

Abstract
SARS‐CoV‐2 infection is causing a pandemic disease that is reflected in challenging public health problems worldwide. Human leukocyte antigen (HLA)‐based epitope prediction and its association with disease outcomes provide an important base for treatment design. A bioinformatic prediction of T cell epitopes and their restricted HLA Class I and II alleles was performed to obtain immunogenic epitopes and HLA alleles from the spike protein of the severe acute respiratory syndrome coronavirus 2 virus. Also, a correlation with the predicted fatality rate of hospitalized patients in 28 states of Mexico was done. Here, we describe a set of 10 highly immunogenic epitopes, together with different HLA alleles that can efficiently present these epitopes to T cells. Most of these epitopes are located within the S1 subunit of the spike protein, suggesting that this area is highly immunogenic. A statistical negative correlation was found between the frequency of HLA‐DRB1*01 and the fatality rate in hospitalized patients in Mexico.
Highlights
First HLA association study for COVID‐19 in Mexico
An epitope prediction for HLA Class I and II provided a list of highly immunogenic epitopes from the S protein of SARS‐CoV2 with potential use for vaccine development.
A multi‐level approach revealed a correlation of HLA‐DRB1*01 frequency with fatality in Mexican hospitalized patients at ecological level.
1. INTRODUCTION
The coronavirus disease (COVID‐19) was declared as a pandemic by the World Health Organization (WHO) in March of 2020. 1 It is estimated that by June the 10th of 2020 there were over 6.19 million confirmed cases and 370,000 deaths worldwide.
COVID‐19 is a disease generated by the novel severe acute respiratory syndrome‐coronavirus‐2 (SARS‐CoV‐2), with a wide range of clinical manifestations, like fever (88.7%), cough (67.8%), fatigue (38.1%), and acute respiratory distress syndrome in severe cases. 2 Interestingly, the molecular and clinical manifestations of the disease vary between asymptomatic, mild‐symptomatic, and severe patients, requiring hospitalization in some cases to prevent fatal outcomes. 3
Currently, the SARS‐CoV‐2 genome has been characterized as a new betacoronavius, which shares around 87% of genomic identity with bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21 viruses. 4 A recent analysis by Zhou et al. 5 reported that there is a 96.2% identity with BatCoVRaTG13 and a 79.5% identity with SARS‐CoV. 2 The genomic characterization of the virus not only provides information about its taxonomy and probable origin but also offers opportunities to perform deeper analysis using bioinformatics tools.
The angiotensin‐converting enzyme‐2 (ACE‐2) receptor and the transmembrane serine protease 2 are essential components of the human host for the virus entry into the upper respiratory epithelial cells. The virus recognizes ACE‐2 through the viral spike glycoprotein (S), and this event leads to the virus–cell membrane fusion. 6 The S glycoprotein is found as a homotrimer of three identical monomers, each one of which is divided into two subunits: S1 and S2. The first subunit folds in four domains: A, B, C, and D. The B domain possesses a receptor‐binding domain that recognizes ACE2, hence it is important for viral entry. 7 The S2 subunit sequence has two tandem domains, namely, HR1 and HR2, that play an essential role in the viral fusion to the membrane. 8 Furthermore, analysis of the spike protein showed that it is conserved among SARS‐CoV and SARS‐CoV‐2 with 76.3% of identity and 87.3% of similarity. 9
Several studies focused on viral diseases have shown that clinical severity is closely associated with some individual factors, such as genetic background and immune response. The human leukocyte antigen (HLA) is responsible for the antigen presentation to T cells and, therefore, a key component for adaptive immune response initiation. The HLA genes are the most polymorphic genes in the human genome, and these polymorphisms influence the ability to present different sets of epitopes to T cells. Some HLA molecules are more efficient than others presenting certain antigens, which may lead to a better induction of immune responses. This fact has already been proven for some viral diseases like A H1N1 influenza 10 and HIV. 11
It has been previously reported an association between SARS‐CoV infection and HLA‐B*07:03, 12 HLA‐Cw*08:01, 13 HLA‐B*46:01, and HLA‐B*54:01. Specifically, it has been reported that the individuals who are HLA‐B*46:01 positive have a higher risk of severe infection, 14 whereas the frequency of HLA‐DRB2*03:01 is lower among patients with COVID‐19. 12
Mexico is one of the top 10 countries with higher mortality, and its number of cases and deaths keeps increasing significantly. 15 Some of the most common haplotypes reported in Mexico′s less affected states are HLA A*02‐B*35‐DRB1*08‐DBQ1*04, A*68‐B*39‐DRB1*04‐DBQ1*03:02, and A*02‐B*15‐DRB1*08‐DBQ1*04, according to the Allele Frequency Net Database website (www.allelefrequencies.netwww.allelefrequencies.net). 16
On the other hand, up to now, Mexico City is the region with the highest number of reported cases. The studies regarding allele frequency in this city have reported that its haplotype is largely composed of Native American haplotypes, specifically 63.85 ± 1.55% American, 28.53 ± 3.13% European, and a less apparent 7.61 ± 1.96% African. 17 Individually, some studies have reported that the most frequent alleles in Mexican population are HLA‐A*02, ‐A*24, ‐A*68, ‐B*35, ‐B*39, ‐B*51, ‐DRB1*04, ‐DRB1*08, ‐DRB1*07, ‐DQB1*0302, ‐DQB1*0301, and ‐DQB1*0201. 18 Nonetheless, there are no studies related to the HLA association with the susceptibility or the resistance against COVID‐19 in the Mexican population. The understanding of the relationship between viral infection, HLA, and disease susceptibility is important to drive towards vaccine development and molecular epidemiology research that can contribute to novel therapies.
