

SARS‐CoV‐2 exploits many strategies to subvert innate immune responses allowing the virus to replicate and disseminate within the host. The extent to which the virus replicates within the host, and the efficacy of the host innate immune response to eradicate the infection and trigger effective adaptive immune responses, but not hyper‐responses of innate immunity strongly determines the disease outcome. Understanding the innate immune factors that exacerbate vascular complications following infection will be crucial to control severe disease.
Keywords: COVID‐19, endothelia, immunology, inflammation, SARS‐CoV‐2
Summary
Innate immune sensing of viral molecular patterns is essential for development of antiviral responses. Like many viruses, SARS‐CoV‐2 has evolved strategies to circumvent innate immune detection, including low cytosine–phosphate–guanosine (CpG) levels in the genome, glycosylation to shield essential elements including the receptor‐binding domain, RNA shielding and generation of viral proteins that actively impede anti‐viral interferon responses. Together these strategies allow widespread infection and increased viral load. Despite the efforts of immune subversion, SARS‐CoV‐2 infection activates innate immune pathways inducing a robust type I/III interferon response, production of proinflammatory cytokines and recruitment of neutrophils and myeloid cells. This may induce hyperinflammation or, alternatively, effectively recruit adaptive immune responses that help clear the infection and prevent reinfection. The dysregulation of the renin–angiotensin system due to down‐regulation of angiotensin‐converting enzyme 2, the receptor for SARS‐CoV‐2, together with the activation of type I/III interferon response, and inflammasome response converge to promote free radical production and oxidative stress. This exacerbates tissue damage in the respiratory system, but also leads to widespread activation of coagulation pathways leading to thrombosis. Here, we review the current knowledge of the role of the innate immune response following SARS‐CoV‐2 infection, much of which is based on the knowledge from SARS‐CoV and other coronaviruses. Understanding how the virus subverts the initial immune response and how an aberrant innate immune response contributes to the respiratory and vascular damage in COVID‐19 may help to explain factors that contribute to the variety of clinical manifestations and outcome of SARS‐CoV‐2 infection.
Introduction
The emergence in Wuhan, China, of a novel severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) triggered an epidemic of the coronavirus disease 2019 (COVID‐19). As of 9 September 2020, the confirmed 27 761 748 cases, including 902–306 deaths, have been reported worldwide (worldometers.info/coronavirus). At the end of January 2020, the World Health Organization (WHO) declared COVID‐19 a pandemic and a global health emergency.
The family Coronaviridae is subdivided into Torovirinae and Coronavirinae, that contains the genera Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. The human coronaviruses (HCoV) belong to the αlpha‐CoV (HCoV‐229E and HCoV‐NL63) and beta‐CoV (Middle East respiratory syndrome coronavirus‐MERS‐CoV, SARS‐CoV, HCoV‐OC43 and HCoV‐HKU1) (Table 1 [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11]). In comparison with most HCoVs that cause mild upper respiratory tract infections, SARS‐CoV, MERS‐CoV and SARS‐CoV‐2 induce severe pneumonia [12]. The clinical presentation of COVID‐19 ranges from mild ‘flu‐like’ symptoms to severe respiratory failure and death, although between 17·9 and 57% of SARS‐CoV‐2 infections are asymptomatic, depending on the population [13]. Common symptoms include fever, cough, fatigue, shortness of breath, headache and pneumonia. In addition, some patients develop gastrointestinal problems [14] and neurological manifestations, including headache, dizziness, hyposmia and hypogeusia. Age and co‐morbidities, i.e. hypertension, chronic obstructive pulmonary disease, diabetes, obesity and cardiovascular disease, predispose to more severe manifestations, including severe respiratory failure, septic shock, coagulation dysfunction, strokes, cardiovascular problems [15] and neurological manifestations [16]. Although the origin and transmission of SARS‐CoV‐2 is unclear, genome sequencing reveals marked similarities with SARS‐CoV [17]. However, in comparison, SARS‐CoV‐2 spreads more quickly than SARS‐CoV, probably due to the 10–20%‐fold higher in infectivity and transmissibility during the initial non‐symptomatic period (4–5 days). In some cases, transmission has been reported after development of initial symptoms despite the presence of antibodies [18], indicating that both neutralizing antibodies and T cell responses are necessary to prevent reinfection and for protection [19]. This is further supported by studies showing that programmed cell death 1 (PD‐1)+CD57+ T cell exhaustion, depletion or inactivation is associated with viral persistence in severe cases [20].
