

Abstract

By combining cyclic voltammetry (CV) and shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS), the adsorption behavior of two alkynes, propargyl alcohol (PA) and 2-methyl-3-butyn-2-ol (MeByOH), undergoing hydrogenation on Pt basal plane single-crystal electrodes is investigated. It is found that PA and MeByOH give rise to strong surface sensitivities in relation to both hydrogenation activity and molecular fragmentation into adsorbed species such as CO. For PA, irreversible adsorption is strongly favored for Pt{100} and Pt{110} but is weak in the case of Pt{111}. It is suggested that the presence of the primary alcohol substituent is key to this behavior, with the order of surface reactivity being Pt{100} > Pt{110} > Pt{111}. In contrast, for MeByOH, strong irreversible adsorption is observed on all three basal plane Pt surfaces and we propose that this reflects the enhanced activity of the alkyne moiety arising from the inductive effect of the two methyl groups, coupled with the decreased activity of the tertiary alcohol substituent toward fragmentation. Pt{111} also exhibits singular behavior in relation to MeByOH hydrogenation in that a sharp Raman band at 1590 cm–1 is observed corresponding to the formation of a di-σ/π-bonded surface complex as the alkyne adsorbs. This band frequency is some 20 cm–1 higher than the analogous broadband observed for PA and MeByOH adsorbed on all other basal plane Pt surfaces and may be viewed as a fingerprint of Pt{111} terraces being present at a catalyst surface undergoing hydrogenation. Insights into the hydrogenation activity of different Pt{hkl} surfaces are obtained using quantitative comparisons between Raman bands at hydrogenation potentials and at 0.4 V vs Pd/H, the beginning of the double-layer potential region, and it is asserted (with support from CV) that Pt{110} is the most active plane for hydrogenation due to the presence of surface defects generated via the lifting of the (1 × 2) to (1 × 1) clean surface reconstruction following flame annealing and hydrogen cooling. Our findings are also consistent with the hypothesis that Pt{111} planes are most likely to provide semihydrogenation selectivity of alkynes to alkenes, as reported previously.
Keywords: semihydrogenation, substituent effect, SHINERS, platinum single crystal, electrochemistry
1. Introduction
Unsaturated alcohols are important reagents for the synthesis of fine chemicals and their selective hydrogenation plays a critical role in the pharmacology, perfumery, and food industries. Semihydrogenation of acetylenic bonds selectively to alkenes is a particular challenge in this context and is achieved via heterogeneous catalysis, typically using supported precious metal nanoparticles. The strong adsorption of unsaturated alcohols on metal surfaces through the unsaturated carbon–carbon bonds is key to this application and is also exploited in their widespread use as corrosion inhibitors.1−3 Understanding this interaction under relevant conditions is therefore of significant interest and the adsorption behavior of unsaturated alcohols on numerous transition metals has been investigated in both the gas and liquid phases.2,3
Propargyl alcohol (PA, or 2-propyn-1-ol) (Figure 1a) is one such compound that has found considerable attention, and whose adsorption behavior on Pt has been shown to resemble that of acetylene, which adsorbs through a nondissociative process involving a partial rupture of the C≡C bond.4,5 Electron energy loss spectroscopy (EELS) and Auger spectroscopy indicate that PA is adsorbed with the unsaturated carbon–carbon bond parallel to the Pt surface.5 Furthermore, electrochemical methods have been used to study the adsorption and redox behavior of PA in acid solutions, and a nondissociative adsorption on Pt was also postulated based on voltammetric measurements.6 Different products of the electrochemical reduction of PA on Pt have been reported, with propane, propylene, and ethane being observed previously,7,8 while electro-oxidation of PA was shown to produce the corresponding aldehyde.9−13 PA also strongly adsorbs on Pd in acid solutions,14−16 generating similar products to Pt during electroreduction but CO2 as the sole electro-oxidation product.14 The adsorption of PA on Ru16 and Au17,18 has also been studied.
Figure 1.

Schematic structures of (a) propargyl alcohol (PA) and (b) 2-methyl-3-butyn-2-ol (MeByOH).
Studies of PA also include the interaction of adsorbed PA with CO, methanol, and CO2.19,20 In particular, PA has attracted much attention in the context of mitigating global greenhouse gas emissions, offering the prospect of converting captured CO2 into high-value chemicals by cyclization with PA.21−25 Noble-metal free metal–organic frameworks have also demonstrated great potential for the cyclization of PA with CO2 at atmospheric pressure and room temperature.26,27
The growing application of PA is the driving force for studies of its adsorption on various metal surfaces, with the aim of improving catalyst activity and selectivity, using surface-modifying adsorbates as a common route for improving selective heterogeneous catalysts.28−30 The concept of intrinsic selectivity at the metal surface has also been explored.31,32 Different crystallite topographies (e.g., terraces, corners, edges, and kinks) lead to differences in reactivity and selectivity, due to their highly specific interactions with the adsorbates.33
The closely related molecule 2-methyl-3-butyn-2-ol (MeByOH) (Figure 1b) has also found attention as an unsaturated alcohol, differing from PA only by the presence of two methyl groups at the C1 position. The steric bulk of these methyl groups coupled with their electron induction effect in mitigating the alcohol-induced polarization of alkyne substituent electrons is expected to influence the alkyne bond orientation and reactivity as the molecule approaches metal surfaces during catalysis, impacting activity and selectivity. The catalytic hydrogenation of MeByOH has been used to demonstrate the distinct selectivity of cubic, octahedral, and cuboctahedral particles of Pt.31 The results indicate that semihydrogenation of the alkyne occurs preferentially on terrace sites, while further hydrogenation to the fully saturated alcohol occurs over edge sites. Our previous work has also found that a strong association of alkynes with Pt defect sites to produce a long-lived di-σ/π-alkene surface complex allows for deep hydrogenation of this intermediate to the alkane.33
Previously, researchers have combined cyclic voltammetry (CV) and surface-enhanced Raman spectroscopy (SERS) to probe the changes at the catalyst surface with modifications.34−37 With these techniques, not only can the distribution of different surface sites be quantitatively determined, but also the adsorbed species can be identified by monitoring their molecular vibrations. SERS has also been shown to be a powerful means for determining the orientation of the surface-adsorbed molecules; due to surface selection rules, the geometry of the adsorbed molecules and the bond orientation relative to the surface play a significant role in determining the relative intensity of individual SERS bands.39,40 Moreover, by electrochemically evolving hydrogen at the electrode surface, one can controllably recreate the conditions of liquid phase catalytic hydrogenation, thus allowing the origins of catalytic selectivity to be explored.
The breakthrough of shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) has significantly expanded the reach of SERS, allowing observation of surface-enhanced Raman signals on non-SERS-active substrates such as well-defined single-crystal surfaces.38 It was demonstrated that Au nanoparticles (NPs) coated with a thin shell of SiO2 (referred to here as Au@SiO2) deposited on a single-crystal electrode could be used to observe SERS even in the absence of intrinsic roughness and without requiring coinage metals such as Cu, Ag, or Au to be used as the working electrode. Hence, weakly SERS-active substrates such as Pt may be endowed with SERS activity because of the ‘hotspots’ created around Au@SiO2 NPs on the surface. Previously, we combined SHINERS with the electrochemical methods to monitor the vibrational modes of alkynes and their intermediates adsorbed on Pt single-crystal surface in situ.33
Here, we exploit the techniques of CV and in situ electrochemical SHINERS to investigate the adsorbed intermediates formed during the hydrogenation of PA and MeByOH on Pt{hkl} single crystals.
2. Experimental Section
2.1 Chemicals
HAuCl4 (Johnson Matthey, Assay 41.79 wt %), sodium citrate dehydrate (Sigma-Aldrich, >99%), (3-aminopropyl)trimethoxysilane (APTMS, Alfa Aesar, 97%), sodium silicate (Sigma-Aldrich, reagent grade), H2SO4 (BDH, Aristar grade), perchloric acid (BDH, Aristar grade), propargyl alcohol (Sigma-Aldrich, 99%), and 2-methyl-3-butyn-2-ol (Alfa Aesar, 98%) were used as received and all aqueous solutions were prepared using ultrapure water (18.2 MΩ cm).