So far, the control of the COVID‐19 pandemic remains a challenge, resulting in thousands of new cases and deaths reported daily. It is necessary to find prophylaxis and specific treatments to contain this uncontrolled infection and to reduce the global morbidity and mortality. The generation of a vaccine that targets this virus remains as the primary solution, 19 however, the lack of knowledge regarding the immune response, such as the HLA‐virus interactions, makes it a challenging task.
In addition, the genetic variations among different populations and their possible link with SARS‐CoV‐2 viral responses remain unknown. In this report, we analyze which epitopes of the SARS‐CoV‐2 spike protein are highly immunogenic and able to be presented by HLA Class I and II in different populations using bioinformatic tools. We also demonstrate an ecological correlation between HLA allele frequency and the predicted fatality rate in hospitalized patients of 28 Mexican states.
2. METHODS
2.1. Study design
A bioinformatic epitope prediction of the S glycoprotein was performed. This gave information about the most immunogenic peptide‐HLA matches and the HLA alleles that are more likely to present these epitopes efficiently. Also, an ecological study was made to look for correlations between the HLA allele frequencies and the predicted fatality rate of hospitalized patients with COVID‐19 to May 29th, 2020.
2.2. Bioinformatic epitope prediction
Bioinformatic analyses were performed to predict HLA Class I and II epitopes using the sequence of the SARS‐CoV‐2 spike protein. The sequence for the SARS‐CoV‐2 S glycoprotein was obtained from the GenBank with the accession number QHR63290.2 in FASTA format. This sequence was then submitted to the TepiTool server from the IEDB Analysis Resource database (http://tools.iedb.org/tepitool/). 20 The epitope prediction was performed for the 27 most frequent HLA‐A and ‐B alleles that cover for most populations (Table S1). 21 Once the total epitope list was obtained, it was submitted to the T cell Class I pMHC immunogenicity predictor server (http://tools.iedb.org/immunogenicity/) to get the immunogenicity score, which is predicted according to the amino acid residues of the peptide. 22
The peptide‐HLA pairs with a positive immunogenicity score and a predicted IC50 level lower than the established cut‐off (Table S2) from the complete list were chosen, 23 considering that the lower the IC50 value, the higher the binding affinity. The 10 more immunogenic peptide‐MHC combinations from this list were selected.
The epitopes for HLA Class II molecules were also predicted using the same sequence as before and submitting it to the IEDB MHC Class II epitope prediction tool (http://tools.iedb.org/mhcii/) using the IEDB recommended 2.2 algorithm and the most common HLA‐DP, DQ, and DR alleles (Table S1). 24 The predicted epitopes with an SMM‐predicted IC50 value higher than 50 were excluded and the sequences were ordered by the percentile rank. 25 The MHC‐II prediction tools use a core of nine amino acids to predict the best peptide binding affinity, even when the Class II molecules bind peptides with 15 amino acid length, so the 10 SMM cores with the minor percentile rank—what means the highest affinity binding—were selected.
2.3. Structural modeling
To provide a graphical representation of the location of the epitopes, we used the structural model the full‐length SARS‐CoV‐2 spike glycoprotein (ID:6VSB_1_1_1). The full‐length SARS‐CoV‐2 structural model is available at CHARMM‐GUI13 COVID19 Archive. 26
The three‐dimensional (3D) structure was obtained and analyzed using PyMOL® software (Schrödinger LLC. Molecular Graphics System [PyMOL] Version 1.80 LLC, New York, NY, 2015). The basic local alignment search tool online (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to assess the position of the predicted peptides in the glycoprotein and the protein sequence was adjusted manually using the PyMOL tools.
2.4. Analysis of HLA alleles frequency and fatality rates
We selected 28 states of Mexico considering the homogeneity in epidemiological reports and registered the allele frequency of the main capital city of each state. All the states were included except for Mexico State, Baja California Sur, and Tamaulipas because no information was found.
We used the Allele Frequency Net Database (http://www.allelefrequencies.net/default.asp) and searched for populations in North America′s geographical region and used Mexico′s (132) database. The total state samples reported on the databases was of 5840.
For HLA Class I, the subgroup alleles was not reported for 26 of the states. However, Mexico City Mestizo and Veracruz Xalapa did contain subgroup data.
In the selection of the Class II molecules, the HLA‐DPA1*03:01, DPB1*04:02, HLA‐DPA1*01:03, DPB1*02:01, HLA‐DPA1*02:01, DPB1*01:01, and HLA‐DQA1*05:0 alleles were not found in the database of any population. All the frequency data is summarized in Table S3 organized per city.