Table 1.
HCoVs – cell receptors and co‐factors aiding viral entry
| Virus | Date emergence and clinical manifestations | Primary cell receptor | Tissue expression of receptor | Receptors and co‐factors augmenting viral entry | Refs. |
|---|---|---|---|---|---|
| HCoV‐229E | 1965 | APN (CD13) | Renal and GI epithelia, synaptic membranes, pericytes, myeloid cells, fibroblast‐like cells, neurones | TMPRSS2 | [1, 2, 3] |
| URTI, common cold | TMPRSS11D | ||||
| HCoV‐OC43 | 1967 | 9‐O‐acetylated sialic acid | Human epithelial cells, neurones | IFIT2/IFIT3 | [4, 5] |
| URTI, common cold | HLA‐1 | ||||
| SARS‐CoV | 2002 | ACE2 | Respiratory, intestinal epithelial cells, endothelial cells, renal tubules, cerebral neurones, alveolar macrophages, DCs | Cathepsin L, TMPRSS2/11D | [6, 7] |
| Severe acute respiratory syndrome (SARS) | DC‐SIGN (CD206), DC‐SIGNR | ||||
| HCoV‐NL63 | 2004 | ACE‐2 | Pneumocytes, intestinal epithelial cells, endothelial cells, renal tubules, cerebral neurones, alveolar macrophages, DCs | Heparan sulphate proteoglycans | [8] |
| Bronchitis URI, common cold | |||||
| HCoV‐HKU1 | 2005 | 9‐O‐acetylated sialic acid | HUMAN alveolar type II cells | HLA‐C | [9] |
| Pneumonia common in children | |||||
| MERS‐CoV | 2012 | DPP4 (CD26) | Intestinal, alveolar, renal, hepatic and prostate cells activated leucocytes | Furin | [10] |
| SARS | |||||
| SARS‐CoV‐2 | 2019 | ACE2 | Respiratory, intestinal epithelial cells, endothelial cells, renal tubules, cerebral neurones, alveolar macrophages, DCs | Furin | [11] |
| SARS, severe disease associated with ageing and co‐morbidities | TMPRSS2 |
ACE 2 = angiotensin‐converting enzyme 2; APN = aminopeptidase N; DCs = dendritic cells; DC‐SIGN = dendritic cell‐specific intercellular adhesion molecule‐3‐grabbing non‐integrin (CD209); DC‐SIGNR = DC‐SIGN receptor; DPP4 = dipeptidyl peptidase 4; GI = gastrointestinal; HLA‐C = human leucocyte antigen C; IFIT = interferon‐induced proteins with tetratricopeptide repeats; TMPRSS = type II transmembrane serine proteases; URTI = upper respiratory tract infection.