2.2. Nanoparticle Synthesis
Fifty-five nanometers diameter Au seeds were prepared as follows: 100 mL of 0.01% (by mass) HAuCl4 aqueous solution was refluxed at 112 °C. After 30 min, 600 mL of a trisodium citrate solution (38 mM) was added and the reaction mixture was then refluxed for another 30 min. Au@SiO2 NPs were prepared by modification of the method reported by Tian et al.38 The detailed procedure can be found elsewhere.41 Briefly, 0.4 mL of a freshly prepared 1.0 mM aqueous solution of (3-aminopropyl)-trimethoxysilane (APTMS) was added to 30 mL of the Au seeds under vigorous stirring. 3.2 mL of 0.54 wt % sodium silicate solution (pH = 10.28) was added to the mixture. The reaction mixture was kept at 70 °C for 13 min to form pinhole-free Au@SiO2 NPs with an ultrathin shell (2 nm).42 Characterization of the synthesized SHINERS particles was performed by transmission electron microscopy (TEM) using a JEOL 2100 instrument with a LaB6 filament operating at 200 kV. The samples were prepared by dispersing the centrifuged Au@SiO2 NP droplet in ethanol and dropping the suspension onto a lacey carbon film over a 300-mesh copper grid. An example TEM image showing the NPs is given in the Supporting Information (Figure S1) in which the SiO2 shell can be clearly observed.
2.3. Electrochemical Apparatus
A three-electrode electrochemical cell, described in detail previously,36 was used for single-crystal electrochemical measurements. Briefly, it consisted of two compartments, one housing the Pt{hkl} single-crystal working and Pt mesh counter electrodes, the other containing a Pd/H reference electrode. These compartments were joined by two bridges, one above and one below the fill level of the electrolyte, so as to allow solution and gas to flow between the compartments. The cell was fitted with three inlets, one to admit nitrogen to purge the electrolyte of oxygen, a second to admit hydrogen to charge the reference Pd/H electrode, and a third, also connected to a nitrogen supply, to maintain a static inert atmosphere above the electrolyte. Voltammetric data were recorded in degassed 0.1 M aqueous sulfuric acid at a sweep rate of 50 mV s–1 unless otherwise stated, and all potentials throughout this paper are quoted against the Pd/H reference electrode (E = 0.05 V vs RHE). Pt{hkl} single-crystal electrodes were prepared using the Clavilier bead method and cleaned by flame annealing prior to use.43 Adsorption experiments with PA and MeByOH were performed by dipping the clean Pt{hkl} crystal into a small amount of pure PA or MeByOH liquid for 60 s, followed by rinsing the electrode with ultrapure water to remove the excess organic liquid. Estimation of the fractional surface covered by organic species after initial adsorption and following different potential treatments was achieved by integrating the charge under the CVs from 0 to 0.4 V, which is associated with hydrogen adsorption/desorption at available Pt sites, and dividing this by the charge measured over the same potential region on the pristine Pt{hkl} electrode.
In situ SHINERS measurements were performed in a custom-built spectroelectrochemical cell, also described previously.44 The Pt{hkl} single-crystal electrode was modified with Au@SiO2 NPs by allowing a small droplet of the NPs suspended in water to dry out on the electrode surface to leave a film of NPs behind. The SHINERS spectra were recorded at the surface of the Pt electrode in the presence of 0.1 M H2SO4 solution containing 0.1 M PA or MeByOH. To create hydrogenation conditions, hydrogen gas was evolved at the working electrode by holding the potential at −0.05 V. Hydrogen bubbles forming on the surface of the electrode, which might affect the detection of SHINERS signals, were removed by flowing the electrolyte through the cell using a peristaltic pump. Spectral acquisition was started a few seconds after changing the applied potential, after which time the signal was considered to be stable. To electrochemically clean the surface of the electrode without roughening the surface, potential cycling was performed between −0.1 and 0.8 V at 100 mV s–1 in 0.1 M H2SO4 prior to all SHINERS experiments.44
2.4. Raman Spectroscopic Measurements
Raman spectroscopic measurements were performed using a LabRam Raman microscope (Horiba JobinYvon Ltd.) fitted with a HeNe laser (λ = 633 nm, output power 16 mW), with data recording and processing performed using the proprietary LabSpec software (Horiba JobinYvon).44
3. Results
3.1. CV Studies of PA and MeOH Adsorption on Pt{hkl} Electrodes
PA adsorption on the three basal plane Pt single-crystal electrodes was investigated in 0.1 M H2SO4 using cyclic voltammetry. CVs were performed over different potential ranges to explore how oxidizing and reducing environments affected the surface coverage of chemisorbed species.
Figure 2a shows the CVs of the Pt{111} single-crystal electrode before (black line) and after (pink line) PA adsorption. In the latter case, an upper anodic limit of 0.4 V was used initially to quantify the adsorption behavior alone, in the absence of any unwanted electro-oxidation. The clean crystal CV exhibits the characteristic features that are typical of the Pt{111} electrode, with symmetric hydrogen adsorption/desorption currents observed between 0 and 0.3 V and sulfate adsorption/desorption occurring between 0.3 and 0.5 V (including the pair of sharp “butterfly” peaks at 0.44 V, indicative of the large Pt{111} terraces). The effect of PA adsorption is to slightly reduce the charge associated with these electrosorption processes due to blocking of the surface sites. Quantitative comparison of the two CVs indicates that only approximately 10% of the Pt{111} electrode surface area is blocked as a result of PA adsorption, suggesting that the interaction with PA and this crystal surface is rather weak.
Figure 2.

CVs of PA adsorption on (a) Pt{111}, (b) Pt{110}, and (c) Pt{100} single-crystal electrodes in 0.1 M H2SO4: Clean Pt{111} before PA adsorption (black line), after PA adsorption (pink line), after oxidative cycling (blue line), and after hydrogenation (red line). Scan rate: 50 mV·s–1.
A common observation when small organic molecules come into contact with Pt electrodes is for them to partially fragment to form CO and other small organic species, which adsorb strongly to the Pt surface sites.42,45,46 To assess how much of the Pt{111} surface was blocked with CO and other molecular fragments capable of being anodically stripped from the electrode, as a result of PA adsorption and decomposition, the working electrode potential was cycled rapidly (at 200 mV s–1 for 3 cycles) between 0 and 0.8 V. While some studies have suggested that CO may persist on surfaces above this potential range,47 our previous work has indicated that, under the conditions of low CO coverage expected here, 0.8 V is a sufficiently positive potential to oxidatively remove completely any adsorbed CO,33 and this was confirmed by the absence of any further CO stripping currents after this potential treatment. Figure 2a (blue line) shows the effect of this cycling procedure, which indicates the recovery of most of the electrochemically active surface area after such a treatment. However, only a small pair of peaks at 0.44 V was observed compared to the CV prior to PA adsorption, indicating that the long-range order of the Pt{111}-sulfate ion adlayer surface was still disrupted by some remaining adsorbed PA, in addition to any other organic species that could not be removed by this anodic treatment. Finally, to explore the effect of hydrogenation, hydrogen gas was subsequently evolved on the surface by performing cyclic voltammetry on the same PA-decorated electrode in the potential range −0.1 and 0.8 V (Figure 2a, red line). The result was further recovery of the Pt{111} long-range order with the pair of sharp peaks at 0.44 V becoming more pronounced, indicating that the Pt{111} electrode was almost as clean as the pristine surface.
We note that the above approach to investigating the site occupation of the Pt surface, in which oxidative potentials are applied prior to the hydrogenation treatment, may lead to complications in which partially oxidized organic species might persist on the surface and then be susceptible to hydrogenation. However, by cycling the potential up to 0.8 V, one can ensure that the surface is at least free from CO as well as potentially other fragmented organics, so that the hydrogenation behavior of the intact adsorbed alkyne can then be probed on a more pristine surface. The alternative approach of applying hydrogenation conditions first was not found to be as informative as the above procedure, so was not explored further.
To investigate the surface structure sensitivity of PA adsorption, the same tests were performed on Pt{110} and Pt{100} single-crystal electrodes. As shown in Figure 2b,c, PA adsorbs much more extensively on these two crystal surfaces, with close to 50% coverage on Pt{110} and 90% on Pt{100}, based on the integrated charge between 0 and 0.4 V. In the case of Pt{110} (Figure 2b), a significant fraction of the surface sites are released by oxidative removal of the CO/molecular fragments (blue line) and after hydrogenation the pristine surface is almost completely recovered (red line). Similarly, the Pt{100} surface is largely recovered by oxidative cycling (Figure 2c, blue line), but hydrogenation had no further effect on the surface.