2.5. Fatality rate
We used national public data reporting all individuals with a result for SARS‐CoV‐2 in Mexico to July 8th, 2020 (SARS‐CoV‐2 Mexico database). This database is compiled by the Ministry of Health (available at https://www.gob.mx/salud/documentos/datos-abiertos-152127). We considered the following information: age, sex, state of birth, date of birth (if applicable), and type of healthcare facility where the patients were assisted—IMSS, ISSSTE, SSA, private hospital, and others. There is also information about comorbidities—diabetes, hypertension, obesity, asthma, immunosuppression, chronic kidney disease, and cardiovascular disease, as well as smoking status and hospitalization status. The registration options were yes, no, not known, or not specified. Finally, it is specified whether the patients were attended in sentinel units. The primary care sentinel institutions test for SARS‐CoV‐2 to one of every 10 patients with an acute respiratory infection, while the nonsentinel institutions perform tests according to physician criteria. The 100% of patients with severe acute respiratory infection who require hospitalization are tested in both institutions, sentinel, and nonsentinel. 27
The total database contained 684,804 records. We only included records of Phase 3 (614,370). We excluded 60,520 patients who were admitted for hospitalization after July 1st to allow the presentation of the outcome “death,” since the median from hospitalization to death was 7 days. Of the 553,850 remaining records, 307,421 had a negative or pending result, 173,724 were not hospitalized, 123 did not have information of the state of birth, and 1026 were indigenous people. We excluded 448 pregnant women because the immune response is expected to be different. 28 Finally, nine records were eliminated because the date of death was before the admission date. Hence, our final sample was 71,099 records.
2.6. Statistical analysis
To create a predictive model of the hospitalized fatality rate—number of deaths caused by COVID‐19, we performed a stepwise approach with all the variables reported in the SARS‐CoV‐2 Mexico database in a Poisson model. All the variables that were significantly associated with death were kept in the model: age, sex, diabetes, hypertension, obesity, chronic kidney disease, type of healthcare, being a sentinel unit or not, and admission date. We explored if a multilevel model, using state of birth as a second level, would be a better fit for the data, but the LR test was not significant (p = 1). Hence, the state of birth variable was included in the Poisson model. Afterward, the predictive risk of death in each state was calculated. Then, a factorial analysis was performed with the 21 HLA types to determine groups that explained the variance between them and selected the representative HLA allele of each factor as the one with the maximum correlation within the factor. We selected seven factors that explained 85.2% of the variance and selected the HLA with the highest correlation within each factor as follows: Factor 1 HLA‐A*68:01, Factor 2 HLA‐A*11:01, Factor 3 HLA‐DRB1*07:01, Factor 4 HLA‐A*01:01, Factor 5 HLA‐B*57:01, Factor 6 HLA‐DRB1*01:01, and Factor 7 HLA‐B*58:01 (Table S4). Afterward, a Spearman's rank correlation was performed between the seven HLA allele frequencies and the risk of death at state level. A p < .05 was considered statistically significant. The analyses were performed in Stata v14 and figure were created using Graphpad Prism version 6.0®.
3. RESULTS
To assess the best Spike protein epitope‐HLA Class I matches, its sequence was analyzed looking for epitope predictions in the most frequent HLA‐A and HLA‐B alleles. The 10 most immunogenic peptides with a higher affinity binding to its restricted HLA are shown in Table 1.
Table 1.
HLA Class I epitope prediction
| Peptide/protein residues (predicted immunogenicity score) | HLA Restriction | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | GTHWFVTQR/1096‐1104 | HLA‐A*31:01 | HLA‐A*68:01 | HLA‐A*11:01 | HLA‐A*03:01 | |||
| (0.3513) | ||||||||
| Predicted IC50 | 9.5 | 14.5 | 29.6 | 379.3 | ||||
| 2 | RSFIEDLLF/813‐821 | HLA‐B*58:01 | HLA‐B*57:01 | HLA‐A*32:01 | ||||
| (0.2744) | ||||||||
| Predicted IC50 | 7.5 | 24.6 | 62.7 | |||||
| 3 | FIAGLIAIV/1218‐1224 | HLA‐A*02:03 | HLA‐A*02:06 | HLA‐A*02:01 | HLA‐A*68:02 | |||
| (0.272) | ||||||||
| Predicted IC50 | 3.2 | 6.3 | 8.5 | 13.7 | ||||
| 4 | EVFNATRFA/338‐346 | HLA‐A*68:02 | ||||||
| (0.2182) | ||||||||
| Predicted IC50 | 12 | |||||||
| 5 | QYIKWPWYI/1205‐1213 | HLA‐A*23:01 | HLA‐A*24:02 | |||||
| (0.2162) | ||||||||
| Predicted IC50 | 4.3 | 6.9 | ||||||
| 6 | NTQEVFAQV/775‐783 | HLA‐A*68:02 | ||||||
| (0.1788) | ||||||||
| Predicted IC50 | 5.2 | |||||||
| 7 | WTAGAAAYY/256‐264 | HLA‐A*26:01 | HLA‐A*68:01 | HLA‐A*01:01 | HLA‐A*30:02 | |||
| (0.1525) | ||||||||
| Predicted IC50 | 9.9 | 27.4 | 31.1 | 36.4 | ||||
| 8 | SANNCTFEY/160‐168 | HLA‐B*35:01 | ||||||
| (0.1327) | ||||||||
| Predicted IC50 | 14.1 | |||||||
| 9 | YLQPRTFLL/267‐275 | HLA‐A*02:01 | HLA‐A*02:03 | HLA‐A*02:06 | HLA‐B*08:01 | HLA‐A*23:01 | HLA‐A*24:02 | HLA‐A*32:01 |
| (0.1305) | ||||||||
| Predicted IC50 | 4.1 | 7.8 | 9.1 | 23.9 | 125.3 | 201.3 | 202.7 | |
| 10 | VVFLHVTYV/1057‐1065 | HLA‐A*02:03 | HLA‐A*02:06 | HLA‐A*02:01 | HLA‐A*68:02 | |||
| (0.1278) | ||||||||
| Predicted IC50 | 9.3 | 11.9 | 21.2 | 24.5 | ||||
Abbreviation: HLA, human leukocyte antigen.
