

Visual Abstract

Keywords: chronic rejection, mast cells, basophils, interferon alpha, IgE
Abstract
Background and objectives
Active antibody-mediated rejection is the main cause of kidney transplant loss, sharing with SLE the alloimmune response and the systemic activation of the IFN-α pathway. IgE-mediated immune response plays a key role in the development of SLE nephritis and is associated with IFN-α secretion. The aim of our study was to investigate IgE-mediated immune response in antibody-mediated rejection.
Design, setting, participants, & measurements
This was a cross-sectional study of 56 biopsy-proven antibody-mediated rejection study participants, 80 recipients with normal graft function/histology (control), 16 study participants with interstitial fibrosis/tubular atrophy, and six participants with SLE. We evaluated graft IgE deposition, tryptase (a mast cell marker), and CD203 (a specific marker of activated basophils) by immunofluorescence/confocal microscopy. In addition, we measured serum concentration of human myxovirus resistance protein 1, an IFN-α–induced protein, and anti-HLA IgE.
Results
We observed a significantly higher IgE deposition in tubules and glomeruli in antibody-mediated rejection (1766±79 pixels) and SLE (1495±43 pixels) compared with interstitial fibrosis/tubular atrophy (582±122 pixels) and control (253±50 pixels). Patients with antibody-mediated rejection, but not control patients and patients with interstitial fibrosis/tubular atrophy, presented circulating anti-HLA IgE antibodies, although with a low mean fluorescence intensity. In addition, immunofluorescence revealed the presence of both mast cells and activated basophils in antibody-mediated rejection but not in control and interstitial fibrosis/tubular atrophy. The concentration of circulating basophils was significantly higher in antibody-mediated rejection compared with control and interstitial fibrosis/tubular atrophy. MxA serum levels were significantly higher in antibody-mediated rejection compared with control and correlated with the extent of IgE deposition.
Conclusions
Our data suggest that IgE deposition and the subsequent recruitment of basophils and mast cells within the kidney transplant might play a role in antibody-mediated rejection.
Introduction
IgE antibodies have long been recognized as the antigen-specific triggers of allergic reactions. However, recent advances have clarified the evolving pathogenic role for IgE from allergies to other immune-mediated disorders. The presence of self-reactive IgE has now been shown in several autoimmune diseases such as SLE, bullous pemphigoid, and chronic urticaria (1), and they appear to be present in a significant subgroup of patients. Their appearance is often associated with a worse disease outcome, although the exact mechanisms by which IgE autoantibodies exert their detrimental effects are still largely unclear (2).
In SLE, an autoimmune disease characterized by a substantial production of self-reactive antibodies (3), the total IgE and antinuclear IgE autoantibody levels are elevated and seem to be significantly associated with disease severity and, in particular, with the development of lupus nephritis in an atopy-independent manner (3). Indeed, despite their role in allergies, IgE antibodies are not associated with a higher rate of atopic reactions in these patients (4). The deposition of IgE immune complexes at the glomerular and tubulointerstitial levels suggests that they may play a direct pathogenic role in the development of kidney damage featuring SLE (5). The IgE immune complexes may induce a significant tissue injury through the recruitment and activation of the two main cell types expressing the IgE receptor, mast cells and basophils, as shown in several studies on lupus nephritis (6). There is increasing evidence that, other than their role in allergy, basophils and mast cells can amplify the humoral immune response and directly cause tissue damage through the release of proinflammatory and profibrotic cytokines and growth factors. In addition, several studies evidenced a novel Th2 axis of SLE pathophysiology connecting IgE, basophils, plasmacytoid dendritic cells, and IFN-α signature in which autoreactive IgE is the main actor (6–9). Indeed, Henault et al. (10) clearly demonstrated that IgE induces IFN-α secretion from plasmacytoid dendritic cells that, in turn, can promote, among other effects, autoantibodies production by plasma cells.
Active antibody-mediated rejection, one of the main threats to kidney allograft outcomes, might be defined as an alloimmune disease characterized, similarly to SLE, by a complex process that involves the interplay of different cellular and molecular pathways leading to the production of an array of pathogenic antibodies (11). Antibody ligation to HLA or non-HLA antigens expressed on endothelial cells can activate the complement system and induce the recruitment of leukocytes and natural killer cells, leading to endothelial damage, loss of vascular integrity, and coagulation cascade activation (12).
Antibody-mediated rejection also shares with SLE the development of autoantibodies that may play a role in antibody-mediated kidney damage (13). Indeed, Thaunat et al. (14) clearly demonstrated a breakdown of B cell tolerance within the graft in patients with antibody-mediated rejection leading to the production of antibodies directed against intracellular and autoantigens. Interestingly, our research group suggested a possible pathogenic link between antibody-mediated rejection and SLE, reporting an association between the diagnosis of antibody-mediated rejection and the activation of the IFN-α/β signaling pathway in PBMCs, one of the SLE immunologic signatures (13). On this basis, in this study, we investigated whether IgE-associated immune response might play a role in the pathogenesis of antibody-mediated rejection.