To more easily compare these changes, the relative surface coverages associated with each of the treatments are plotted in Figure 3a. In each case, this was calculated based on the relative change in the integrated charge in the hydrogen adsorption/desorption potential region, which gives a good indication of the availability of the Pt sites. The red bars show the initial fraction of the sites blocked in total by exposure to PA and its molecular fragments formed upon adsorption, while the blue and yellow bars represent the fraction of the blocked sites after the oxidative and hydrogenation treatments, respectively.
Figure 3.

Fraction of the sites blocked by adsorption of (a) PA and (c) MeByOH calculated by charge integration of CV data, before (red bars) and after oxidative (blue bars) and hydrogenation (yellow bars) treatments. Data plotted in (b, d) show the fraction of the sites released by CO/molecular fragment oxidation (blue) and the sites released by hydrogenation (yellow) normalized to the initial charge blocked.
Together these results indicate that PA adsorption is highly surface sensitive, with binding affinity increasing in the order Pt{111} < Pt{110} < Pt{100}. How this initial coverage is separated into CO/oxidizable material and reducible material across the different Pt surfaces is also interesting. First, we note that the amount of the CO/oxidizable material generated (Figure 3a, difference between the red and blue bars) broadly scales with the total amount of the material initially adsorbed (Figure 3a, red bars) so follows the same trend as above. In contrast, the absolute amount of PA removed by hydrogenative treatment (Figure 3a, difference between blue and yellow bars) is most substantial for the Pt{110} case, which we tentatively attribute to a higher number of surface defects present due to the well-documented reconstruction that occurs on this surface to form a defective mixture of 1 × 1 and 1 × 2 phases.48 We will return to this observation later in the context of the recorded Raman spectra. The material remaining on the surface after both oxidative and hydrogenative treatments (Figure 3a, yellow bars) is believed to be irreversibly bound organic material that cannot be easily oxidized below 0.8 V and is not susceptible to hydrogenation under our experimental conditions. The precise identity of these adspecies is not clear but, based on the behavior of acetylene and PA on Pt surfaces18,49,50 we postulate that these may be adsorbed oxygenated organic fragments, larger and more strongly bound than CO, that result from the partial decomposition of PA as it adsorbs on the Pt surface. Further analysis of these is beyond the scope of the present work, but we note that the fraction of the surface occupied by this remaining material is relatively small and appears to scale with the initial total surface coverage. To normalize for the influence of initial uptake of PA at each Pt{hkl} electrode, the fractions of sites released by the oxidative and hydrogenation treatments are plotted in Figure 3b, in this case divided by the initial PA blocking of the electrode surface.
It is evident from Figure 3b that a clear trend emerges for PA adsorption whereby the proportion of the CO/oxidizable material formed increases in the order Pt{111} ≈ Pt{110} < Pt{100}. This is in accordance with expectations based on previous studies of primary alcohols such as methanol, ethanol, ethylene glycol, and glycolic acid51−53 adsorbed on the Pt{hkl} electrodes with the strongest “CO poisoning” observed for Pt{100} and the least for Pt{111}. Interestingly, precisely the opposite trend is observed for the relative amounts of the material corresponding to hydrogenative stripping. It is worth mentioning briefly the possible influence of the anion here. Clearly, sulfate and hydrogen are both displaced by the irreversible adsorption of the organic molecule following dipping into the neat liquid. Hence, it is difficult to envisage a displacement of the organic molecule by sulfate anions following an oxidative potential sweep (more likely is the subsequent filling of the vacated sites by sulfate anions). This may change for a more strongly adsorbing anion such as iodide. In the case of PA on Pt{111}, the blocking of anion adsorption (as well as hydrogen adsorption) is less marked. It could be that the sulfate anions are successfully competing for the sites relative to the alkyne in this case. However, this would require a substantial increase in the binding of the sulfate anions as a function of potential and a decrease in adsorption strength of alkyne relative to Pt{110} and Pt{100}. Electro-oxidation of small organics has been correlated rather precisely with the oxidative charge with little role of the anions,54 so we do not believe that the anion is impacting the observed behavior. Similar behavior was observed in perchlorate electrolytes, confirming this assertion.
To explore the substituent effects on the adsorption and hydrogenation behavior of unsaturated alcohols, the same CV experiments as above were performed on MeByOH, the methyl-substituted analogue of PA. We first consider the electrochemical behavior of the Pt{111} electrodes after exposure to MeByOH, and the CVs obtained are shown in Figure 4a. In stark contrast to PA, MeByOH exhibits a strong affinity for Pt{111}, with more than 80% of the surface becoming blocked by MeByOH before any electrochemical treatment (Figure 4a, pink line). After cycling between 0 and 0.8 V to remove CO and similar oxidizable material, only approximately 10% of the surface area was recovered (Figure 4a, blue line), indicating a relatively limited amount of fragmentation of the molecule upon adsorption on Pt{111}. However, hydrogenation of the adsorbed molecules occurs readily, with a pair of sharp peaks emerging at 0.44 V and recovery of more than 95% of the surface after evolving hydrogen gas (Figure 4a, red line).
Figure 4.

CVs of adsorption on (a) Pt{111}, (b) Pt{110}, and (c) Pt{100} single-crystal electrodes in 0.1 M sulfuric acid: before (pink) and after (blue) oxidation, after hydrogenation (red), and the clean Pt{111} (black). Scan rate: 50 mV·s–1.
Qualitatively similar behavior was exhibited on the Pt{110} and Pt{100} electrodes (Figure 4b,c), although again, quantitative differences were observed as demonstrated by the fraction of the blocked sites calculated from the integrated charges, as shown in Figure 3c. As observed with PA, the initial coverage (Figure 3c, red bars) is higher for Pt{100} than for Pt{110}, while more of the surface is recovered by hydrogenation for Pt{110} (Figure 3c, difference between the blue and yellow bars). The CO/oxidizable molecular fragment coverage (Figure 3c, difference between the red and blue bars) also appears to scale with initial coverage for Pt{110} and Pt{100}, but this trend is not followed by Pt{111}, which, combined with the large fraction of the surface released by hydrogenation, points to a rather unique interaction between MeByOH and Pt{111}. As discussed above in the context of PA, when the fraction of the sites released by each treatment is normalized to the total amount of surface blocking by adsorbed MeByOH, there is a negative correlation between the proportion of the hydrogenative product released and the amount of the CO/oxidizable molecular fragments generated (Figure 3d). However, it should be noted that in comparison with PA, a lower proportion of CO and oxidizable material is formed (and hence a greater fraction of hydrogenative products) at Pt{hkl}, consistent with the expected lower reactivity of tertiary alcohols relative to primary alcohols toward fragmentation.55
3.2. In Situ SHINERS Studies of PA and MeByOH Adsorption on the Pt{hkl} Electrodes
It is clear from the above CV measurements that PA and MeByOH exhibit different adsorption and hydrogenation behaviors on the Pt{hkl} surfaces. To further study this, in situ SHINERS experiments were carried out by passing electrolyte solutions containing 0.1 M alkyne in 0.1 M H2SO4 over the electrode surfaces in a flow cell described previously.56
We first focus on the interaction of PA with a Pt{111} single-crystal electrode decorated with Au@SiO2 NPs utilized as the working electrode. Prior to introducing the alkyne, control measurements were performed in 0.1 M H2SO4 and the typical spectra are shown in Figure S2 (Supporting Information), in which the expected Raman bands associated with Pt–O and Pt–H are observed under oxidative and hydrogenation conditions, respectively. To probe the behavior of PA on Pt{111}, SHINERS experiments were performed in 0.1 M PA/0.1 M H2SO4 at two different potentials corresponding to hydrogen evolution conditions (−0.05 V) and the beginning of the double-layer potential region (0.4 V), and the Raman spectra obtained are shown in Figure 5a,b.
Figure 5.

SHINERS spectra of 0.1 M PA in 0.1 M H2SO4 adsorbing on (a, b) Pt{111}, (c, d) Pt{110}, and (e, f) Pt{100} single-crystal electrode surfaces. Measurements were performed at (a, c, e) hydrogen evolution potentials and (b, d, f) 0.4 V. The spectra are vertically offset for ease of comparison.
