

Abstract
The ongoing pandemic caused by a novel coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2), affects thousands of people every day worldwide. Hence, drugs and vaccines effective against all variants of SARS‐CoV‐2 are crucial today. Viral genome mutations exist commonly which may impact the encoded proteins, possibly resulting to varied effectivity of detection tools and disease treatment. Thus, this study surveyed the SARS‐CoV‐2 genome and proteome and evaluated its mutation characteristics. Phylogenetic analyses of SARS‐CoV‐2 genes and proteins show three major clades and one minor clade (P6810S; ORF1ab). The overall frequency and densities of mutations in the genes and proteins of SARS‐CoV‐2 were observed. Nucleocapsid exhibited the highest mutation density among the structural proteins while the spike D614G was the most common, occurring mostly in genomes outside China and United States. ORF8 protein had the highest mutation density across all geographical areas. Moreover, mutation hotspots neighboring and at the catalytic site of RNA‐dependent RNA polymerase were found that might challenge the binding and effectivity of remdesivir. Mutation coldspots may present as conserved diagnostic and therapeutic targets were found in ORF7b, ORF9b, and ORF14. These findings suggest that the virion's genotype and phenotype in a specific population should be considered in developing diagnostic tools and treatment options.
Keywords: coldspot, coronavirus, genetic variability, mutation, mutation hotspot, SARS‐CoV‐2, virus bioinformatics
1. INTRODUCTION
Coronavirus disease 2019 (COVID‐19) presented with pneumonia‐like symptoms surfaced from a seafood market at Wuhan, Hubei Province in China in December 2019, and has since spread across the globe. 1 According to the WHO, it has affected 213 countries and territories with 23,057,288 people infected and 800,906 deaths worldwide. 2 Mitigation of this public health crisis can be accomplished through effective public health safety protocols, vaccines, and targeted viral treatment. The scientific community has then been in haste to develop vaccines and therapeutic drugs to combat the COVID‐19.
COVID‐19 is caused by a novel coronavirus, the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2). 3 , 4 It is a positive‐sense RNA virus, like SARS‐CoV and Middle East respiratory syndrome coronavirus, with a genome size of 29,903 nucleotides. 5 Figure 1 shows the comparison of the genes and proteins between SARS‐CoV‐2 and SARS‐CoV (2003). Most of its genome codes for ORF1ab (~72%) which is involved in viral replication and pathogenesis, while other ORFs code for structural proteins (spike [S], envelope [E], membrane glycoprotein [M], and nucleocapsid [N]). Genes for accessory proteins are also present in SARS‐CoV‐2 as with SARS‐CoV, however, some of the proteins coding for these accessory proteins (ORF3a, ORF7b, ORF8, and ORF10) are yet to be identified for their function. 6 , 7 Mutations in coronaviruses are expected to have mutation rates ranging between 10−5 and 10−3 substitutions per nucleotide site per cell infection (s/n/c). 8 , 9 Accordingly, information can be obtained from SARS‐CoV‐2 genomes coming from initial cases in Wuhan, China up to the recent submissions.
Figure 1.
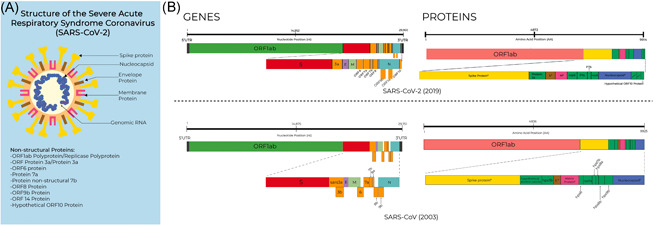
SARS‐CoV‐2 structure, and comparison of the genomes and proteomes of SARS‐CoV‐2 (2019) and SARS‐CoV (2003). (A) Structure of the SARS‐CoV‐2, the etiologic agent of COVID‐19. Information on these proteins is publicly available from the COVID‐19 UniProt Resource (https://covid-19.uniprot.org/). (B) Comparison of the genomes and proteomes of SARS‐CoV‐2 and SARS‐CoV (2003). COVID‐19, coronavirus disease 2019; SARS‐CoV, severe acute respiratory syndrome coronavirus
Determining mutation hotspots and coldspots in SARS‐CoV‐2 may provide insights on their effects on the properties (i.e., virulence, infectivity, and severity) and characteristics of SARS‐CoV‐2. Hence, drugs, vaccines, and diagnostics effective against SARS‐CoV‐2 variants are crucial today in containing the COVID‐19 pandemic. This study provides an overview of mutation characteristics at the coding and noncoding regions of the SARS‐CoV‐2 genome, as well as the mutations in the translated proteins.
2. MATERIALS AND METHODS
2.1. Collection of SARS‐CoV‐2 genomes
Publicly available genomes from 31 countries submitted to the National Center for Biotechnology Information (NCBI) nucleotide database and the GISAID EpiCoV™ database by January 19, 2020 to May 15, 2020, were collected for the study (Table S1). Gathering of 151 publicly available “complete” and/or “partial” (genome length > 29,700 nucleotides = complete; genome length < 29,700 nucleotides = partial) genomes of SARS‐CoV‐2 (reference sequence NC_045512, GenBank) was conducted from March 12, 2020 to May 15, 2020. There were two data collection points in this study: genomes from both databases that were submitted from December 2019 to March 2020 (86 genomes) and December 2019 to May 2020 (65 additional genomes). Manual grouping of these sequences according to three geographic areas was made for ease of analysis. China‐derived samples were classified under the “China,” United States‐derived samples were classified under the “USA,” while the genome sequences from other countries besides United States or China were classified under the “Others.” The overall data set containing all the samples from China, United States, and Others is referred as the “Total.”
2.2. Nucleotide and amino acid variant detection
Each genome sequence was aligned to NC_045512 using the MAFFT. 10 , 11 The default parameters as presented in the web tool were used for the multiple sequence alignment. The nucleotide variants from the reference sequence (NC_045512) were manually annotated and were re‐evaluated using the “Low Frequency Variant Detection” tool of the CLC Genomics Workbench 20.0.3. (QIAGEN Bioinformatics, Aarhus, Denmark). Mutations from both the coding and noncoding regions were recorded.
Using the nucleotide mutations, the resulting amino acid mutations throughout the proteome of SARS‐CoV‐2 were determined. The amino acid changes were automatically annotated using the “Map Reads to Reference” tool and a subsequent run in the “Low Frequency Variant Detection” tool in the CLC Genomics Workbench 20.0.3. The resulting proteome from each SARS‐CoV‐2 genome was created and edited using CLC Genomics Workbench 20.0.3. The whole proteome was then aligned for phylogenetic analysis, and for identification of the resulting amino acid mutations.
2.3. Construction of phylogenetic trees
A phylogenetic tree based on the translated protein‐coding genes of SARS‐CoV‐2 was constructed using the same command‐line in IQ‐TREE version 1.6.12 and was also edited, and visualized using MEGA X. 12 , 13 , 14 , 15 The phylogenetic tree was constructed using an ultrabootstrap method considering 1000 and considered 151 genomes for the construction of the said tree. 12 , 13 , 14 The resulting tree was edited and visualized using MEGA X. 15
2.4. Data analysis
Mutation hotspots were identified as genome sites with two or more occurring mutations; on the other hand, mutation coldspots are those with no occurring mutations. The characterization of nucleotide mutations was done in terms of the nature of the nucleotide substitution (transition or transversion) and insertion and deletions (indel). The mutation densities (Equation 1) in the genome and proteome of SARS‐CoV‐2 were determined.
| (1) |
Amino acid substitutions were characterized according to the nature of change that occurred (e.g., leucine to isoleucine would be classified under “Similar Change,” serine to phenylalanine would be classified under “Polar <> Neutral,” aspartic acid to serine would be classified under “Charged <> Polar,” while glutamic acid to glycine would be “Charged <> Neutral”). Furthermore, amino acid substitutions leading to residues with similar nature were classified as “Similar Change,” while those substitutions that did not produce amino acids with similar nature were classified under “Dissimilar Change.”
3. RESULTS
The mutations in the genome and proteome of SARS‐CoV‐2 are described per geographic area (China, United States, and Others) in two time points (December 2019–March 2020; December 2019–May 2020). This section starts with a presentation of the phylogenetic data according to the nucleotide sequences and amino acids of SARS‐CoV‐2. Then, the nucleotide substitution types (transversions, transitions, and InDels) were identified per geographic area in the two time points. This was followed by a presentation of the amino acid substitutions due to nucleotide mutations. Finally, remarkable mutations and mutation patterns in the proteins of SARS‐CoV‐2 (S glycoprotein, ORF8, and N) were reported.
