

Abstract
Infection with the SARS‐CoV‐2 virus has rapidly become a global pandemic for which we were not prepared. Several clinical trials using previously approved drugs and drug combinations are urgently under way to improve the current situation. A vaccine option has only recently become available, but worldwide distribution is still a challenge. It is imperative that, for future viral pandemic preparedness, we have a rapid screening technology for drug discovery and repurposing. The primary purpose of this research project was to evaluate the DeepNEU stem‐cell based platform by creating and validating computer simulations of artificial lung cells infected with SARS‐CoV‐2 to enable the rapid identification of antiviral therapeutic targets and drug repurposing. The data generated from this project indicate that (a) human alveolar type lung cells can be simulated by DeepNEU (v5.0), (b) these simulated cells can then be infected with simulated SARS‐CoV‐2 virus, (c) the unsupervised learning system performed well in all simulations based on available published wet lab data, and (d) the platform identified potentially effective anti‐SARS‐CoV2 combinations of known drugs for urgent clinical study. The data also suggest that DeepNEU can identify potential therapeutic targets for expedited vaccine development. We conclude that based on published data plus current DeepNEU results, continued development of the DeepNEU platform will improve our preparedness for and response to future viral outbreaks. This can be achieved through rapid identification of potential therapeutic options for clinical testing as soon as the viral genome has been confirmed.
Keywords: antiviral, DeepNEU, drug discovery and repurposing, pandemic preparedness, SARS‐CoV‐2, unsupervised learning
DeepNEU is a machine‐learning platform that uses genomic data to simulate induced pluripotent stem cells (aiPSCs) and differentiated cell types like lung cells. In this study, simulated lung cells (aiLUNG) were infected with simulated SARS‐CoV‐2 virus and used for rapid identification of antiviral targets and drug repurposing. Potentially effective anti‐SARS‐CoV‐2 two drug combinations were identified for urgent clinical studies.
Significance statement.
The current results showed that validated DeepNEU v5.0 platform of this study accurately derived aiLUNG cells from aiPSC simulations. The aiLUNG simulations could be exposed to simulated SARS‐CoV‐2 virus and reproduce the genotypic and phenotypic profile associated with the infection. This study also demonstrated that the aiLUNG‐COVID‐19 simulations can be used to rapidly repurpose novel and known drug combinations with anti‐SARS‐CoV‐2 therapeutic potential for animal and human trial validation. The rational process described in this article required the existence of a validated genome for the viral genome(s) in question. While DeepNEU requires continued development and validation, it is very likely that the most important application of this technology will be in improving disease preparedness for future outbreaks.
1. INTRODUCTION
Infection with the SARS‐CoV‐2 virus and the resultant COVID‐19 disease has rapidly become the most lethal global pandemic when compared to the SARS and H1N1 outbreaks in 2003 and 2009, respectively. 1 SARS‐CoV‐2 is essentially a new virus to the human host, although it shares >80% genetic homology with the SARS‐CoV‐2 virus responsible for the 2003 outbreak. 1 As a result, we as a population have little or no innate immunity to this new pathogen. While we still have few effective therapies for SARS‐CoV‐2 Infection, multiple vaccines will continue to become available which should reduce disease severity and facilitate herd immunity. A few brave physicians have made the decision to treat SARS‐CoV‐2 infected patients with off label approved drugs with encouraging but preliminary results. Many clinical trials with previously approved drugs and drug combinations are currently underway to improve this therapeutic dilemma. A vaccine option has only recently become available, but worldwide distribution is still a challenge.
In the last two decades, our world has experienced outbreaks of Ebola, SARS, H1N1, and now SARS‐CoV‐2. As the World Health Organization (WHO) has clearly stated, new and lethal viral pathogens are certain to emerge in the future and if we disregard this warning, we are likely to experience similar pandemics in the future. It is imperative that we are prepared to act as early as possible when or ideally before future viral outbreaks.
The advent of reprogramming human induced pluripotent stem cells (iPSCs) from donors' somatic cells has created new opportunities to study and understand the underlying pathophysiology of human diseases, including a growing number of viral infections, like Zika Virus (ZIKV), hepatitis C virus (HCV), and Influenza virus (H1N1). 2 , 3 Unfortunately, cellular reprogramming to produce iPSCs remains a challenge due to the high costs, demanding resource needs and the tendency of iPSCs to revert to their original somatic genotypes over time. Importantly, the limited access to donor cells also remains a major concern especially for developing new drug therapies for viral and other infectious diseases. To overcome the technological and ethical limitations still associated with iPSC models, we have created DeepNEU. The DeepNEU platform is a validated hybrid deep‐machine learning system with elements of fully connected recurrent neural networks (RNNs), cognitive maps (CMs), support vector machines, and evolutionary systems (GA). Previously the DeepNEU platform has been used to generate artificially induced pluripotent stem cells (aiPSCs), neural stem cells, cardiomyocytes, and skeletal muscle cells. 4 , 5
The purpose of the present research was to evaluate an updated version of our machine‐learning platform DeepNEU v5.0 for creating computer simulations of artificially induced type 1 (AT1) and type 2 (AT2) alveolar lung cells (aiLUNG) derived from artificially induced human pluripotent stem cells (aiPSCs). These uninfected aiLUNG cells were then exposed to infection with simulated SARS‐CoV‐2 virus. Finally, the aiLUNG‐COVID19 simulations were applied to drug repurposing of a small group of approved drugs with well‐known mechanisms of action. The genomic and phenotypic profiles of wild‐type aiLUNG cells and aiLUNG‐COVID‐19 simulations were validated based on the currently available experimental wet lab and other data. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Ideally, in the future, this new technology would be implemented as soon as the new viral genome has been identified and validated. For example, the SARS‐CoV‐2 genome and cell receptor data were published in early March 2020 (GenBank accession number: MT126808.1).
2. METHODS
The DeepNEU platform is a literature validated hybrid deep‐machine learning system with elements of fully connected RNNs, CMs, and evolutionary systems (GA). 4 , 5 The detailed methodology for simulation development and validation used in the current experiments has been described previously. 4 , 5 The current DeepNEU database (v5.0) contains all the information found in the previous version (v3.6) plus important information upgrades in the form of new gene, protein, and phenotypic relationship data. For example, the previous DeepNEU database version (3.6) contained 3187 gene/proteins or phenotypic concepts and 31 027 nonzero relationships while the current version (5.0) contains 4206 gene/proteins or phenotypic concepts and 37 223 nonzero relationships. This represents more than 1200 new relationships specifically relevant to the SARS‐CoV‐2 viral genome. Each gene/protein and phenotypic concept in DeepNEU v5.0 has on average ~9 gene/protein or phenotypic inputs and outputs.