Both of these spectra exhibit similar bands, some of which are readily identified as being due to PA adsorbed on the Pt surface or in bulk solution, and the proposed band assignments are given in the Supporting Information (Table S1). The most prominent feature of these spectra is a broad and intense band observed at 1570 cm–1, which is assigned to the alkenic stretch of a di-σ/π-bonded surface complex formed as the alkyne adsorbs on the Pt.33,57,58 The red shift in frequency of the band compared to a normal nonconjugated alkene stretch (∼1640 cm–1) is due to a strong interaction of the double bond with the Pt surface.59 At 0.4 V, this band was slightly more intense than at hydrogen evolution potentials, suggesting only marginal loss of the steady-state surface concentration of this species by hydrogenation. Other important bands include the C–H stretching vibrations appearing at 2930 cm–1 and a weak band at 2122 cm–1, attributed to the C≡C stretch of the intact alkyne, although the associated C–C≡C skeletal stretching peak (usually at 314 cm–1) is absent. The appearance of a weak band at ∼2000 cm–1 confirms the presence of adsorbed CO, in agreement with expectations based on the CV data, and the bands below 500 cm–1 are consistent with Pt–C stretches on the surface, either from the adsorbed CO or other fragmented organic species.60 In addition, the bands at 1268 cm–1 (in plane O–H deformation vibration, for primary and secondary alcohols), 1148 cm–1 (saturated tertiary alcohol C–O stretch or C–O stretch of secondary alcohol), 1000 cm–1 (characteristic stretch of primary alcohol C–C–O), 875 cm–1 (secondary alcohol C–C–O stretch), and 678 cm–1 (alcohol O–H out of plane deformation) may be ascribed to vibrations of primary, secondary, and tertiary alcohol molecular decomposition products.61 We note at this point that the majority of these alcohol bands are absent for the tertiary alcohol containing MeByOH, as shown below in Section 3.2. Hence, surface reactivity at both the alkyne and alcohol substituents is indicated via spectroscopy in the case of PA.
Qualitatively similar spectra were also observed for Pt{110} and Pt{100} surfaces in the presence of PA, although the relative intensity of some of the bands was found to vary (Figure 5c–5f). Most notably, the band at 1570 cm–1 associated with the di-σ/π-bonded surface complex is more intense than on Pt{111}, particularly at 0.4 V. However, one must be careful when comparing absolute intensities of SHINERS bands of different Pt{hkl} electrodes due to the likely different Raman enhancement factors. Hence, to aid quantitative comparison of the relative coverages of the di-σ/π-bonded surface complex as a function of potential and Pt surface structure, the ratios of the integrated signal intensity of the normalized 1570 cm–1 band at 0.4 V and under hydrogenation conditions are plotted for each surface in Figure 6. In this way, intensity variations due to different surface structures/different SHINERS particle distributions may be eliminated. From this figure, it is clear that the steady-state surface coverage change of the di-σ/π-bonded surface complex at 0.4 V compared to hydrogen evolving potentials is highest for Pt{110} (a value of unity signifies that no change in the intensity has occurred after hydrogenation). Furthermore, the Pt{110} surface appears to be most active toward hydrogenation of the complex, since it also exhibits the most significant decrease in the 1570 cm–1 band intensity under hydrogen evolution conditions. This observation is consistent with the CV coverage data plotted in Figure 3a, in which the Pt{110} surface exhibits the most substantial recovery of the surface sites after hydrogenation. The smallest change is found with Pt{100}, again in accordance with the CV data in that only a negligible amount of sites were released after hydrogenation of the PA residues on this surface.
Figure 6.
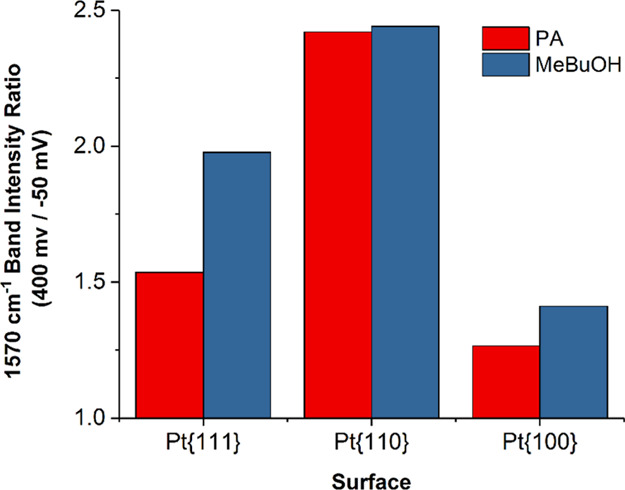
Ratio of the integrated intensities of the di-σ/π-bonded surface complex SHINERS band at 1570 cm–1 at 400 and −50 mV for PA and MeByOH.
Based on our CV and SHINERS data, combined with the known behavior of primary alcohols, we propose the pathways summarized in Scheme 1 for the adsorption of PA on the Pt{hkl} electrodes. Either or both of the two reactive centers of the PA molecule, namely, the alkyne group and/or the primary alcohol position, may interact with the Pt surface and undergo different sequences of elementary steps leading to chemisorption. Interaction of the primary alcohol group with Pt leads directly to fragmentation due to the known electrochemical activity of the C–H bond in the C1 position (Scheme 1, route (i)). Furthermore, primary alcohol reactivity on Pt is highly surface sensitive, with adsorption/decomposition on the Pt{100} surfaces being strongly favored, whereas for Pt{111} this pathway is disfavored.51−53 Again, this would be consistent with more adsorbed CO derived from the primary alcohol substituent being formed on Pt{100} compared with Pt{111}, as reported in the CV data from Figures 2 and 3. Alternatively, PA may interact with Pt via the alkyne group to form the di-σ/π-bonded surface complex (Scheme 1, route (ii)), which as noted above is favored on the defect sites and is therefore prevalent on the reconstructed Pt{110} surfaces. Third, we consider the possibility that strong tilting of the C1 carbon of the σ/π-bonded surface complex toward the Pt surface may allow the additional involvement of the alcohol group in the Pt surface interaction (Scheme 1, route (iii)), thus leading to other chemisorbed species. It is also feasible that this dual interaction of both functional groups with the Pt surface occurs via route (i), as indicated by the diagonal arrows in Scheme 1.
Scheme 1. Proposed Adsorption and Fragmentation Pathways of PA on Pt Surfaces Based on Refs12, 59.

In situ SHINERS was next performed for MeByOH solutions on the three Pt{hkl} surfaces and the spectra are presented in Figure 7. Broadly similar spectra to those observed in the presence of PA were observed, albeit with a number of important differences. Strong bulk alkyne peaks are observed at 2116 cm–1 (C≡C stretch) and 710 cm–1 (CH deformation vibration of alkyne) together with the peaks at 1435 and 1374 cm–1, which may be ascribed to the asymmetric and symmetric vibrations of the methyl groups (these are of course absent from the PA spectrum because it does not contain any methyl groups). There are some vibrations (1173, 1050, and 884 cm–1) that may be interpreted as being associated with the tertiary alcohol substituent, for example, including the 1294 cm–1 band, which we assign either to a tertiary alcohol in plane O–H deformation or an alkyne C≡C–H wagging vibrational overtone.61
Figure 7.

SHINERS spectra of 0.1 M MeByOH in 0.1 M H2SO4 adsorbing on (a, b) Pt{111}, (c, d) Pt{110}, and (e, f) Pt{100} single-crystal electrode surfaces. Measurements were performed at (a, c, e) hydrogen evolution potentials and (b, d, f) at 0.4 V. The spectra are vertically offset for ease of comparison.