Materials and Methods
Study Population
This study is a single-center, patient-control, observational study. Fifty-six kidney transplant recipients with biopsy-proven chronic active antibody-mediated rejection according to Banff 2013 criteria (15) with circulating donor-specific antibodies (cutoff of >3000 mean fluorescence intensity) were enrolled in the study after providing written informed consent (patient group). In addition, we enrolled, after obtaining written informed consent, 152 kidney transplant recipients with stable kidney function and no significant histologic changes with or without circulating anti-HLA antibodies who received graft biopsies per protocol (de novo appearance of circulating anti-HLA antibodies) or for the appearance of urine abnormalities (microhematuria and/or proteinuria; control group), and 59 transplant recipients with clinical and histologic evidence of interstitial fibrosis/tubular atrophy (IFTA; IFTA group). Sixteen of 59 IFTA and 80 of 152 control transplant recipients matched to the patients by propensity score were used for further analysis. We also enrolled a group of six patients with classes 3 (n=4) and 4 (n=2) lupus nephropathy as a positive control for IgE tissue immunofluorescence. The study was carried out according to the principles of the Declaration of Helsinki, and it was approved by the local ethics committee (protocol no. 670/C.E.). The clinical and research activities reported are consistent with the Principles of the Declaration of Istanbul as outlined in the “Declaration of Istanbul on Organ Trafficking and Transplant Tourism.”
Confocal Microscopy
Paraffin-embedded sections (3–4 µm) from kidney biopsies were used to measure IgE deposition by indirect immunofluorescence and confocal microscopy analysis. Deparaffinization of kidney biopsy sections was performed through immersion in pure xylene for 15 minutes and rehydration using graded alcohol series for a few minutes: absolute alcohol (6 minutes), 95%–90% alcohol (1 minute), 70% alcohol (1 minute), and 50% alcohol (1 minute). The slides were then washed with water and allowed to dry. Antigenic epitope demasking was performed using a pressure-induced method by pressure cooker using EDTA buffer (0.01 M, pH 8).
After antigen unmasking in EDTA buffer, sections were washed in PBS and incubated with 5% BSA in PBS for 1 hour at room temperature. Sections were incubated overnight at 4°C with a primary antibody against IgE (1:500, ab195580 rabbit monoclonal anti-human IgE; Abcam, Cambridge, United Kingdom), followed by incubation for 1 hour with the appropriate secondary antibody (1:200 Alexa Fluor 488 goat anti-rabbit; Molecular Probes, Eugene, OR). The sections were counterstained with To-pro-3 (1:3000; Molecular Probes), mounted in Gel/Mount (Biomeda, Foster City, CA), and sealed with nail varnish. Negative controls were performed by omitting the primary antibodies. Specific fluorescence was acquired with a Leica TCS SP2 confocal laser-scanning microscope (Leica, Wetzlar, Germany).
The number of CD203c+ cells and their spatial relationship with IgE deposits were investigated in kidney biopsies using double staining. Sections were incubated with a primary antibody against IgE and the appropriate secondary antibody, as described above, and then with a mouse monoclonal anti-CD203 antibody (1:100, ab118445; Abcam) followed by incubation with the appropriate secondary antibody (1:200, Alexa Fluor 546 goat anti-mouse I; Molecular Probes).
In addition, paraffin-embedded sections of kidney biopsies were used for double-staining IgE/tryptase. Briefly, slides were deparaffinized, hydrated, boiled in citrate buffer (0.01 M, pH 6), and blocked with 5% goat serum (Sigma, Milan, Italy). After incubation with a primary antibody against IgE followed by incubation for 1 hour with the secondary antibody, as previously described, sections were incubated with a mouse monoclonal antimast cell tryptase antibody (1:100, ab2378; Abcam) and then with the appropriate secondary antibody (1:200, Alexa Fluor 546 goat anti-mouse I; Molecular Probes), and they were counterstained with To-pro-3 (1:3000) before mounting in Gel Mount (Biomeda Corp). The primary antibody was omitted for negative controls.
Images were acquired with the Leica TCS SP2 confocal laser-scanning microscope. Appropriate single-stained controls were used with each antibody to assure that no fluorochrome spectral crossbleeding was present. Average intensity value of immunofluorescence signal was quantified using Adobe Photoshop CS6 software. LIF file format images were converted to tagged image file format. The average signal intensity for each individual channel was measured using the histogram function. Individual channels were gray scaled prior to measurement.
The count of CD203c+ and tryptase+ cells was performed in at least ten high-power (×630) fields/sections by two independent observers blinded to the origin of the slides. The final counts were the mean of the two measures, and interobserver variability was <20%.
Circulating Basophils, Immunoglobulin E, and Human Myxovirus Resistance Protein 1
All blood samples obtained from enrolled patients were analyzed by an automated hematology analyzer in order to determine the basophil concentration. Serum IgE antibody levels were measured by immunoCAP total IgE (Thermo Scientific, Monza, Italy) according to the manufacturer’s instruction.
IFN-regulated resistance GTP-binding protein MxA serum levels were measured using a commercially available ELISA kit (LS-F7459; LifeSpan Biosciences, Seattle, WA) according to the manufacturer’s instructions in all of the patients whose serum sample was available. The OD at ʎ=450 nm was measured on a multimode microplate reader (2030, VictorX3; PerkinElmer). The value of 1000 ng/ml was attributed to the sample with the highest observed OD value.
Measurement of Circulating Histocompatibility Leukocyte Antigen–Specific Immunoglobulin E Antibodies and Human Myxovirus Resistance Protein 1
HLA-specific IgE was measured in a subgroup of randomly selected transplant recipients (n=10 antibody-mediated rejection, n=5 control, and n=5 IFTA) and six patients with SLE used as controls. We worked with a Luminex methodology using Immucor systems (Immucor Lifecodes Single Antigen). The anti-IgE conjugate used is a commercial product (SouthernBiotech Rat Anti-Mouse IgE-PE; lot H021-PC72Y), and it was diluted as per the final concentration (10 µg/ml) used by the kit producer for IgG test (CE-IVD; product of Immucor Lifecodes Single Antigen). In addition, to avoid any IgG interference, before testing for anti-HLA IgE, we treated the sera with a specific protocol of IgG inactivation with IdeS Protease, a specific antibody cysteine protease that recognizes all human IgG subclasses, cleaving specifically at single recognition site below the hinge region.