Although the most immunogenic peptide from this list is GTHWFVTQR, the match with the highest affinity was between the peptide FIAGLIAIV and HLA‐A*02:03. Of note, here we analyzed the most frequent Class I A and B alleles, so this analysis reveals epitopes that can be used for vaccine development and the HLA alleles that best present epitopes of this particular protein.
The best epitopes and HLA Class II alleles were also predicted, as shown in Table 2. The prediction tool for HLA Class II uses a core of nine amino acids to predict the binding efficiency of peptides to the pocket of the molecules, even if this core is in the middle of different peptides of 15 amino acids. Interestingly, among this whole set of peptides, only seven HLA molecules resulted with a high binding affinity: HLA‐DPA1*01:03/DPB1*02:01, HLA‐DPA1*02:01/DPB1*01:01, HLA‐DPA1*03:01/DPB1*04:02, HLA‐DQA1*05:01/DQB1*03:01, HLA‐DRB1*01:01, HLA‐DRB1*07:01, and HLA‐DRB1*09:01.
Table 2.
HLA Class II epitope prediction
| SMM core | Peptides | HLA‐restriction | Percentile rank | |
|---|---|---|---|---|
| a | FELLHAPAT | LSFELLHAPATVCGP | HLA‐DRB1*01:01 | 0.03 |
| VLSFELLHAPATVCG | HLA‐DRB1*01:01 | 0.03 | ||
| VVLSFELLHAPATVC | HLA‐DRB1*01:01 | 0.03 | ||
| SFELLHAPATVCGPK | HLA‐DRB1*01:01 | 0.09 | ||
| VVVLSFELLHAPATV | HLA‐DRB1*01:01 | 0.09 | ||
| FELLHAPATVCGPKK | HLA‐DRB1*01:01 | 0.71 | ||
| b | VVVLSFELL | QPYRVVVLSFELLHA | HLA‐DPA1*03:01/DPB1*04:02 | 0.24 |
| PYRVVVLSFELLHAP | HLA‐DPA1*03:01/DPB1*04:02 | 0.25 | ||
| YRVVVLSFELLHAPA | HLA‐DPA1*03:01/DPB1*04:02 | 0.25 | ||
| PYRVVVLSFELLHAP | HLA‐DPA1*02:01/DPB1*01:01 | 0.3 | ||
| QPYRVVVLSFELLHA | HLA‐DPA1*02:01/DPB1*01:01 | 0.3 | ||
| YQPYRVVVLSFELLH | HLA‐DPA1*02:01/DPB1*01:01 | 0.3 | ||
| YRVVVLSFELLHAPA | HLA‐DPA1*02:01/DPB1*01:01 | 0.3 | ||
| PYRVVVLSFELLHAP | HLA‐DPA1*01:03/DPB1*02:01 | 0.36 | ||
| QPYRVVVLSFELLHA | HLA‐DPA1*01:03/DPB1*02:01 | 0.36 | ||
| YQPYRVVVLSFELLH | HLA‐DPA1*01:03/DPB1*02:01 | 0.36 | ||
| YRVVVLSFELLHAPA | HLA‐DPA1*01:03/DPB1*02:01 | 0.36 | ||
| RVVVLSFELLHAPAT | HLA‐DPA1*02:01/DPB1*01:01 | 0.63 | ||
| VVVLSFELLHAPATV | HLA‐DPA1*02:01/DPB1*01:01 | 0.68 | ||
| RVVVLSFELLHAPAT | HLA‐DPA1*03:01/DPB1*04:02 | 0.85 | ||
| YQPYRVVVLSFELLH | HLA‐DPA1*03:01/DPB1*04:02 | 2.2 | ||
| c | FLVLLPLVS | FVFLVLLPLVSSQCV | HLA‐DRB1*01:01 | 0.24 |
| MFVFLVLLPLVSSQC | HLA‐DRB1*01:01 | 0.24 | ||
| VFLVLLPLVSSQCVN | HLA‐DRB1*01:01 | 1.3 | ||
| FLVLLPLVSSQCVNL | HLA‐DRB1*01:01 | 1.8 | ||
| d | VLSFELLHA | RVVVLSFELLHAPAT | HLA‐DRB1*01:01 | 0.24 |
| e | GYQPYRVVV | GYQPYRVVVLSFELL | HLA‐DPA1*02:01/DPB1*01:01 | 0.3 |
| f | FGAGAALQI | SGWTFGAGAALQIPF | HLA‐DRB1*09:01 | 0.33 |
| TSGWTFGAGAALQIP | HLA‐DRB1*09:01 | 0.34 | ||
| GWTFGAGAALQIPFA | HLA‐DRB1*09:01 | 0.35 | ||
| WTFGAGAALQIPFAM | HLA‐DRB1*09:01 | 0.67 | ||
| WTFGAGAALQIPFAM | HLA‐DQA1*05:01/DQB1*03:01 | 1.6 | ||
| g | FVFLVLLPL | MFVFLVLLPLVSSQC | HLA‐DPA1*03:01/DPB1*04:02 | 0.34 |
| FVFLVLLPLVSSQCV | HLA‐DPA1*03:01/DPB1*04:02 | 0.36 | ||
| MFVFLVLLPLVSSQC | HLA‐DPA1*01:03/DPB1*02:01 | 5.2 | ||
| h | RVVVLSFEL | GYQPYRVVVLSFELL | HLA‐DPA1*01:03/DPB1*02:01 | 0.36 |
| GYQPYRVVVLSFELL | HLA‐DPA1*03:01/DPB1*04:02 | 6.2 | ||
| i | FTISVTTEI | AIPTNFTISVTTEIL | HLA‐DRB1*07:01 | 0.4 |
| PTNFTISVTTEILPV | HLA‐DRB1*07:01 | 0.51 | ||
| IPTNFTISVTTEILP | HLA‐DRB1*07:01 | 0.52 | ||
| TNFTISVTTEILPVS | HLA‐DRB1*07:01 | 0.52 | ||
| NFTISVTTEILPVSM | HLA‐DRB1*07:01 | 2.5 | ||
| FTISVTTEILPVSMT | HLA‐DRB1*07:01 | 2.6 | ||
| j | TNFTISVTT | IAIPTNFTISVTTEI | HLA‐DRB1*07:01 | 0.47 |
Abbreviation: HLA, human leukocyte antigen.