SARS‐CoV‐2 is a positive‐sense RNA (29 903 nucleotides) enveloped virus of 60–140 nm diameter [21]. The envelope is studded with homotrimers spike proteins of 8–12 nm length that are heavily decorated with N‐glycans (Fig. 1) [22, 23]. Similar to other HCoVs, the SARS‐CoV‐2 genome encodes for four structural proteins: the spike (S), membrane (M), envelope (E) and the nucleocapsid (N) protein. The 5′ end of the genome is comprised of open reading frame (ORFa/ab), encoding two large polyproteins including the replicase protein crucial for self‐generation of the non‐structural proteins (nsp), while ORFs 2–10 encode the viral structural proteins – spike, envelope, membrane and nucleocapsid, and the accessory proteins (Fig. 1b). Differences between the structural, non‐structural and accessory proteins of SARS‐CoV‐2 and other coronaviruses help to explain the high infectivity rate and the range of pathologies observed [12, 15, 16]. While knowledge of SARS‐CoV‐2 is rapidly emerging, parallels with SARS‐CoV, as well as ongoing sequencing data and antigenic typing, will be crucial to understand the dynamics of the pandemic. SARS‐CoV‐2 cell entry is similar to SARS‐CoV, being mediated by the binding of the receptor‐binding domain (RBD) of the S1 protein to the angiotensin‐converting enzyme‐2 (ACE‐2), although other receptors, such as CD147 and CD‐specific intercellular adhesion molecule‐3‐grabbing integrin (SIGN), have been reported (Table 1). Docking of the RBD to the receptor and the action of furin, a serine protease that separates the S1 and S2 proteins, exposes a second binding domain on S2 allowing membrane fusion. Binding of the S protein to ACE‐2 requires priming by cell proteases, primarily transmembrane protease, serine 2 (TMPRSS2); however, TMPRSS2 is expressed by a subset of ACE2+ cells supporting the notion that the virus probably uses other host enzymes such as TMPRSS4, lysosomal cathepsins and neuropilin‐1 [24] to augment the impact of furin and expose the RDB, thus promoting SARS‐CoV‐2 entry [11]. The structural proteins M, E and N are crucial for stability of the viral genome and viral replication. The nsp and accessory proteins [25], encoded by 10 ORFs, have differing functions during viral replication (Table 2) [26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63] and many also act to deviate the innate immune response, thus augmenting viral replication and spread. The degree to which the innate immune system is suppressed and evaded clearly determines the viral load and the host’s outcome to infection, the clinical symptoms and the severity of the disease.
Fig. 1.

SARS‐CoV‐2 structure and genome. (a) SARS‐CoV‐2 is a positive‐sense RNA enveloped virus with the spike (S), membrane (M), envelope (E) proteins embedded in the lipid envelope, while the nucleocapsid (N) protein is associated with the RNA. (b) The 5′ end of the genome is comprised of open reading frame (ORF)a/ab encoding two large polyproteins, including the replicase protein crucial for self‐generation of the non‐structural proteins (nsp), while ORFs 2–10 encode the viral structural proteins (S, M, E and N) and accessory proteins. (c) The homotrimers spike proteins of 8–12 nm length are heavily decorated with N‐glycans moieties that can be recognized by antibodies, C‐type lectins and mannose‐binding proteins that aid viral attachment to permissible cells, activate the complement system and may be recognised by macrophages and antibodies (d).
Table 2.
Immune evasions strategies of genome and encoded proteins of SARS‐CoV and (by inference) SARS‐CoV‐2
| Gene | Protein | Function | Impact on immune response action | Comments | Refs. |
|---|---|---|---|---|---|
| Reduced CpG | Decreased activity of ZAP and APOBEC3G. | ZAP expressed in immune cells | [26] | ||