3.1. Nucleotide and amino acid‐based phylogenetic analyses of SARS‐CoV‐2 show three major clades of SARS‐CoV‐2 and a minor clade (P6810S ORF1ab)
The phylogenetic analysis of mutations in different regions was analyzed using MAFFT software and three major clades were identified. As shown in Figure 2, the L3606F (ORF1ab) is characterized by the color pink, P4715L (ORF1ab)/D614G (S) is highlighted by the color green, and L84S (ORF8)/S2839S (ORF1ab) is denoted by the color blue. The largest among these clades were the L84S (ORF8), having 43 samples. This was caused by a transition substitution in the ORF8 gene (T28144C) leading to an L84S substitution in the ORF8 protein. L84S (ORF8) had four subclades; two of these subclades had subclades as well (Figure 2B). The second‐largest major clade was the P4715L (ORF1ab)/D614G (S) having two subclades. These subclades were identified as R203K and G204R (N), and the G57H (ORF3a)/T265I (ORF1ab)/S3384L (ORF1ab). Lastly, the L3606F (ORF1ab) major clade contained 19 samples; 52.63% of these samples were from the “Others” geographic area, 36.84% were from the United States, while 10.53% were from China. This clade also has a subclade represented by the V378I mutation (ORF1ab; Figure 2B).
Figure 2.
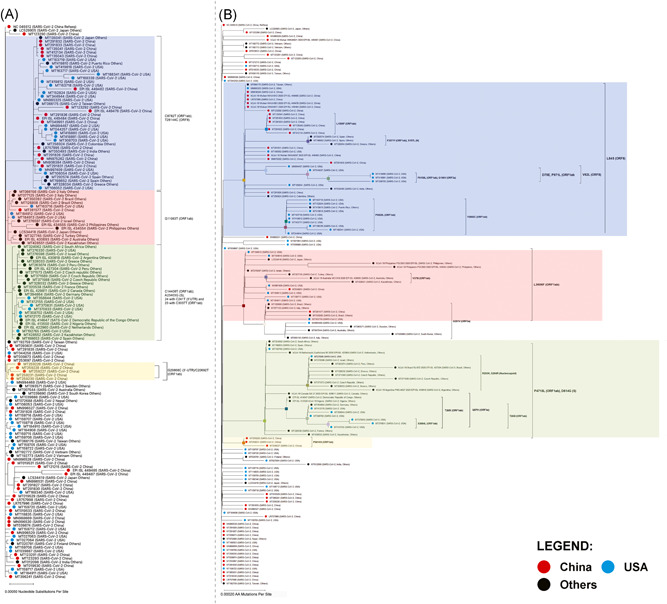
Phylogenetic tree of 151 SARS‐CoV‐2 from genomes collected from March 12, 2020 to April 2020 from NCBI GenBank™ and GISAID EpiCoV®. (A) Phylogenetic tree based on the genomes of SARS‐CoV‐2. (B) Phylogenetic tree based on the proteins of SARS‐CoV‐2. Individual viral samples are represented as dots. Samples under the geographic cluster “China” are colored red, blue for sequences under the geographic cluster “USA,” while for the “Others” geographic cluster, these are colored black. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; NCBI, National Center for Biotechnology Information
A transversion substitution (29868G>C) in the 3ʹ‐untranslated region (UTR) of the SARS‐CoV‐2 genome was identified which defined the occurrence of a nucleotide‐based clade. This clade also contained a subclade bearing a missense mutation in the 2ʹ‐O‐ribose methyltransferase (nsp16) of ORF1ab (20692C>T; P6810S). Overall, the mutations classifying this clade were identified in five China‐derived samples, while P6810S has not been identified in current literature.
3.2. The proportion of transitions, transversion, and indels in SARS‐CoV‐2 genome is similar among the geographical areas
The genomic mutation profile of SARS‐CoV‐2 was evaluated, and the distribution of the mutations across the viral genomes from different geographical areas is summarized in Figure 3A. Overall, in total, 674 nucleotide mutations were identified using genome samples collected from December 2019–May 2020 (Table 1).
Figure 3.
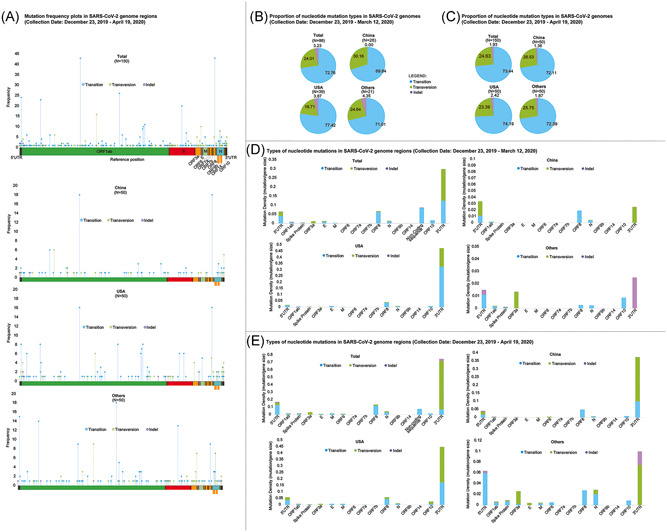
Characterization of nucleotide mutations in SARS‐CoV‐2. SARS‐CoV‐2 genomes were identified independently, and mutations were considered to occur spontaneously. Mutations were identified by identifying substitutions in the SARS‐CoV‐2 reference genome NCBI GenBank™ accession ID: NC_045512. (A) Nucleotide mutation frequency plot in total (overall), and in geographical clusters: China, United States, and Others. (B) Proportion of the nucleotide mutation types in SARS‐CoV‐2 genomes submitted on December 23, 2019–March 11, 2020, and (C) December 23, 2019–April 19, 2020. These were grouped as total, China, United States, and other. (D and E) Mutation density profiles of total SARS‐CoV‐2 genomes and clustered geographically: China, United States, and Others between the two time points. Mutation markers are colored according to the type of nucleotide change, that is, transition (blue), transversion (green), indel (violet). The maximum genome coverage of read‐mapped genomes for variant detection is indicated (e.g., N = 150 in total for overall data set). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; NCBI, National Center for Biotechnology Information
Table 1.
Summary of detected nucleotide and amino acid mutations in SARS‐CoV‐2
| Genome region | Protein/peptide chain | Domain | Nucleotide position | Reference | Allele | Frequency | Relative frequency | AA position | Amino acid mutation | Type of AA change/mutation |
|---|---|---|---|---|---|---|---|---|---|---|
| 5ʹ‐UTR | N/A | N/A | 4 | A | T | 1 | 0.67 | N/A | N/A | N/A |
| 26 | A | ‐ | 2 | 1.33 | ||||||
| 28 | C | T | 2 | 1.33 | ||||||
| 31 | A | T | 1 | 0.67 | ||||||
| 34 | A | T | 1 | 0.67 | ||||||
| 35 | A | T | 2 | 1.33 | ||||||
| 36 | C | T | 2 | 1.33 | ||||||
| 75 | C | A | 1 | 0.67 | ||||||
| 104 | T | A | 1 | 0.67 | ||||||
| 111 | T | C | 1 | 0.67 | ||||||
| 112 | T | G | 1 | 0.67 | ||||||
| 119 | C | G | 1 | 0.67 | ||||||
| 120 | T | C | 1 | 0.67 | ||||||
| 124 | G | A | 1 | 0.67 | ||||||
| 186 | C | T | 2 | 1.33 | ||||||
| 241 | C | T | 24 | 16.00 | ||||||
| 254 | C | T | 2 | 1.33 | ||||||
| ORF1ab | Leader protein/nsp1 | Leader protein/nsp2 | 270 | A | G | 1 | 0.01 | 2 | E>G | Charged<>Neutral |
| 313 | C | T | 3 | 2.00 | 16 | Silent | Silent | |||
| 490 | T | A | 4 | 0.03 | 75 | D>E | Similar Charge | |||
| 508 | TGGTCATGTTATGGT | ‐ | 2 | 0.01 | 82 | G82_V86del | Deletion | |||
| 514 | T | C | 1 | 0.67 | 83 | Silent | Silent | |||
| 565 | T | C | 1 | 0.67 | 100 | Silent | Silent | |||
| 614 | G | A | 1 | 0.01 | 117 | A>T | Polar<>Neutral | |||
| 618 | A | G | 1 | 0.01 | 118 | Y>C | Polar<>Neutral | |||
| 654 | G | A | 1 | 0.01 | 130 | G>E | Charged<>Neutral | |||
| 686 | AAGTCATTT | ‐ | 1 | 0.01 | 141 | Deletion | Deletion | |||
| 721 | T | C | 1 | 0.67 | 152 | Silent | Silent | |||
| nsp2 | nsp3 | 884 | C | T | 1 | 0.01 | 207 | R>C | Charged<>Polar | |
| 1059 | C | T | 6 | 0.04 | 265 | T>I | Polar<>Neutral | |||
| 1076 | C | T | 1 | 0.01 | 271 | P>S | Polar<>Neutral | |||
| 1102 | C | T | 1 | 0.67 | 279 | Silent | Silent | |||
| 1385 | C | T | 1 | 0.01 | 374 | H>Y | Charged<>Neutral | |||
| 1397 | G | A | 3 | 0.02 | 378 | V>I | Similar Charge | |||
| 1431 | ATG | ‐ | 1 | 0.01 | 389 | Deletion | Deletion | |||