To track down and illustrate the specific location of the peptides in the SARS‐CoV‐2 spike glycoprotein, the corresponding 3D model was obtained. In this model, the different predicted epitopes (Tables 1 and 2) were searched in the protein structure considering its subunits and domains (Figure 1). Notably, HLA Class I peptides WTAGAAAYY, SANNCTFEY, and YLQPRTFLL—7, 8, and 9—are located in the A domain, which is highly conserved among other coronavirus species, 8 suggesting that these could also be epitopes for other coronaviruses. On the other hand, it was found that the Class II epitopes FELLHAPAT, VVVLSFELL, FLVLLPLVS, VLSFELLHA, and FTISVTTEI—a, b, c, d, and h—and the HLA Class I EVFNATRFA—4 are preferentially found in the B domain.
Figure 1.
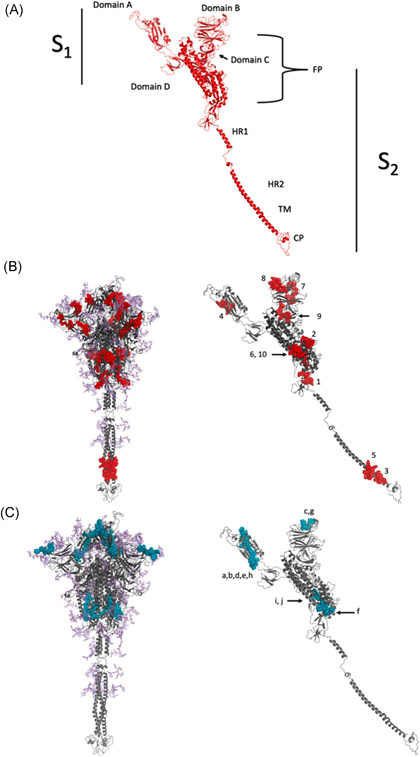
Localization analysis of immunogenic peptides of SARS‐CoV‐2 spike glycoprotein by three‐dimensional modeling. (A) Structure of the SARS‐CoV‐2 spike glycoprotein with S1–S2 subunits. The S1 domains consist of A, B, C, and D. The S2 subunit consists of the fusion peptides and domains HR1 and HR2. (B) The predicted epitopes for HLA Class I are shown in red (C) and the suggested peptides for HLA Class II in blue. The peptides are marked individually, listed from 1 to 10 for Class I and a–j for Class II, corresponding to the immunogenicity Tables 1 and 2. HLA, human leukocyte antigen; SARS‐CoV‐2, severe acute respiratory syndrome‐coronavirus‐2
3.1. HLA allele analysis and correlation with a predicted fatality rate in hospitalized patients
After factorial analysis, we found a significant negative correlation between the frequency of the HLA‐DRB1*01:01 allele and the predicted fatality rate in hospitalized patients (R = −0.44, p = .02; Figure 2). No other significant correlations were observed (Table 3).
Figure 2.
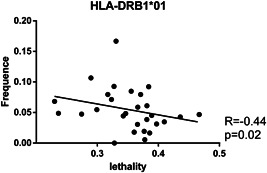
Spearman's correlation of HLA‐DRB1*01:01 frequency and fatality rate. The correlation is shown as a dot plot graph with the regression tendency line. The frequency of this HLA allele in Mexico was obtained and a correlation was performed with the predicted risk of death associated with SARS‐CoV‐2 infection in hospitalized patients. According to the bioinformatic prediction, the HLA‐DRB1*01:01 molecule can efficiently present eleven of the S protein predicted epitopes (LSFELLHAPATVCGP, VLSFELLHAPATVCG, VVLSFELLHAPATVC, SFELLHAPATVCGPK, VVVLSFELLHAPATV, FELLHAPATVCGPKK, FVFLVLLPLVSSQCV, MFVFLVLLPLVSSQC, VFLVLLPLVSSQCVN, FLVLLPLVSSQCVNL, and RVVVLSFELLHAPAT; Table 2). HLA, human leukocyte antigen; SARS‐CoV‐2, severe acute respiratory syndrome‐coronavirus‐2
Table 3.
