

Abstract
Epithelial ovarian cancer (EOC) has one of the highest deaths to incidence ratios among all cancers. High grade serous ovarian carcinoma (HGSOC) is the most common and deadliest EOC histotype due to the lack of therapeutic options following debulking surgery and platinum/taxane-based chemotherapies. For recurrent chemosensitive HGSOC, poly(ADP)-ribose polymerase inhibitors (PARPi; olaparib, rucaparib, or niraparib) represent an emerging treatment strategy. While PARPi are most effective in homologous recombination DNA repair-deficient (HRD) HGSOCs, recent studies have observed a significant benefit in non-HRD HGSOCs. However, all HGSOC patients are likely to acquire resistance. Therefore, there is an urgent clinical need to understand PARPi resistance and to introduce novel combinatorial therapies to manage PARPi resistance and extend HGSOC disease-free intervals. In a panel of HGSOC cell lines, we established matched olaparib sensitive and resistant cells. Transcriptome analysis of the matched olaparib-sensitive versus -resistant cells revealed activation of Wnt signaling pathway and consequently increased TCF transcriptional activity in PARPi-resistant cells. Forced activation of canonical Wnt signaling in several PARPi-sensitive cells via WNT3A reduced olaparib and rucaparib sensitivity. PARPi resistant cells were sensitive to inhibition of Wnt signaling using the FDA-approved compound, pyrvinium pamoate, which has been shown to promote downregulation of β-catenin. In both an HGSOC cell line and a patient-derived xenograft model, we observed that combining pyrvinium pamoate with olaparib resulted in a significant decrease in tumor burden. This study demonstrates that Wnt signaling can mediate PARPi resistance in HGSOC and provides a clinical rationale for combining PARP and Wnt inhibitors.
Keywords: PARP inhibitor, wnt signaling, ovarian cancer, therapy resistance, DNA repair
INTRODUCTION:
Epithelial ovarian cancer (EOC) is the deadliest gynecological malignancy and has a high death to incidence ratio (63 deaths:100 cases) (1). The most common EOC histotype is high grade serous ovarian carcinoma (HGSOC) and over 80% of HGSOC are diagnosed at late stages (III/IV) (1). About 80% of HGSOC patients respond to first-line therapy, which includes surgical debulking and platinum/taxane-based chemotherapies. However, HGSOC recurs in a majority of patients, who are subsequently treated with additional chemotherapeutic regimens (2). As tumors acquire chemoresistance, disease-free intervals are shortened with each subsequent recurrence, making the identification of effective and novel therapeutic strategies an urgent clinical need.
The Cancer Genome Atlas revealed approximately 50% of HGSOC tumors have mutations or deficiencies in the homologous recombination (HR) DNA repair pathway (3). For example, mutations or epigenetic silencing of BRCA1/2 are detected in 30% of HGSOC cases. HR-deficient cancers can be targeted in a synthetic lethal fashion using poly(ADP)-ribose polymerase (PARP) inhibitors (2,4–6). There are three PARP inhibitors (PARPi; olaparib, rucaparib, and niraparib) that are FDA-approved for recurrent HGSOC. Notably, PARPi offer a clinical benefit in patients with and without HR-deficient HGSOC tumors (7,8). Nevertheless, in a similar fashion as first-line chemotherapies, PARPi treated patients will likely recur with resistant disease.
Tumorigenesis and chemoresistance in numerous cancer types, including ovarian cancer, can be attributed to Wnt signaling. Canonical Wnt signaling is mediated through ligand (i.e., WNT3A) stimulation of frizzled (FZD) and lipoprotein receptor-related protein (LRP) receptors. WNT-receptor interactions promote sequestration of the β-catenin degradation complex leading to β-catenin accumulation. Increased nuclear β-catenin leads to its interaction with T-cell factor (TCF) and lymphoid enhancer factor (LEF) transcriptional activators and upregulation of TCF/LEF target genes (e.g. FOSL1). Previously, we established β-catenin-dependent TCF transcriptional activation is antagonized by non-canonical Wnt (β-catenin-independent) signaling, and in HGSOC primary tumors the loss of non-canonical Wnt signaling was associated with worse overall survival (9). Hyperactivation of Wnt signaling has been attributed to chemotherapeutic resistance in a variety of epithelial-derived cancers (10–12). Based on these observations, we investigated how aberrant activation of canonical β-catenin-dependent Wnt signaling regulates PARPi response and/or resistance.
In this study, we examined HGSOC models of acquired PARPi resistance. We observed that hyperactivation of the canonical Wnt signaling pathway was necessary and sufficient to promote olaparib and rucaparib resistance. In PARPi-resistant HGSOC cell lines, we observed an increase in TCF transcriptional activity compared to parental cells. We screened a panel of Wnt signaling inhibitors and observed an FDA-approved compound (pyrvinium pamoate, Pyr. Pam.) induced cell death in PARPi-resistant cells. PARPi-resistant cells had increased DNA repair capacity by both non-homologous end-joining (NHEJ) and HR, independent of BRCA2 reversion mutations. Pyrvinium pamoate effectively inhibited both NHEJ and HR-mediated repair in PARPi-resistant cells. In an ex vivo culture of a primary HGSOC tumor, PARPi and pyrvinium pamoate reduced proliferation. Utilizing HGSOC cell lines and HGSOC patient-derived xenograft (PDX) mouse models, we demonstrated that combining olaparib with pyrvinium pamoate significantly inhibited the rate of tumor growth, dissemination, and overall tumor burden in vivo.
MATERIALS AND METHODS:
Cell lines and culture conditions.
HGSOC cell lines were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cell lines PEO1, OVCAR10, OVCA433, and TOV-21G were obtained from the Gynecologic Tumor and Fluid Bank (GTFB) at the University of Colorado. UWB1.289 were obtained from the American Tissue Culture Collection. TOV-21G used as positive control for wildtype BRCA2. Viral packaging cells (293FT) were cultured in DMEM supplemented with 10% FBS at 37°C supplied with 5% CO2. Cells lines were authenticated at The University of Arizona Genomics Core using short tandem repeat DNA profiling. Regular Mycoplasma testing was performed using MycoLookOut Mycoplasma PCR detection (Sigma).
Gynecologic Tissue and Fluid Bank (GTFB).
The University of Colorado has an Institutional Review Board approved protocol (COMIRB #07-935) in place to collect tissue from gynecologic patients with both malignant and benign disease processes. All participants are counseled regarding the potential uses of their tissue and sign a consent form approved by the Colorado Multiple Institutional Review Board.
Retrovirus and lentivirus transduction.
Retrovirus production and transduction were performed as described previously (13). Lentivirus was packaged using the Virapower Kit from Life Technologies (Carlsbad, CA) following the manufacturer’s instructions as described previously (9). WNT3A construct was previously described (14). Cells transduced with virus encoding a puromycin resistance gene were selected in 1 µg/ml puromycin.
Reverse-transcriptase quantitative PCR (RT-qPCR).
RNA was isolated from cells with the RNeasy Mini Kit followed by on-column DNase digest (Qiagen). mRNA expression was determined using SYBR green Luna Universal One-step RT-PCR kit (New England Biolabs) with a BioRad CFX96 thermocycler. β−2-microglobulin (B2M), Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and 18s were used as internal controls as stated in figured legends. All primer sequences are in supplementary table 2.
TCF transcriptional reporter.
Using FuGENE6 transfection reagent (Promega), cells were transfected with either TOP-FLASH or FOP-FLASH. M50 Super 8x TOPFlash and FOPFlash were gifts from Randall Moon (Addgene # 12456/12457). Cells were incubated for 72 hours and subsequently lysed and analyzed using Luciferase Assay Kit (Promega), and luminescence was measured with a Promega GloMax.