| ORF1a | nsp1 | Mediates RNA replication and processing. Involved in RNA degradation | Modulates calcineurin/ NFAT pathway. Cleaves host RNA. Inhibits cyclophilins and immunophilins. Blocks ATF2/c‐Jun, IRF3 and IRF7, NF‐κB, decreases STAT‐1 phosphorylation. Interferes with RIG‐1 pathway | Associated with immune pathogenicity and long‐term cytokine dysregulation. Promotes host RNA degradation | [27, 28, 29] |
| nsp2 | Replicase essential for proofreading replication | May bind to prohibitin 1 and 2 involved in apoptosis, mitochondrial biogenesis and intracellular signalling. Modulates host survival signalling | Associated with pathogenicity. Deletion attenuates viral growth and RNA synthesis | [30, 31] | |
| nsp3 | Papain like protease (PLpro). Processes pp1a and pp1ab | De‐ubiquitinates and deISGylates host proteins. Blocks IFN‐α, IFN‐β, CXCL10 and CCL5. Inhibits TLR‐7 signalling by removing Lys63‐linked polyubiquitination of TRAF3 and 6. | Forms DMV and replication process evade innate immune recognition | [32, 33, 34] | |
| Complexes with nsp4 and nsp6 | Antagonizes IRF3, stabilizes IκBα, thereby blocking NF‐κB signalling | ||||
| Interacts with STING‐TRAF–TBK1 complex | |||||
| nsp4 | Complexes with nsp3 and nsp6 to form the DMV. May anchor RTC to ER | Helps replication process evade innate immune recognition | [33] | ||
| nsp5 | Chymotrypsin‐like protease (3CLpro) | Induces apoptosis and growth arrest via caspase‐3 and caspase‐9 | [35] | ||
| nsp6 | Complexes with nsp3 and nsp4 to form DMV | Helps replication process evade innate immune recognition. Activates autophagosome | [33] | ||
| nsp7 | Complexes with nsp8 and nsp12 for viral replication | Nsp7–nsp8 form the primase complex | [36] | ||
| nsp8 | Complexes with nsp7 and nsp 12 for viral replication | Nsp7–nsp8 form the primase complex | [36] | ||
| nsp9 | Involved in viral genomic RNA reproduction but exact role unclear | Interacts with nsp8 | [37, 38] | ||
| nsp10 | Complexes with nsp 1, 7 and 14. Multi‐functional co‐factor in replication | Interacts with the oxidoreductase system causing cytopathic effect | Activator of nsp14 function | [39, 40, 41] | |
| Aids RNA capping, thus evades RIG‐1 and MDA‐5 recognition | Forms a complex with nsp16 | ||||
| nsp11 | Peptide resulting from cleavage of pp1a at nsp10/11 junction | Not known | |||
| ORF1b (nsps in addition to 1–11) | nsp12 | RNA‐dependent | Targeting mitochondria limits host cellular responses | [36] | |
| RNA polymerase (RdRp) | |||||
| nsp13 | Helicase key for efficient replication of viral genome | Caps RNA, thus evades RIG‐I and MDA‐5 signalling | Failure to trigger IFIT1 | [42] | |
| nsp14 |
Exons 3′–5′ exonuclease play crucial role in viral RNA synthesis and capping. Complexes with nsp10 |
Involved in the capping through its function as a guanine‐N7 methyltransferase helping nsp16 evade RIG‐1 and MDA‐5 recognition | [43] | ||
| nsp15 | Uridylate‐specific endoribonuclease (EndU) | Limits exposure of viral dsRNA to the sensors MDA‐5, PKR and OAS/RNaseL. Inhibits poly U, thereby evading MDA‐5 thus antagonizing IFN‐a/β production | [44] | ||
| nsp16 | 2′‐O‐ribose methyl transferase involved in RNA capping. Complexes with nsp10 | Caps RNA, thus evades RIG‐I and MDA‐5 signalling | Failure to trigger IFIT1 | [41, 45, 46] | |
| ORF2 | Spike | Heavily glycosylated with 22 glycans | Masks immunogenic protein epitopes | [22, 23, 47, 48, 49] | |
| ACE/ACE‐2 interaction | Induced misbalanced in RAS that triggers inflammation | ||||
| Requires priming to expose membrane fusion | Masks immunogenic protein epitopes | ||||
| ORF3a | ORF3a | Interact with SARS‐CoV M, S, E and 7a proteins | Activates PERK pathway, triggers apoptosis through expression of ATF4 and CHOP. Down‐regulates and degrades type 1 IFNR | Expressed on cell surface. Induces fibrinogen, stress pathways, necrotic cell death, activates inflammasome | [50, 51, 52] |
| Forms viroporins | |||||