Correlation between the representative HLA alleles (7) resulted from factorial analysis and fatality rate in Mexico states (n = 26)
| R | p | |
|---|---|---|
| F1: HLA‐A*68 | .15 | .45 |
| F2: HLA‐A*11:01 | −.3 | .12 |
| F3: HLA‐DRB1*07:01 | .11 | .6 |
| F4: HLA‐A*02:01 | .05 | .79 |
| F5: HLA‐B*57:01 | .35 | .07 |
| F6: HLA‐DRB1*01:01 | −.44 | .02 |
| F7: HLA‐B*58:01 | −.14 | .49 |
Note: Spearman's rank correlation
4. DISCUSSION
Determining HLA interactions with epitopes for optimal presentation is crucial for understanding the immunological response to SARS‐CoV‐2. Here, we present a group of epitopes of the spike protein that can be efficiently presented to CD8 and CD4 T cells and are probably related to the virus′s immune‐mediated elimination. These peptides can be either used for the peptide‐based design of vaccines or in further analysis of the immunogenicity and structure of this relevant protein.
COVID‐19 vaccine development includes five clinical‐Phase I vaccine candidates, 11 preclinical‐vaccine candidates, and 26 research‐stage vaccine candidates. 29 , 30 Recently, the full proteome of the SARS‐CoV‐2 has been characterized through in silico analysis to show the prediction of the most immunogenic epitopes from each viral protein for 438 MHC alleles—either Class I or Class II. 31 , 32 This knowledge has been considered in the design of two of the Phase I‐vaccine candidates, which are LV‐SMENP‐DC and pathogen‐specific aAPC. Nevertheless, most of the other vaccine candidates have been designed based on the spike protein of the SARS‐CoV‐2 due to better immunogenic and protective potential. The S protein is the main target for COVID‐19 vaccine development. Even though the S gene sequences of SARS‐CoV‐2 have a 93.2% nucleotide sequence identity to the bat coronavirus RaTG13 and less than a 75% nucleotide sequence identity with the SARS‐CoV, three out of the five Phase I‐vaccine candidates—which are mRNA‐1273, Ad5‐nCoV, and INO‐4800s, have been designed using this protein as the main target. 29 , 30
Remarkably, our structural analysis of the protein shows a higher abundance of epitopes in the A and B domains of the S1 subunit of the virus, indicating that, in the case of this part of the protein being processed by the host cells, it could represent a highly immunogenic region. In this analysis, we did not look for B cell epitopes in the structure of the protein. We cannot confirm that the specific target of the presented epitopes could interfere with its viral function, as would be the case of neutralizing antibodies.
HLA peptide groove sequence determines which epitopes from an antigen are presented to the immune system to elicit an effective response. The high rate of polymorphisms in the HLA locus can indicate a different ability to respond to certain antigens by different individuals. Furthermore, some HLA alleles can be more efficient in presenting certain antigens, thus also in protecting from certain infections. 11 Our analysis from the most representative HLA alleles revealed those that present more effectively the spike protein antigens of SARS‐CoV‐2, hence, one can hypothesize that their presence in an individual might confer an enhanced ability to defend against the virus.
To assess this, we analyzed the frequency of these alleles and their relation to the disease dynamics in different states of Mexico. Although it would be interesting to extrapolate these results to several countries with different epidemiological behaviors of the disease, epidemiological reports would be highly heterogeneous and data at an individual level associated with risk of death would be needed to adjust the fatality rate.
While there is a myriad of factors related to the lethality of the disease, little is known about the involvement of the immune system in this regard. It has been proposed that many patients develop an exaggerated immune response against the infection, accompanied by a cytokine releasing syndrome 33 or autoinflammatory syndromes. 34 Also, Grifoni et al. 35 showed that T helper cell responses (initiated by HLA Class II molecules) seem to be protective against the infection through a strong correlation with the production of virus‐specific antibodies, and also that they are highly represented by S‐protein specific clones.
A significant negative correlation was found between the frequency of the Class II HLA‐DRB1*01 allele and the fatality rate in hospitalized patients from the states that were included. Remarkably, this correlation was weak, suggesting that other important factors apart from HLA could be involved in the protection. Therefore, it is plausible that the correlation we found based on bioinformatic predictions, would mean that these alleles could show some degree of protection against lethal outcomes of the disease. Although, the frequency of this specific allele is low in the different states, so the overall effect in fatality rates might be small. Thus, further experimental studies are needed to reinforce these outcomes.
HLA‐DRB1*01 alleles have been previously associated with multiple sclerosis resistance. 36 Nevertheless, its role in the susceptibility to viral diseases remains poorly understood. A recent report demonstrated, using molecular docking, that this molecule can interact with the VYQLRARSV epitope from the ORF‐7a protein of the SARS‐CoV‐2 virus. 37 Our results revealed an ecological negative correlation of this allele and that it can present a set of epitopes. Previous reports have identified that this allele can present at least nine epitopes of the M protein and 11 of the N protein (Table S4), revealing that this molecule can be highly relevant for SARS‐CoV‐2 immunity.
A remarkable characteristic of this study is that we narrowed it to the S protein, which has been the most used target for vaccine development. Considering that we did not include other viral proteins, we made an exhaustive bibliographic review that allowed us to compile a total of 77T cell epitopes for the M protein and 87 for the N protein that were already evaluated experimentally and included an analysis of the HLA alleles used for its prediction. As shown in Table S4, the HLA‐A*26:01, HLA‐A*03:01, HLA‐A*11:01, HLA‐A*31:01, HLA‐A*32:01, HLA‐A*68:01, HLA‐B*57:01, HLA‐B*58:01, HLA‐A*01:01, HLA‐A*02:01, HLA‐A*02:03, HLA‐A*02:06, HLA‐A*68:02, HLA‐A*23:01, HLA‐A*24:02, HLA‐B*35:01, HLA‐A*30:02, and HLA‐DRB1*01:01 alleles—which resulted in our epitope prediction—can also be effective presenting peptides of other proteins like M and N.