The most striking difference is found in the case of MeByOH on Pt{111}, which at hydrogen evolution potentials (Figure 7a) exhibits a sharp and intense peak at 1590 cm–1 superimposed on the broad 1500–1650 cm–1 band associated with the di-σ/π-bonded surface complex. While we cannot be certain as to the precise origin of this sharp band, based on the Raman shift we postulate that this reflects one specific molecular orientation of the di-σ/π-bonded surface complex, which is favored under hydrogenation conditions and which causes the alkenic stretch of the complex to more strongly satisfy the surface selection rules for intense Raman scattering. In addition, we tentatively suggest that the positive frequency shift of around 20 cm–1 as compared with the analogous band in PA on Pt{111} could be ascribable to a change in the interaction of the alkenic bond of the di-σ/π-bonded surface complex with the surface, as might be expected from a configuration more tilted away from the surface. By contrast, the broadbands in this spectral region exhibited on Pt{110} and Pt{100}, as well as Pt{111} at 0.4 V, reflect a multiplicity of various orientations of the di-σ/π-bonded complex that coexist on the surface. Together with the CV behavior, this further supports a unique interaction between MeByOH and Pt{111}. The absence of this behavior in the case of PA suggests that the strong interaction is brought about by the presence of the methyl substituents, suggesting an inductive, hydrophobic, or steric effect. We note that comparable Raman spectra were previously observed in the case of MeByOH adsorption on polycrystalline Pt under hydrogen evolution conditions, as well as at 0.4 V when the defect sites of polycrystalline Pt were blocked with polyvinylpyrrolidone (PVP), and the sharp Raman band was attributed to the di-σ/π-bonded surface complex in a tilted arrangement on the Pt{111} facets.33 Our present observations suggest that in ref (33), the molecule was indeed binding specifically to Pt{111} terraces present on the polycrystalline surface. Hence, the observation of such a Raman band for MeByOH under hydrogenation conditions is proposed as a method of fingerprinting the presence of the Pt{111} terraces in any particular Pt hydrogenation catalyst.
We also note an increase in the intensity of the C–H stretching bands for all Pt{hkl} surfaces in the 2820–2980 cm–1 range for MeByOH (Figure 7) compared to PA (Figure 5), which reflects the presence of the methyl substituents in MeByOH that are not present in PA. Moreover, this band appears to undergo an increase in the intensity when hydrogen evolution potentials are applied compared to 0.4 V, and this effect is most pronounced in the case of Pt{111}. This suggests that the favored orientation(s) under hydrogenation conditions bring the methyl groups either closer to the surface, or again help satisfy the relevant surface selection rules, resulting in a stronger Raman signal. Furthermore, adsorbed CO is seen as a product of MeByOH adsorption on Pt{hkl} as signified by the presence of the peaks at 2000 and 490 cm–1 (Pt–C stretch) with the most intense CO stretch peak occurring for potentials at which hydrogen is not being generated. Souto et al. proposed that the formation of CO from tertiary alcohols may arise from fragmentation of the molecule via the formation of acetyl surface species, which subsequently collapse to produce adsorbed CO.54
Quantitative comparison of the integrated change in the di-σ/π-bonded surface complex peak intensity between 0.4 V and hydrogenation potentials is shown for each of the Pt surfaces in Figure 6. Notably, the changes in the intensity observed for MeByOH on Pt{110} and Pt{100} are comparable to those measured for PA on these surfaces. In contrast, a much more substantial change is observed for the case of MeByOH on Pt{111} than for PA, which is in keeping with the large fraction of the surface sites released by hydrogenation in the CV measurements and is again consistent with a unique interaction of MeByOH with this surface.
In Scheme 2, a summary of the adsorption and fragmentation mechanism for MeByOH on Pt{hkl} is proposed based on the CV and SHINERS data. Unlike the pathways shown for PA in Scheme 1, we propose that MeByOH is limited to a single route of interaction with Pt via the alkyne functionality, as the tertiary alcohol lacks the electrochemically active C–H bond at the C1 position that facilitates route (i) in Scheme 1. We also consider the possibility that the steric presence of the methyl groups in MeByOH may lock the di-σ/π-bonded surface complex in a specific orientation in which the OH group is pointed toward the Pt surface, which may be particularly stable in the case of Pt{111} due to the more compact atomic structure.
Scheme 2. Proposed Adsorption and Fragmentation Pathways of MeByOH on the Pt Surfaces Based on Ref59.

4. Discussion
Before we discuss the different adsorption and reaction behaviors of PA and MeByOH on the Pt surfaces, it is worth briefly considering the stability of these molecules in acidic solutions even in the absence of a solid catalyst. We note that tertiary α-acetylenic alcohols such as MeByOH may be susceptible to the Meyer–Schuster or Rupe rearrangements to form the corresponding α,β-unsaturated aldehyde or ketone. These reactions are acid-catalyzed but according to the literature62,63 carboxylic acids are much more effective than mineral acids at facilitating these processes, and if the latter are used in isolation, they are required at high concentrations with elevated temperature. Therefore, the relatively modest conditions used during the SHINERS measurements (0.1 M H2SO4 at room temperature) would not be considered aggressive enough to facilitate these rearrangements, and we saw no evidence in the Raman spectra to suggest otherwise. It is also worth noting that the PA and MeByOH used in our experiments were not 100% pure and contained impurities between 1 and 2%. At the dilution level used in our SHINERS experiments (0.1 M alkyne), any such impurities would be unlikely to contribute significantly to the observed response, and this is supported by the absence of any erroneous Raman bands. In the case of the neat liquid adsorption experiments, we cannot rule out the involvement of impurities, but given the strong adsorption behavior of alkynes in general on Pt, we do not believe these to be substantial.
In terms of their interaction with Pt, the first key difference between the two molecules investigated in this study is that MeByOH generally has a strong affinity for all basal Pt surfaces, while PA binds strongly to Pt{110} and Pt {100} but only very weakly to Pt{111}. This is evident from both the electrochemical response and the SHINERS data. We propose two possible reasons for this: (i) the presence of the methyl groups in MeByOH inductively increases the electron density on the C≡C bond, improving electron availability for binding to Pt; (ii) the presence of the methyl groups makes MeByOH more hydrophobic than PA, causing the equilibrium between the aqueous phase and surface-adsorbed phase to be shifted in favor of the latter. The differing hydrogenation behavior of the two molecules on the different surfaces suggests that the electronic effect (i) may be more likely, although the specific interactions with regard to the differing behaviors of PA and MeByOH toward Pt{111} are not explained solely on the basis of these two factors.
The surface sensitivity of PA adsorption suggests that the lower coordinate sites on the Pt{110} and Pt{100} planes compared to the close-packed Pt{111} arrangement provide a more active surface for adsorption and decomposition, as evidenced by the more substantial surface blocking observed in the initial electrochemistry, the majority of which in all cases could be attributed to the adsorbed CO/oxidizable material. We note that a similar reactivity trend was also observed by Wain et al. for the molecule ethyl pyruvate on Pt{hkl}.64 The surface sensitivity in the case of MeByOH is more complicated, most notably because of the unusually strong interaction of this molecule with Pt{111,} which leads to a unique binding mode of the di-σ/π-bonded complex that appears to be very stable under hydrogenation conditions. It remains uncertain whether this is a precursor to hydrogenation or a spectator species, but the fact that the vast majority of the surface sites can be easily recovered by hydrogenative treatment (according to the electrochemistry data) would suggest that the former is more likely. The fact that the peak frequency associated with the di-σ/π-bonded surface complex of MeByOH is upshifted by ∼20 cm–1 under hydrogenation conditions on Pt{111} compared to other surfaces is an indication that the C=C bond in this particular intermediate is less activated toward deep hydrogenation to the alkane. Such behavior is not observed for PA, which may be attributed to the primary alcohol substituent. Indeed, the strong interaction of the primary alcohol group with the Pt electrode surface may be a key factor in its binding and since this is most pronounced for Pt{100} surfaces in accordance with previous studies,51−53 the absence of strong binding of PA on Pt{111} may simply reflect the relatively poor reactivity of the primary alcohol compared with all other Pt{hkl} surfaces. It is clear that differences in reactivity of PA/MeByOH toward Pt strongly correlate with the expected variation in reactivity between primary and tertiary alcohols.55 This opens up the possibility that the interaction between PA and Pt is not exclusively via the alkyne C≡C bond, but binding may also occur via the alcohol group (Scheme 1). Presumably, the presence of the methyl substituents attached to the alcohol carbon atom would sterically hinder such an interaction in the case of MeByOH, suggesting that it is forced to bind exclusively via the alkyne (Scheme 2). We stress that the reaction of the alcohol group in PA (Scheme 1, route (i)) is electrochemical in nature and occurs via the C–H bond, which in our case is facilitated by the stability of the released proton in the aqueous electrolyte; in the absence of such a driving force, for example, in nonaqueous solvents, PA may also be limited to reaction via the alkyne group alone.
A further general observation in the context of CO is that the intensity of the CO band in all of the SHINERS spectra is found to decrease upon switching from 0.4 V to hydrogen evolution potentials. This is unexpected given that CO binds very strongly to Pt and is unlikely to be substantially displaced by evolving hydrogen. However, these spectra represent a dynamic system in which organic species are being adsorbed and desorbed from solution, so the competitive displacement of CO by these reacting species is more likely.