Sensitivity and specificity are indicated on the instructions for use by the IgG kit producer.
Statistical Analyses
Our data were first tested for normal distribution using the Kolmogorov–Smirnov test for normality. Results are expressed as mean ± SD or median and interquartile range for continuous data and as integers, frequencies, and percentages for categorical data. None of the variables analyzed presented >10% missing data. When present, missing data were imputed using median or most frequent for numerical and categorical variables, respectively. Demographics and clinical and histologic characteristics of patients were compared by one-way ANOVA, Kruskal–Wallis test, unpaired t test, Mann–Whitney U test for continuous data, and chi-square statistic for categorical data, as appropriate. All analyses were performed using SPSS for Windows, release 17.0 (SPSS, Inc., North Sydney, Australia); P=0.05 was considered statistically significant.
Results
The main demographic, clinical, and histologic features of patients with antibody-mediated rejection, patients with normal transplants (control), and patients with IFTA are summarized in Table 1.
Table 1.
Characteristics of kidney transplant recipients included in this study
| Variables | Antibody-Mediated Rejection | Control | Interstitial Fibrosis/Tubular Atrophy |
|---|---|---|---|
| No. of patients | 56 | 80 | 16 |
| Age, yr | 47±12 | 52±14 | 51±14 |
| Women, % | 36 | 31 | 44 |
| Transplantation vintage, yr | 5 (1–9) | 7 (4–12) | 6 (2–8) |
| Serum creatinine, mg/dl | 2.2 (1.7–3.5) | 1.1 (1.0–1.3) | 2.3 (1.5–3.3) |
| Proteinuria, g/24 h | 2.2 (1.5–5.7) | 0.1 (0.1–0.2) | 0.8 (0.3–2.0) |
| Cyclosporin, % of patients | 20 | 21 | 25 |
| Tacrolimus, % of patients | 75 | 79 | 75 |
| Mycophenolate mofetil, % of patients | 43 | 46 | 44 |
| Mycophenolic Ac., % of patients | 29 | 23 | 19 |
| mTOR inhibitors | 9 | 24 | 31 |
| Chronic glomerulopathy Banff score | |||
| 0 | 21 (37.5%) | 80 (100%) | 16 (100%) |
| 1 | 5 (9%) | ||
| 2 | 9 (16%) | ||
| 3 | 21 (37.5%) | ||
| Peritubular capillaritis Banff score | |||
| 0 | 36 (64%) | 80 (100%) | 16 (100%) |
| 1 | 8 (14%) | ||
| 2 | 12 (22%) | ||
| 3 | 0 (0%) | ||
| Glomerulitis Banff lesion score | |||
| 0 | 26 (46%) | 80 (100%) | 16 (100%) |
| 1 | 13 (24%) | ||
| 2 | 12 (21%) | ||
| 3 | 5 (9%) | ||
| Interstitial fibrosis Banff lesion score | |||
| 0 | 0 (0%) | 65 (81%) | 0 (0%) |
| 1 | 22 (39°%) | 15 (19%) | 0 (0%) |
| 2 | 29 (52%) | 12 (75%) | |
| 3 | 5 (9%) | 4 (25%) | |
| Tubular atrophy Banff lesion score | |||
| 0 | 0 (0%) | 67 (84%) | 0 (0%) |
| 1 | 23 (41%) | 13 (16%) | 1 (6%) |
| 2 | 28 (50%) | 12 (75%) | |
| 3 | 5 (9%) | 3 (19%) | |
| c4d positive | 64% | 0% | 0% |
| Anti-HLA antibodies, % | |||
| Positive | 100 | 15 | 0 |
| Negative | 0 | 66 | 100 |
| Not available | 0 | 19 | 0 |
Data are expressed as mean ± SD for age; median (interquartile range) for transplantation vintage, serum creatinine, and proteinuria, and percentage for other parameters. Ac., acid.
IgE immune deposits, absent in the normal grafts (Figure 1A), were markedly higher in antibody-mediated rejection biopsies in the tubulointerstitial area, in particular within the basement membrane of proximal tubules and peritubular capillaries (Figure 1B). The fluorescence pattern was similar to the one present in SLE kidney biopsies (Figure 1C). We observed only a slightly higher tubulointerstitial IgE deposition in IFTA biopsies (Figure 1D) and almost an absence in control ones (Figure 1D).
Figure 1.