Other studies have reported an association between HLA I alleles and several SARS‐CoV outcomes within specific populations: HLA‐B*07:03 with infection rate in China 12 ; or HLA‐B*46:01 13 , 14 with severity and HLA‐Cw*08 with infection in Taiwan. 38 Besides, HLA‐DR*03*01 has been associated with a lower frequency of SARS‐CoV infection. 38
Several limitations need to be acknowledged. First, the association of the frequency of the HLA allele and fatality rate is ecological and cannot be applied at an individual level. Other studies need to be conducted to explore if the association persists at an individual level in hospitalized patients. Second, the predictive model of the fatality case was conducted using only data from hospitalized patients. Given that different comorbidities can lead to hospitalization, we cannot exclude the possibility of collider bias. That is, the conditioning of analysis on hospitalization can produce biased associations between the risk factors and the outcome “fatality rate” in this case. Third, we do not rule out the possibility of misclassification since the information on comorbidities is self‐reported. However, our aim was not to make an inference of the fatality rate at an individual‐level factor, but rather to create a predictive model that was as less biased as possible. Fourth, there may be other state characteristics that are associated with death, such as the health infrastructure or the number of available specialized medical staff that are not considered in the model. Finally, the HLA allele frequencies do not include minorities like the indigenous population, who might have different HLA alleles frequencies.
CONFLICT OF INTERESTS
All authors declare not to have any conflict of interests.
AUTHOR CONTRIBUTIONS
José P. Romero‐López conceived the idea and directed the project. Martha Carnalla‐Cortés and Nancy López‐Olmedo performed all the epidemiological analysis and correlations. Diana L. Pacheco‐Olvera performed the molecular modeling and epitope localization. Julia Moreno‐Manjón, Jacqueline Oliva‐Ramírez, and Brian Bernal‐Alferes participated in manuscript writing and revision. Ethel García‐Latorre and María L. Domínguez‐López revised the manuscript. Arturo Reyes‐Sandoval and LuisJiménez‐Zamudio participated in the discussion of the results. The authors did not receive any funding for this study.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
Martha Carnalla‐Cortés, Diana L. Pacheco‐Olvera, Juan M. Ocampo‐Godínez, Julia Moreno‐Manjón, and Brian Bernal‐Alferes receive grants from Consejo Nacional de Ciencia y Tecnología (CONACyT), Ethel García‐Latorre and María L. Domínguez‐López receive scholarships from EDI and COFAA‐IPN. Jacqueline Oliva‐Ramírez, Ethel García‐Latorre, María L. Domínguez‐López, Nancy López‐Olmedo, and Arturo Reyes‐Sandoval receive a scholarship from Sistema Nacional de Investigadores‐CONACyT.
Romero‐López JP, Carnalla‐Cortés M, Pacheco‐Olvera DL, et al. A bioinformatic prediction of antigen presentation from SARS‐CoV‐2 spike protein revealed a theoretical correlation of HLA‐DRB1*01 with COVID‐19 fatality in Mexican population: An ecological approach. J Med Virol. 2021;93:2029–2038. 10.1002/jmv.26561
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Sohrabi C, Alsafi Z, O'Neill N, et al. World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID‐19). Int J Surg. 2020;76:71‐76. 10.1016/j.ijsu.2020.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guan W, Ni Z, Hu Y. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382:1708‐1720. 10.1056/NEJMoa2002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siddiqi HK, Mehra MR. COVID‐19 illness in native and immunosuppressed states: a clinical–therapeutic staging proposal. J Heart Lung Transplant. 2020;39(5):405‐407. 10.1016/j.healun.2020.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;0(0):271‐280. 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tortorici MA, Veesler D. Structural insights into coronavirus entry. Adv Virus Res. 2019;105:93‐116. 10.1016/bs.aivir.2019.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade‐long structural studies of SARS. J Virol. 2020;94:1‐9. 10.1128/jvi.00127-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baruah V, Bose S. Immunoinformatics‐aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019‐nCoV. J Med Virol. 2020;92:495‐500. 10.1002/jmv.25698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Falfán‐Valencia R, Narayanankutty A, Reséndiz‐Hernández JM, et al. An increased frequency in HLA class I alleles and haplotypes suggests genetic susceptibility to influenza A (H1N1) 2009 pandemic: a case‐control study. J Immunol Res. 2018;2018:1‐12. 10.1155/2018/3174868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miura T, Brockman MA, Schneidewind A, et al. HLA‐B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare Gag variants associated with reduced viral replication capacity and strong cytotoxic T‐lymphotye recognition. J Virol. 2009;83(6):2743‐2755. 10.1128/jvi.02265-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ng MHL, Lau KM, Li L, et al. Association of human‐leukocyte‐antigen class I (B*0703) and class II (DRB1*0301) genotypes with susceptibility and resistance to the development of severe acute respiratory syndrome. J Infect Dis. 2004;190(3):515‐518. 10.1086/421523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen YMA, Liang SY, Shih YP, et al. Epidemiological and genetic correlates of severe acute respiratory syndrome coronavirus infection in the hospital with the highest nosocomial infection rate in Taiwan in 2003. J Clin Microbiol. 2006;44(2):359‐365. 10.1128/JCM.44.2.359-365.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin M, Tseng HK, Trejaut JA, et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med Genet. 2003;4:1‐7. 10.1186/1471-2350-4-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Download Today′s Data on the Geographic Distribution of COVID‐19 Cases Worldwide. https://coronavirus.gob.mx/wp-content/uploads/2020/04/Lineamiento_de_vigilancia_epidemiologica_de_enfermedad_respiratoria-_viral.pdf. Accessed June 10, 2020.