| 1548 | G | A | 1 | 0.01 | 428 | S>N | Similar Charge | |||
| 1623 | T | C | 1 | 0.01 | 453 | I>T | Polar<>Neutral | |||
| 1691 | A | G | 1 | 0.01 | 476 | I>V | Similar Charge | |||
| 1895 | G | T | 1 | 0.01 | 544 | V>L | Similar Charge | |||
| 2091 | C | T | 1 | 0.01 | 609 | T>I | Polar<>Neutral | |||
| 2269 | A | T | 1 | 0.67 | 668 | Silent | Silent | |||
| 2277 | T | C | 1 | 0.01 | 671 | I>T | Polar<>Neutral | |||
| 2388 | C | T | 1 | 0.01 | 708 | T>I | Polar<>Neutral | |||
| 2416 | C | T | 2 | 1.33 | 717 | Silent | Silent | |||
| 2446 | T | C | 1 | 0.67 | 727 | Silent | Silent | |||
| 2472 | C | T | 7 | 4.67 | 736 | Silent | Silent | |||
| 2717 | G | A | 1 | 0.01 | 818 | G>S | Polar<>Neutral | |||
| nsp3 | nsp3 chain | 2875 | G | A | 1 | 0.67 | 870 | Silent | Silent | |
| 2971 | G | T | 1 | 0.01 | 902 | M>I | Similar Charge | |||
| Nsp3 N‐terminal/DUF3655 (domain with unknown function) | 3037 | C | T | 23 | 15.33 | 924 | Silent | Silent | ||
| 3099 | C | T | 2 | 0.01 | 945 | T>I | Polar<>Neutral | |||
| 3177 | C | T | 4 | 0.03 | 971 | P>L | Similar Charge | |||
| nsp3 chain | 3259 | G | T | 1 | 0.01 | 998 | Q>H | Charged<>Polar | ||
| Macro domain | 3299 | T | C | 1 | 0.67 | 1012 | Silent | Silent | ||
| 3333 | TTG | ‐ | 1 | 0.01 | 1023 | Deletion | ||||
| 3411 | C | T | 1 | 0.01 | 1049 | A>V | Similar Charge | |||
| 3518 | G | T | 1 | 0.01 | 1085 | V>F | Similar Charge | |||
| 3738 | C | T | 1 | 0.01 | 1158 | P>L | Similar Charge | |||
| nsp3 chain | 3778 | A | G | 1 | 0.67 | 1171 | Silent | Silent | ||
| Nsp3 ss‐polyA binding domain | 4234 | C | T | 1 | 0.67 | 1323 | Silent | Silent | ||
| 4402 | T | C | 6 | 4.00 | 1379 | Silent | Silent | |||
| PL2Pro domain | 4780 | C | T | 1 | 0.67 | 1505 | Silent | Silent | ||
| Papain‐like viral protease | 4946 | T | C | 1 | 0.01 | 1561 | S>P | Polar<>Neutral | ||
| 5062 | G | T | 6 | 0.04 | 1599 | L>F | Similar Charge | |||
| 5084 | A | G | 1 | 0.01 | 1607 | I>V | Similar Charge | |||
| Papain‐like viral protease/peptidase C16 | 5572 | G | T | 1 | 0.01 | 1769 | M>I | Similar Charge | ||
| 5608 | A | G | 1 | 0.67 | 1781 | Silent | Silent | |||
| 5784 | C | T | 1 | 0.01 | 1840 | T>I | Polar<>Neutral | |||
| 5845 | A | T | 1 | 0.01 | 1860 | K>N | Charged<>Polar | |||
| Nucleic acid‐binding domain | 6026 | C | T | 1 | 0.01 | 1921 | P>S | Polar<>Neutral | ||
| 6031 | C | T | 1 | 0.67 | 1922 | Silent | Silent | |||
| 6035 | A | G | 1 | 0.01 | 1924 | S>G | Polar<>Neutral | |||
| 6040 | C | T | 2 | 1.33 | 1925 | Silent | Silent | |||
| 6312 | C | A | 2 | 0.01 | 2016 | T>K | Charged<>Polar | |||
| nsp3 chain | 6501 | C | T | 1 | 0.01 | 2079 | P>L | Similar Charge | ||
| 6636 | C | T | 1 | 0.01 | 2124 | T>I | Polar<>Neutral | |||
| 6695 | C | T | 1 | 0.01 | 2144 | P>S | Polar<>Neutral | |||
| 6819 | G | T | 2 | 0.01 | 2185 | S>I | Polar<>Neutral | |||
| 6968 | C | A | 1 | 0.01 | 2235 | L>I | Similar Charge | |||
| 6996 | T | C | 2 | 0.01 | 2244 | I>T | Polar<>Neutral | |||
| 7016 | G | A | 1 | 0.01 | 2251 | G>S | Polar<>Neutral | |||
| 7105 | C | T | 1 | 0.67 | 2280 | Silent | Silent | |||
| 7488 | C | T | 1 | 0.01 | 2408 | T>I | Polar<>Neutral | |||
| 7866 | G | T | 1 | 0.01 | 2534 | G>V | Similar Charge | |||
| 8001 | A | C | 1 | 0.01 | 2579 | D>A | Charged<>Neutral | |||
| 8388 | A | G | 1 | 0.01 | 2708 | N>S | Similar Charge | |||
| nsp4 | nsp4 | 8653 | G | T | 1 | 0.01 | 2796 | M>I | Similar Charge | |
| 8728 | A | G | 1 | 0.67 | 2821 | Silent | Silent | |||
| 8782 | C | T | 43 | 28.67 | 2839 | Silent | Silent | |||
| 8945 | A | G | 1 | 0.01 | 2894 | N>D | Charged<>Polar | |||
| 8987 | T | A | 1 | 0.01 | 2908 | F>I | Similar Charge | |||
| 9034 | A | G | 1 | 0.67 | 2923 | Silent | Silent | |||
| 9157 | T | C | 1 | 0.67 | 2964 | Silent | Silent | |||
| 9274 | A | G | 1 | 0.67 | 3003 | Silent | Silent | |||
| 9430 | C | T | 1 | 0.67 | 3055 | Silent | Silent | |||
| 9474 | C | T | 1 | 0.01 | 3070 | A>V | Similar Charge | |||
| 9477 | T | A | 3 | 0.02 | 3071 | F>Y | Similar Charge | |||
| 9491 | C | T | 1 | 0.01 | 3076 | H>Y | Charged<>Neutral | |||
| 9534 | C | T | 1 | 0.01 | 3090 | T>I | Polar<>Neutral | |||
| 9561 | C | T | 1 | 0.01 | 3099 | S>L | Polar<>Neutral | |||
| 9924 | C | T | 1 | 0.01 | 3220 | A>V | Similar Charge | |||
| 10015 | C | T | 1 | 0.67 | 3250 | Silent | Silent | |||
| 3C‐like proteinase | Peptidase C30 domain | 10036 | C | T | 1 | 0.67 | 3257 | Silent | Silent | |
| 10232 | C | T | 2 | 0.01 | 3323 | R>C | Charged<>Polar | |||
| nsp6 | nsp6 | 10507 | C | T | 1 | 0.67 | 3414 | Silent | Silent | |
| 11075 | ‐ | TTT | 1 | 0.01 | 3605_3606 | F_LinsF | Insertion | |||
| 11083 | G | C | 1 | 0.01 | 3606 | L>F | Similar Charge | |||
| 11083 | G | T | 16 | 0.11 | 3606 | L>F | Similar Charge | |||
| 11101 | A | G | 1 | 0.67 | 3612 | Silent | Silent | |||
| 11410 | G | A | 2 | 1.33 | 3715 | Silent | Silent | |||
| 11750 | C | T | 1 | 0.01 | 3829 | L>F | Similar Charge | |||
| 11752 | C | T | 1 | 0.67 | 3829 | Silent | Silent | |||
| nsp7 | nsp7 | 11764 | T | A | 1 | 0.01 | 3833 | N>K | Charged<>Polar | |
| 11916 | C | T | 3 | 0.02 | 3884 | S>L | Polar<>Neutral | |||
| 11937 | G | A | 1 | 0.01 | 3891 | C>Y | Polar<>Neutral | |||
| nsp8 | nsp8 | 11956 | C | T | 1 | 0.67 | 3897 | Silent | Silent | |
| 12102 | C | T | 1 | 0.01 | 3946 | S>L | Polar<>Neutral | |||
| 12115 | C | T | 1 | 0.67 | 3950 | Silent | Silent | |||
| 12473 | C | T | 1 | 0.67 | 4070 | Silent | Silent | |||
| 12478 | G | A | 2 | 0.01 | 4071 | M>I | Similar Charge | |||
| nsp9 | nsp9 | 12534 | C | T | 1 | 0.01 | 4090 | T>I | Polar<>Neutral | |
| nsp10 | nsp10 | 13072 | C | T | 1 | 0.67 | 4269 | Silent | Silent | |
| 13225 | C | G | 1 | 0.01 | 4321 | F>L | Similar Charge | |||
| 13226 | T | C | 1 | 0.67 | 4321 | Silent | Silent | |||
| RNA‐dependent RNA polymerase | RNA‐dependent RNA polymerase N‐terminal | 13620 | C | T | 1 | 0.67 | 4452 | Silent | Silent | |
| 13730 | C | T | 2 | 0.01 | 4489 | A>V | Similar Charge | |||
| 14408 | C | T | 26 | 0.17 | 4715 | P>L | Similar Charge | |||
| RdRp chain | 14657 | C | T | 1 | 0.01 | 4798 | A>V | Similar Charge | ||