2.1. The DeepNEU simulations
The initial goal of this project was to create computer simulations (aiPSCs) of human induced pluripotent stem cells (iPSC) and lung (aiLUNG) cell models then validate these models using the results published by References 4, 5, 7, 12, 13, 15, 20, 22, 23 and others as described above. Briefly, for the aiPSC models, the input or initial state vector of dimension N was set to all zeros except for transcription factors OCT4, KLF4, SOX2, and cMYC (also known as OKSM). These four factors were given a value of +1 indicating that they were turned on for the first iteration. These values were not locked on so that after the first iteration all values were determined by evolving system behavior. The lung cell models (aiLUNG) were created through direct conversion of the aiPSCs to aiLUNG cells using overexpression of NK2 Homeobox 1 (NKX‐2.1), and Wnt Family Member 5A (Wnt5a) in the presence of a simulated lung cell medium. 12 , 20 Once validated with published peer reviewed data, the aiLUNG simulations were exposed to simulated SARS‐CoV‐2 infection by turning on extracellular Spike‐RBP (RNA Binding Domain) in the presence of active Transmembrane Serine Protease 2 (TMPRSS2). Finally, several potential factors and combinations of factor inhibitors were evaluated regarding their ability to reduce the production and release of new SARS‐CoV‐2 viral particles. A summary of the 12 key simulations generated in the present study are presented in Table 1 below.
TABLE 1.
Summary of evaluated single and double drug combinations
| Model | Status | Cocktail |
|---|---|---|
| aiPSC‐WT | Pluripotent uninfected | Fibroblast + OKSM + Dox |
| aiLUNG (ie, wild type) | Differentiated uninfected | aiPSC + NKX‐2.1 + WNT5a + LUNG medium |
| aiLUNG + SARS‐CoV‐2 | Differentiated infected and untreated | aiLUNG + initial viremia + active TMPRSS2 |
| aiLUNG + SARS‐CoV‐2 + HCQ | Differentiated infected and treated (1 drug) | aiLUNG + viremia + active TMPRSS2 + HCQ |
| aiLUNG + SARS‐CoV‐2 + RdRPi | Differentiated infected and treated (1 drug) | aiLUNG + viremia + active TMPRSS2 + RdRP inhibitor |
| aiLUNG + SARS‐CoV‐2 + PLproi | Differentiated infected and treated (1 drug) | aiLUNG + viremia + active TMPRSS2 + PLpro inhibitor |
| aiLUNG + SARS‐CoV‐2 + 3CLproi | Differentiated infected and treated (1 drug) | aiLUNG + viremia + active TMPRSS2 + 3CLpro inhibitor |
| aiLUNG + SARS‐CoV‐2 + HCQ + RdRPi | Differentiated infected and treated (2 drug) | aiLUNG + viremia + active TMPRSS2 + HCQ + RdRP inhibitor |
| aiLUNG + SARS‐CoV‐2 + HCQ + PLproi | Differentiated infected and treated (2 drug) | aiLUNG + viremia + active TMPRSS2 + HCQ + PLpro inhibitor |
| aiLUNG + SARS‐CoV‐2 + HCQ + 3CLproi | Differentiated infected and treated (2 drug) | aiLUNG + viremia + active TMPRSS2 + HCQ + 3CLpro inhibitor |
| aiLUNG + SARS‐CoV‐2 + RdRPi + PLproi | Differentiated infected and treated (2 drug) | aiLUNG + viremia + active TMPRSS2 + RdRP inhibitor + PLpro inhibitor |
| aiLUNG + SARS‐CoV‐2 + RdRPi + 3CLproi | Differentiated infected and treated (2 drug) | aiLUNG + viremia + active TMPRSS2 + RdRP inhibitor + 3CLpro inhibitor |
| aiLUNG + SARS‐CoV‐2 + PLproi + 3CLproi | Differentiated infected and treated (2 drug) | aiLUNG + viremia + active TMPRSS2 + PLpro inhibitor + 3CLpro inhibitor |
Abbreviations: 3CLpro, 3 chymotrypsin Like protease; aiLUNG‐COVID‐19, aiLUNG + SARS‐CoV‐2; Dox, doxycycline; HCQ, hydroxychloroquine; OKSM, OCT4, KLF4, SOX2, cMYC; PLpro, papain like protease; RdRP, RNA dependent RNA polymerase.
The final predictions from the aiPSC and aiLUNG simulations regarding the expression or repression of genes and proteins and presence or absence of phenotypic features were directly compared with published data as outlined above. Model prediction values ≥0 were classified as expressed or upregulated for genes/proteins or present in the case of phenotypic features while values < 0 were classified as downregulated, not expressed, or absent. All experiments in this study except for the earliest screening run were conducted in triplicate (N = 3) using different initial input vectors.
Statistical analysis of the aiPSC and aiLUNG predictions vs the published data used the unbiased binomial test. This test provides an exact probability, can compensate for prediction bias, and is ideal for determining the statistical significance of experimental deviations from an actual distribution of observations that fall into two outcome categories (eg, agree vs disagree). A P value <.05 is considered significant and is interpreted to indicate that the observed relationship between aiPSC predictions and actual outcomes is unlikely to have occurred by chance alone.
2.2. DeepNEU platform specification
The current DeepNEU database (Version 5.0) contains 4206 gene/protein or phenotypic concepts and 37 223 nonzero relationships resulting in a large amount of information flowing into and out of each node in the fully connected recurrent network. On average, each node in the network initially has ~9 inputs and ~9 outputs. An updated analysis of all positive and negative network connections revealed a bias toward positive outputs. The pretest probability of a positive outcome prediction is .661 and the pretest probability of a negative prediction is therefore .339. This system bias was used when applying the binomial test to all simulation outcomes.
3. RESULTS
3.1. The aiPSC simulations
In this study, we began by programming DeepNEU (v5.0) to simulate iPSCs (ie, aiPSCs) using a defined set of reprogramming factors 4 , 5 in the presence of ascorbic acid and doxycycline. Following the protocol that we have established, we turned on the key transcription factors that were previously reported to successfully induce pluripotency in iPSC derived from human fibroblasts. Briefly, OCT4, KLF4, SOX2, and cMYC (OKSM) were turned on as were ascorbic acid and doxycycline. 4 , 5
The unsupervised aiPSC model converged quickly (20 iterations) to a new system wide steady state without evidence of overtraining after 1000 iterations. The aiPSC simulations expressed the same human ESC specific surface antigen and genomic profiles. The expression profile of several factors (N = 15) consistent with the signature of undifferentiated human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSC) includes OCT3/4, SOX2, NANOG, growth and differentiation factor 3 (GDF3), reduced expression 1 (REX1), fibroblast growth factor 4 (FGF4), embryonic cell‐specific gene 1 (DPPA5/ESG1), developmental pluripotency‐associated 2 (DPPA2), DPPA4, and telomerase reverse transcriptase (hTERT). In a previous study, the expression levels of OCT3/4, SOX2, NANOG, SALL4, E‐CADHERIN (CDH1), and hTERT as determined by Western blotting were also similar in iPSC and hESC, including stage‐specific embryonic antigen 3/4 (SSEA‐3/4), tumor‐related antigen (TRA‐1‐81), alkaline phosphatase (ALP), and NANOG protein. 24 Importantly, all the undifferentiated ESC/iPSC makers mentioned above regarding iPSC were also upregulated/expressed in the aiPSC simulations. The probability that all (N = 15) of these aiPSC‐WT outcomes were correctly predicted by chance alone using the binomial test is .0021. These results are presented in Figures S1 and S2.