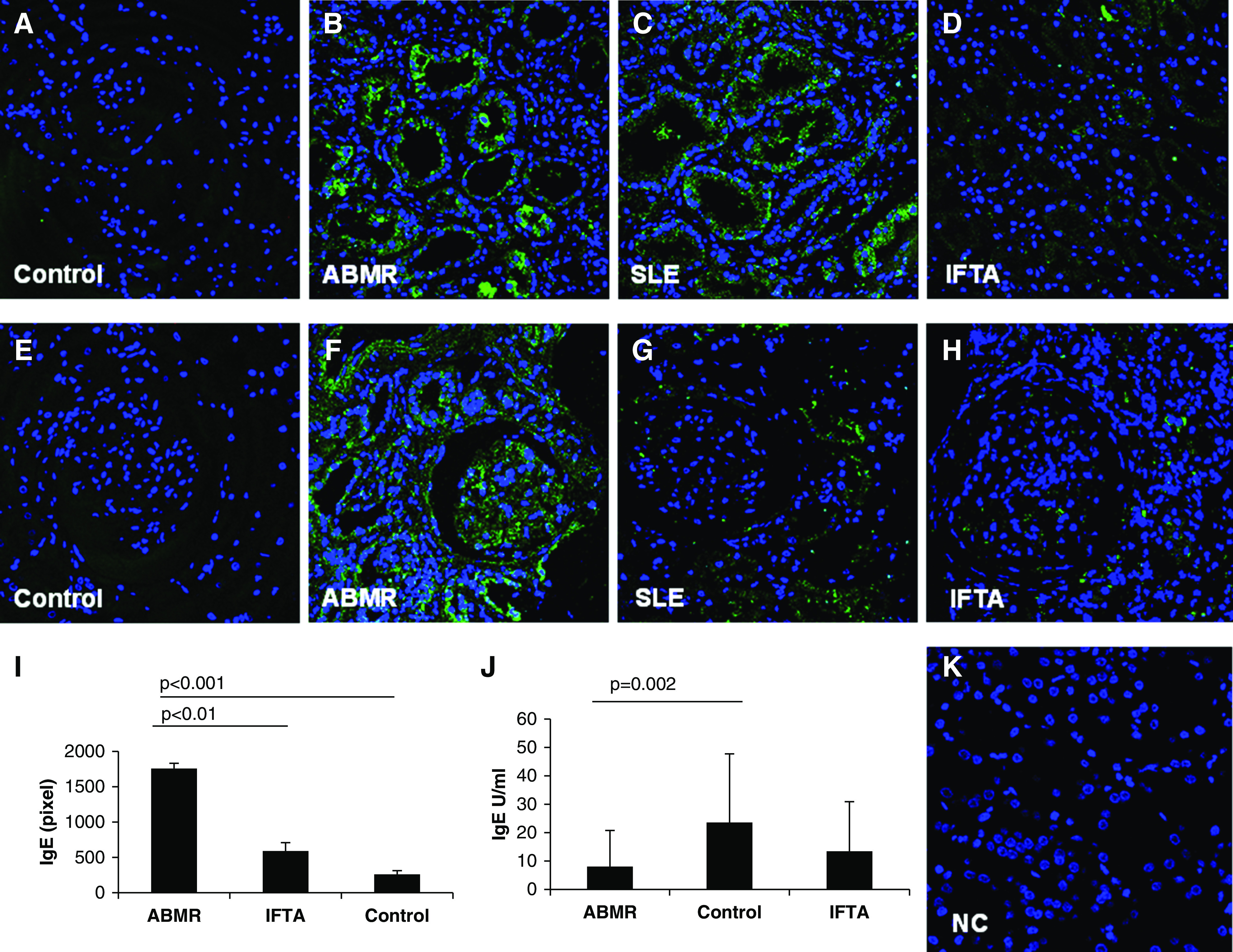
Deposition of IgE antibodies in graft biopsies. IgE deposition (green) in tubulointerstitial (A–D) and glomerular areas (E–H) was investigated by confocal laser microscopy in antibody-mediated rejection (ABMR; n=30), control (n=40), and interstitial fibrosis/tubular atrophy (IFTA) graft biopsies (n=16). Patients with SLE (n=6) were used as positive controls for IgE tissue immunofluorescence. Nuclei are highlighted with To-pro-3 in blue. Negative control was obtained as described in Materials and Methods (K). Overall quantification of mean florescence intensity, performed as described in Materials and Methods and expressed as percentage of pixels per area fraction, demonstrated a statistically significant higher IgE deposition in ABMR biopsies compared with IFTA and control graft tissues (I). Serum total IgE levels, expressed in units per milliliter, were significantly lower in patients with ABMR compared with control graft recipients (J).
At the glomerular level, IgE deposits were absent in control graft biopsies (Figure 1E). Diffuse deposits of IgE were observed only in patients with antibody-mediated rejection (Figure 1F), whereas we detected only a weak IgE-specific signal in SLE and IFTA tissue samples (Figure 1, G and H, respectively). The overall quantification of specific IgE immunofluorescence confirmed a statistically significant higher IgE immune complex deposition in patients with antibody-mediated rejection and patients with SLE compared with patients with normal kidneys and patients with IFTA grafts (Figure 1I). Quantitative data of IgE deposition are reported in Table 2. Furthermore, we evaluated total circulating IgE levels, and we observed significantly lower numbers of this class of Ig in patients with antibody-mediated rejection compared with control graft recipients (Figure 1J). Finally, we investigated the presence of donor HLA-specific IgE by a modified Immucor-Lifecodes Luminex protocol in a subgroup of patients with antibody-mediated rejection, control patients, and patients with IFTA. We observed the presence of specific anti-HLA IgE antibodies at low mean fluorescence intensity only in patients with antibody-mediated rejection (162±18 mean fluorescence intensity), but not in both control groups. Interestingly, IgE antibodies presented the same specificities of anti-HLA IgG (7602±2503 mean fluorescence intensity) in each patient studied (Table 2).
Table 2.