Colony formation assay.
Cell lines were seeded and treated with increasing olaparib doses. Cell medium and olaparib were changed every two days for 12 days. Colonies were fixed (10% methanol/10% acetic acid) and stained with 0.4% crystal violet. Crystal violet was dissolved in fixative and absorbance was measured at 570nm.
3D Cell Culture.
3D Matrigel cell culture was performed as described previously (13). Cells were seeded on growth factor reduced Matrigel (Corning) and cultured in growth medium with 3% growth factor reduced Matrigel. Cells were treated with DMSO or olaparib. Media was changed every 3 to 4 days for 12 days. Images were taken using an Olympus DP73. Acini diameter was measured using ImageJ.
Reagents and antibodies.
Olaparib, rucaparib, WNT-C59, and PRI724 were obtained from Selleckchem. Pyrvinium pamoate was obtained from Sigma Aldrich. The following antibodies were obtained from the indicated suppliers: BRCA2 (Bethyl, Cat#A303-434A, 1:2000), Vinculin (Cell Signaling Technology, Cat#13901, 1:1000), WNT3A (R&D systems, Cat# MAB9025-100, 1:1000), γH2Ax (Ser139) (EMD Millipore, Cat# 05-636, 1:1000), mouse anti-β-Actin (Abcam, Cat# ab6276, 1:10,000), Rabbit anti-β-Actin (Abcam, Cat# ab8227, 1:10,000), Rad51 (Abcam, Cat# ab176458). Anti-rat HRP (Jackson ImmunoResearch, Cat# 112-035-062, 1:5000)
Immunoblotting.
Protein was extracted with RIPA buffer (150 mM NaCl, 1% TritonX-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0) supplemented with Complete EDTA-free protease inhibitors (Roche), NaF and Na3VO4. Protein was separated on a SDS-PAGE and transferred to PVDF membrane. Primary antibody incubation was performed overnight at 4°C. Secondary goat anti-rabbit (IRDye 680RD or IRDye 800CW, LI-COR, Cat # 92568071 or Cat # 926-32211, 1:20,000) and goat anti-mouse (IRDye 680RD or IRDye 800CW, LI-COR, Cat # 926-68070 or Cat# 925-32210, 1:20,000) antibodies were applied for one hour at room temperature. Blots were visualized using the Licor Odyssey Imaging System. For WNT3A immunoblotting, antibodies were diluted in 5% milk/TBST (50 mM Tris pH7.5, 150 mM NaCl, 0.1% Tween20) blocking buffer and HRP chemiluminescent signal was detected with SuperSignal West Femto (Thermo scientific) and visualized using the G:Box (SYNGENE).
RNA-Sequencing.
RNA was isolated from PEO1 olaparib-sensitive (n=2) and four PEO1 olaparib-resistant clones using RNeasy columns with on-column DNase digest (Qiagen). RNA quality was confirmed using an Agilent Tapestation and all RNA used for library preparation had a RIN>9. Libraries were created using Illumina TruSEQ stranded mRNA library prep (#RS-122-2102). Strand-specific pair-ended libraries were pooled and run on HiSeq4000 (Illumina). Library creation and sequencing were performed at the Genomics Core at the University of Colorado Anschutz Medical Campus. HISAT2 (15) was used for alignment against GRCh37 version of the human genome. Samples were normalized using TPM (Transcripts per Million) measurement and gene expression using the GRCh37 gene annotation was calculated using home-made scripts. The analysis was performed by the Division of Translational Bioinformatics and Cancer Systems Biology at the University of Colorado School of Medicine. RNA-sequencing has been deposited to NCBI: GSE117765.
Comet Assay.
Protocol as Trevigen’s protocol. Cells were seeded in 10 cm dishes and treated with 3 μM olaparib. 24 hours after plating, cells were collected and plated in Low Melt Agarose on slides. Cells were lysed and then electrophoresed in a neutral buffer (100 mM Tris, pH 9.0, 300 mM sodium acetate). Cells were stained with Sybr Gold. Images were taken using a Nikon DS-Ri2. Tail moment was measured using ImageJ.
Two-plasmid functional DNA repair assay.
Two-plasmid functional assays were performed to assess distal non-homologous end joining (distal NHEJ), microhomology end-joining (mh-NHEJ), and homology directed repair (HDR) (16). Cells were transfected with pimEJ5GFP (distal NHEJ) or EJ2GFP (mh-NHEJ) or pDRGFP (HDR). Cells were transfected with I-SceI, which introduces DNA double-strand breaks in the plasmids. After 72 hours, cells were collected and examined via a flow cytometer to quantify GFP positive cells. pimEJ5GFP and EJ2GFP were gifts from Jeremy Stark (Addgene # 44026/44025). pDRGFP and pCBASceI were gifts from Maria Jasin (Addgene # 26475/26477).
γH2Ax resolution assay.
Cells were plated in a 6-well plate and irradiated with 5 Gy. Irradiated cells were collected and utilized for protein extraction. Protein was used for immunoblot against γH2Ax and β-actin. Immunoblot signal was visualized on a Licor Odyssey and fluorescence signal was measured with the ImageStudio v4.0 software. γH2Ax signal was normalized to β-actin, graphed in Prism Graphpad, and followed by a linear regression analysis.
Cell Viability Assay.
HGSOC cells were seeded in a 96-well plate and treated for 72 hours. Cells were washed with PBS and incubated with MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide, Sigma Aldrich, St. Louis, MO). Formazan was dissolved in DMSO and absorbance (590 nm/620 nm) was measured with a Molecular Devices SpectraMax M2 microplate reader.
Annexin V/Propidium Iodide assay.
Phosphatidylserine externalization was detected using an Annexin V/propidium iodide (PI) staining kit (Life Technologies) following the manufacturer’s instructions. Annexin V/PI positive cells were detected using a Gallios Flow Cytometer (Flow Cytometry Core, University of Colorado) and analyzed with FlowJo software module.
Animal Models.
The protocols were approved by the Institutional Animal Care and Use Committee (IACUC). The PDX-1009 model was derived from ascites-associated disease isolated from a primary and untreated stage IIIC high grade serous carcinoma. The patient received six cycles of carboplatin + taxol and the disease was considered chemosensitive. The patient recurred nine months after cessation of therapy with chemoresistant disease and succumbed to her disease two months after disease progression. For the recurrent olaparib-insensitive HGSOC model, patient-derived ascites (2.9 × 106 cells) were intraperitoneally injected. After 7 days, tumor-bearing mice were randomized and treated with vehicle control (10% cyclodextrin) or olaparib (50 mg/kg, daily) for 21 days. Mice were monitored for 2 months, and recurred tumor and ascites were collected and used for analysis and subsequent PDX studies. For olaparib and Pyr. Pam. combination experiment, the sample size of 9 mice per group was determined based on the data shown from in vitro experiments. Intraperitoneal xenograft was performed as described previously (17). A lentiviral GFP/Luciferase was transduced into PEO1-WNT3A cells and GFP positive cells were sorted twice as previously described (18). Briefly, 3.6 X 106 GFP/luciferase-expressing PEO1-WNT3A cells were injected into the peritoneal cavity of 6-8-week-old female immunocompromised mice (n= 9-10 per group). PEO1-WNT3A cells were utilized for the in vivo evaluation because PEO1-OR cells failed to form IP tumors. 4 weeks after injection, tumors were visualized by injecting luciferin (i.p.: 4 mg/mice in PBS) and imaged with an In Vivo Imaging System (IVIS). Mice were randomized into 4 groups based on luciferase activity and weight. For both the PEO1-WNT3A and PDX models, mice were treated with vehicle control (10% cyclodextrin), olaparib (daily, 50 mg/kg), pyrvinium pamoate (daily, 0.5 mg/kg), or olaparib/pyrvinium pamoate. Mice were IP injected daily for 21 days and were imaged twice a week. Images were analyzed using Live Imaging 4.0 software. At end of the experiments, tumors were surgically dissected and tumor burden was calculated based on tumor weight. Peritoneal tumor nodules were quantified.