| 14786 | C | T | 1 | 0.01 | 4841 | A>V | Similar Charge | |||
| 14805 | C | T | 6 | 4.00 | 4847 | Silent | Silent | |||
| 14849 | T | G | 1 | 0.01 | 4862 | L>R | Charged<>Neutral | |||
| 14856 | A | T | 1 | 0.67 | 4864 | Silent | Silent | |||
| 14858 | T | A | 1 | 0.01 | 4865 | V>D | Charged<>Neutral | |||
| RdRp catalytic | 15324 | C | T | 4 | 2.67 | 5020 | Silent | Silent | ||
| 15418 | G | T | 1 | 0.01 | 5052 | A>S | Polar<>Neutral | |||
| 15597 | T | C | 1 | 0.67 | 5111 | Silent | Silent | |||
| 15607 | T | C | 1 | 0.67 | 5115 | Silent | Silent | |||
| RdRp Chain | 15910 | G | T | 1 | 0.01 | 5216 | D>Y | Charged<>Neutral | ||
| 15960 | C | T | 1 | 0.67 | 5232 | Silent | Silent | |||
| Helicase | CoVi ZnBD | 16272 | T | G | 1 | 0.67 | 5336 | Silent | Silent | |
| 16293 | C | T | 1 | 0.67 | 5343 | Silent | Silent | |||
| 16325 | G | C | 1 | 0.01 | 5354 | C>S | Similar Charge | |||
| 16467 | A | G | 1 | 0.67 | 5401 | Silent | Silent | |||
| Helicase chain | 16877 | C | T | 1 | 0.01 | 5538 | T>I | Polar<>Neutral | ||
| 17000 | C | T | 1 | 0.01 | 5579 | T>I | Polar<>Neutral | |||
| (+) RNA virus Helicase ATP‐binding domain | 17141 | C | A | 1 | 0.01 | 5626 | A>D | Charged<>Neutral | ||
| 17247 | T | C | 2 | 1.33 | 5661 | Silent | Silent | |||
| 17249 | C | T | 1 | 0.01 | 5662 | A>V | Similar Charge | |||
| 17280 | G | T | 1 | 0.67 | 5672 | Silent | Silent | |||
| 17373 | C | T | 5 | 3.33 | 5703 | Silent | Silent | |||
| 17376 | A | G | 1 | 0.67 | 5704 | Silent | Silent | |||
| 17410 | C | T | 1 | 0.01 | 5716 | R>C | Similar Charge | |||
| 17423 | A | G | 1 | 0.01 | 5720 | Y>C | Polar<>Neutral | |||
| 17470 | C | T | 1 | 0.67 | 5736 | Silent | Silent | |||
| (+) RNA virus Helicase C‐terminal domain | 17747 | C | T | 9 | 0.06 | 5828 | P>L | Similar Charge | ||
| 17825 | C | T | 1 | 0.01 | 5854 | T>I | Polar<>Neutral | |||
| 17858 | A | G | 10 | 0.07 | 5865 | Y>C | Polar<>Neutral | |||
| 17894 | C | T | 1 | 0.01 | 5877 | A>V | Similar Charge | |||
| Guanine‐N7 methyltransferase | Guanine‐N7 methyltransferase | 18060 | C | T | 11 | 7.33 | 5932 | Silent | Silent | |
| 18115 | C | T | 1 | 0.01 | 5951 | H>Y | Charged<>Neutral | |||
| 18126 | T | C | 1 | 0.67 | 5954 | Silent | Silent | |||
| 18603 | T | C | 1 | 0.67 | 6113 | Silent | Silent | |||
| 18736 | T | C | 3 | 0.02 | 6158 | F>L | Similar Charge | |||
| 18744 | C | T | 1 | 0.67 | 6160 | Silent | Silent | |||
| 18788 | C | T | 1 | 0.01 | 6175 | T>I | Polar<>Neutral | |||
| 18814 | C | T | 1 | 0.67 | 6184 | Silent | Silent | |||
| 18975 | T | A | 1 | 0.67 | 6273 | Silent | Silent | |||
| 18996 | T | C | 1 | 0.67 | 6244 | Silent | Silent | |||
| 18998 | C | T | 2 | 0.01 | 6245 | A>V | Similar Charge | |||
| 19065 | T | C | 1 | 0.67 | 6267 | Silent | Silent | |||
| 19175 | A | C | 1 | 0.01 | 6304 | D>A | Charged<>Neutral | |||
| 19610 | C | T | 1 | 0.01 | 6449 | T>I | Polar<>Neutral | |||
| N‐Endo, uridylate‐specific endoribonuclease | N‐Endo, uridylate‐specific endoribonuclease | 19684 | G | T | 1 | 0.01 | 6474 | V>L | Similar Charge | |
| 20268 | A | G | 1 | 0.67 | 6668 | Silent | Silent | |||
| 20281 | T | C | 1 | 0.01 | 6673 | F>L | Similar Charge | |||
| 20298 | ATT | ‐ | 1 | 0.01 | 6679 | Deletion | Deletion | |||
| 20437 | A | T | 1 | 0.01 | 6725 | S>C | Similar Charge | |||
| 20449 | A | T | 1 | 0.01 | 6729 | N>Y | Polar<>Neutral | |||
| 2ʹ‐O‐Ribose methyltransferase | 2′‐O‐Ribose methyltransferase | 20692 | C | T | 3 | 0.02 | 6810 | P>S | Polar<>Neutral | |
| 20936 | C | T | 1 | 0.01 | 6891 | T>M | Polar<>Neutral | |||
| 20995 | G | A | 1 | 0.01 | 6911 | G>S | Polar<>Neutral | |||
| 21137 | A | G | 1 | 0.01 | 6958 | K>R | Similar Charge | |||
| 21147 | T | C | 1 | 0.67 | 6961 | Silent | Silent | |||
| 21316 | G | A | 1 | 0.01 | 7018 | D>N | Charged<>Polar | |||
| 21384 | ‐ | T | 1 | 0.01 | 7041 | Insertion | Insertion | |||
| 21386 | C | T | 1 | 0.01 | 7041 | S>F | Polar<>Neutral | |||
| 21387 | ‐ | TT | 1 | 0.01 | 7042 | Insertion | Insertion | |||
| 21426 | T | G | 1 | 0.67 | 7054 | Silent | Silent | |||
| S | Spike protein S1/surface glycoprotein S1 | Spike protein S1/surface glycoprotein S1 chain | 21595 | C | T | 1 | 0.67 | 11 | Silent | Silent |
| Spike protein S1 N‐terminus | 21644 | T | A | 1 | 0.01 | 28 | Y>N | Polar<>Neutral | ||
| 21691 | C | T | 1 | 0.67 | 43 | Silent | Silent | |||
| 21707 | C | T | 2 | 0.01 | 49 | H>Y | Charged<>Neutral | |||
| 21711 | C | T | 1 | 0.01 | 50 | S>L | Polar<>Neutral | |||
| 21784 | T | A | 1 | 0.01 | 74 | N>K | Charged<>Polar | |||
| 21830 | G | T | 1 | 0.01 | 90 | V>F | Similar Charge | |||
| 21906 | A | G | 1 | 0.01 | 115 | Q>R | Charged<>Polar | |||
| 21991 | TTA | ‐ | 1 | 0.01 | 145 | Deletion | Deletion | |||
| 22033 | C | A | 1 | 0.01 | 157 | F>L | Similar Charge | |||
| 22104 | G | T | 1 | 0.01 | 181 | G>V | Similar Charge | |||
| 22151 | A | G | 1 | 0.01 | 197 | I>V | Similar Charge | |||
| 22224 | C | G | 1 | 0.01 | 221 | S>W | Polar<>Neutral | |||
| 22303 | T | G | 1 | 0.01 | 247 | S>R | Charged<>Polar | |||
| 22432 | C | T | 1 | 0.67 | 290 | Silent | Silent | |||
| 22468 | G | T | 1 | 0.67 | 302 | Silent | Silent | |||
| Spike receptor binding domain/spike protein S1 C‐terminal domain | 22785 | G | T | 1 | 0.01 | 408 | R>I | Charged<>Neutral | ||
| Spike receptor binding domain | 23185 | C | T | 1 | 0.67 | 541 | Silent | Silent | ||
| 23271 | C | T | 1 | 0.01 | 570 | A>V | Similar Charge | |||
| Spike protein S1/surface glycoprotein S1 chain | 23403 | A | G | 26 | 0.17 | 614 | D>G | Charged<>Neutral | ||
| 23520 | C | T | 1 | 0.01 | 653 | A>V | Similar Charge | |||
| 23613 | C | T | 1 | 0.01 | 684 | A>V | Similar Charge | |||
| Spike protein S2/surface glycoprotein S2 | Spike protein S2 chain | 23876 | G | A | 1 | 0.01 | 772 | V>I | Similar Charge | |
| 23929 | C | T | 2 | 1.33 | 789 | Silent | Silent | |||
| 23952 | T | G | 1 | 0.01 | 797 | F>C | Polar<>Neutral | |||
| Spike protein S2′ chain/fusion peptide 1 | 24022 | T | C | 1 | 0.67 | 820 | Silent | Silent | ||
| 24023 | C | T | 1 | 0.67 | 821 | Silent | Silent | |||
| Spike protein S2′ chain/heptad repeat 1 | 24325 | A | G | 3 | 2.00 | 921 | Silent | Silent | ||