Principal outcomes of the study
| Kidney Tissue | Antibody-Mediated Rejection | Control | Interstitial Fibrosis/Tubular Atrophy | P Value |
|---|---|---|---|---|
| IgE, pixels | 1766±79 | 253±50 | 582±122 | 0.01 |
| IgE, U/ml | 9±13 | 24±24 | 14±17 | 0.002 |
| Anti-HLA IgE antibodies, MFI | 162±18 | 0 | 0 | |
| CD203c+, n cells per field | 21±2 | 1±0.4 | 3±0.6 | 0.01 |
| Basophils, 103/μl | 0.05±0.03 | 0.02±0.02 | 0.01±0.01 | 0.01 |
| Triptase+, n cells per field | 5±0.6 | 0.3±0.6 | 1.3±0.6 | <0.001 |
| MxA, pg/ml | 180±156 | 53±27 | — | <0.001 |
MFI, mean fluorescence intensity; MxA, human myxovirus resistance protein 1; —, not analyzed.
We investigated the presence of basophils, the main IgE-responsive cells, within the kidney grafts and their spatial relationship with IgE deposits performing a double immunofluorescence for IgE (green in Figure 2) and CD203c (red in Figure 2), a marker of basophil activation. As expected, we detected a significant tubulointerstitial infiltration of these cells in SLE biopsies (Figure 2A). Interestingly, antibody-mediated rejection grafts also presented evidence of higher tubulointerstitial basophils compared with IFTA biopsies and control grafts (Figure 2, B–D). Noteworthy, these cells were strictly associated with the IgE deposits (Figure 2, A–D). The relative signal quantification of the kidney-infiltrating IgE+CD203c+ cells demonstrated a statistically significant higher number of these cells in antibody-mediated rejection compared with the two other study groups (Figure 2E). At the peripheral level, total circulating basophils were significantly higher in patients with antibody-mediated rejection compared with patients with control grafts and patients with IFTA (Figure 2F).
Figure 2.

Evaluation of graft-infiltrating and circulating basophils. The number of graft-infiltrating activated basophils cells (CD203c expression; red), along with IgE deposition (green), was investigated by confocal microscopy in ABMR (n=30), control (n=40), and IFTA (n=16) biopsies; patients with SLE (n=6) were used as positive controls for tissue immunofluorescence (A–D). Nuclei are highlighted with To-pro-3 in blue. The arrows indicate colocalization of IgE (green) and CD203c (red). Double-positive cells (yellow) are expressed as mean n cells per field ± SD (E). The absolute number of circulating basophils subset was detected on blood samples obtained from patients with ABMR (n=40), control patients (n=80), and IFTA graft recipients (n=16) and expressed as mean n 10e3/µl ± SD (F).
We then investigated the presence of graft-infiltrating mast cells, a class of immune cells responsive to IgE, using tryptase as a specific marker and their colocalization with IgE deposits. Although the mast cell recruitment within the grafts seems to be less conspicuous compared with basophil infiltration, our data demonstrate a significantly higher number of tryptase+ cells in grafts with antibody-mediated rejection compared with control and IFTA grafts (Figure 3).
Figure 3.

Analysis of graft-infiltrating mast cells. Graft-infiltrating mast cells (tryptase expression; red), along with IgE deposition (green), were visualized by confocal microscopy in ABMR (n=30), IFTA (n=16), and control (n=35) biopsies; patients with SLE (n=6) were used as positive controls for tissue immunofluorescence (A–D). Nuclei are highlighted with To-pro-3 in blue. The white rectangles indicate colocalization of IgE (green) and tryptase (red; B). The number of tryptase-positive cells (yellow), counted by two observers blinded to the origin of the slides and expressed as mean n cells per field ± SD, was significantly higher in ABMR biopsies compared with IFTA and control graft tissues (E).
MxA serum concentration was significantly higher in patients with antibody-mediated rejection compared with control graft recipients with normal graft function and histology with or without donor-specific anti-HLA antibodies (Figure 4A). Interestingly, MxA serum levels were directly and significantly correlated with the extent of IgE deposition (R2=0.32, P<0.001). Finally, we applied ROC curve analysis to evaluate the sensitivity and specificity of serum MxA as a marker of antibody-mediated rejection. The area under the curve was 0.793, and a value of serum MxA of 68 pg/ml allowed discrimination of patients with antibody-mediated rejection with a specificity of 64% and a sensitivity of 89% (Figure 4B).
Figure 4.

Analysis of MxA (human myxovirus resistance protein 1) serum levels. MxA serum levels were evaluated by ELISA test in patients with ABMR (n=52) and in an independent set of anti-HLA+ (n=12) and anti-HLA− control transplant recipients (n=63; A). Receiver operating characteristic curve analysis was used to evaluate the sensitivity and specificity of MxA as a marker of ABMR (B). AUC, area under the curve.
Discussion
In this study, we demonstrate for the first time a significant deposition of IgE within the graft of patients with active antibody-mediated rejection. In the last decade, an increasing number of reports suggested IgE as a possible contributor to the pathogenesis of several chronic inflammatory and autoimmune disorders, including rheumatoid arthritis and SLE, opening brand new opportunities for therapeutic strategies. A phase 1 trial (NCT01716312) evaluating the safety of omalizumab in SLE has already established a proof of concept of these approaches in humans (8). Interestingly, in our study, the pattern of IgE kidney deposition (mainly tubular and interstitial) was similar in kidney transplant recipients with antibody-mediated rejection and patients with SLE and did not differ from the one reported in other investigations on the role of this Ig in the pathogenesis of lupus nephritis. In SLE, IgE presents the feature of autoantibodies, and several studies demonstrated their specificity for dsDNA. In the transplantation setting, it should be reasonable to expect that these Igs may react with donor antigens. Indeed, in our patients with antibody-mediated rejection, we observed the presence of circulating anti-HLA IgE antibodies, although at a very low mean fluorescence intensity compared with IgG. Our results are similar to the recent observation of Farkas et al. (16) that reported the presence of anti-HLA IgE with a low mean fluorescence intensity in a small group of broadly sensitized patients who had previously rejected their kidney allografts. The very low mean fluorescence intensity that we observed might be also due to the competition with anti-HLA IgG. Indeed, in our patients, IgE shared the same HLA specificities with IgG.