An added complication in the context of surface sensitivity studies at single-crystal electrodes is the issue of defects, which, as discussed above, are more prevalent in the case of Pt{110} due to the known surface reconstruction.33,48 This likely leads to a correlation between defect density and hydrogenation activity in that, with the exception of MeByOH on Pt{111}, which appears to be a special case, Pt{110} is associated with the largest total surface area recovered by hydrogenation in the electrochemistry data (Figure 3a,c) as well as the largest decrease in the steady-state coverage of the di-σ/π-bonded complex Raman band upon exposure to hydrogenation conditions (Figure 6). Moreover, in ref (33) it was shown that the transformation of alkyne → alkene → alkane was facile at defects but strongly inhibited at Pt{111} terrace sites and furthermore that decoration of defect sites by both bismuth adatoms and/or PVP could lead to enhanced selectivity toward the alkenic product. It is worth noting here the equivalence of the electrochemically induced hydrogenation conditions used in this work and the H2-saturated liquid environments commonly employed in catalytic hydrogenation. On this basis, we propose that the above observations of surface structure sensitivity may in principle be extended more broadly to selective hydrogenation over Pt catalysts, but further work would be required to confirm this.
5. Conclusions
The electrochemical adsorption and hydrogenation of PA and MeByOH under well-defined conditions on Pt single-crystal electrodes have been investigated using an in situ spectroelectrochemical approach combined with CV. Surface structure sensitivity is observed in both the electrochemistry and the SHINERS spectra of PA and MeByOH with a strong propensity for CO formation over hydrogenation exhibited on the Pt{100} surfaces compared to Pt{111}, especially using PA as the reactant. Generally, these spectroscopic and electrochemical observations are consistent with each other and are in keeping with the idea that defect sites lead to more adsorption of the alkenic surface complex. For example, it is found that under hydrogen evolution conditions, the largest relative change (i.e. loss of the alkenic complex) is observed for Pt{110}. This is in accordance with it being the most catalytically active surface for hydrogenation. We propose that such a catalytic property relates directly to the surface density of the defect sites and indicates a higher level of activation of the alkyne bond compared to Pt{111}, thus rendering the Pt{111} sites the most likely to engender semihydrogenation selectivity over deep hydrogenation. MeByOH is different, primarily in its interaction with Pt{111}, leading to stronger adsorption compared to PA as measured by the electrochemistry, as well as different SHINERS behaviors. The Pt{110} and Pt{100} responses in the presence of MeByOH appear to be broadly similar to those with PA. However, the sharp “alkenic” peak, unique to MeByOH, observed on Pt{111} under hydrogen evolution conditions points to a very specific binding interaction in this case with less pronounced C=C bond activation compared to the other basal planes. Taken together, our observations suggest that the avoidance of all adsorption sites except Pt{111} terraces is the most effective route to generating superior selective catalysts for alkyne semihydrogenation.
Acknowledgments
The authors acknowledge the financial support from the U.K. National Measurement System, part of the Department for Business, Energy & Industrial Strategy. S.G. is also funded by the EPSRC National Facility for Photoelectron Spectroscopy (HarwellXPS), operated by Cardiff University and UCL under contract number PR16195.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c02967.
Raman band assignments, TEM image of Au@SiO2 NPs, Raman spectra of Au@SiO2 deposited on the Pt electrode surface (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Epelboin I.; Keddam M.; Takenouti H. Use of Impedance Measurements for the Determination of the Instant Rate of Metal Corrosion. J. Appl. Electrochem. 1972, 2, 71–79. 10.1007/BF00615194. [DOI] [Google Scholar]
- Sánchez M. A.; Podestá J. J.; Arvia A. J. Use of the Coulostatic Technique in the Study of Corrosion and Inhibition of Steel in 0.5 M H2SO4 in the Presence of Propargyl Alcohol and 2-Mercapto-Benzothiazol. An. Quim. 1989, 85, 8–14. [Google Scholar]
- Şahin M.; Bilgiç S. The Effect of Crotyl Alcohol on the Corrosion of Austenitic Chromium-nickel Steel. Appl. Surf. Sci. 1999, 147, 27–32. 10.1016/S0169-4332(98)00374-2. [DOI] [Google Scholar]
- Delgado A. B.; Castro Luna A. M.; Triaca W. E.; Arvia A. J. The Adsorption and the Potentiodynamic Electro-Oxidation of Acetylene on Platinized Platinum. J. Electrochem. Soc. 1982, 129, 1493. 10.1149/1.2124190. [DOI] [Google Scholar]
- Gui J. Y.; Kahn B. E.; Lin C. H.; Lu F.; Salaita G. N.; Stern D. A.; Zapien D. C.; Hubbard A. T. Studies of Adsorbed Unsaturated Alcohols at Well-defined Pt(111) Electrode Surfaces by Cyclic Voltammetry Assisted by Vibrational Spectroscopy (EELS). J. Electroanal. Chem. Interfacial Electrochem. 1988, 252, 169–188. 10.1016/0022-0728(88)85080-0. [DOI] [Google Scholar]
- Gonçalves R. S.; Léger J. M.; Lamy C. Electrochemical Studies of the Adsorption of Propargyl Alcohol on a Smooth Platinum Electrode in Acid Medium. Electrochim. Acta 1989, 34, 433–438. 10.1016/0013-4686(89)87022-7. [DOI] [Google Scholar]
- Horányi G.; Inzelt G.; Torkos K. Reductive Cleavage of C-OH Bonds in Allyl Position-Formation of Gaseous Products in the Course of the Cathodic Reduction of Some Simple Unsaturated Alcohols. J. Electroanal. Chem. 1979, 101, 101–108. 10.1016/S0022-0728(79)80082-0. [DOI] [Google Scholar]
- Horányi G. Electrocatalytic Reductive Splitting of C-O and C-OH Bonds at Platinum Electrodes. Electrochim. Acta 1986, 31, 1095–1103. 10.1016/0013-4686(86)80120-7. [DOI] [Google Scholar]
- Pastor E.; Wasmus S.; Iwasita T.; Arévalo M. C.; González S.; Arvia A. J. Spectroscopic Investigations of C3 Primary Alcohols on Platinum Electrodes in Acid Solutions.: Part I. n-Propanol. J. Electroanal. Chem. 1993, 350, 97–116. 10.1016/0022-0728(93)80199-R. [DOI] [Google Scholar]
- Pastor E.; Schmidt V. M.; Iwasita T.; Arévalo M. C.; González S.; Arvia A. J. The Reactivity of Primary C3-Alcohols on Gold Electrodes in Acid Media. A Comparative Study Based on DEMS Data. Electrochim. Acta 1993, 38, 1337–1344. 10.1016/0013-4686(93)80067-A. [DOI] [Google Scholar]
