3168. doi: 10.1016/j.bmcl.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matralis A. N., Malik A., Penzo M., Moreno I., Almela-Armedariz M. J., Camino I., Crespo-Fernandez B., Saadeddin A., Ghidelli-Disse S., Rueda L., Calderon F., Osborne S. A., Drewes G., Boesche M., Fernandez-Alvaro E., Hernando J. I. M., Baker D. A. J. Med. Chem. 2019;62:9217. doi: 10.1021/acs.jmedchem.9b01099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo M., de las Heras-Duena L., Mata-Cantero L., Diaz-Hernandez B., Vazquez-Muniz M. J., Ghidelli-Disse S., Drewes G., Fernandez-Alvaro E., Baker D. A. Sci. Rep. 2019;9:7005–7018. doi: 10.1038/s41598-019-42801-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lededoer M. A., Ledford B. and Maltois F., WO2004022555, 2004.
- Tummalapalli S. R., Bhat R., Chojnowski A., Prorok M., Kreiss T., Goldberg R., Canan S., Hawryluk N., Mortensen D., Khetani V., Zeldis J., Siekierka J. J., Rotella D. P. ACS Med. Chem. Lett. 2018;9:210. doi: 10.1021/acsmedchemlett.7b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Chojnowski A., Gaskill K., DeMartini W., Goldberg R. L., Siekierka J. J. Mol. Biochem. Parasitol. 2011;176:90. doi: 10.1016/j.molbiopara.2010.12.008. [DOI] [PubMed] [Google Scholar]
- Yang H., Li J., Wu Z., Li W., Liu G., Tang Y. Chem. Res. Toxicol. 2017;30:1355. doi: 10.1021/acs.chemrestox.7b00083. [DOI] [PubMed] [Google Scholar]
- Yang H., Sun L., Li W., Liu G., Tang Y. Front. Chem. 2018;6:30. doi: 10.3389/fchem.2018.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T. E. H., Goodman J. M., Gutsell S., Russell P. J. Toxicol. Sci. 2018;165:213. doi: 10.1093/toxsci/kfy144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.