| 24351 | C | T | 1 | 0.01 | 930 | A>V | Similar Charge | |||
| 24370 | C | T | 1 | 0.67 | 936 | Silent | Silent | |||
| Spike protein S2′ chain | 24694 | A | T | 2 | 1.33 | 1044 | Silent | Silent | ||
| 24789 | C | T | 1 | 0.01 | 1076 | T>I | Polar<>Neutral | |||
| 24862 | A | G | 1 | 0.67 | 1100 | Silent | Silent | |||
| Spike protein S2′ chain/heptad repeat 2 | 25156 | C | T | 1 | 0.67 | 1198 | Silent | Silent | ||
| ORF3a | ORF3a protein | ORF3a protein | 25433 | C | T | 1 | 0.01 | 14 | T>I | Polar<>Neutral |
| 25533 | T | A | 1 | 0.67 | 47 | Silent | Silent | |||
| 25534 | G | T | 1 | 0.01 | 48 | V>F | Similar Charge | |||
| 25563 | G | T | 5 | 0.03 | 57 | Q>H | Charged<>Polar | |||
| 25672 | C | A | 1 | 0.01 | 94 | L>I | Similar Charge | |||
| 25687 | G | T | 1 | 0.01 | 99 | A>S | Polar<>Neutral | |||
| 25771 | C | A | 1 | 0.01 | 127 | L>I | Similar Charge | |||
| 25775 | G | T | 1 | 0.01 | 128 | W>L | Similar Charge | |||
| 25806 | A | T | 1 | 0.67 | 138 | Silent | Silent | |||
| 25810 | C | G | 1 | 0.01 | 140 | L>V | Similar Charge | |||
| 25979 | G | T | 3 | 0.02 | 196 | G>V | Similar Charge | |||
| 26048 | T | G | 1 | 0.01 | 219 | L>W | Similar Charge | |||
| 26088 | C | T | 1 | 0.67 | 232 | Silent | Silent | |||
| 26144 | G | T | 10 | 0.07 | 251 | G>V | Similar Charge | |||
| E | Envelope protein | Envelope protein | 26326 | C | T | 2 | 1.33 | 28 | Silent | Silent |
| 26354 | T | A | 1 | 0.01 | 37 | L>H | Charged<>Neutral | |||
| M | Membrane glycoprotein | Membrane glycoprotein | 26526 | G | T | 1 | 0.01 | 2 | A>S | Polar<>Neutral |
| 26530 | A | G | 1 | 0.01 | 3 | D>G | Charged<>Neutral | |||
| 26729 | T | C | 5 | 3.33 | 69 | Silent | Silent | |||
| 26849 | G | T | 1 | 0.01 | 109 | M>I | Similar Charge | |||
| 27046 | C | T | 1 | 0.01 | 175 | T>M | Polar<>Neutral | |||
| ORF6 | ORF6 protein | ORF6 protein | 27225 | G | T | 1 | 0.01 | 8 | Q>H | Charged<>Polar |
| 27299 | T | C | 1 | 0.01 | 33 | I>T | Polar<>Neutral | |||
| 27384 | T | C | 1 | 0.67 | 61 | Silent | Silent | |||
| ORF7a | ORF7a protein | SARS coronavirus X4 like | 27635 | C | T | 1 | 0.01 | 81 | S>L | Polar<>Neutral |
| ORF8 | ORF 8 protein | ORF 8 protein | 27925 | C | T | 1 | 0.01 | 11 | T>I | Polar<>Neutral |
| 27964 | C | T | 1 | 0.01 | 24 | S>L | Polar<>Neutral | |||
| 28077 | G | C | 5 | 0.03 | 62 | V>L | Similar Charge | |||
| 28144 | T | C | 43 | 0.29 | 84 | L>S | Polar<>Neutral | |||
| N | Nucleocapsid phosphoprotein | Nucleocapsid phosphoprotein | 28311 | C | T | 1 | 0.01 | 13 | P>L | Similar Charge |
| 28378 | G | T | 2 | 1.33 | 35 | Silent | Silent | |||
| 28409 | C | T | 1 | 0.01 | 46 | P>S | Polar<>Neutral | |||
| 28657 | C | T | 3 | 2.00 | 128 | Silent | Silent | |||
| 28688 | T | C | 3 | 2.00 | 139 | Silent | Silent | |||
| 28792 | A | T | 1 | 0.67 | 173 | Silent | Silent | |||
| 28854 | C | T | 2 | 0.01 | 194 | S>L | Polar<>Neutral | |||
| 28857 | G | T | 1 | 0.01 | 195 | R>I | Charged<>Neutral | |||
| 28863 | C | T | 3 | 0.02 | 197 | S>L | Polar<>Neutral | |||
| 28878 | G | A | 1 | 0.01 | 202 | S>N | Similar Charge | |||
| 28881 | G | A | 10 | 0.07 | 203 | R>K | Similar Charge | |||
| 28882 | G | A | 10 | 0.07 | 204 | G>R | Charged<>Neutral | |||
| 28883 | G | C | 10 | 0.07 | 204 | G>R | Charged<>Neutral | |||
| 28887 | C | T | 1 | 0.01 | 205 | T>I | Polar<>Neutral | |||
| 28896 | C | G | 2 | 0.01 | 208 | A>G | Similar Charge | |||
| 28916 | G | A | 1 | 0.01 | 215 | G>S | Polar<>Neutral | |||
| 28985 | G | T | 1 | 0.01 | 238 | G>C | Polar<>Neutral | |||
| 29029 | T | C | 1 | 0.67 | 252 | Silent | Silent | |||
| 29095 | C | T | 5 | 3.33 | 274 | Silent | Silent | |||
| 29140 | G | T | 1 | 0.01 | 289 | Q>H | Charged<>Polar | |||
| 29148 | T | C | 1 | 0.01 | 292 | I>T | Polar<>Neutral | |||
| 29230 | C | T | 1 | 0.67 | 319 | Silent | Silent | |||
| 29301 | A | T | 1 | 0.01 | 343 | A>V | Similar Charge | |||
| 29303 | C | T | 2 | 0.01 | 344 | P>S | Polar<>Neutral | |||
| 29527 | G | A | 2 | 1.33 | 418 | Silent | Silent | |||
| Between N and ORF10 non coding region | N/A | N/A | 29540 | G | A | 2 | 1.33 | |||
| ORF10 | ORF10 protein | ORF10 protein | 29563 | C | T | 1 | 0.67 | 2 | Silent | Silent |
| 29573 | G | A | 1 | 0.01 | 6 | V>I | Similar Charge | |||
| ORF10, stem loop | N/A | N/A | 29635 | C | T | 1 | 0.67 | N/A | N/A | N/A |
| 3′ UTR: 3′ UTR | N/A | N/A | 29695 | A | G | 1 | 0.67 | N/A | N/A | N/A |
| 29700 | A | G | 3 | 2.00 | ||||||
| 29736 | G | T | 3 | 2.00 | ||||||
| 29742 | G | A | 1 | 0.67 | ||||||
| 29742 | G | T | 2 | 1.33 | ||||||
| 29750 | CGATCGAGTG | ‐ | 1 | 0.67 | ||||||
| 29751 | G | C | 2 | 1.33 | ||||||
| 29786 | G | C | 1 | 0.67 | ||||||
| 29844 | A | G | 1 | 0.67 | ||||||
| 29845 | T | G | 2 | 1.33 | ||||||
| 29846 | T | A | 1 | 0.67 | ||||||
| 29847 | T | G | 1 | 0.67 | ||||||
| 29848 | T | G | 1 | 0.67 | ||||||
| 29861 | G | A | 2 | 1.33 | ||||||
| 29864 | G | A | 1 | 0.67 | ||||||
| 29867 | T | A | 2 | 1.33 | ||||||
| 29868 | G | A | 2 | 1.33 | ||||||
| 29868 | G | C | 5 | 3.33 | ||||||
| 29870 | C | A | 5 | 3.33 | ||||||
| 29873 | A | T | 1 | 0.67 |
Abbreviations: E, envelope; M, membrane glycoprotein; N, nucleocapsid; S, spike; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; UTR, untranslated region.
Generally, mutation frequencies among the geographical areas followed 3:1 transition to transversion ratio (Figure 3B,C), in which the C>T substitution was most common (44.7%), followed by T>C (13.95%). Interestingly, ORF3a and 3ʹ UTR genes had higher transversion density than transition similar between the two timepoints; while G>T transversion (10.83%), was the third most frequently occurring nucleotide change. Altogether, approximately similar proportions of nucleotide change types were observed between genomes among the geographical areas collected from December to March 2020 versus December–May 2020 (Figures 3B,C). These findings may suggest that the genomic mutation characteristics of SARS‐CoV‐2 from the earlier timepoint may not be significantly varied from the later period (e.g., between March and May 2020).