- Pastor E.; Wasmus S.; Iwasita T.; Arévalo M. C.; González S.; Arvia A. J. DEMS and in-situ FTIR Investigations of C3 Primary Alcohols on Platinum Electrodes in Acid Solutions.: Part II. Allyl Alcohol. J. Electroanal. Chem. 1993, 353, 81–100. 10.1016/0022-0728(93)80288-S. [DOI] [Google Scholar]
- Pastor E.; Wasmus S.; Iwasita T.; Arévalo M. C.; González S.; Arvia A. J. Spectroscopic Investigations of C3-Primary Alcohols on Platinum Electrodes in Acid Solutions.: Part III. Propargyl Alcohol. J. Electroanal. Chem. 1994, 371, 167–177. 10.1016/0022-0728(93)03250-S. [DOI] [Google Scholar]
- Rodríguez J. L.; Souto R. M.; González S.; Pastor E. DEMS Study on the Adsorption and Reactivity of Benzyl Alcohol on Palladium and Platinum. Electrochim. Acta 1998, 44, 1415–1422. 10.1016/S0013-4686(98)00264-3. [DOI] [Google Scholar]
- Arévalo M. C.; Rodríguez J. L.; Pastor E. Adsorption, Oxidation and Reduction Reactions of Propargyl Alcohol on Palladium as Studied by Electrochemical Mass Spectroscopy. J. Electroanal. Chem. 1999, 472, 71–82. 10.1016/S0022-0728(99)00269-7. [DOI] [Google Scholar]
- Arévalo M. C.; Rodríguez J. L.; Pastor E. Elucidation of the Reaction Pathways of Allyl Alcohol at Polycrystalline Palladium Electrodes. J. Electroanal. Chem. 2001, 505, 62–71. 10.1016/S0022-0728(01)00457-0. [DOI] [Google Scholar]
- Souto R. M.; Rodríguez J. L.; Pastor G.; Pastor E. Investigation of the Electrochemical Reactivity of Benzyl Alcohol at Platinum and Palladium Electrodes. Electrochim. Acta 2000, 45, 1645–1653. 10.1016/S0013-4686(99)00390-4. [DOI] [Google Scholar]
- Schmiemann U.; Müller U.; Baltruschat H. The Influence of the Surface on the Adsorption of Ethene, Ethanol and Cyclohexene as Studied by DEMS. Electrochim. Acta 1995, 40, 99–107. 10.1016/0013-4686(94)00243-T. [DOI] [Google Scholar]
- Arévalo M. C.; Rodríguez J. L.; Castro-Luna A. M.; Pastor E. Adsorption, Oxidation and Reduction of Crotyl Alcohol on Platinum: A DEMS and in situ FTIRS Study. Electrochim. Acta 2006, 51, 5365–5375. 10.1016/j.electacta.2006.02.007. [DOI] [Google Scholar]
- Arévalo M. C.; Pastor E.; González S.; Arvia A. J. Coadsorption Phenomena and Adsorbate Competition in Surface Electrochemical Reactions Involving Carbon Monoxide and Other Organic Residues. Electrochim. Acta 1991, 36, 2183–2187. 10.1016/0013-4686(91)85227-X. [DOI] [Google Scholar]
- Arévalo M. C.; Gomis-Bas C.; Pastor E.; González S.; Arvia A. J. Multiple Adsorbate Interactions between Reduced CO2 Adsorbates and either Allyl Alcohol or Propargyl Alcohol Residues on Platinum in 0.5 M Sulphuric Acid. Electrochim. Acta 1992, 37, 1083–1091. 10.1016/0013-4686(92)85226-B. [DOI] [Google Scholar]
- Jiang H.-F.; Wang A. Z.; Liu H. L.; Qi C. R. Reusable Polymer-Supported Amine-Copper Catalyst for the Formation of α-Alkylidene Cyclic Carbonates in Supercritical Carbon Dioxide. Eur. J. Org. Chem. 2008, 13, 2309–2312. 10.1002/ejoc.200701165. [DOI] [Google Scholar]
- Cui M.; Qian Q.; He Z.; Ma J.; Kang X.; Hu J.; Liu Z.; Han B. Synthesizing Ag Nanoparticles of Small Size on a Hierarchical Porosity Support for the Carboxylative Cyclization of Propargyl Alcohols with CO2 under Ambient Conditions. Chem. Eur. J. 2015, 21, 15924–15928. 10.1002/chem.201502479. [DOI] [PubMed] [Google Scholar]
- Song Q.; Yu B.; Li X.; Ma R.; Diao Z.; Li R.; Li W.; He L. Efficient Chemical Fixation of CO2 Promoted by a Bifunctional Ag2WO4/Ph3P System. Green Chem. 2014, 16, 1633–1638. 10.1039/c3gc42406e. [DOI] [Google Scholar]
- Yuan Y.; Xie Y.; Zeng C.; Song D.; Chaemchuen S.; Chen C.; Verpoort F. A Recyclable AgI/OAc– Catalytic System for the Efficient Synthesis of α-Alkylidene Cyclic Carbonates: Carbon Dioxide Conversion at Atmospheric Pressure. Green Chem. 2017, 19, 2936–2940. 10.1039/C7GC00276A. [DOI] [Google Scholar]
- Wu Y.; Zhao Y.; Li R.; Yu B.; Chen Y.; Liu X.; Wu C.; Luo X.; Liu Z. Tetrabutylphosphonium-based Ionic Liquid Catalysed CO2 Transformation at Ambient Conditions: A Case of Synthesis of α-Alkylidene Cyclic Carbonates. ACS Catal. 2017, 7, 6251–6255. 10.1021/acscatal.7b01422. [DOI] [Google Scholar]
- Hou S.; Dong J.; Jiang X.; Jiao Z.; Zhao B. A Noble-Metal-Free Metal-Organic Framework (MOF) Catalyst for the Highly Efficient Conversion of CO2 with Propargylic Alcohols. Angew. Chem., Int. Ed. 2019, 58, 577–581. 10.1002/anie.201811506. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Lan X.; Zhang Y.; Li M.; Bai G. Copper(I) Iodide Cluster-based Lanthanide Organic Frameworks: Synthesis and Application as Efficient Catalysts for Carboxylative Cyclization of Propargyl Alcohol with CO2 under Mild Conditions. Dalton Trans. 2019, 48, 11063–11069. 10.1039/C9DT01859J. [DOI] [PubMed] [Google Scholar]
- Rajaram J.; Narula A. P. S.; Chawla H. P. S.; Dev S. Semihydrogenation of Acetylenes: Modified Lindlar Catalyst. Tetrahedron 1983, 39, 2315–2322. 10.1016/S0040-4020(01)91960-X. [DOI] [Google Scholar]
- Griffiths S. P.; Johnston P.; Wells P. B. Enantioselective Hydrogenation: Chemistry of the Modification of Platinum by Cinchonidine. Appl. Catal., A 2000, 191, 193–204. 10.1016/S0926-860X(99)00319-1. [DOI] [Google Scholar]
- Okamoto M.; Hirao T.; Yamaai T. Polymers as Novel Modifiers for Supported Metal Catalyst in Hydrogenation of Benzaldehydes. J. Catal. 2010, 276, 423–428. 10.1016/j.jcat.2010.10.008. [DOI] [Google Scholar]
- Crespo-Quesada M.; Yarulin A.; Jin M.; Xia Y.; Kiwi-Minsker L. Structure Sensitivity of Alkynol Hydrogenation on Shape- and Size-Controlled Palladium Nanocrystals: Which Sites are most Active and Selective?. J. Am. Chem. Soc. 2011, 133, 12787–12794. 10.1021/ja204557m. [DOI] [PubMed] [Google Scholar]
- Schmidt E.; Vargas A.; Mallat T.; Baiker A. Shape-Selective Enantioselective Hydrogenation on Pt Nanoparticles. J. Am. Chem. Soc. 2009, 131, 12358–12367. 10.1021/ja9043328. [DOI] [PubMed] [Google Scholar]
- Attard G. A.; Bennett J. A.; Mikheenko I.; Jenkins P.; Guan S.; Macaskie L. E.; Wood J.; Wain A. J. Semi-hydrogenation of Alkynes at Single Crystal, Nanoparticle and Biogenic Nanoparticle Surfaces: the Role of Defects in Lindlar-type Catalysts and the Origin of Their Selectivity. Faraday Discuss. 2013, 162, 57–75. 10.1039/c3fd00007a. [DOI] [PubMed] [Google Scholar]
- Attard G. A.; Griffin K. G.; Jenkins D. J.; Johnston P.; Wells P. B. Enantioselective Hydrogenation of Ethyl Pyruvate Catalysed by Pt/Graphite: Superior Performance of Sintered Metal Particles. Catal. Today 2006, 114, 346–352. 10.1016/j.cattod.2006.02.075. [DOI] [Google Scholar]
- Bennett J. A.; Attard G. A.; Deplanche K.; Casadesus M.; Huxter S. E.; Macaskie L. E.; Wood J. Improving Selectivity in 2-Butyne-1,4-diol Hydrogenation Using Biogenic Pt Catalysts. ACS Catal. 2012, 2, 504–511. 10.1021/cs200572z. [DOI] [Google Scholar]