Immunohistochemistry.
Tumor specimens were fixed in 10% buffered formalin, paraffin embedded and sectioned by the Histopathology Core (The University of Colorado Cancer Center). The Histopathology core stained tumor sections for Ki67 (Thermo Scientific, cat. # RM-9106), cleaved caspase 3 (Cell Signaling, cat. # 9661), and β-catenin (Cell Marque, cat. # 224M-15). H-score was calculated for each tumor section as previously described (9).
Ex vivo Cultures.
A primary HGSOC tumor was obtained from the GTFB at the University of Colorado (COMIRB #07-935). The tumor was sectioned with Krumdieck Tissue Slicer. Tumor sections were cultured with pBABE-puro-gLuc. gLuc-tagged tissue sections were treated with olaparib (1 μM), Pyr Pam (1 μM), or in combination. Following a 72 hour incubation, gLuc activity was measured with Gaussia Luciferase kit (NEB) and analyzed on a Promega GloMax reader.
Statistical Analysis.
Statistical analyses and p-value calculations were performed using GraphPad Prism 7 (GraphPad) for Mac OS. Quantitative data are expressed as mean ± SEM unless otherwise stated. Analysis of variance (ANOVA) with Fisher’s Least Significant Difference (LSD) was used to identify significant differences in multiple comparisons. Combination index was calculated using CompuSyn. For all statistical analyses, the level of significance was set at 0.05.
RESULTS:
PARPi are effective in homologous recombination deficient (HRD) tumors, however, PARPi convey a significant clinical benefit in most HGSOC patients (7,8). Despite this initial benefit, acquired PARPi resistance eventually occurs, highlighting a need to better understand the underlying resistance mechanisms. We focused on olaparib, which has moderate to high PARP1/2 trapping activity and has been in clinical use longer than any other PARPi (19). We established olaparib-resistant HGSOC cell lines (PEO1-olaparib resistant, PEO1-OR; and OVCA433-olaparib resistant, OVCA433-OR) through step-wise escalation of olaparib. PEO1 cell lines were utilized because they are established HGSOC cells lines that have mutated BRCA2 (20,21). The OVCA433 cell line is TP53 mutated and BRCA-wildtype. Olaparib resistance was confirmed with a dose-response colony formation assay (Fig. 1A and Sup. Fig. 1A). In PEO1-OR cells, we confirmed cross-resistance to another PARPi, rucaparib (Sup. Fig. 1B). We isolated four clonal populations of PEO1-OR cells and confirmed >25-fold increase in half maximal inhibitory concentrations in all four PEO1-OR clones (Fig. 1B). BRCA reversion mutations have been shown to lead to PARPi resistance (22), and so, using an N-terminal BRCA2 antibody we confirmed that BRCA2 protein was not restored in PEO1-OR clonal populations (Sup. Fig. 1C), suggesting an alternative mechanism of acquired olaparib resistance.
Figure 1. Olaparib resistant high grade serous ovarian cancer cells have increased Wnt activation.
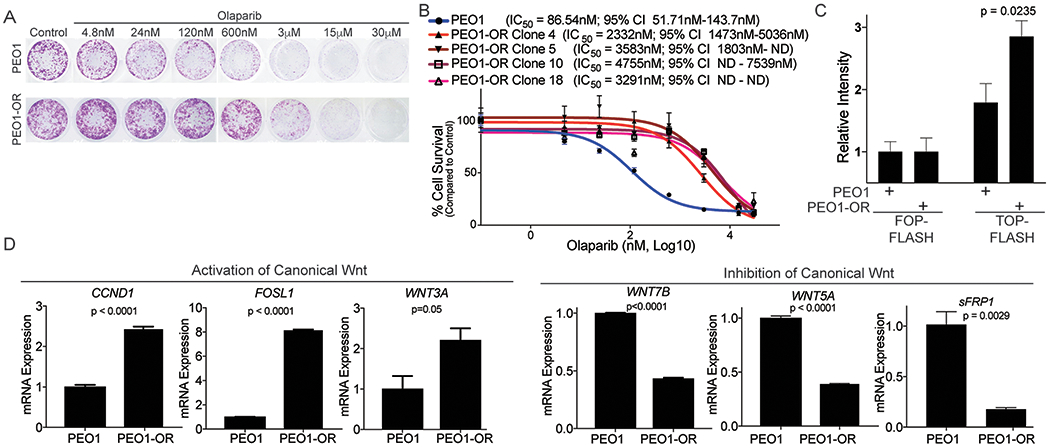
A) PEO1 (TP53 and BRCA2-mutated) were treated in a step-wise fashion with increasing doses of olaparib. PEO1 sensitive and resistant (PEO1-OR) cells were plated in a 24-well plate and treated with increasing doses of olaparib for 12 days. Cells were stained with crystal violet. B) Four clonal olaparib resistant populations were established and olaparib resistance was confirmed with a dose response colony formation assay. Dose response curves are graphed and IC50 with 95% confidence interval indicated. C) PEO1 and PEO1-OR were transfected with TCF transcriptional reporter (TOP-FLASH) or a control reporter (FOP-FLASH). Luciferase activity was measured. D) RNA from PEO1 and PEO1-OR was extracted and utilized for a quantitative reverse-transcriptase PCR (qRT-PCR) against CCND1, FOSL1, WNT3A, WNT7B, WNT5A, and SFRP1. Beta-2-microglobulin (B2M) was used as an internal control. Experiments performed in triplicate. Statistical test used to calculate p-values, unpaired two-tailed t-test. Error bars = SEM.
To examine an alternative mechanism of resistance, we evaluated the transcriptome of PARPi resistant cells. RNA-sequencing (RNA-seq) of the PEO1-OR clones was performed to identify differentially regulated gene expression and pathways compared to matched olaparib-sensitive cells. Principal component analysis and hierarchical clustering of sequences found replicates segregated together (Sup. Fig. 1D–E). Comparing sensitive cells to the PEO1-OR clones there were 1,819 differentially regulated genes (FDR<15%, p<0.00001; Sup. Table 1). Gene Set Enrichment Analysis, pathway enrichment (KEGG), and transcription factor analysis (23,24) of differentially expressed genes was performed to identify putative pathways mediating olaparib resistance. A majority of differentially expressed genes are regulated by TCF3 and LEF1 transcription factors (Table 1). KEGG pathway analysis revealed several enriched pathways including MAPK signaling, focal adhesion, and Wnt signaling (Table 2). Enrichment of TCF3/LEF1 targets and Wnt signaling is consistent with activation of canonical, β-catenin-dependent Wnt signaling in PEO1-OR cells. In olaparib-resistant cell lines, we examined TCF transcriptional activity through a reporter assay (TOP/FOP-FLASH). PEO1-OR and OVCA433-OR cells had a significant increase in TOP-FLASH activity (Fig. 1C and Sup. Fig. 1F). In PEO1-OR cells, we validated several genes from the RNA-seq analysis that were associated with activation (FOSL1, CCND1, WNT3A) or inhibition (WNT5A, WNT7B, SFRP1) of canonical Wnt signaling. Wnt target genes and activators were upregulated and Wnt signaling inhibitors were repressed in PEO1-OR cells (Fig. 1D). In PEO1-OR cells, we observed a significant increase in WNT3A, a potent driver of canonical Wnt signaling. Consistently, in OVCA433-OR cells, Wnt target genes were also upregulated (Sup. Fig. 1G). We observed that increased Wnt signaling was associated with PARPi resistance and could potentially be functioning to promote or maintain resistance.