3.2. The wild‐type (uninfected) aiLUNG simulations
Once validated the aiPSC simulations were used to create the wild‐type lung cell simulations (aiLUNG) by activating NKX2.1 and Wnt5a in the presence of a simulated lung cell medium as described previously. 12 , 20 The unsupervised aiLUNG simulations converged quickly (24 iterations) to a new system wide steady state without evidence of overtraining after 1000 iterations.
Several previous studies have contributed to our current understanding of the expression/activity profile of genotypic and phenotypic factors consistent with the signature of wild‐type human lung cells 4 , 5 , 7 , 12 , 13 , 15 , 20 , 22 , 23 . These genotypic factors (N = 9) including Aquaporin 5 (AQP5), Forkhead Box J1 (FOXJ1), HOP Homeobox (HOPX), Oligomeric Mucus/Gel‐Forming (Mucin5AC), NK2 Homeobox 1 (NKX‐2.1/TITF1), Tumor Protein P63 (p63), Podoplanin (PDPN/T1a), Surfactant Protein C (SFTPC/SPC), SRY‐Box Transcription Factor 9 (Sox9) and phenotypic factors (N = 6), Alveolar type 1 cells (ATI), Alveolar type 2 cells (ATII), Alveolar type 1 precursor cells (ATI Precursor), Alveolar type 2 precursor cells (ATII Precursor), Alveolar type 1 sacular cells (ATI Sacular), and Alveolar type 2 sacular cells (ATII Sacular) were used in this study to validate the aiLUNG simulations.
The aiLUNG simulations produced a similar expression profile when compared with actual human wild‐type lung cell specific factors outlined above (Figure 1A), consistent with previous studies. 4 , 5 , 7 , 12 , 13 , 15 , 20 , 22 , 23 The probability that all (N = 15) of these aiLUNG outcomes were correctly predicted by chance alone using the binomial test is .0021. Importantly, the data also indicate that the generation of aiLUNG cells from aiPSC produces a heterogenous population of alveolar cell precursors and more mature alveolar cells (Figure 1B), consistent with previous study. 25
FIGURE 1.
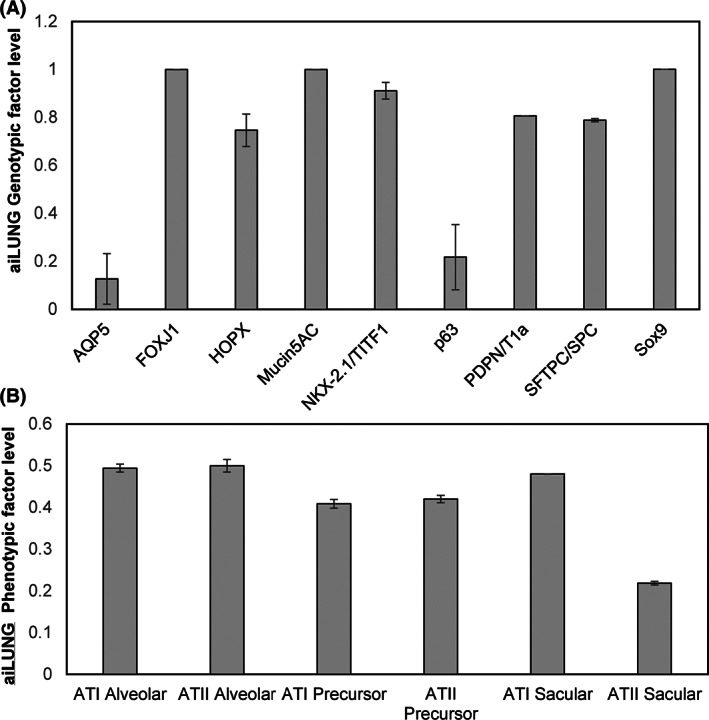
DeepNEU simulation of differentiated aiLUNG‐WT cells. A, Expression of genotypic markers of differentiated aiLUNG cells. B, Expression of phenotypic features of differentiated aiLUNG. The vertical y‐axis represents the semiquantitative levels of genotypic and phenotypic factors that are estimated regarding an arbitrary base line where 0 = base line and 1 = maximum expression. The y‐axis represents the expression level of each factor relative to arbitrary baseline. Data represent mean of three experiments ± 99% confidence interval. ATI Alveolar, alveolar type 1 cells; ATII Alveolar, alveolar type 2 cells; ATI Precursor, Alveolar type 1 precursor cells; ATII Precursor, alveolar type 2 precursor; ATI Sacular, alveolar type 1 sacular cells; ATII Sacular, alveolar type 2 sacular cells
3.3. Simulation of SARS‐CoV‐2‐infected aiLUNG cells (aiLUNG‐COVID‐19)
Once validated against current published wet lab data, the aiLUNG cells were exposed to simulated SARS‐CoV‐2 virus. For this simulated infection, the concept of SARS‐CoV‐2 viremia was activated (turned on). First, the SARS‐CoV‐2 viremia activates a viral life cycle consisting of (a) interaction of the viral Spike protein with its receptor protein Angiotensin‐converting enzyme 2 (ACE2), (b) endocytosis of the virus‐ACE2 complex, (c) intracellular uncoating of viral single stranded RNA, (d) transcription and translation of the viral genome, (e) assembly of new viral particles, and (f) exocytosis of new viral particles which completes the cycle by contributing to the level of viremia. 26 The SARS‐CoV‐2 genome consists of four structural genes and at least six nonstructural genes. 27 The structural genes (N = 4) are Spike (S), Nucleocapsid (N), Envelope (E) and Membrane (M) and produce S, N, E, and M proteins (N = 4), respectively. 27 The nonstructural coding genes (N = 6) and proteins (N = 13) are orf1a/b polyprotein (orf1a/b), orf3a protein (orf3a), orf6 protein (orf6), orf7a protein (orf7a), orf8 protein (orf8), and orf10 protein (orf10). 9 , 16 , 26 , 27 Other important nonstructural proteins (NSPs) include NSP1, NSP2, NSP3, Papain Like protease (PLpro), NSP5/3Chymotrypsin Like protease (3CLpro), NSP12/RdRP/Replicase, and NSP13/Helicase. 9 , 16 , 26 , 27 The 17 gene or protein expression profile was compared with the uninfected aiLUNG simulations to assess the validity of the simulated SARS‐CoV2 infection. All gene or protein factors were expressed or upregulated in the aiLUNG‐SARS‐CoV‐2 vs aiLUNG simulations. This genotypic expression data are summarized in Figure 2A. The probability that all (N = 17) of these aiLUNG‐COVID‐19 simulation outcomes were correctly predicted by chance alone using the binomial test is .0009.