Despite their role in allergies, IgE antibodies are not associated with a higher rate of atopy in patients with SLE (4). In antibody-mediated rejection, we noticed that the presence of new autoreactive IgE is remarkably independent on allergic episodes too. Thus, IgE exerts its detrimental effects through atopy-independent pathways. The potential functional contributions of IgE autoantibodies to pathologic expressions of SLE and other autoimmune diseases have been emerging in the last few years. Their presence has been associated with the perpetuation of autoimmunity because self-reactive IgE promotes adaptive immune responses against self through both T cell and B cell activation. Several studies suggested a novel Th2 axis in SLE pathophysiology that might connect IgE, basophils, plasmacytoid dendritic cells, and IFN-α signature in which autoreactive IgE plays a pivotal role (6–9). In this setting, Henault et al. (10) clearly demonstrated that IgE induces IFN-α secretion from plasmacytoid dendritic cells that, in turn, can promote, among other effects, autoantibodies production by plasma cells.
Type 1 IFNs have pleiotropic roles in the immune system by modulating the activation of monocytes, dendritic cells, T cells, NK cells, and B cells. They are key drivers of antiviral immunity but also autoimmunity because they trigger B cell activation, proliferation, and plasma cells’ differentiation into autoantibody-producing cells (17). Unabated secretion of type 1 IFNs is central to SLE. The levels of IgE anti-DNA autoantibodies in SLE correlate with disease activity, and in vitro experiments have shown that IgE synergizes with IgG to trigger the secretion of IFN-α and other proinflammatory mediators. Accordingly, when IgE antibodies were neutralized by a blocking anti-IgE antibody, IFN-α produced by PBMCs in response to patient’s serum was significantly reduced, supporting the notion that IgE autoantibodies have an active role in the disease (10). Indeed, the involvement of the IFN pathway in SLE pathogenesis has now also been clinically validated in two phase 2 proof of concept studies using inhibitors that block either IFN-α or the type 1 IFN receptor (3).
Studying the transcriptomics of PBMCs, we have previously demonstrated that an IFN-α signature characterizes antibody-mediated rejection, supporting the hypothesis of a similarity between this condition sustained by the presence of donor-specific alloantibodies and the autoimmune humoral response featuring lupus nephritis (13). In this study, we demonstrated unequivocally that patients with antibody-mediated rejection present a strong IgE deposition at the graft level, further supporting an important pathogenic similarity between antibody-mediated rejection and SLE. In antibody-mediated rejection, as well as in lupus nephritis, IgE immune complexes deposition is associated with a higher infiltration of plasmacytoid dendritic cells and with an enhanced IFN-α signaling. This is suggested by significantly higher levels of circulating MxA, a specific marker of IFN-α signaling. Thus, it is conceivable that maintenance of the alloimmune response and the appearance of autoantibodies in this setting, as demonstrated by previous studies, might be due to this pathway. The reduction of total circulating IgE that we observed in our patients might strengthen the hypothesis of a significant IFN-α response in this setting because non-autoreactive IgE has a protective effect against type 1 IFN in SLE, known as the “IgE paradox” (8).
A further pathway activated by IgE and leading to the amplification of antibody response is represented by the activation of basophils (18). These cells express a specific IgE receptor on their membrane and are activated by IgE binding. For a long time, basophils were neglected and only considered to be circulating cells of minor importance. In addition, the low number of these cells in peripheral blood (<0.5% of total leukocytes) made their purification difficult in clinical laboratories. The consequent lack of satisfactory in vitro studies clearly hampered research on basophils’ functions for many years. We now appreciate that, after they are activated, basophils can directly and indirectly activate B cells, induce their proliferation, and boost antibody production (19). Lupus nephritis is characterized by a higher number of kidney-infiltrating basophils and by circulating basophils showing elevated l-selectin and CD203c, both associated with disease activity, suggesting a possible role for these cells in SLE-induced kidney injury (20). In line with this hypothesis, Charles et al. (6) demonstrated that basophils depletion can ameliorate lupus nephritis by decreasing autoantibody titer. Interestingly, Colvin and Dvorak (21) demonstrated >40 years ago a higher infiltration of basophils in acute rejection of kidney grafts. In the same period, Egido et al. (22) observed that, following kidney transplantation, basophils from six of 23 recipients showed microscopic signs of degranulation after contact with donor lymphocytes. Our finding of higher basophils infiltration in patients with antibody-mediated rejection opens up a completely different perspective on the role of these cells in the pathogenesis of graft rejection. Noteworthy, we clearly demonstrated significant basophils activation in antibody-mediated rejection biopsies. Indeed, CD203c, the basophils’ marker used in this study, is highly specific for activated basophils (23). Thus, these cells infiltrating the graft present the same phenotype of circulating basophils in SLE. However, the number of these cells at the peripheral level represents the main difference between our observation and previous studies on the role of basophils in SLE. We observed a significantly higher number of circulating basophils, whereas several reports observed a lower number in peripheral basophils in SLE (20). The reduction of basophil numbers in lupus was explained by the presence of ongoing immunosuppressive therapy. Also, graft recipients are in the same conditions, although qualitatively, the immunosuppressive therapy in the two settings is quite different. Indeed, SLE therapy is mainly on the basis of corticosteroids, whereas in our patients, the main drugs are calcineurin inhibitors.