Among the SARS‐CoV‐2 genomic regions, the UTRs yielded the highest mutation density, with 7.5 × 10−3 mutation density at the 5ʹ‐UTR and 2.5 × 10−2 mutation density at the 3ʹ‐UTR among all geographical areas, for both timepoints (Figure 3D,E). Notably, indels were found mostly at the UTRs. As shown in Figure 4B, no UTR mutations were common among all areas, while mutations common between United States and Others are at 5ʹ‐UTR (241C>T) and 3ʹ‐UTR (29742G>T and 29870C>A); and between China and Others, 26delA and 28C>T at the 5ʹ‐UTR were common. Overall, the UTRs are consistently densely mutated suggesting that these genome regions are mutation prone regions of the SARS‐CoV‐2 genome.
Figure 4.
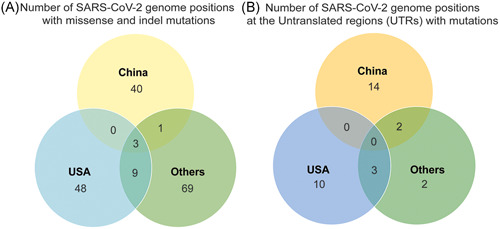
Commonly occurring hotspots were identified per geographical clusters (China, United States, and Others) and results were summarized as the number of SARS‐CoV‐2 genome positions with (A) missense and indel mutations at the protein‐coding regions and (B) detected mutations at the untranslated regions (UTRs), mainly, 5ʹ and 3ʹ‐UTR
3.3. Most amino acid substitutions in SARS‐CoV‐2 genomes from the United States and Others geographic areas resulted to residues with a similar nature (“Similar Change”) for both time points
The impact of overall genomic mutation characteristics in the viral proteins were then investigated from the genomic data and the description of these will be according to geographic area and will be magnified towards the differences between the two time points. Most of the nucleotide mutations in the SARS‐CoV‐2 genome (62.01%) lead to missense mutation in their proteins. Genome reference positions or nucleotide mutation hotspots 11083 (ORF1ab; nsp6), 26144 (ORF3a), and 28144 (ORF8) were common among all geographical areas (Figure 4A).
Most of the amino acid substitutions in China were “Polar ←→ Neutral” changes (66.67%) for the first time point, while this proportion decreased at the second time point (57.14%), with an addition of deletion mutations (1.43%). There was also an increase in substitutions where residues had a “Similar Change” in nature (e.g., valine ←→ isoleucine; 18.52% ‐ 12/2019‐03/2020; 31.43% ‐ 12/2019‐04/2020). These data could be seen in Figure 5B. Furthermore, the mutation hotspots based on mutation densities also changed in China, where mutations in the Spike glycoprotein, Protein 3a, Membrane protein, ORF6 protein, and ORF10 protein appeared in the second time point (Figure 5C).
Figure 5.
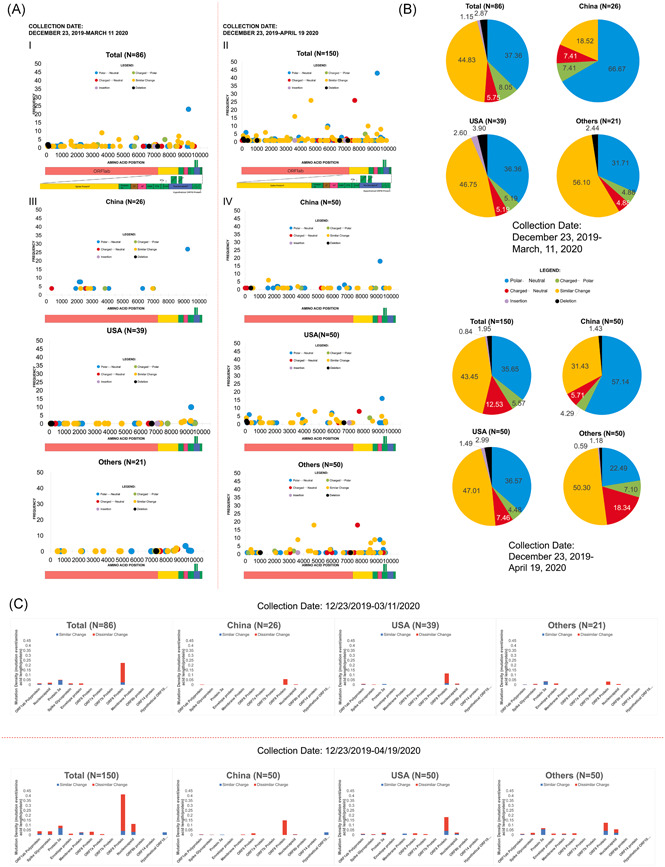
Characterization of amino acid mutations in SARS‐CoV‐2. Collection dates refer to the collection dates according to the annotated date of collection from GISAID or NCBI GenBank. (A) Comparison of the amino acid mutations according to the nature of the change in charge between earlier and overall data, and across different geographic clusters (China, United States, and Others). (B) Proportion of the nature of the change in amino acid charge and Indels that occurred in the total sample population, and for the geographic clusters between earlier and overall data. (C) Comparison of the mutation density profiles between earlier and overall data. Red indicates mutation density values resulting in amino acids having dissimilar nature to the reference, while blue indicates the mutation density values that resulted in amino acids having similar nature to the reference. The maximum genome coverage of read‐mapped genomes for variant detection are indicated (e.g., N = 150 in overall total for overall data set). SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; NCBI, National Center for Biotechnology Information
In the United States, the proportions of the type of amino acid substitutions did not change drastically (Figure 5). “Polar ←→ Neutral” mutations were almost similar between the two time points (36.36%12/2019‐03/2020; 36.57% ‐ 12/2019‐04/2020), while “Similar change” mutations changed minimally (46.75%12/2019‐03/2020; 47.01% ‐ 12/2019‐04/2020).). “Similar change” mutations had the highest frequency among the mutation types in United States samples. Mutation density presented in bar graphs show that there was an appearance of amino acid substitutions in the M and ORF7a proteins (Figure 5C).
For the Others geographic area, there is a great change in the proportions of mutations that are “Polar ←→ Neutral,”“Charged ←→ Polar,” and “Charged ←→ Neutral” (Figure 5B). The proportion of “Polar ←→ Neutral” mutations in the earlier time point was higher than that of the second time point (31.71% → 22.49%) as shown in Figure 5B. The proportion of “Charged ←→ Polar” and “Charged ←→ Neutral” mutations increased between the two time points (4.88% → 7.10% “Charged ←→ Polar”; 4.88% → 18.34% “Charged ←→ Neutral”). Appearance of mutations in the M protein and ORF6 protein occurred in the Others geographic area according to the mutation density graphs (Figure 5C), with the appearance of “Similar Change” substitutions in the second time point for the ORF8 protein and N protein as compared to the initial time point (Figure 5C).
The total count of amino acid substitutions in the proteins of SARS‐CoV‐2 was 381. From the data that was collected at the earlier timepoint, most of the mutations in the proteins were classified under “Similar Change” (44.83%), while insertions were the least frequent (1.15%). In addition of data from the later study timepoint, “Similar Change” mutations were most frequent however with decreased proportion (43.45%); and insertions was also the least frequent then at 0.84% proportion. The breakdown of the mutations in the SARS‐CoV‐2 proteins based on the collected genomes are shown in Figure 5A.
Overall, there was an observed shift in the proportions of the different classes of amino acid mutations between the two collection periods and geographic areas. There was an increase in proportion of “Similar Change” mutations in China between the two collection periods, while deletion mutations emerged at later time (Figure 5A). In comparison with United States, the proportion of the classes of amino acid mutations were generally unchanged. Prominent mutations have been found and further evaluated in this study in a spacio‐temporal perspective, which involve both structural and nonstructural proteins of SARS‐CoV‐2.
3.4. The D614G substitution in the spike glycoprotein is the most frequently occurring mutation among the structural proteins and occurred mostly in the Others geographic area
In samples from China, the D614G substitution did not occur, in both time points (Figure 5A), however, in the United States samples, there was an increase in the frequency of the D614G mutation (n D614G = 1 → n D614G = 8; Figure 5A). The same pattern was seen in the Others geographic area (n D614G = 4 → n D614G = 18). The mutation density of the spike glycoprotein increased in all of the geographic areas (China, United States, and Others areas, based on Figure 5C).
The D614G substitution in the Spike glycoprotein (S) occurred five times in the sample population from the data collected at earlier time and occurred 26 times from the overall total data. This mutation occurred with the P4715L (ORF1ab) mutation (Figures 2B and 5A). The D614G is a result of a transition mutation in the S gene of SARS‐CoV‐2 (23403A>G) and classified as “Charged←→Neutral” aa mutation. The mutation density S based on earlier data was 0.01414 mutation events/aa length of S glycoprotein, while this value approximately doubled based on the overall data. In addition, four other hotspots in the spike protein were detected in this study (Table 1). These data may suggest that the S variant occurred outside of China and is more observed in separate countries and in the United States.