FIGURE 2.
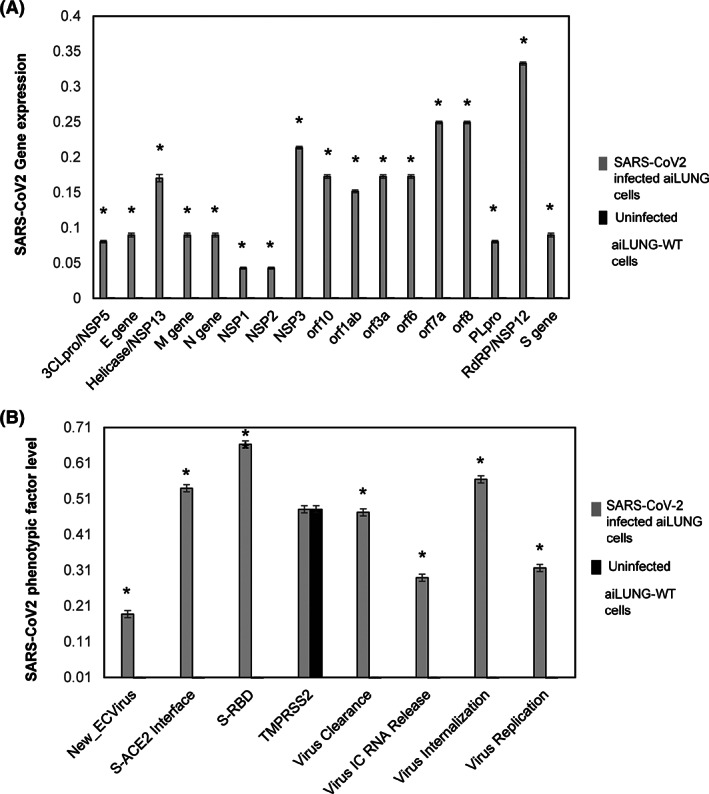
DeepNEU successful simulation of SARS‐CoV‐2‐infected aiLUNG cells. A, Expression of (N = 17) SARS‐CoV‐2 genotypic features (genes/proteins) in aiLUNG cells. B, Expression of viral phenotypic features (N = 8) strength in SARS‐CoV‐2 infected aiLUNG cells. The vertical y‐axis represents the semiquantitative levels of genotypic and phenotypic factors that are estimated regarding an arbitrary base line where 0 = base line and 1 = maximum expression, The y‐axis represents the expression level of each factor relative to arbitrary baseline. Data represent mean of three independent experiments ± 99% confidence interval. *P < .05
A SARS‐CoV‐2 infection phenotypic profile was also developed from the published literature outlined above. These phenotypic features (N = 8) include: New Extracellular Virus release, Spike‐ACE2 Interface, Spike‐RBD, TMPRSS2, Virus Clearance, Virus Intracellular RNA release, Virus Internalization, and Virus Replication. These data are summarized in Figure 2B. The presence of all phenotypic features of SARS‐CoV‐2 infection was correctly predicted by the aiLUNG‐COVID‐19 when compared with the aiLUNG simulations. The probability that all (N = 8) of these aiLUNG‐COVID‐19 outcomes were predicted correctly by chance alone using the binomial test is .036.
To summarize, the probability that all 17 genotypic and all eight phenotypic features of simulated aiLUNG SARS‐CoV‐2 infection (N = 25) were accurately predicted by chance alone using the binomial test is .00003.
3.4. Application of the validated aiLUNG simulations to potential therapeutic target identification and anti‐COVID‐19 drug repurposing
Comparison of all predictions for >4100 genotypic and phenotypic factors from the aiLUNG‐COVID‐19 and aiLUNG simulations revealed a subset of these factors that stood out as viral genes that could be potential therapeutic targets. To that end, we have evaluated the effect of using single or double drug combinations that either block the expression or function of SARS‐CoV‐2 coding genes. Based on the two tailed Mann‐Whitney U test, the estimated P values for comparing these aiLUNG‐COVID‐19 vs aiLUNG factors were highly significant at P = .00001. This inclusive subset of potential therapeutic targets (N = 17) included: 3 Chymotrypsin Like protease (3CLpro)/NSP5, E gene, Helicase/NSP13, M gene, N gene, NSP1, NSP2, NSP3, orf10, orf1ab, orf3a, orf6, orf7a, orf8, Papain Like protease (PLpro), RdRP/NSP12 and S gene was selected for further evaluation. In preliminary experiments, inhibition of each of these 17 viral genes was added to the aiLUNG simulations in an iterative manner in order to assess changes in gene expression. These interventions resulted in (a) variable improvement in the anti‐COVID‐19 gene expression profile in general and (b) a reduction in the release of new SARS‐CoV‐2 virus particles specifically. Inhibiting six of these gene products, namely orf1ab, PLpro, orf7a8, RdRP/Nsp12, 3CLpro/NSP5, orf8, and one known drug, hydroxychloroquine (HCQ), stood out as being particularly effective potential single anti‐COVID‐19 therapeutic options. HCQ was included in the initial experiments because (a) it has multiple COVID‐19 relevant cellular targets, 28 (b) it is already approved for other indications including malaria and inflammatory diseases, and (c) early anti‐COVID‐19 results from at least one small trial appear promising. 29 In fact, during our initial screening experiments based on genotypic features (Figure 3A), HCQ proved to be the second most effective anti‐COVID‐19 single drug option while simulated inhibition of orf1ab was most effective. Importantly, orf1ab had been included as a potential therapeutic target because it represents ~70% of the SARS‐CoV‐2 genome. A summary of treatment effects on the COVID‐19 phenotypic features is presented in Figure 3B.
FIGURE 3.

Predictions of anti‐COVID‐19 effects of selected drugs from preliminary screening experiments. Anti‐COVID19 efficacy of selected drugs on (A) viral genes expression and (B) viral phenotypic features expression levels in COVID‐19 infected aiLUNG cells, respectively. The legend scale ranges from a maximum negative change (−1) to maximum positive change (+1) relative to a qualitative baseline condition (= 0). Data represent mean of three independent experiments ± 99% confidence interval
Three potential therapeutic targets were excluded from further study at this time because there are no identified or approved small molecule candidates. The excluded factors are orf1ab, orf8, and orf7a8 leaving three factors, 3CLpro, PLpro, and RdRP plus HCQ for further evaluation.