Table 1.
Transcription factor enrichment in 1,819 PEO1-OR associated differentially regulated genes.
| Rank | Transcription Factor | # Genes in Gene Set | # Genes in Overlap | % of Overlap | p-value | FDR q-value |
|---|---|---|---|---|---|---|
| 1 | TCF3 | 2485 | 339 | 13.64% | 8.48E-100 | 5.22E-97 |
| 2 | SP1 | 2940 | 361 | 12.28% | 3.07E-93 | 9.44E-91 |
| 3 | MAZ | 2274 | 293 | 12.88% | 1.99E-79 | 4.09E-77 |
| 4 | LEF1 | 1972 | 255 | 12.93% | 6.04E-69 | 9.29E-67 |
| 5 | UNKNOWN | 1890 | 244 | 12.91% | 1.04E-65 | 1.28E-63 |
| 6 | NFAT | 1896 | 221 | 11.66% | 1.31E-51 | 1.34E-49 |
| 7 | AP4 | 1524 | 189 | 12.40% | 6.21E-48 | 5.45E-46 |
| 8 | FOXO4 | 2061 | 223 | 10.82% | 1.15E-46 | 8.84E-45 |
| 9 | TATA1 | 1296 | 158 | 12.19% | 4.07E-39 | 2.78E-37 |
| 10 | AP1 | 1121 | 142 | 12.67% | 5.00E-37 | 3.08E-35 |
Table 2.
KEGG pathway analysis from 1,819 PEO1-OR associated differentially regulated genes.
| Rank | KEGG | # Genes in Gene Se | # Genes in Overlap | % of Overlap | p-value | FDR q-value |
|---|---|---|---|---|---|---|
| 1 | Pathways In Cancer | 328 | 62 | 18.90% | 6.12E-26 | 1.14E-23 |
| 2 | Focal Adhesion | 201 | 41 | 20.40% | 6.90E-19 | 6.42E-17 |
| 3 | Axon Guidance | 129 | 29 | 22.48% | 5.97E-15 | 3.70E-13 |
| 4 | Cell Adhesion Molecules Cams | 134 | 29 | 21.64% | 1.72E-14 | 8.01E-13 |
| 5 | MAPK Signaling Pathway | 267 | 41 | 15.36% | 2.22E-14 | 8.27E-13 |
| 6 | Regulation Of Actin Cytoskeleton | 216 | 35 | 16.20% | 3.46E-13 | 1.07E-11 |
| 7 | Leukocyte Transendothelial Migration | 118 | 25 | 21.19% | 1.80E-12 | 4.78E-11 |
| 8 | WNT Signaling Pathway | 151 | 28 | 18.54% | 2.72E-12 | 6.32E-11 |
| 9 | T Cell Receptor Signaling Pathway | 108 | 23 | 21.30% | 1.23E-11 | 2.53E-10 |
| 10 | Small Cell Lung Cancer | 84 | 19 | 22.62% | 2.45E-10 | 4.55E-09 |
PARPi resistant cells have increased Wnt signaling, therefore we wanted to know if Wnt hyperactivation was sufficient to drive resistance. To examine the impact on increased Wnt/TCF activation on olaparib response, the open reading frame of a Wnt ligand (WNT3A) was transduced into a panel of HGSOC cells including PEO1, UWB1.289 (BRCA1-mutated), and OVCAR10 (BRCA-wildtype) cells. We utilized a tagless WNT3A expression construct to limit tag-dependent Wnt activity (14). WNT3A expression was confirmed to be significantly upregulated through quantitative PCR (qPCR) and immunoblot (Fig. 2A–B and Sup. Fig. 2A–B). In PEO1-WNT3A, TCF transcriptional activity was significantly increased (Fig. 2C), and canonical Wnt target genes (CCND1 and FOSL1) were upregulated (Fig. 2D). Next, olaparib sensitivity was assessed in PEO1-WNT3A cells via dose response in 2D colony formation and 3D acini formation. The overexpression of WNT3A promoted olaparib insensitivity in both 2D and 3D culture conditions (Fig. 2E–H). We also found that PEO1-WNT3A were resistant to an independent PARPi, rucaparib (Sup. Fig. 2C). Notably, in both the 2D and 3D olaparib dose response assays, PEO1-WNT3A cells were still moderately sensitive to olaparib suggesting an attenuation of olaparib’s effect and not complete resistance. Furthermore, olaparib sensitivity was assessed in additional WNT3A overexpressing HGSOC cell lines. UWB1.289-WNT3A and OVCAR10-WNT3A cells were more resistant than matched controls (Sup. Fig. 2D–E). UWB1.289 cells are BRCA1-mutated highlighting that Wnt activation is able to convey olaparib resistance independent of which BRCA gene is mutated. The Wnt-dependent decrease in olaparib sensitivity suggests that increased Wnt signaling is sufficient to promote resistance.
Figure 2. Activation of Wnt signaling contributes to olaparib resistance.

A) PEO1 cells were transduced with either lentiviral control (Control) or a construct specific for WNT3A. mRNA expression of WNT3A was evaluated in PEO1-Control and PEO1-WNT3A. Beta-2-microglobulin (B2M) was used as an internal control. B) Same as A, but protein was extracted, separated on a SDS-PAGE, and immunoblotted against WNT3A. Loading control = β-actin. Densitometry was performed on WNT3A blot by normalizing to β-actin C) PEO1 control or PEO1-WNT3A cells were transfected with a TCF reporter (TOP-FLASH) and control (FOP-FLASH). 72 hours after TOP/FOP-FLASH transfection luciferase activity was measured. D) RNA was extracted from PEO1 control and PEO1-WNT3A cells and used for qRT-PCR against FOSL1 and CCND1. B2M was used as an internal control. E) PEO1 control and PEO1-WNT3A cells were plated in a 24-well plate, treated with increasing dose of olaparib for 12 days, and remaining cells were stained with crystal violet. F) Same as E, quantification of crystal violet staining. G) PEO1 control and PEO1-WNT3A single suspensions were plated onto growth factor reduced matrigel, treated with increasing dose of olaparib, and cultured for 12 days. H) Same as G, acini diameter was measured and graphed as a percent of vehicle control. Experiments performed in triplicate. Statistical test used to calculate p-values, unpaired two-tailed t-test or ANOVA. Error bars = SEM.
Since Wnt signaling was activated in olaparib-resistant HGSOC cells and Wnt ligand over-expression was sufficient to reduce HGSOC cell sensitivity to olaparib, we examined the impact of pharmacologic inhibition of Wnt signaling. Wnt signaling was blocked by targeting distinct components of Wnt signaling using a porcupine inhibitor (WNT-C59) to block Wnt ligand secretion and two β-catenin inhibitors (PRI724 and Pyrvinium pamoate). PRI724 blocks β-catenin transcriptional activation by steric inhibition of β-catenin and one of its co-activators, CBP (CREB binding protein). Pyrvinium pamoate (Pyr. Pam.) promotes β-catenin downregulation by activating the β-catenin degradation complex (25). WNT-C59 did not reduce PEO1 or PEO1-OR cell viability at the highest concentration tested (50 μM) (Fig. 3A–B). OVCA433 and OVCA433-OR were also utilized to determine the impact of the Wnt inhibitors on HGSOC viability (Sup. Fig. 3A). Complementary to the MTT assay, we measured double strand DNA content as a surrogate for cell number. Similar to the MTT findings, we observed that all of the HGSOC cells were acutely sensitive to Pyr. Pam. (Sup. Fig. 3B–C), Notably, the PEO1-OR cells were significantly more sensitive to Pyr. Pam. compared to PEO1 (IC50=56.7 nM versus IC50= 315 nM, p<0.0001) (Fig. 3C). These data provide a rationale for using Pyr. Pam. to inhibit both PARPi sensitive and resistant HGSOC cell viability.