3.5. ORF7b protein coldspots and ORF8 protein hotspots are conserved among all geographical areas
Among the geographical areas, no mutations were found in ORF6, ORF7a/7b, ORF9b, ORF10, and ORF14 proteins by the earlier study timepoint, hence considered as coldspots at that period (Figures 3D and 5B). On the other hand, at the later time point, only ORF7b, ORF9b, and ORF14 proteins were identified as mutation coldspots (Figures 3E and 5B). Note that it may be due to limitations in annotation of various viral genome regions that no mutations were detected in ORF9b and ORF14 proteins, as the study based the identification of genes and proteins from publicly available annotation to reference sequence (NCBI GenBank™ Accession ID: NC_045512). All in all, the ORF7b gene/protein was observed to have no mutations in all geographical region and between the study timepoints, therefore this gene may be potentially conserved in SARS‐CoV‐2.
Prominently, ORF8 protein presented the highest mutation density among nonstructural proteins (0.223 mutations/aa site in overall total), similar in all geographical areas similar in two timepoints (Figures 3D,E and 5B). Collectively at the later timepoint, its mutation density almost doubled. Along with the increased in mutation densities in other notable sites: doubled in nsp3 (0.072 mutations/aa site by March‐0.136 mutations/aa site by May), and quadrupled in the RNA‐dependent‐RNA polymerase (RDRP) (0.0139 mutations/aa site by March‐0.0515 mutations/aa site by May). The recurrence of ORF8 mutations were attributed to L84S which consistently was the most frequently occurring in China and United States (Figures 3A and 5A). In Others, however, the most recurrent mutation varied that was G251V in protein 3a in earlier timepoint, while P4715L in RDRP by the later timepoint (Figures 3A and 5A). This may suggest that the distinctive abundance of ORF8 mutations is generally similar among different areas, as its collective frequency increases over time.
3.6. The Nucleocapsid Phosphoprotein (N) exhibited the highest mutation density among the structural proteins of SARS‐CoV‐2
For both time points, N had the highest mutation density (0.02148 for earlier data; 0.1122 for overall data). Twelve nucleotide sites considered as hotspots in N, comprising 48% of the mutations in N (Table 1). Mutation densities of the other structural proteins are shown in Figure 5B. Interestingly, 10 SARS‐CoV‐2 samples had a substitution mutation in nucleotide positions 28881–28883 (GGG>AAC). This nucleotide mutation led to two amino acid substitutions (R203K and G204R). The earliest recorded SARS‐CoV‐2 genome having this mutation was from Florida, USA (February 28, 2020; accession ID: MT276330) while the other nine genomes that have this mutation come from the Others geographic area (Israel, Peru, Brazil, Greece, Czech Republic, and Argentina). However, the order of mutation densities of structural proteins among geographic areas varied, with the Others geographic area having N as the third highest mutation density for the overall data (Figure 5C). These suggest that the mutation in the N protein did not occur initially in China but occurred first from the United States.
4. DISCUSSION
4.1. Presence of a novel mutation and a high frequency mutation in SARS‐CoV‐2
Nsp16 is responsible for the messenger RNA capping of the coronavirus genome, primarily to protect from host recognition. 16 According to the crystal structure of nsp16, the domain of P6810 in nsp16 is unknown, however, it is characterized as part of a bend in nsp16. Proline exhibits conformational rigidity projected to result to a kink; its substitution may cause a change in the steric conformation of the aforementioned bend. 16 In addition, one of the immediate surrounding amino acids of nsp16–nsp10 complex that is proximal to P6810 is a tryptophan at aa position 7029 of ORF1ab. 16 Substitution of serine (P6810S) might exhibit an enhanced interaction for hydrogen bonding with tryptophan. 17 , 18 There is a need to further investigate this mutation to determine its significance in host evasion. It is important also to further evaluate its prevalence in the Chinese population, and in the global population to fully understand its implications in the function of nsp16.
The increased recurrence of L84S mutation may suggest that this variant might be favorable for virus' survival across geographical regions. 6 , 19 The subclades of L84S have mutations that may affect viral replication, immune evasion, viral release, and virion assembly. 1 , 20 , 21 , 22 , 23 Further research may ascertain the changes in the function of ORF8 due to this mutation, in virus replication, as well as potential changes in immune evasion and viral release.
4.2. Comparison of mutations in different SARS‐CoV‐2 studies reveal similarities and differences in mutation patterns
Observations in this study are consistent with the general pattern where transitions are more prevalent over transversions, perhaps due to steric considerations. 24 , 25 Interestingly, mutations in ORF3a (modulating host immune response), and 3′‐UTR (RNA stability and translation) consists largely of transversions, suggesting that these regions may be more erroneous than other regions and more prone to random substitution of transversions. 24 This might suggest that there are changes in virulence and replication stability across global regions.
Differences in findings may be observed based on previously published literature, using the mutation landscape of SARS‐CoV‐2. A study by Pachetti et al. 25 described that a mutation in RDRP (nt14408) increased in count, 7 (February) to 10 (cumulative by March). This was consistent with this study's findings with greater recurrence; 4 occurrences (March) to 26 (cumulative by May). In addition, another research by Kim et al. 26 also described the low frequency of mutations in E, M, and ORF7a, similar by this study's result. Other studies such as this described high frequency of mutations in ORF1ab and may be attributed to the relatively high genome length of the region. To address this, this study normalized the factor of gene length and presented the data through mutation densities of each gene in SARS‐CoV‐2. Discrepancies in mutation frequencies between this study and that of Tiwari and Mishra 27 may be attributed to the following reasons: (1) In this study, a single frequency of a mutation is already considered a valid mutation. In contrast to Tiwari and Mishra's 27 study, mutations should occur at least three times before these were considered as legitimate mutations. (2) Since the samples considered in this study were collected at a later time during the pandemic, thus providing more time and opportunity for the virus to accumulate mutations. In contrast to Tiwari and Mishra's study where samples were collected earlier into the pandemic, less time for the virus to accumulate mutations. 27
4.3. Implications of identified mutations in SARS‐CoV‐2 to treatment options and diagnostics
Remdesivir is currently at Phase 3 of COVID‐19 clinical trials, which is known to inhibit RDRP. 28 The active component of remdesivir (GS‐441524; adenosine nucleotide analog) binds to RDRP catalytic site and halts nucleic acid elongation. 29 The missense mutation (D722Y) occurred at the catalytic site along with neighboring variants (V472D and L469S), a change from an acidic to a nonpolar residue, may potentially result to increase in hydrophobicity at the region, leading to a more elusive conformation. This potential impact may significantly influence the RDRP conformation which might challenge the effectivity of remdesivir. 30 Hence, SARS‐CoV‐2 RDRP mutations, especially considering regional variability, should be further investigated on their potential effect on RDRP structure and function to support the use of remdesivir.
The absence of D614G mutation in China while it was abundant in the Others geographic area suggest potentially variable effectiveness of vaccines and neutralization factors that target the RBD among different geographic areas. Alternatively, relatively conserved regions in Spike heptad 1‐heptad two repeats, may present as potential drug or vaccine targets, inhibiting viral entry. As shown in this study, mutations in the Spike glycoprotein could confer alterations in its domains which may be involved in epitope recognition (i.e., RBD, S1‐N terminal domain) of neutralizing antibodies (nAbs). 31 , 32 Hence, binding of the potential nAb with putative SARS‐CoV‐2 epitopes may be hindered. Further studies should be done to evaluate putative effectiveness of neutralizing monoclonal antibodies against SARS‐CoV‐2.
The changes in the mutation frequencies and densities in N imply that the evolution of the genes and proteins of N over time in different landmasses is beneficial for the adaptation of SARS‐CoV‐2 as it spreads globally. 32 Currently, the WHO, and the Centers for Disease Control and Prevention recommend the use of N1 and N2 genes in COVID‐19 surveillance. 33 Recent publications have criticized the use of these genes in COVID‐19 diagnosis using reverse transcriptase‐polymerase chain reaction (RT‐PCR) because of its relatively high mutation index. 34 , 35 There are variants that fall in the forward primer for N3, and in the reverse primer of N1, (nt 28688). 36 This was a hotspot mutation in the genome and proteome of SARS‐C‐oV‐2, as observed in this study. These support that the variations in N may pose difficulties in diagnosis using N‐targeted primers for quantitative RT‐PCR.
The SARS‐CoV‐2 genomes used in this study are assumed to have come from individuals undergoing COVID‐19 testing and before any of them received antiviral treatment. Since SARS‐CoV‐2 genomes from individuals who have received antiviral treatment are not currently available, comparisons on the mutation patterns between these two groups cannot be determined yet, but speculations can be made. Mutations in the virus can exist and persist in the absence of selective pressure, therefore the diversity of mutations is high and no variants exist with unusually high frequencies. This is likely the phenomenon we have observed, with a few exceptions like L84S (ORF8), D614G (S), and L3606F (ORF1ab). However, antiviral drugs can serve as selective pressure against certain types of mutations in the viruses, possibly reducing the overall diversity of the virus, but at the same time, increasing the frequencies of a select few virus variants that are resistant to the antiviral drug. These variants may be more dominant in the population and this may affect the overall patterns and frequencies of mutations in SARS‐CoV‐2.