Next, we evaluated all single drug candidates (N = 4) and all possible (N = 6) double drug combinations which included: (a) HCQ + PLproI, (b) HCQ + RdRPI, (c) HCQ + 3CLproI, (d) PLproI + RdRPi, (e) PLproI + 3CLproI, and (f) RdRPi + 3CLproI in triplicate. The most effective single agent in the final group was HCQ when compared with the untreated aiLUNG‐COVID‐19 simulations (two‐tailed, unpaired t test P < .001). All six combinations were effective against SARS‐CoV‐2 infection using the 17 viral target profile outlined above (Figure 4A,B) and the data indicate that HCQ + a PLproi and HCQ + 3CLproii and PLpeoi + 3CLproi are the most effective of the double drug combinations evaluated. Importantly three of the four most effective combinations included HCQ.
FIGURE 4.
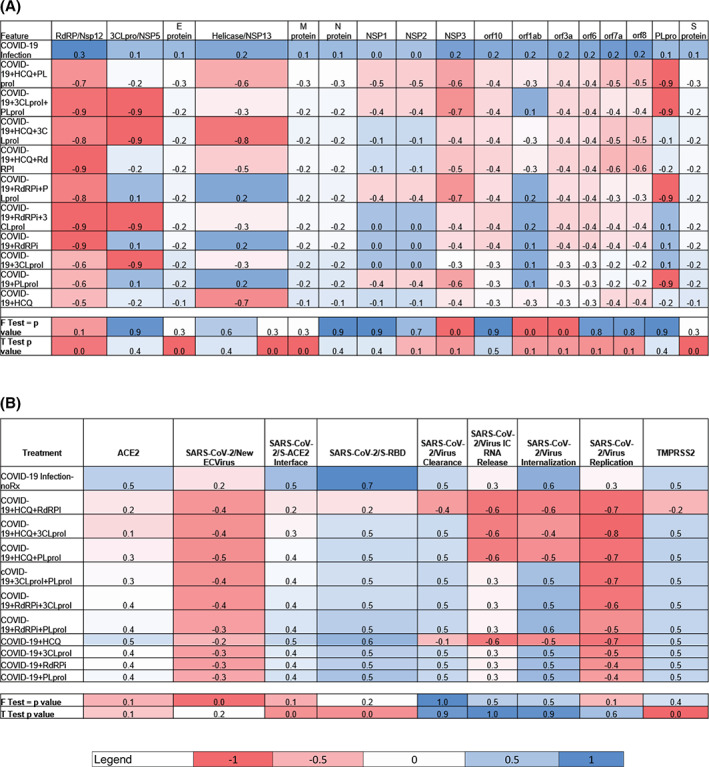
Final predictions of anti‐COVID‐19 effects of selected drugs experiments. Anti‐COVID19 efficacy of selected drugs on (A) viral genotypic and (B) viral phenotypic features expression/activity levels in COVID‐19 infected aiLUNG cells, respectively. The legend scale ranges from the maximum negative change (−1) to the maximum positive change (+1) relative to a qualitative baseline condition (0). Data represent mean of three independent experiments ± 99% confidence interval
Finally, a statistical analysis of these data using the paired two‐tailed t test (Figure 5) confirmed this assessment regarding anti‐COVID‐19 efficacy. Further analysis indicated that based on the genotypic profile, the double drug combinations (N = 6) were generally more effective than the single drugs (N = 4) (two‐tailed t test P < .005). Overall, the phenotypic profile was not significantly different between single and double drug combinations (P < .29). However, the single phenotypic concept New Extracellular Virus was able to distinguish between groups in favor of the double drug combinations (P < .01).
FIGURE 5.
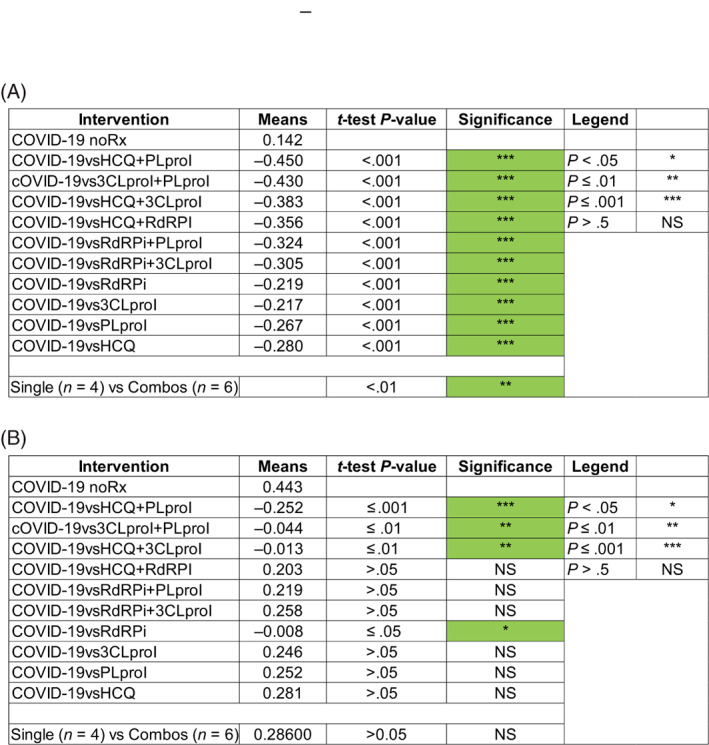
Statistical analysis of final DeepNEU predictions of anti‐COVID‐19 efficacy for repurposed drugs. The paired two‐tailed t test was used to evaluate the effects of each treatment on viral (A) genotypic profile and (B) genotypic profiles compared with the aiLUNG (wild‐type) profiles. The data from three independent experiments were used to conduct this analysis. The level of significance was set at P < .05 for all comparisons. The analysis also indicated that overall, the two drug combinations (N = 6) outperformed the single drug treatments (N = 4) based on the genotypic profiles (P < .01). Based on the phenotypic profiles, no significant differences were seen (P > .05)
4. DISCUSSION
The main purpose of this research was to generate and then validate potentially useful stem cell‐based computer simulations of SARS‐CoV‐2 infection in simulated lung cells (aiLUNG) with the same genotypic/phenotypic markers as iLUNG cells that were produced in wet lab experiments. 12 , 20 Additionally, the phenotypic and genotypic features of SARS‐CoV‐2 infection that were simulated by DeepNEU v5.0 are in agreement with the recent findings as outlined above. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 The data from the present experiments indicate that SARS‐CoV‐2 viral infection can indeed be accurately simulated in aiLUNG using the DeepNEU (v5.0) machine‐learning platform. We believe that, as of this writing, these are the first and only results of this kind in the published literature. Notably, these results are consistent with and extend our earlier research. In this regard, the current aiPSC‐WT simulation results are remarkably consistent with results from the aiPSC‐WT models that we reported previously. 4 , 5 The updated (v5.0) aiPSC simulations also (a) rapidly (<30 iterations) achieved new system wide steady states, (b) showed no evidence of overfitting after 1000 iterations, and (c) accurately reproduced wet lab results from the peer‐reviewed literature 24 based on the bias adjusted binomial test.