Figure 3. Pyrvinium pamoate significantly inhibits cell viability and synergizes with olaparib to induce apoptosis in PEO1-OR cells.
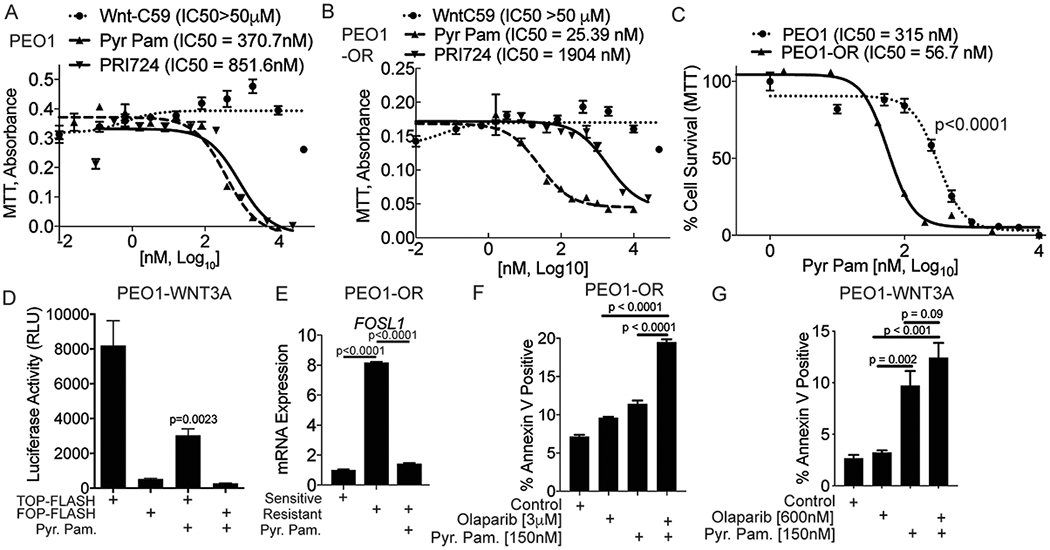
A) PEO1 cells were treated with increasing doses of three different Wnt inhibitors (Wnt-C59, Pyr. Pam., and PRI724). Cells were treated for 48 hours and a MTT assay was used to examine changes in cell viability. IC50 values are indicated for each inhibitor. B) Same as A, but used PEO1-OR cells. C) PEO1 and PEO1-OR were incubated for 48 hours with increasing doses of Pyr. Pam. and a MTT assay was used to examine changes in cell viability. IC50 values indicated. D) PEO1-WNT3A cells were transfected with a TCF reporter (TOP-FLASH) and control (FOP-FLASH). Cells were treated with Pyr. Pam. (370 nM) and luciferase activity was measured. E) PEO1-OR cells were treated with vehicle control or Pyr. Pam. (370 nM) and compared to untreated PEO1 cells. RNA was extracted and RT-qPCR was performed against FOSL1. B2M was used as an internal control. P-values calculated with ANOVA. F) PEO1-OR cells were treated with vehicle control (DMSO), olaparib (3 μM), Pyr. Pam. (150 nM), or in combination. Cells were treated for 72 hours and subjected to an apoptosis assay (AnnexinV-positive). G) Same as F, but examined PEO1-WNT3A cells. Experiments performed in triplicate. Unless noted, statistical test used to calculate p-values, unpaired two tailed t-test. Error bars = SEM.
Pyr. Pam. is an FDA approved anthelminthic that functions to promote β-catenin degradation, but can also inhibit other oncogenic proteins (26). In vitro Pyr. Pam. has been reported to inhibit colorectal, ovarian, and breast cancers (26–28). In PEO1-OR cells, we confirmed that Pyr. Pam. inhibited TCF transcriptional activity and repressed FOSL1 expression (Fig. 3D–E). Apoptosis was measured via phosphatidylserine externalization and cell permeability (Annexin/PI) in both PEO1-OR and PEO1-WNT3A cells treated with a combination of olaparib and Pyr. Pam.. Examination of the single agent treatment confirmed that both the PEO1-OR and PEO1-WNT3A cells exhibited an attenuated apoptotic response to olaparib and combining olaparib and Pyr. Pam. promoted more apoptosis compared to single agent alone (Fig. 3F–G). These data suggest that treating olaparib resistant HGSOC in combination (olaparib and Pyr. Pam.) could promote increased tumor cell apoptosis.
We next examined the mechanism of Wnt signaling mediated PARPi insensitivity. PARPi resistance independent of a BRCA-reversion mutation has been attributed to the restoration of DNA replication fork (RF) stability and/or increased DNA repair capacity (29,30). Given that ectopic expression of WNT3A resulted in decreased olaparib sensitivity, we examined RF stability following WNT3A overexpression in PEO1 cells. RF stability was assessed as previously described (31). Although significant, WNT3A overexpression only marginally rescued hydroxyurea-induced RF degradation (9.21 vs. 10.43 microns for PEO1 and PEO1-WNT3A, respectively, Sup. Fig 4A). We next examined DNA damage and repair capacity through functional assays.
We evaluated DNA damage in PEO1 and PEO1-OR cells through single-cell electrophoresis (Comet assay). In PEO1 cells, olaparib treatment resulted in a significant (p < 0.0001) increase in tail moment (Fig. 4A–B). However, treatment of PEO1-OR cells with the same dose of olaparib failed to increase the tail moment suggesting altered DNA repair response (Fig. 4A–B). We examined DNA damage repair response through γH2Ax resolution and by utilizing two-plasmid functional assays. As previously demonstrated (32), we utilized a marker of DNA damage (Serine 139 phosphorylated histone H2x, γH2Ax) and examined the rate of γH2Ax resolution as a DNA repair read-out. PEO1, PEO1-WNT3A, and PEO1-OR cells were irradiated (5 Gy) and γH2Ax levels were examined via immunoblot. Over an 8-hour time course, both PEO1-WNT3A and PEO1-OR cells resolved γH2Ax 2.5X and 2.1X faster than PEO1 cells, respectively (Fig. 4C–D). These data suggest that WNT3A expression is potentially contributing to more efficient DNA repair in a similar fashion as the PEO1-OR cells.
Figure 4. PEO1-OR cells have increased DNA repair capacity, which is inhibited by Pyr. Pam.