In SLE, IgE immune deposits have been associated with a significant progression of kidney damage (20). It is well known that these Igs can directly interact with and activate mast cells. Tissue mast cells are notorious for their role as potential mediators of kidney damage. They express the high-affinity IgE receptor (FcεRI), and IgE crosslinking results in the release of proinflammatory and fibrogenic molecules, including tryptase, that are stored in their cytoplasmic granules (9). Tryptase can proteolytically activate a G protein cell surface receptor known as protease-activated receptor 2 (PAR2). We have previously demonstrated that PAR2 expression is significantly upregulated in IgA nephropathy and that it is directly and significantly associated with tubulointerstitial fibrosis. Interestingly, the activation of PAR2 on tubular cells in vitro leads to a higher expression of two key profibrotic molecules, plasminogen activator inhibitor 1 and TGF-β (24). A link between mast cells and interstitial fibrosis was also reported in kidney transplantation. Goto et al. (25) observed a significant correlation between the number of infiltrating mast cells and the extent of extracellular matrix deposition in kidney allograft. With a completely different approach, Mengel et al. (26) confirmed this observation. These authors reported a significantly higher number of mast cell transcripts in graft biopsies characterized by prominent scarring (26). In addition, they report that high levels of mast cells’ transcripts were significantly associated with worse transplant outcome (26). Our observation would suggest a potential mechanism of mast cells recruitment and activation specific for patients with antibody-mediated rejection. A more intense activation of mast cells in antibody-mediated graft damage might explain, in part, the particularly progressive feature of this condition.
The main limitations of this study are represented by the small sample size and by the lack of a mechanistic exploration of the observed phenomenon. Regarding the first point, we would consider this study as a proof of concept of a pathogenic hypothesis suggested by a previous observation. In addition, active antibody-mediated rejection may be diagnosed even if donor-specific anti-HLA antibodies are absent. It would have been interesting to evaluate the presence of IgE graft deposition in this condition. A small group of our control graft recipients presented a variable titer of circulating donor-specific anti-HLA antibodies, however, in none of them did we observe IgE deposition. In addition, we do not provide any mechanistic information, although it is largely documented in the existing literature. Finally, further testing would be needed to validate the anti-IgE reagents before our observation could be implemented as a clinical laboratory test.
In conclusion, our observation would suggest a novel concept of antibody-mediated rejection pathogenesis connecting IgE, basophils, mast cells, and type 1 IFN signature, in which self-reactive IgE seems to be the orchestrator. These findings potentially open new therapeutic perspectives, although our study should be considered as a hypothesis-generating investigation that warrants further validation in a large observational cohort study.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by Italian Ministry of Health grant Ricerca Finalizzata 2009 RF/2009 1471624 (to G. Grandaliano).
Acknowledgments
We thank Dr. Paolo Azzone, technical sales manager of Immucor Systems, who provided insight and expertise to assist the Luminex Assay for detection of HLA-specific IgE antibodies. We thank Dr. Samantha De Tullio (Clinical Biochemistry Unit, Department of Clinical and Experimental Medicine, University of Foggia) for her technical assistance in handling the Luminex instrument. We acknowledge Prof. Gaetano Corso (Clinical Biochemistry Unit, Department of Clinical and Experimental Medicine, University of Foggia) for his helpful suggestions during the drafting of our protocol for serum IgG depletion prior to performing anti-HLA IgE antibody assay.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
See related editorial, “IgE in Antibody-Mediated Rejection: A Novel Pathogenic Mechanism?” on pages 1392–1393.