To more accurately simulate SARS‐CoV‐2 infection, we next created aiLUNG cells by exposing the validated aiPSC simulations to NKX2.1 and WNT5a in the presence of doxycycline and ascorbic acid. The direct generation of human lung cells from iPSC is well documented in the peer‐reviewed literature. 12 , 20 Using the same approach, the simulations evolved quickly to a new steady state that accurately reproduced the genotypic and phenotypic features of differentiated wild‐type human lung cells (aiLUNG). We then created the aiLUNG‐COVID‐19 viral infection simulations by activating and adding a simulated SARS‐CoV‐2 viremia to the aiLUNG initial input vector and allowing the system to evolve to a new stable steady state. Importantly, in the aiLUNG‐COVID‐19 simulations, the genotypic and phenotypic features of differentiated human lung cells were preserved while the same simulations also reproduced the genotypic and phenotypic features of the SARS‐CoV‐2 infection life cycle. It is also notable that the aiLUNG simulations can also be used to investigate other viral diseases that primarily affect lung cells like Influenza A and B, respiratory syncytial virus, parainfluenza, and adenovirus.
Once the aiLUNG and aiLUNG‐COVID‐19 simulations were created and validated against the available peer‐reviewed wet lab research, they were applied in two specific areas including therapeutic target identification and drug repurposing. In the case of a viral pandemic for which no approved therapies are available, the rational evaluation of currently licensed drugs to identify potentially effective therapies or simple combination therapies may represent the most efficient path to improved outcomes in combination with early and widespread testing. In the future, rapid drug repurposing is likely to become vitally important when we are forced to deal with an outbreak of a much more lethal virus. For example, the NIPAH virus (named after Sungai Nipah, a village in the Malaysian Peninsula where pig farmers became ill with encephalitis) has been associated with a human death rate of ~70% 30 compared with the 1% to 2% death rate seen so far with COVID‐19 infection. It would make remarkable good sense to begin planning for the next outbreak now.
In addition to disease modeling, DeepNEU has also been designed to be used as a tool for rapid and cost‐effective therapeutic target identification and drug discovery. The use of stem cells for targeted drug discovery has been well reported in the peer‐reviewed literature. 31 , 32 , 33 Our previously published approach to this issue has three simple components. 5 First, we compared all predictions from the aiLUNG‐COVID‐19 and. aiLUNG simulations. The statistical analysis identified several potentially important therapeutic targets. We used the Mann‐Whitney test to estimate the level of significance because not all the data were normally distributed. The exact P values for the selected potential therapeutic targets from the aiLUNG‐COVID‐19 vs aiLUNG simulations were all highly significant at .0001. To further explore these findings, the second step was modified in a rational manner from Reference 5. Instead of creating individual data sets and regression models for many single and double drug combinations, we used the aiLUNG‐COVID‐19 vs aiLUNG simulations to evaluate the anti‐COVID‐19 effects of inhibition or activation of each individual potential therapeutic targets. The results of this iterative process were then combined with data from recently published papers that identified potential repurposing targets for treating COVID‐19 infection 34 , 35 , 36 , 37 with drugs currently approved for other indications or in late clinical testing. The final group of selected targets represents a rationally defined smaller subset derived from iterative simulated testing and the published data. Based on these combined results, the three genotypic features PLpro, 3CLpro, RdRP and the multiple targets affected by HCQ were identified as potential therapeutic targets for SARS‐CoV‐2 infection warranting further investigation. HCQ is approved for several inflammatory diseases in multiple countries including Canada and the US. Idarubicin, approved for treating certain cancers, is a RdRP inhibitor. Remdesivir is a potent RdRP inhibitor currently in multiple clinical trials against COVID‐19 with promising early results. 28 , 36 , 38 In the third step, we used the validated aiLUNG COVID‐19 and iLUNG simulations to evaluate the efficacy of the final subset of 10 treatment options based on genotypic and phenotypic profiles outlined above. The aiLUNG‐COVID‐19 simulations with HCQ locked on and/or PLpro, 3CLpro, and RdRP locked off were used to simulate target activation and inhibition, respectively. The final 10 treatment options studied included (a) the four single agents defined above and (b) their six combinations (ie, HCQ + RdRP inhibition, HCQ + PLpro inhibition, HCQ + 3CLpro inhibition, RdRP inhibition + PLpro inhibition, RdRP inhibition + 3CLpro inhibition, and PLpro inhibition + 3CLpro inhibition). The data presented in Figures 3, 4, and 5 indicate that while all single agent have some beneficial effect, HCQ appears to be the most effective single agent studied. Regarding the double drug combinations, HCQ + a PLpro inhibitor and HCQ +3CLproi inhibitor and PLpro inhibitor+3CLpro inhibitor are the most effective of the double drug combinations and all these combinations outperform HCQ alone. This short list of double drug combinations can be further stratified. Since HCQ and the RdRP inhibitor Idarubicin are already approved for other indications, it would make sense to further evaluate this combination first, followed by HCQ and Remdesivir which is currently in multiple clinical trails. As of this writing, we are not aware of any other published AI based methods for creating differentiated aiLUNG cells for rapid identification of potentially effective double drug combinations for treating SARS‐CoV‐2 lung infection.
Although we were focused on repurposing small molecules, we had also postulated that DeepNEU had the potential to identify targets for vaccine development. In order to evaluate this possibility, we revisited the three anti‐COVID‐19 targets that had no small molecule candidates. These three genotypic options were orf1ab, orf7, and orf7/8. We were particularly interested in the polyprotein orf1ab which represents ~70% of the SARS‐CoV‐2 genome and was the single most efficacious target we studied. The essential requirement for the identified factors to become potential vaccine targets is that they must be immunogenic. In fact, a recent computational analysis identified several SARS‐CoV‐2 antigenic proteins. While orf8a was the most antigenic of the proteins considered, it was rapidly degraded without its companion protein orf8b. ORF1ab was also determined to be antigenic 39 and polyclonal antibodies have successfully been raised against full length orf1ab recombinant protein in rabbits (Abnova, Catalog #: PAB11368). More recently, a multiepitope vaccine based on orf1ab sequences has been designed that is antigenic and capable of activating B cells and T cells. 40 We conclude that if antigenicity/immunogenicity can be documented for a specific target, then DeepNEU disease specific simulations may also be an important tool capable of identifying potential vaccine targets.