- Attard G. A.; Gillies J. E.; Harris C. A.; Jenkins D. J.; Johnston P.; Price M. A.; Watson D. J.; Wells P. B. Electrochemical Evaluation of the Morphology and Enantioselectivity of Pt/Graphite. Appl. Catal., A 2001, 222, 393–405. 10.1016/S0926-860X(01)00845-6. [DOI] [Google Scholar]
- Li J.; Yang Z.; Ren B.; Liu G.; Fang P.; Jiang Y.; Wu D.; Tian Z. Surface-Enhanced Raman Spectroscopy Using Gold-Core Platinum-Shell Nanoparticle Film Electrodes: toward a Versatile Vibrational Strategy for Electrochemical Interfaces. Langmuir 2006, 22, 10372–10379. 10.1021/la061366d. [DOI] [PubMed] [Google Scholar]
- Li J.; Huang Y.; Ding Y.; Yang Z.; Li S.; Zhou X.; Fan F.; Zhang W.; Zhou Z.; Wu D.; Ren B.; Wang Z. L.; Tian Z. Shell-Isolated Nanoparticle-Enhanced Raman Spectroscopy. Nature 2010, 464, 392–395. 10.1038/nature08907. [DOI] [PubMed] [Google Scholar]
- Moskovits M.; Suh J. S. Surface Geometry Change in 2-Naphthoic Acid Adsorbed on Silver. J. Phys. Chem. A 1988, 92, 6327–6329. 10.1021/j100333a030. [DOI] [Google Scholar]
- Suh J. S.; Kim J. Three Distinct Geometries of Surface-Adsorbed Carboxylate Groups. J. Raman Spectrosc. 1998, 29, 143–148. 10.1002/(sici)1097-4555(199802)29:23.0.co;2-#. [DOI] [Google Scholar]
- Guan S.Determination of the Role of Nanoparticle Active Sites in Catalytic Hydrogenation Reactions by Cyclic Voltammetry and Novel in-situ Surface Spectroscopy. Ph.D. Thesis, Cardiff University, 2014. [Google Scholar]
- Guan S.; Donovan-Sheppard O.; Reece C.; Willock D. J.; Wain A. J.; Attard G. A. Structure Sensitivity in Catalytic Hydrogenation at Platinum Surfaces Measured by Shell-Isolated Nanoparticle Enhanced Raman Spectroscopy (SHINERS). ACS Catal. 2016, 6, 1822–1832. 10.1021/acscatal.5b02872. [DOI] [Google Scholar]
- Clavilier J.; El-Achi K.; Rodes A. In situ Probing of Step and Terrace Sites on Pt(S)-[n(111) × (111)] Electrodes. Chem. Phys. 1990, 141, 1–14. 10.1016/0301-0104(90)80014-O. [DOI] [Google Scholar]
- Rees N. V.; Taylor R. J.; Jiang Y.; Morgan I. R.; Knight D. W.; Attard G. A. In situ surface-Enhanced Raman Spectroscopic Studies and Electrochemical Reduction of α-Ketoesters and Self Condensation Products at Platinum Surfaces. J. Phys. Chem. C 2011, 115, 1163–1170. 10.1021/jp106495b. [DOI] [Google Scholar]
- Sexton B. A. Methanol Decomposition on Platinum (111). Surf. Sci. 1981, 102, 271–281. 10.1016/0039-6028(81)90321-6. [DOI] [Google Scholar]
- Greeley J.; Mavrikakis M. Competitive Paths for Methanol Decomposition on Pt(111). J. Am. Chem. Soc. 2004, 126, 3910–3919. 10.1021/ja037700z. [DOI] [PubMed] [Google Scholar]
- Marković N. M.; Ross P. N. Surface Science Studies of Model Fuel Cell Electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. 10.1016/S0167-5729(01)00022-X. [DOI] [Google Scholar]
- Attard G.; Barnes C.. Surfaces; Oxford University Press, 1998; p 10. [Google Scholar]
- Berná A.; Kuzume A.; Herrero E.; Feliu J. M. Ethylene Adsorption and Oxidation on Pt(hkl) in Acidic Media. Surf. Sci. 2008, 602, 84–94. 10.1016/j.susc.2007.09.046. [DOI] [Google Scholar]
- Schmiemann U.; Baltruschat H. The Adsorption of Ethene at Pt Single Crystal Electrodes. Desorption Products and Observation of Multiple Adsorption States by DEMS. J. Electroanal. Chem. 1992, 340, 357–363. 10.1016/0022-0728(92)80311-Q. [DOI] [Google Scholar]
- Grozovski V.; Climent V.; Herrero E.; Feliu J. M. The Role of the Surface Structure in the Oxidation Mechanism of Methanol. J. Electroanal. Chem. 2011, 662, 43–51. 10.1016/j.jelechem.2011.02.032. [DOI] [Google Scholar]
- Arán-Ais R.; Santos N. A.; Villulas H. M.; Feliu J. M. Oxidation of Ethanol and its Derivatives on Well Defined Pt Single Crystal Electrodes Vicinal to Pt(111): a Comparative Study. ECS Trans. 2013, 53, 11–22. 10.1149/05328.0011ecst. [DOI] [Google Scholar]
- Colmati F.; Tremiliosi-Filho G.; Gonzalez E. R.; Berná A.; Herrero E.; Feliu J. M. Surface Structure Effects on the Electrochemical Oxidation of Ethanol on Platinum Single Crystal Electrodes. Faraday Discuss. 2009, 140, 379–397. 10.1039/B802160K. [DOI] [PubMed] [Google Scholar]
- Souto R. M.; Rodríguez J. L.; Pastor E. Revealing Structural Effects, part II: the Influence of Molecular Structure on the Adsorption of Butanol Isomers on Platinum. Chem. Eur. J. 2005, 11, 3309–3317. 10.1002/chem.200400478. [DOI] [PubMed] [Google Scholar]
- Takky D.; Beden B.; Leger J. M.; Lamy C. Evidence for the Effect of Molecular Structure on the Electrochemical Reactivity of Alcohols: Part I. Electrooxidation of the Butanol Isomers on Noble Metal Electrodes in Alkaline Medium. J. Electroanal. Chem. Interfacial Electrochem. 1983, 145, 461–466. 10.1016/S0022-0728(83)80101-6. [DOI] [Google Scholar]
- Taylor R. J.; Jiang Y.; Rees N. V.; Attard G. A.; Jeffery E. L.; Willock D. J. Enantioselective Hydrogenation of α-Ketoesters: an in situ Surface-Enhanced Raman Spectroscopy (SERS) Study. J. Phys. Chem. C 2011, 115, 21363–21372. 10.1021/jp208403g. [DOI] [Google Scholar]
- Camplin J. P.; Eve J. K.; McCash E. M. RAIRS of Small Alkynes on Pd(100). Phys. Chem. Chem. Phys. 2000, 2, 4433–4440. 10.1039/b004344n. [DOI] [Google Scholar]
- Avery N. R.; Sheppard N. The Use of Thermal Desorption and Electron Energy Loss Spectroscopy for the Determination of the Structures of Unsaturated Hydrocarbons Chemisorbed on Metal Single-Crystal Surfaces I. Alk-1-enes on Pt(111). Proc. R. Soc. London, Ser. A 1986, 405, 1–25. 10.1098/rspa.1986.0037. [DOI] [Google Scholar]
- Bron M.; Holze R. Spectroelectrochemical Investigation of the Adsorption and Oxidation of Unsaturated C4-Alcohols. Surf. Sci. 2000, 457, 178–184. 10.1016/S0039-6028(00)00369-1. [DOI] [Google Scholar]
- Zhang P.; Chen Y.; Cai J.; Liang S.; Li J.; Wang A.; Ren B.; Tian Z. An Electrochemical in situ Surface-Enhanced Raman Spectroscopic Study of Carbon Monoxide Chemisorption at a Gold Core-Platinum Shell Nanoparticle Electrode with a Flow Cell. J. Phys. Chem. C 2009, 113, 17518–17526. 10.1021/jp906697b. [DOI] [Google Scholar]
- Socrates G.Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; John Wiley & Sons, 2004; p 94. [Google Scholar]
- Swaminathan S.; Narayanan K. V. Rupe and Meyer-Schuster rearrangements. Chem. Rev. 1971, 71, 429–438. 10.1021/cr60273a001. [DOI] [Google Scholar]
- Engel D. A.; Dudley G. B. The Meyer-Schuster Rearrangement for the Synthesis of α,β-Unsaturated Carbonyl Compounds. Org. Biomol. Chem. 2009, 7, 4149–4158. 10.1039/b912099h. [DOI] [PubMed] [Google Scholar]
- Wain A. J.; O’Connell M. A.; Attard G. A. Insights into Self-Poisoning during Catalytic Hydrogenation on Platinum Surfaces Using ATR-IR Spectroelectrochemistry. ACS Catal. 2018, 8, 3561–3570. 10.1021/acscatal.8b00492. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.