A) PEO1 and PEO1-OR cells treated with vehicle control or olaparib (3 μM) for 72 hours and a single cell suspension was fixed in agarose. Fixed cells were subjected to electrophoresis and DNA was labeled. Representative images of cells. B) Same as A, DNA tails (at least 100 cells) were measured with ImageJ and graphed as tail moment. C) PEO1, PEO1-OR, and PEO1-WNT3A cells were irradiated (5 Gy) and incubated for the indicated time. Protein was extracted, separated by a SDS-PAGE, and immunoblotted against of serine 139 phosphorylated histone H2x (γH2Ax). Loading control = β-actin. D) Densitometric analysis of C. Relative γH2Ax levels calculated based on β-actin levels graphed over time analyzed with a linear regression. The slopes are graphed with standard error. E) Two-plasmid functional assay performed to assess distal non-homologous end joining. PEO1 and PEO1-OR clones were stably transfected with pimEJ5GFP and subsequently transfected with I-SceI restriction enzyme. After 72 hours, transfected cells were collected and examined via a flow cytometer to quantify GFP positive cells. Representative gating strategy is shown. F) Quantification of E. P-values calculated with ANOVA. G) Same as F, but utilized pDR-GFP to measure homology directed repair in PEO1 and PEO1-OR clones. P-values calculated with ANOVA. H) PEO1-OR clone 4 (C4) cells were treated with Pyr. Pam. (150 nM) 24 hours after I-SceI transfection and distal NHEJ assessed. I) PEO1-OR clone 4 cells were treated with Pyr. Pam (+ 150 nM and ++ 370 nM) 24 hours after I-SceI transfection and HR assessed. Experiments performed in triplicate. Unless noted statistical test used to calculate p-values, unpaired two tailed t-test. Error bars = SEM.
We subsequently employed a functional two-plasmid DNA repair system to assess specific functional DNA repair pathways: HR, distal NHEJ, and microhomology NHEJ (mh-NHEJ). Briefly, a unique restriction enzyme, I-SceI, introduces DNA double-strand breaks in a unique GFP-mutated plasmid and DNA repair by the specific pathway leads to the restoration of a GFP open-reading frame (16). To limit heterogeneity, PEO1-OR clonal populations were analyzed for three DNA repair pathways. Cells were incubated for 72 hours after I-SceI transfection and the percentage of GFP-positive cells was measured. Only one of the four PEO1-OR clones had a significant increase in mh-NHEJ-mediated repair compared to the sensitive control (Sup. Fig. 4B). In contrast, all four PEO1-OR clones had a significant increase in distal-NHEJ (Fig. 4E–F) and three of the four PEO1-OR clones had a significant increase in HR-mediated repair compared to sensitive cells (Fig. 4G). We confirmed HR-mediated repair in PEO1-OR clones by irradiating (IR, 5 Gy) cells and evaluating Rad51-foci formation, which is a functional HR readout (33). IR-induced Rad51-positive cells were quantified for each PEO1-OR clone and we found a significant increase in IR-induced Rad51 positive cells in three of the four PEO1-OR clones compared to PEO1 parental cells (Sup. Fig. 4C–D). The functional DNA repair assays demonstrate that DNA damage repair is increased through both NHEJ and HR mechanisms.
Utilizing the functional repair assays, we next wanted to determine whether inhibiting Wnt signaling with Pyr. Pam. could alter DNA damage response. We observed that Pyr. Pam. treatment significantly inhibited both distal-NHEJ and HR-mediated DNA repair (Fig. 4H–I). Taken together, these data suggest PARPi resistant cells have increased DNA damage repair capacity independent of BRCA2 reversion mutations, which is attenuated by a Wnt inhibitor.
We next assessed the response to olaparib and/or Pyr. Pam by utilizing an ex vivo model of a primary BRCA1-mutated (c.3664G>T) chemonaïve tumor. Primary tumors were sectioned with a Krumdieck tissue slicer, which produces uniform tissue slices that can be utilized for short-term culture. The HGSOC primary tumor was sectioned and tagged with a secreted luciferase (Guassia Luciferase; gLuc). gLuc activity has been utilized to measure proliferation and is directly correlated to cell number (34,35). gLuc-tagged tumor sections were treated with olaparib and/or Pyr. Pam.. Based on the BRCA-status, we expected the tumor to be responsive to olaparib. We observed that olaparib, Pyr. Pam., and combination treated tumor sections had significantly reduced gLuc activity compared to the vehicle control (Fig. 5A). The gLuc activity in the combination treated tumors was significantly lower compared to olaparib alone suggesting an increased benefit to combining Wnt and PARP inhibition in an olaparib-sensitive tumor. We next evaluated Pyr. Pam. activity in in vivo models of olaparib resistance.
Figure 5. Combination of olaparib and Pyr. Pam. significantly inhibited HGSOC tumor progression.

A) A primary high grade serous ovarian cancer was utilized for ex vivo culture. Tumor sections were tagged with a secreted luciferase and subsequently treated with olaparib and Pyr. Pam. for 48 hours. Cell culture media was refreshed and luciferase activity was measured after 4 hours. B) GFP/luciferase PEO1-WNT3A cells were injected into the intraperitoneal cavity of immunocompromised mice. Tumors were allowed to establish for 4 weeks. Mice were imaged and randomized based on luminescence intensity on Day 0. Mice were treated daily for 21 days with vehicle control (n = 9), olaparib (n = 9, 50 mg/kg), Pyr. Pam. (n = 10, 0.5 mg/kg), and olaparib/Pyr. Pam. (n = 9, 0.5 mg/kg). Mice were imaged twice a week. Mice were imaged and sacrificed on Day 22. The rate of luminescence change over the course of treatment graphed. C) Images of tumors derived from all groups. Scale bar = 1 cm. D) The weight of total tumor burden indicated as tumor weight. E) The number of tumor nodules from each group. F) Immunohistochemical (IHC) analysis of treated tumors from (B) against Ki67, Cleaved (Cl.) caspase 3, and β-catenin. Representative images of consecutive images. Scale bar = 100 micron. G) Histology score (% of section x intensity) for Ki67 H) Same as G, but for Cl. Caspase 3. I) Same as G, but for β−catenin. J) Olaparib resistant PDX tumor cells were i.p. injected into NSG mice and tumors were allowed to establish for 2 weeks. Mice were treated daily for 21 days with vehicle control (n = 10), olaparib (n = 10, 50 mg/kg), Pyr. Pam. (n = 10, 0.5 mg/kg), and olaparib/Pyr. Pam. (n = 10, 0.5 mg/kg). The volume of tumor cells isolated from peritoneal cavity was quantified. Statistical test used to calculate p-values, Analysis of variance (ANOVA). Error bars = SEM.
We examined the anti-tumor properties of combining olaparib with Pyr. Pam. in an in vivo intraperitoneal (IP) model of HGSOC. The in vitro results suggest an increase in Wnt signaling leads to decreased PARPi sensitivity and increased DNA damage repair. Consistent with previous reports, PEO1 cells fail to efficiently form IP tumors (36) and notably PEO1-OR cells also failed to establish tumors (data not shown). However, PEO1-WNT3A cells did form IP tumors, therefore, we tagged PEO1-WNT3A with GFP/luciferase and injected the cells into the peritoneal cavity of nude SCID gamma (NSG) mice. Tumors were allowed to establish for four weeks and then treated with vehicle control, olaparib, Pyr. Pam., or in combination. In PEO1-WNT3A cells, in vitro treatment with olaparib was able to reduce 2D and 3D albeit to a lesser extent than PEO1 cells, therefore we predicted PEO1-WNT3A tumors would still demonstrate a level of olaparib sensitivity. Measured by total flux (photons/sec), we observed that all treatments resulted in a significant reduction in tumor growth rate compared to control mice (Fig. 5B and Sup. Fig. 5A). However, compared to controls upon examination of tumor weight the olaparib and Pyr. Pam. treated tumors were not significantly different. Only the olaparib/Pyr. Pam. treated tumors resulted in a significant decrease in tumor weight compared to vehicle control treated tumors (12.7 vs. 3.978 mg, p=0.0365, Fig. 5C–D). Dissemination of tumors was measured by quantifying GFP-positive tumor nodules during necropsy. There was not a significant difference between the number of control and olaparib treated tumors. Only mice treated with Pyr. Pam. and olaparib/Pyr. Pam. demonstrated a significant reduction in the number of tumor nodules (Fig. 5E). With respect to toxicity, although not significant, we did observe that mice in all treatment groups had reduced body weight (Sup. Fig. 5B). Immunohistochemistry (IHC) against Ki67 (proliferation), cleaved caspase 3 (apoptosis), and β-catenin was performed (Fig. 5F). IHC staining of tumors was blindly scored and a histology score (H-Score) was calculated for each tumor section (37). Olaparib and combination treated tumors had a significant decrease in Ki67 compared to control tumors (Fig. 5G). Notably, only the combination had an increase in cleaved caspase 3 compared to control tumors (Fig. 5H), suggesting that WNT3A-mediated olaparib insensitivity is potentially mediated through an attenuated apoptotic response. Pyr. Pam. inhibits Wnt signaling by promoting β-catenin degradation. We noted that Pyr. Pam. and combination treated tumors had significantly lower β-catenin expression (Fig. 5I). These data suggest that PEO1-WNT3A tumors were less responsive to olaparib and that inhibiting Wnt signaling led to a more robust anti-tumor effect.