In conclusion, this study highlights the importance of the characterization of both nucleotide and amino acid mutation landscape in SAR‐CoV‐2 to identify hotspots and coldspots that may be significant in the effectivity of diagnostic tools and treatment options for COVID‐19, over the different areas worldwide as the pandemic continues.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests regarding this study.
AUTHOR CONTRIBUTIONS
Christian Luke D. C. Badua, Karol Ann T. Baldo, and Paul Mark B. Medina designed this study. Christian Luke D. C. Badua and Karol Ann T. Baldo equally contributed to data collection, data analysis, technical graphics and processing, and writing the paper. Paul Mark B. Medina contributed to critical evaluation of the figures and results, and the critical review of the manuscript. All authors contributed to revising the manuscript and approving of the final version submitted.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors would like to acknowledge and give their sincere gratitude towards the scientific community involved in providing, curating, and disseminating data for SARS‐CoV‐2 genomes, particularly GISAID and NCBI. The authors would also like to acknowledge QIAGEN for their generous action of providing free access for a limited time to their Genomics Workbench Application. In addition, the University of the Philippines College of Medicine MD‐PhD Batch 10, Baldo, J. Asi, R. J., and Batulan, R. Q., are to be acknowledged for their aid in checking the readability and comprehensibility of the paper. Finally, the authors would like to acknowledge all the healthcare workers and frontline‐support workers involved in the response to the COVID‐19 pandemic, in our institution, and in our country. The authors of this study declared that this study has received no financial support.
Badua CLDC, Baldo KAT, Medina PMB. Genomic and proteomic mutation landscapes of SARS‐CoV‐2. J Med Virol. 2021;93:1702‐1721. 10.1002/jmv.26548
Christian Luke D. C. Badua and Karol Ann T. Baldo contributed equally to this study and are co‐first authors.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are publicly available in NCBI GenBank at https://www.ncbi.nlm.nih.gov/nucleotide/ and in GISAID EpicCoV at https://www.gisaid.org/.
REFERENCES
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . 2020. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200824-weekly-epi-update.pdf?sfvrsn=806986d1_4. Accessed August 27, 2020.
- 3. Gorbalenya AE, Baker SC, Baric RS, et al. Severe acute respiratory syndrome‐related coronavirus: the species and its viruses – a statement of the Coronavirus Study Group [published online ahead of print Febraury 11, 2020]. bioRxiv. 2020. 10.1101/2020.02.07.937862 [DOI] [Google Scholar]
- 4. Chu H, Chan JF‐W, Yuen TT‐T, et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS‐CoV‐2 and SARS‐CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID‐19: an observational study. Lancet Microbe. 2020;1(1):e14‐e23. 10.1016/s2666-5247(20)30004-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Enjuanes L. Coronavirus Replication and Reverse Genetics. Berlin, NY: Springer; 2005. [Google Scholar]
- 7. Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS‐CoV‐2. Gene Reports. 2020;19:100682. 10.1016/j.genrep.2020.100682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cotten M, Watson SJ, Zumla AI, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio. 2014;5(1):e01062‐13. 10.1128/mbio.01062-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peck KM, Lauring AS. Complexities of viral mutation rates. J Virol. 2018;92(14):e01031‐17. 10.1128/jvi.01031-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017;20(4):1160‐1166. 10.1093/bib/bbx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuraku S, Zmasek CM, Nishimura O, Katoh K. aLeaves facilitates on‐demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013;41(W1):W22‐W28. 10.1093/nar/gkt389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nguyen L‐T, Schmidt HA, Haeseler AV, Minh BQ. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2014;32(1):268‐274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kalyaanamoorthy S, Minh BQ, Wong TKF, Haeseler AV, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods. 2017;14(6):587‐589. 10.1038/nmeth.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoang DT, Chernomor O, Haeseler AV, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2017;35(2):518‐522. 10.1093/molbev/msx281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547‐1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim Y, Jedrzejczak R, Endres M, Godzik A, Joachimiak A. Crystal structure of the methyltransferase‐stimulatory factor complex of NSP16 and NSP10 from SARS CoV‐2 [published online ahead of print April 20, 2020]. bioRxiv. 10.2210/pdb6w61/pdb [DOI] [Google Scholar]
- 17. National Center for Biotechnology Information . PubChem Database. 2020. L‐Proline, CID=145742, https://pubchem.ncbi.nlm.nih.gov/compound/Proline. Accessed June 7, 2020.
- 18. National Center for Biotechnology Information . PubChem Database. 2020. Serine, CID=5951, https://pubchem.ncbi.nlm.nih.gov/compound/Serine. Accessed June 7, 2020.
- 19. Maitra A, Sarkar MC, Raheja H, et al. Mutations in SARS‐CoV‐2 viral RNA identified in Eastern India: possible implications for the ongoing outbreak in India and impact on viral structure and host susceptibility. J Biosci. 2020;45(1):76. 10.1007/s12038-020-00046-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, Jameel S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand‐independent downregulation of the type 1 interferon receptor. PLoS One. 2009;4(12):e8342. 10.1371/journal.pone.0008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang C, Narayanan K, Ito N, Peters CJ, Makino S. Severe acute respiratory syndrome coronavirus 3a protein is released in membranous structures from 3a protein‐expressing cells and infected cells. J Virol. 2006;80(1):210‐217. 10.1128/jvi.80.1.210-217.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rota PA. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394‐1399. 10.1126/science.1085952 [DOI] [PubMed] [Google Scholar]
- 23. Tseng Y‐T, Wang S‐M, Huang K‐J, Wang C‐T. SARS‐CoV envelope protein palmitoylation or nucleocapid association is not required for promoting virus‐like particle production. J Biomed Sci. 2014;21(1):34. 10.1186/1423-0127-21-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao Z, Boerwinkle E. Neighboring‐nucleotide effects on single nucleotide polymorphisms: a study of 2.6 million polymorphisms across the human genome. Genome Res. 2002;12(11):1679‐1686. 10.1101/gr.287302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pachetti M, Marini B, Benedetti F, et al. Emerging SARS‐CoV‐2 mutation hot spots include a novel RNA‐dependent‐RNA polymerase variant. J Transl Med. 2020;18(1):179. 10.1186/s12967-020-02344-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim JS, Jang JH, Kim JM, Chung YS, Yoo CK, Han MG. Genome‐wide identification and characterization of point mutations in the SARS‐CoV‐2 genome. Osong Public Health Res Perspect. 2020;11(3):101‐111. 10.24171/j.phrp.2020.11.3.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tiwari M, Mishra D. Investigating the genomic landscape of novel coronavirus (2019‐nCoV) to identify non‐synonymous mutations for use in diagnosis and drug design. J Clin Virol. 2020;128:104441. 10.1016/j.jcv.2020.104441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ou J, Zhou Z, Dai R, et al. Emergence of RBD mutations in circulating SARS‐CoV‐2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein [published online ahead of print March 23, 2020]. bioRxiv. 10.1101/2020.03.15.991844 [DOI] [Google Scholar]
- 29. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. 10.1016/s0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS‐CoV‐2. Natl Sci Rev. 2020;7(6):1012‐1023. 10.1093/nsr/nwaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang L, Jackson C, Mou H, et al. The D614G mutation in the SARS‐CoV‐2 spike protein reduces S1 shedding and increases infectivity [published online ahead of print June 12, 2020]. bioRxiv. 10.1101/2020.06.12.148726 [DOI] [Google Scholar]
- 32. Tian X, Li C, Huang A, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus‐specific human monoclonal antibody. Emerg Microbes Infect. 2020;9(1):382‐385. 10.1080/22221751.2020.1729069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang S, Hillyer C, Du L. Neutralizing antibodies against SARS‐CoV‐2 and other human coronaviruses. Trends Immunol. 2020;41(5):355‐359. 10.1016/j.it.2020.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moosa M, Banerjee P. Subversion of host stress granules by coronaviruses: potential roles of π‐rich disordered domains of viral nucleocapsids. J Med Virol. 2020;92(4):455‐459. 10.1002/jmv.26195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin S, Shen R, He J, Li X, Guo X. Molecular modeling evaluation of the binding effect of ritonavir, lopinavir and darunavir to severe acute respiratory syndrome coronavirus 2 proteases [published online ahead of print February 18, 2020]. bioRxiv. 10.1101/2020.01.31.929695 [DOI] [Google Scholar]
- 36. Zeng W, Liu G, Ma H, et al. Biochemical characterization of SARS‐CoV‐2 nucleocapsid protein. Biochem Biophys Res Commun. 2020;527(3):618‐623. 10.1016/j.bbrc.2020.04.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data that support the findings of this study are publicly available in NCBI GenBank at https://www.ncbi.nlm.nih.gov/nucleotide/ and in GISAID EpicCoV at https://www.gisaid.org/.