- 16. Gonzalez‐Galarza FF, McCabe A, Santos EJM, et al. Allele frequency net database (AFND) 2020 update: gold‐standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2019;48(D1):D783‐D788. 10.1093/nar/gkz1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barquera R, Martínez‐Álvarez JC, Hernández‐Zaragoza DI, et al. Genetic diversity of HLA system in six populations from Mexico City Metropolitan Area, Mexico: Mexico City North, Mexico City South, Mexico City East, Mexico City West, Mexico City Center and rural Mexico City. Hum Immunol. 2019;81:539‐543. 10.1016/j.humimm.2019.07.297 [DOI] [PubMed] [Google Scholar]
- 18. Barquera R, Zúñiga J, Hernández‐Díaz R, et al. HLA class I and class II haplotypes in admixed families from several regions of Mexico. Mol Immunol. 2008;45(4):1171‐1178. 10.1016/j.molimm.2007.07.042 [DOI] [PubMed] [Google Scholar]
- 19. Shi J, Zhang J, Li S, et al. Epitope‐based vaccine target screening against highly pathogenic MERS‐CoV: an In Silico approach applied to emerging infectious diseases. PLoS One. 2015;10(12):1‐16. 10.1371/journal.pone.0144475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paul S, Sidney J, Sette A, Peters B. TepiTool: a pipeline for computational prediction of T cell epitope candidates. Curr Protoc Immunol. 2016;114:1‐24. 10.1002/cpim.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiskopf D, Angelo MA, De Azeredo EL, et al. Comprehensive analysis of dengue virus‐specific responses supports an HLA‐linked protective role for CD8+ T cells. Proc Natl Acad Sci U S A. 2013;110(22):E2046‐E2053. 10.1073/pnas.1305227110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calis JJA, Maybeno M, Greenbaum JA, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol. 2013;9(10):e1003266. 10.1371/journal.pcbi.1003266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paul S, Weiskopf D, Angelo MA, et al. Alleles are associated with peptide‐binding repertoires of different size, affinity, and immunogenicity. J Immunol. 2013;191(12):5831‐5839. 10.4049/jimmunol.1302101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics. 2010;11:568. 10.1186/1471-2105-11-568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM‐align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:8. 10.1186/1471-2105-8-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. CHARMM GUI . www.charmmgui.org/docs/archive/covid19.
- 27. Lineamiento Estandarizado Para La Vigilancia Epidemiológica Y Por Laboratorio De La Enfermedad Respiratoria Viral. ABRIL DE 2020. https://coronavirus.gob.mx/wp-content/uploads/2020/04/Lineamiento_de_vigilancia_epidemiologica_de_enfermedad_respiratoria-_viral.pdf. Accessed June 10, 2020.
- 28. Rasmussen SA, Smulian JC, Lednicky JA, Wen TS, Jamieson DJ. Coronavirus Disease 2019 (COVID‐19) and pregnancy: what obstetricians need to know. Am J Obstet Gynecol. 2020;222(5):415‐426. 10.1016/j.ajog.2020.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thanh Le T, Andreadakis Z, Kumar A, et al. The COVID‐19 vaccine development landscape. Nat Rev Drug Discov. 2020;19(5):305‐306. 10.1038/d41573-020-00073-5 [DOI] [PubMed] [Google Scholar]
- 30. Lee N, McGeer A. The starting line for COVID‐19 vaccine development. Lancet. 2020;395(10240):1815‐1816. 10.1016/S0140-6736(20)31239-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nguyen A, David JK, Maden SK, et al. Human leukocyte antigen susceptibility map for SARS‐CoV‐2. J Virol. 2020;94(13):1‐12. 10.1128/JVI.00510-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barquera R, Collen E, Di D, et al. Binding affinities of 438 HLA proteins to complete proteomes of seven pandemic viruses and distributions of strongest and weakest HLA peptide binders in populations worldwide. HLA. 2020;96(3):277‐298. 10.1111/tan.13956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cao X. COVID‐19: immunopathology and its implications for therapy. Nat Rev Immunol. 2020;20(5):269‐270. 10.1038/s41577-020-0308-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galeotti C, Bayry J. Autoimmune and inflammatory diseases following COVID‐19. Nat Rev Rheumatol. 2020;16:413‐414. 10.1038/s41584-020-0448-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grifoni A, Weiskopf D, Ramirez SI, Smith DM, Crotty S, Sette A. Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell. 2020;181:1489‐1501. 10.1016/j.cell.2020.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mamedov A, Vorobyeva N, Filimonova I, et al. Protective allele for multiple sclerosis HLA‐DRB1*01:01 provides kinetic discrimination of myelin and exogenous antigenic peptides. Front Immunol. 2020;10:3088. 10.3389/fimmu.2019.03088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Joshi A, Joshi BC, Mannan MA, Kaushik V. Epitope based vaccine prediction for SARS‐COV‐2 by deploying immuno‐informatics approach. Informatics Med Unlocked. 2020;19:100338. 10.1016/j.imu.2020.100338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang SF, Chen KH, Chen M, et al. Human‐leukocyte antigen class I CW 1502 and Class II DR 0301 genotypes are associated with resistance to severe acute respiratory syndrome (SARS) infection. Viral Immunol. 2011;24(5):421‐426. 10.1089/vim.2011.0024 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.