PARPi resistant tumors are difficult to obtain (2) so we established a patient-derived xenograft (PDX) model of olaparib-resistant HGSOC (PDX-GTFB1009-olaparib resistant, PDX-GTFB1009-OR). Primary tumor cells were isolated from patient ascites and tumors were propagated through NSG mice. The patient was diagnosed with stage IIIC HGSOC, was chemonaïve when treated with carboplatin + taxol, and after treatment was designated chemosensitive. Subsequently, the chemosensitive PDX-GTFB1009 cells were injected into mice and tumor-bearing mice were treated with control or olaparib and tumors were allowed to recur after treatment. Olaparib-treated tumors that had recurred were collected and used to establish the GTFB1009-OR model. Utilizing this model, we examined the effect of Pyr. Pam. on tumor burden. GTFB1009-OR tumor cells were injected into NSG mice and tumor-bearing mice were treated with control, olaparib, Pyr. Pam., or in combination. Tumors were highly resistant to olaparib (Fig. 5J). In contrast, compared to control or olaparib, Pyr. Pam. treatment alone significantly inhibited tumor burden. Also, the combination treated mice had a significant reduction in tumor burden measured by the volume of tumor cells within the ascites fluid (Fig. 5J and Sup. Fig. 5C). Given the robust Pyr. Pam. response, it was difficult to assess whether the addition of olaparib was beneficial and suggests that Pyr. Pam. alone could be effective against olaparib resistant tumors. Also, the lack of remaining tumor tissue in the Pyr. Pam. and combination group prohibited downstream investigations. As a surrogate for toxicity we measured body weight. While we observed a reduction, it was not significant compared to control treated mice. The in vivo response observed in the xenograft models of HGSOC suggests that targeting Wnt signaling could prove to be an effective next-line therapeutic option following PARPi treatment.
DISCUSSION:
Utilization of PARP inhibitors in the clinic is continuing to expand, which highlights an urgent need to better understand resistance mechanisms. In this report, we established that olaparib resistant HGSOC cells display hyperactivation of Wnt signaling and increased TCF transcriptional activity. Olaparib resistant HGSOC cells demonstrated increased DNA repair capacity, independent of BRCA2 reversion mutations, and pharmacologic inhibition of Wnt signaling attenuated the DNA damage repair response. Moreover, the combination of PARP and Wnt inhibition significantly inhibited tumor burden in HGSOC cell lines and patient-derived xenograft models.
Transcriptome analysis of PEO1-OR cells revealed several significantly enriched signaling pathways and transcription factors, which highlights there are potentially multiple contributing pathways involved in promoting and maintaining PARPi resistance. We noted that olaparib resistant PEO1 and OVCA433 had an increase in TCF transcriptional activation. In the PEO1-OR, TCF3 and LEF1 transcription factors were predicted to regulate a significant proportion of the olaparib resistant-related genes. TCF and LEF transcriptional activation are dependent on Wnt/β-catenin activation. Therefore, we defined the relationship between Wnt signaling and PARPi response. Wnt activation promoted olaparib resistance in three independent cell lines. Wnt signaling is a driver of several major biological processes including proliferation, stemness, epithelial-mesenchymal transition, and DNA damage. Wnt activation has been linked to chemotherapy resistance in a variety of cancer types including prostate, colorectal, and ovarian (38–40).
Beyond TCF/LEF transcription factors, we noted in PEO1-OR cells that the heterodimeric AP-1 transcription factor was identified as a top transcriptional regulator of differentially expressed genes. There is significant crosstalk between Wnt signaling and AP-1 regulation such as Wnt signaling-mediated expression of AP-1 subunits (FOSL1, JUN). Increased AP-1 activity following increased subunit transcription has been reported to mediate a more aggressive phenotype in squamous cell carcinoma (41). Additionally, there is evidence that Wnt and Dishevelled proteins are able to activate both JNK, a canonical AP-1 activator, as well as AP-1 transcriptional complexes (42). Notably, p53, BRCA, NFκB, and AP-1 play critical roles in promoting the expression of DNA repair genes [reviewed in (43)]. HGSOC are often characterized by inactivation of p53 and BRCA, therefore these cancers are potentially more dependent on AP-1 for DNA damage repair. Taken together, therapeutically targeting AP-1 could possibly provide a synthetic lethality in PARPi resistant HGSOC. Future studies will investigate the relationship between Wnt signaling, AP-1 activation, and PARPi resistance.
In this report, several Wnt inhibitors were examined, all of which have distinct mechanisms of action. Inhibitors of porcupine-mediated Wnt ligand secretion and β-catenin/CBP interaction failed to induce cytotoxic effects in the HGSOC cell lines tested, suggesting a potential secretion independent Wnt activity (14). In contrast, HGSOC cells were sensitive to Pyr. Pam. induced β-catenin degradation, suggesting that future studies should focus on β-catenin stability. A caveat is that while Pyr. Pam. is an established Wnt inhibitor, it is also known to inhibit other oncogenic pathways including Akt/PI3K (26). We found Pyr. Pam. significantly reduced TCF transcriptional activity indicating that an inhibitor of the β-catenin and TCF interaction could be an effective strategy to treat PARPi resistant HGSOC. With respect to translatability, clinical studies found that Pyr. Pam. has relatively poor pharmacodynamics properties and limited bioavailability (44). Similarly to platinum-based chemotherapies, Pyr. Pam. could be administered directly into the peritoneal cavity and because of its pharmacodynamic properties, would not result in systemic off-target effects. Toxicities reported by the National Institute of Health are minimal, with a small proportion of Pyr. Pam. treated adults patients experiencing nausea, vomiting, and cramping at large doses when delivered in suspension (45).
BRCA-reversion mutations are a described adaptation that leads to PARPi resistance (46,47). Transcript and protein analysis did not reveal secondary BRCA2 reversion events in PEO1-OR cells. Independent of BRCA-reversion mutations, RF stability has been shown to promote PARPi resistance (29); however, we did not find that the hyperactivation of Wnt signaling induced a robust change in RF stability. We observed that HR and distal NHEJ DNA damage repair were increased in some PEO1-OR cells compared to sensitive. Notably, all of the PEO1-OR clones did show an increase in either HR or distal NHEJ, suggesting that increased DNA repair capacity is potentially being promoted through both Wnt-dependent and independent mechanisms. Distal NHEJ and mhNHEJ are predominantly performed by similar effector proteins, but are distinguished by the DNA polymerases, DNA protein kinase (DNA-PK) and nucleases [reviewed in (48)], indicating that targeting the distal NHEJ pathway in PARPi resistant tumors is possible without inhibiting mhNHEJ.