4.1. Update on the limitations of DeepNEU v5.0
In our recent paper, 5 we identified and discussed several limitations of the DeepNEU platform. First, the issue of incomplete data persists but continues to improve on an almost daily basis. Version 3.6 contained 3781 gene/proteins or phenotypic concepts and 31 027 nonzero relationships while the current version (5.0) contains 4206 gene/proteins or phenotypic concepts and 37 223 nonzero relationships Overall the data in v5.0 represents more than 18% of the human genome compared with <15% in version 3.6. Included in this number is more than 1200 new relationships specifically relevant to the COVID‐19 viral genome. In addition, each gene/protein and phenotypic concept in DeepNEU v5.0 now has on average ~9 gene/protein or phenotypic inputs and outputs compared with ~8 previously.
Second, predictions from advanced computer modeling systems still require wet lab confirmation and this continues to be important for DeepNEU v5.0 as well. A major goal of this project was to make the findings regarding the potential therapeutic benefit of novel anti‐COVID‐19 drug combinations freely available to the global research community for wet lab validation at the very earliest opportunity. We also plan to validate these important predictions and we are currently looking for development partners with the goal of confirming these predictions in animal models of SARS‐CoV‐2 infection. We commit to making any additional information available at the earliest opportunity.
Third, we are now in the process of migrating the upgraded DeepNEU platform to the cloud. Since our last publication, we have become a corporate member of the IBM I3 incubator program and are currently working with IBM to migrate our platform to the IBM Cloud. This has been successfully completed and will permit more rapid simulation development, therapeutic target identification, and drug repurposing for COVID‐19 and other future viral outbreaks. Finally, we continue to evolve our current technology based on a Wise Learning (WL) approach described by Groumpos. 41
5. CONCLUSION/SIGNIFICANCE
The current results from our continued research and development of the DeepNEU platform have confirmed and extended our previous work. 4 , 5 DeepNEU v5.0 accurately derived aiLUNG cells from aiPSC simulations. The aiLUNG simulations could be exposed to simulated SARS‐CoV‐2 virus and reproduce the genotypic and phenotypic profile associated with the infection. We also demonstrated that the aiLUNG‐COVID‐19 simulations can be used to rapidly repurpose novel known drug combinations with anti‐COVID‐19 therapeutic potential for animal and human trial validation. The rational process described in this article has as a prerequisite the existence of a validated genome for the viral genome(s) in question.
While DeepNEU requires continued development and validation, it is very likely that the most important application of this technology will be in improving disease preparedness for future outbreaks.
Determined to improve our future preparedness, WHO has published a list of serious potential outbreaks that could be in our future; the list includes (a) COVID‐19, (b) Crimean‐Congo hemorrhagic fever, (c) Ebola virus disease and Marburg virus disease, (d) Lassa fever, (e) Middle East respiratory syndrome coronavirus (MERS‐CoV) and Severe Acute Respiratory Syndrome (SARS), (f) Nipah, (g) henipaviral diseases, (h) Rift Valley fever, (i) Zika, and (j) Disease X, a previously unknown virus. 39 , 42 The list was provided to specifically drive research and development. All members of this list are transmissible to humans and have no effective therapies, but fortunately most do have identified genomes. 42 , 43 NIPAH viral infection was selected as our next project because of its high lethality to humans. It was discovered in Asia in 1999 and is associated with a ~70% death rate in those infected. 42 , 44 The NIPAH viral genome has been validated and there are currently no effective treatments. 42 , 43 , 44 Small outbreaks continue to occur in Southeast Asia and elsewhere. 30 Importantly, the WHO has gone so far as to identify NIPAH as a prototype for a future pandemic. 42 , 43 It is our intention now to focus our efforts on creating disease simulations and identifying potentially effective therapies for all 10 of the WHO listed diseases. 39 , 42 As of this writing we have an advanced COVID‐19 project and have selected NIPAH infection as our next project for which we now have working disease simulations.
CONFLICT OF INTEREST
W.D. declared leadership position, patent holder, advisory role, and ownership interest with www.123genetix.com. The other author declared no potential conflict of interest.
AUTHOR CONTRIBUTIONS
S.E.: conceptualization, experimental work analysis, manuscript writing, figures preparation. W.D.: conceptualization, experimental work analysis, manuscript writing, figures preparation, performed all computational simulations and COVID‐19 disease modeling.
Supporting information
Fig. S1 DeepNEU simulation of aiLUNG and differentiated aiLUNG‐WT cells. (A) Gene expression in wild type aiPSC simulation. (B) Gene expression of aiLUNG cells differentiated from aiPSC. Data are representative of three independent simulation experiments; error bars indicate ± SEM.
Figure S2: DeepNEU simulation of differentiated aiLUNG‐WT Phenotypic features.
Phenotypic factor levels of aiLUNG cells differentiated from aiPSC. Data are representative of three independent simulation experiments; error bars indicate ± SEM.
Esmail S, Danter W. Viral pandemic preparedness: A pluripotent stem cell‐based machine‐learning platform for simulating SARS‐CoV‐2 infection to enable drug discovery and repurposing. STEM CELLS Transl Med. 2021;10:239–250. 10.1002/sctm.20-0181
DATA AVAILABILITY STATEMENT
All the data generated or analyzed during this study are included in this published article.