References
- 1.Maurer M, Altrichter S, Schmetzer O, Scheffel J, Church MK, Metz M: Immunoglobulin E-mediated autoimmunity. Front Immunol 9: 689, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ettinger R, Karnell JL, Henault J, Panda SK, Riggs JM, Kolbeck R, Sanjuan MA: Pathogenic mechanisms of IgE-mediated inflammation in self-destructive autoimmune responses. Autoimmunity 50: 25–36, 2017. [DOI] [PubMed] [Google Scholar]
- 3.Rönnblom L, Elkon KB: Cytokines as therapeutic targets in SLE. Nat Rev Rheumatol 6: 339–347, 2010. [DOI] [PubMed] [Google Scholar]
- 4.Atta AM, Sousa CP, Carvalho EM, Sousa-Atta ML: Immunoglobulin E and systemic lupus erythematosus. Braz J Med Biol Res 37: 1497–1501, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Atta AM, Santiago MB, Guerra FG, Pereira MM, Sousa Atta ML: Autoimmune response of IgE antibodies to cellular self-antigens in systemic lupus erythematosus. Int Arch Allergy Immunol 152: 401–406, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Charles N, Hardwick D, Daugas E, Illei GG, Rivera J: Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat Med 16: 701–707, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosch X, Lozano F, Cervera R, Ramos-Casals M, Min B: Basophils, IgE, and autoantibody-mediated kidney disease. J Immunol 186: 6083–6090, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augusto JF, Truchetet ME, Charles N, Blanco P, Richez C: IgE in lupus pathogenesis: Friends or foes? Autoimmun Rev 17: 361–365, 2018. [DOI] [PubMed] [Google Scholar]
- 9.Khoryati L, Augusto JF, Shipley E, Contin-Bordes C, Douchet I, Mitrovic S, Truchetet ME, Lazaro E, Duffau P, Couzi L, Jacquemin C, Barnetche T, Vacher P, Schaeverbeke T, Blanco P, Richez C; Fédération Hospitalo-Universitaire ACRONIM : IgE inhibits Toll-like receptor 7- and Toll-like receptor 9-mediated expression of Interferon-α by plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Arthritis Rheumatol 68: 2221–2231, 2016. [DOI] [PubMed] [Google Scholar]
- 10.Henault J, Riggs JM, Karnell JL, Liarski VM, Li J, Shirinian L, Xu L, Casey KA, Smith MA, Khatry DB, Izhak L, Clarke L, Herbst R, Ettinger R, Petri M, Clark MR, Mustelin T, Kolbeck R, Sanjuan MA: Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat Immunol 17: 196–203, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loupy A, Hill GS, Jordan SC: The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol 8: 348–357, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Jin YP, Valenzuela NM, Zhang X, Rozengurt E, Reed EF: HLA class II-triggered signaling cascades cause endothelial cell proliferation and Migration: Relevance to antibody-mediated transplant rejection. J Immunol 200: 2372–2390, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rascio F, Pontrelli P, Accetturo M, Oranger A, Gigante M, Castellano G, Gigante M, Zito A, Zaza G, Lupo A, Ranieri E, Stallone G, Gesualdo L, Grandaliano G: A type I interferon signature characterizes chronic antibody-mediated rejection in kidney transplantation. J Pathol 237: 72–84, 2015. [DOI] [PubMed] [Google Scholar]
- 14.Thaunat O, Graff-Dubois S, Fabien N, Duthey A, Attuil-Audenis V, Nicoletti A, Patey N, Morelon E: A stepwise breakdown of B-cell tolerance occurs within renal allografts during chronic rejection. Kidney Int 81: 207–219, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Haas M, Sis B, Racusen LC, Solez K, Glotz D, Colvin RB, Castro MC, David DS, David-Neto E, Bagnasco SM, Cendales LC, Cornell LD, Demetris AJ, Drachenberg CB, Farver CF, Farris AB 3rd, Gibson IW, Kraus E, Liapis H, Loupy A, Nickeleit V, Randhawa P, Rodriguez ER, Rush D, Smith RN, Tan CD, Wallace WD, Mengel M; Banff meeting report writing committee : Banff 2013 meeting report: Inclusion of C4D-negative antibody-mediated rejection and antibody-associated arterial lesions [published correction appears in Am J Transplant 15: 2784, 2015]. Am J Transplant 14: 272–283, 2014. [DOI] [PubMed] [Google Scholar]
- 16.Farkas AM, Baranyi U, Böhmig GA, Unger L, Hopf S, Wahrmann M, Regele H, Mahr B, Schwarz C, Hock K, Pilat N, Kristo I, Mraz J, Lupinek C, Thalhamer J, Bond G, Kuessel L, Wlodek E, Martin J, Clatworthy M, Pettigrew G, Valenta R, Wekerle T: Allograft rejection is associated with development of functional IgE specific for donor MHC antigens. J Allergy Clin Immunol 143: 335–345.e12, 2019. [DOI] [PubMed] [Google Scholar]
- 17.Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A: Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol 90: 498–504, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sokol CL, Medzhitov R: Emerging functions of basophils in protective and allergic immune responses. Mucosal Immunol 3: 129–137, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Travers J, Rothenberg ME: Eosinophils in mucosal immune responses. Mucosal Immunol 8: 464–475, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pellefigues C, Charles N: The deleterious role of basophils in systemic lupus erythematosus. Curr Opin Immunol 25: 704–711, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colvin RB, Dvorak HF: Letter: Basophils and mast cells in renal allograft rejection. Lancet 1: 212–214, 1974. [DOI] [PubMed] [Google Scholar]
- 22.Egido J, Crespo M, Sanchez MG, Hernando L, Benveniste J: In-vitro basophil degranulation in drug-suspected acute renal failure. Lancet 2: 712–713, 1977. [DOI] [PubMed] [Google Scholar]
- 23.Bühring HJ, Streble A, Valent P: The basophil-specific ectoenzyme E-NPP3 (CD203c) as a marker for cell activation and allergy diagnosis. Int Arch Allergy Immunol 133: 317–329, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Grandaliano G, Pontrelli P, Cerullo G, Monno R, Ranieri E, Ursi M, Loverre A, Gesualdo L, Schena FP: Protease-activated receptor-2 expression in IgA nephropathy: A potential role in the pathogenesis of interstitial fibrosis. J Am Soc Nephrol 14: 2072–2083, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Goto E, Honjo S, Yamashita H, Shomori K, Adachi H, Ito H: Mast cells in human allografted kidney: Correlation with interstitial fibrosis. Clin Transplant 16[Suppl 8]: 7–11, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Mengel M, Reeve J, Bunnag S, Einecke G, Sis B, Mueller T, Kaplan B, Halloran PF: Molecular correlates of scarring in kidney transplants: The emergence of mast cell transcripts. Am J Transplant 9: 169–178, 2009. [DOI] [PubMed] [Google Scholar]