In conclusion, the hyperactivation of Wnt signaling contributes to and partially drives PARPi resistance independent of BRCA-reversion mutations. Inhibition of Wnt signaling reduces DNA repair capacity and significantly inhibits tumor progression in vivo. These findings offer a strong rationale to further examine the role of Wnt signaling in PARP inhibitor response.
Supplementary Material
ACKNOWLEDGEMENTS:
We thank Andrew P. Bradford for critical evaluation of this manuscript. B.G. Bitler is supported by an NIH/NCI grant (R00CA194318), DOD OCRP Pilot Award (OC170228), and The Cancer League of Colorado. M.J. Sikora is supported by NIH/NCI grant (R00CA193734). This work was supported in part by the University of Colorado Cancer Center’s Genomics and Microarray Core Shared Resource funded by NCI grant P30CA046934. Supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Grant support: B.G. Bitler is supported by an NIH/NCI grant (R00CA194318), DOD OCRP Pilot Award (OC170228), and The Cancer League of Colorado. M.J. Sikora is supported by NIH/NCI grant (R00CA193734). This work was supported in part by the University of Colorado Cancer Center’s Genomics and Microarray Core Shared Resource funded by NCI grant P30CA046934. Supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Abbreviations:
- EOC
epithelial ovarian cancer
- HGSOC
high grade serous ovarian cancer
- PARP
poly(ADP) ribose polymerase
- PARPi
PARP inhibitor
- Pyr. Pam.
pyrvinium pamoate
- BRCA2
breast cancer type 2 susceptibility
- HR
homologous recombination
- NHEJ
non-homologous end joining
Footnotes
Competing financial interests: The authors declare no competing financial interests.
REFERENCES:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30 [DOI] [PubMed] [Google Scholar]
- 2.Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol Oncol 2017;147:695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21 [DOI] [PubMed] [Google Scholar]
- 5.McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer Biol Ther 2005;4:934–6 [DOI] [PubMed] [Google Scholar]
- 6.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res 2006;66:8109–15 [DOI] [PubMed] [Google Scholar]
- 7.Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol 2016;140:199–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med 2016;375:2154–64 [DOI] [PubMed] [Google Scholar]
- 9.Bitler BG, Nicodemus JP, Li H, Cai Q, Wu H, Hua X, et al. Wnt5a Suppresses Epithelial Ovarian Cancer by Promoting Cellular Senescence. Cancer Res 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F, et al. Long non-coding RNA UCA1 increases chemoresistance of bladder cancer cells by regulating Wnt signaling. FEBS J 2014;281:1750–8 [DOI] [PubMed] [Google Scholar]
- 11.Nagaraj AB, Joseph P, Kovalenko O, Singh S, Armstrong A, Redline R, et al. Critical role of Wnt/beta-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget 2015;6:23720–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang ZM, Wu JF, Luo QC, Liu QF, Wu QW, Ye GD, et al. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/beta-catenin pathway. Oncogene 2016;35:4787–97 [DOI] [PubMed] [Google Scholar]
- 13.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 2015;21:231–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao DM, Ferguson RL, Sikora MJ. WNT4 and WNT3A activate cell autonomous Wnt signaling independent of secretion. bioRxiv 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12:357–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol 2012;920:379–91 [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res 2016;76:6320–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanna C, Kwok L, Finlay-Schultz J, Sartorius CA, Cittelly DM. Labeling of Breast Cancer Patient-derived Xenografts with Traceable Reporters for Tumor Growth and Metastasis Studies. J Vis Exp 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, et al. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat Commun 2016;7:12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol 2013;229:422–9 [DOI] [PubMed] [Google Scholar]
- 23.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73 [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol 2010;6:829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venerando A, Girardi C, Ruzzene M, Pinna LA. Pyrvinium pamoate does not activate protein kinase CK1, but promotes Akt/PKB down-regulation and GSK3 activation. Biochem J 2013;452:131–7 [DOI] [PubMed] [Google Scholar]
- 27.Xu L, Zhang L, Hu C, Liang S, Fei X, Yan N, et al. WNT pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int J Oncol 2016;48:1175–86 [DOI] [PubMed] [Google Scholar]
- 28.Zhang C, Zhang Z, Zhang S, Wang W, Hu P. Targeting of Wnt/beta-Catenin by Anthelmintic Drug Pyrvinium Enhances Sensitivity of Ovarian Cancer Cells to Chemotherapy. Med Sci Monit 2017;23:266–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016;535:382–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 2017;68:414–30 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choe KN, Nicolae CM, Constantin D, Imamura Kawasawa Y, Delgado-Diaz MR, De S, et al. HUWE1 interacts with PCNA to alleviate replication stress. EMBO Rep 2016;17:874–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams BR, Golding SE, Rao RR, Valerie K. Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One 2010;5:e10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A 1995;92:2298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tannous BA. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc 2009;4:582–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wheeler LJ, Watson ZL, Qamar L, Yamamoto TM, Post MD, Berning AA, et al. CBX2 identified as driver of anoikis escape and dissemination in high grade serous ovarian cancer. Oncogenesis 2018;7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hernandez L, Kim MK, Lyle LT, Bunch KP, House CD, Ning F, et al. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol Oncol 2016;142:332–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarty KS Jr., Szabo E, Flowers JL, Cox EB, Leight GS, Miller L, et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res 1986;46:4244s–8s [PubMed] [Google Scholar]
- 38.Ohigashi T, Mizuno R, Nakashima J, Marumo K, Murai M. Inhibition of Wnt signaling downregulates Akt activity and induces chemosensitivity in PTEN-mutated prostate cancer cells. Prostate 2005;62:61–8 [DOI] [PubMed] [Google Scholar]
- 39.Su HY, Lai HC, Lin YW, Liu CY, Chen CK, Chou YC, et al. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int J Cancer 2010;127:555–67 [DOI] [PubMed] [Google Scholar]
- 40.Xu N, Shen C, Luo Y, Xia L, Xue F, Xia Q, et al. Upregulated miR-130a increases drug resistance by regulating RUNX3 and Wnt signaling in cisplatin-treated HCC cell. Biochem Biophys Res Commun 2012;425:468–72 [DOI] [PubMed] [Google Scholar]
- 41.Ding X, Pan H, Li J, Zhong Q, Chen X, Dry SM, et al. Epigenetic activation of AP1 promotes squamous cell carcinoma metastasis. Sci Signal 2013;6:ra28 1,–13, S0–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Amerongen R Alternative Wnt pathways and receptors. Cold Spring Harb Perspect Biol 2012;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res 2013;41:8403–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith TC, Kinkel AW, Gryczko CM, Goulet JR. Absorption of pyrvinium pamoate. Clin Pharmacol Ther 1976;19:802–6 [DOI] [PubMed] [Google Scholar]
- 45.The Pharmacological Basis of Therapeutics. Goodman LS, Gilman A, editors: New York: Macmillan Publishing Co; 1975. [Google Scholar]
- 46.Christie ELF S; Doig K; Pattnaik S; Dawson SJ; Bowtell DD Reversion of BRCA1/2 Germline Mutations Detected in Circulating Tumor DNA From Patients With High-Grade Serous Ovarian Cancer. J Clin Oncol 2017 [DOI] [PubMed] [Google Scholar]
- 47.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008;451:1111–5 [DOI] [PubMed] [Google Scholar]
- 48.Pannunzio NR, Li S, Watanabe G, Lieber MR. Non-homologous end joining often uses microhomology: implications for alternative end joining. DNA Repair (Amst) 2014;17:74–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.