REFERENCES
- 1. Callaway E, Cyranoski D, Mallapaty S. The coronavirus pandemic in five powerful charts. Nature. 2020;579:482‐483. [DOI] [PubMed] [Google Scholar]
- 2. Majolo F, Marinowic DR, Moura AÁ, Machado DC, da Costa JC. Use of induced pluripotent stem cells (iPSCs) and cerebral organoids in modeling the congenital infection and neuropathogenesis induced by Zika virus. J Med Virol. 2019;91:525‐532. [DOI] [PubMed] [Google Scholar]
- 3. Trevisan M, Sinigaglia A, Desole G, et al. Modeling viral infectious diseases and development of antiviral therapies using human induced pluripotent stem cell‐derived systems. Viruses. 2015;7:3835‐3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Esmail S, Danter WR. DeepNEU: artificially induced stem cell (aiPSC) and differentiated skeletal muscle cell (aiSkMC) simulations of infantile onset POMPE disease (IOPD) for potential biomarker identification and drug discovery. Front Cell Dev Biol. 2019;7:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Danter WR. DeepNEU: cellular reprogramming comes of age–a machine learning platform with application to rare diseases research. Orphanet J Rare Dis. 2019;14:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thevarajan I, Nguyen TH, Koutsakos M, et al. Breadth of concomitant immune responses prior to patient recovery: a case report of non‐severe COVID‐19. Nat Med. 2020;26(4):453‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghaedi M, Le AV, Hatachi G, et al. Bioengineered lungs generated from human iPSCs‐derived epithelial cells on native extracellular matrix. J Tissue Eng Regen Med. 2018;12:e1623‐e1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu Y, Cheng Y, Wu Y. Understanding SARS‐CoV‐2‐mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. 2020;35(3):266‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. von Brunn A, Teepe C, Simpson JC, et al. Analysis of intraviral protein‐protein interactions of the SARS coronavirus ORFeome. PLoS One. 2007;22(5):e459. 10.1371/journal.pone.0000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu W, Li H. COVID‐19: attacks the 1‐beta chain of hemoglobin and captures the porphyrin to inhibit human heme metabolism. ChemRxiv. 2020. Preprint 10.26434/chemrxiv.11938173.v9 [DOI]
- 11. Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID‐19 and potential vaccines: lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol. 2020;38:1‐9. [DOI] [PubMed] [Google Scholar]
- 12. Wong AP, Shojaie S, Liang Q, et al. Conversion of human and mouse fibroblasts into lung‐like epithelial cells. Sci Rep. 2019;9:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Frank DB, Penkala IJ, Zepp JA, et al. Early lineage specification defines alveolar epithelial ontogeny in the murine lung. Proc Natl Acad Sci USA. 2019;116:4362‐4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J, Lau YF, Lamirande EW, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS‐CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS‐CoV infection. J Virol. 2010;84:1289‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barkauskas CE, Chung M‐I, Fioret B, Gao X, Katsura H, Hogan BL. Lung organoids: current uses and future promise. Development. 2017;144:986‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Y, Peng F, Wang R, et al. The deadly coronaviruses: the 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J Autoimmun. 2020;109:102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Báez‐Santos YM, John SES, Mesecar AD. The SARS‐coronavirus papain‐like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. Proteolytic activation of the SARS‐coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 2013;100:605‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rajsbaum R, García‐Sastre A. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol. 2013;21:421‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mitchell A, Drinnan CT, Jensen T, Finck C. Production of high purity alveolar‐like cells from iPSCs through depletion of uncommitted cells after AFE induction. Differentiation. 2017;96:62‐69. [DOI] [PubMed] [Google Scholar]
- 21. Woo B, Baek K‐H. Interplay of deubiquitinating enzymes and cytokines. Cytokine Growth Factor Rev. 2019;48:40‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tamò L, Hibaoui Y, Kallol S, et al. Generation of an alveolar epithelial type II cell line from induced pluripotent stem cells. Am J Physiol Lung Cell Mol Physiol. 2018;315:L921‐L932. [DOI] [PubMed] [Google Scholar]
- 23. Jacob A, Morley M, Hawkins F, et al. Differentiation of human pluripotent stem cells into functional lung alveolar epithelial cells. Cell Stem Cell. 2017;21:472‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 25. Li C, Smith SM, Peinado N, et al. WNT5a‐ROR signaling is essential for alveologenesis. Cell. 2020;9:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shereen MA, Khan S, Kazmi A, Bashir N, Siddique R. COVID‐19 infection: origin, transmission, and characteristics of human coronaviruses. J Adv Res. 2020;24:91‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan JF‐W, Kok K‐H, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J, Cao R, Xu M, et al. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS‐CoV‐2 infection in vitro. Cell Discov. 2020;6:1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gautret P, Lagier J‐C, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID‐19: results of an open‐label non‐randomized clinical trial. Int J Antimicrob Agents. 2020;56:105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30. Sen N, Kanitkar TR, Roy AA, et al. Predicting and designing therapeutics against the Nipah virus. PLoS Negl Trop Dis. 2019;13:e0007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Young W, D'Souza S, Lemischka I, Schaniel C. Patient‐specific induced pluripotent stem cells as a platform for disease modeling, drug discovery and precision personalized medicine. J Stem Cell Res Ther. 2012;10:2. [Google Scholar]
- 32. Elitt MS, Barbar L, Tesar PJ. Drug screening for human genetic diseases using iPSC models. Hum Mol Genet. 2018;27:R89‐R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han C, Chaineau M, Chen CX‐Q, Beitel LK, Durcan TM. Open science meets stem cells: a new drug discovery approach for neurodegenerative disorders. Front Neurosci. 2018;12:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith M, Smith JC. Repurposing therapeutics for Covid‐19: supercomputer‐based docking to the Sars‐Cov‐2 viral spike protein and viral spike protein‐human ACE2 interface. 2020.
- 35. Kao RY, Tsui WH, Lee TS, et al. Identification of novel small‐molecule inhibitors of severe acute respiratory syndrome‐associated coronavirus by chemical genetics. Chem Biol. 2004;11:1293‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS‐CoV‐2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766‐788. 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zumla A, Chan JF, Azhar EI, Hui DS, Yuen K‐Y. Coronaviruses—drug discovery and therapeutic options. Nat Rev Drug Discov. 2016;15:327‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grein J, Ohmagari N, Shin D, et al. Compassionate use of Remdesivir for patients with severe Covid‐19. N Engl J Med. 2020;382:2327‐2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dodds W. Disease now and potential future pandemics The World's Worst Problems. Switzerland: Springer; 2019:31‐44. [Google Scholar]
- 40. Dhama K, Sharun K, Tiwari R, et al. COVID‐19, an emerging coronavirus infection: advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum Vaccin Immunother. 2020;1‐7. 10.1080/21645515.2020.1735227; https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7103671/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Groumpos PP. Deep learning vs. wise learning: a critical and challenging overview. IFAC‐PapersOnLine. 2016;49:180‐189. https://www.sciencedirect.com/science/article/pii/S240589631632537X. [Google Scholar]
- 42. Mehand MS, Al‐Shorbaji F, Millett P, Murgue B. The WHO R&D blueprint: 2018 review of emerging infectious diseases requiring urgent research and development efforts. Antiviral Res. 2018;159:63‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mehand MS, Millett P, Al‐Shorbaji F, Roth C, Kieny MP, Murgue B. World Health Organization methodology to prioritize emerging infectious diseases in need of research and development. Emerg Infect Dis. 2018;24 10.3201/eid2409.171427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chua KB. Introduction: Nipah virus—discovery and origin Henipavirus. Springer; 2012:1‐9. 10.1007/82_2012_218; https://link.springer.com/content/pdf/10.1007%2F978-3-642-29819-6.pdf. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 DeepNEU simulation of aiLUNG and differentiated aiLUNG‐WT cells. (A) Gene expression in wild type aiPSC simulation. (B) Gene expression of aiLUNG cells differentiated from aiPSC. Data are representative of three independent simulation experiments; error bars indicate ± SEM.
Figure S2: DeepNEU simulation of differentiated aiLUNG‐WT Phenotypic features.
Phenotypic factor levels of aiLUNG cells differentiated from aiPSC. Data are representative of three independent simulation experiments; error bars indicate ± SEM.
Data Availability Statement
All the data generated or analyzed during this study are included in this published article.