| ORF4 | Envelope | Essential for viral assembly and budding. Forms viroporins | Induces ROS and activates inflammasome | [53, 54] | |
| ORF5 | Membrane | Important for viral assembly | Inhibits type I interferon production by impeding the formation of TRAF3. TANK. TBK1/IKKε complex | Induces apoptosis | [55, 56, 57] |
| ORF6 | ORF6 | Plays a role in viral pathogenesis, interacts with ORF8. May aid viral virulence | Inhibits STAT‐1 nuclear import | Promotes RNA polymerase activity | [51] |
| ORF7a | ORF7a | Interacts with S protein and p3a | Inhibits BST‐2 glycosylation, leading to a loss of function of BST‐2. SARS‐CoV ORF7a induces caspase‐dependent apoptosis | BST‐2 restricts virion egress by tethering virions to plasma membrane. Interacts with LFA | [58, 59] |
| Not essential for replication | |||||
| ORF7b | ORF7b | Not essential for viral replication but structural component of the virion | It is an integral membrane protein located in the Golgi compartment | [60] | |
| ORF8 | ORF8 | Differs from other HCoVs | Interact and down‐regulates MHC‐I | SARS‐CoV encodes p8a and p8b that induce caspase‐dependent apoptosis and activates UPR | [61] |
| ORF9 | Nucleocapsid | Stabilizes viral RNA | Targets MAVS–RAF3–TRAF6 and antagonizes IFN‐β | [51, 62, 63] | |
| Interacts with stress granules G3BP1 | |||||
| ORF10 | ORF10 | Ubiquitin ligase | Unknown |
ACE 2 = angiotensin‐converting enzyme 2; BST = bone marrow stromal antigen 2; CHOP = C/EBP homologous protein; DMV = double membrane vesicles; ER = endoplasmic reticulum; HCoV = human coronavirus; IFIT = interferon‐induced proteins with tetratricopeptide repeats; IFN = interferon; IFNR = IFN receptor; LFA = lymphocyte function‐associated antigen 1; MAV = mitochondrial anti‐viral signalling protein; MDA = melanoma differentiation‐associated; MHC = major histocompatibility complex antigen; ORF = open reading frame; PERK = PRKR‐like endoplasmic reticulum kinase; RAS = renin–angiotensin system; RIG‐1 = retinoic acid‐inducible gene I; ROS = reactive oxygen species; RTC = replicase–transcriptase complex; STAT = signal transducer and activator of transcription; UPR = unfolded protein response; ZAP = zinc finger anti‐viral protein.
Evading pattern recognition receptors
Following infection, viral RNA is sensed by several classes of pattern recognition receptors (PPRs). The retinoic acid‐like receptors (RLRs) include retinoid inducible gene I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA‐5), Toll‐like receptors (TLR) – classically 3, 7 and 8, that trigger IFN pathways – and cytokine production (Fig. 2). Once engaged these PPRs act downstream via the kinases TANK‐binding kinase‐1 (TBK1) and inhibitor‐κB kinases (IKKs). Such triggering leads to the activation of the transcription factors interferon (IFN)‐regulatory factor‐3 (IRF3) and 7 (IRF7) and nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB). These subsequently induce expression of type I IFNs (IFN‐α/β) and IFN‐stimulated genes (ISGs) (Fig. 2), many of which have potent anti‐viral activities, as well as other proinflammatory mediators; for example, cytokines, chemokines and anti‐microbial peptides that are essential to initiate the host innate and adaptive immune response. In addition, the absent in melanoma 2 (AIM2)‐like receptors and NOD‐like receptors (NLRs) trigger the inflammasome and IL‐1β and IL‐18 production, leading to pyroptosis (Fig. 2). Other PPRs and downstream factors relevant to SARS‐CoV infection subversion of innate immune responses include C‐type lectins and the stimulator of IFN genes (STING). While the cGas/STING pathway is commonly associated with sensing cytosolic DNA, it is also activated following binding of enveloped viruses to host cells and cytosolic viral RNA [64, 65]. Similar to TLRs and RLR, STING engages TBK1 downstream to active IRF3 and/or NF‐κB inducing type I IFN and/or proinflammatory cytokines (Fig. 2).
Fig. 2.
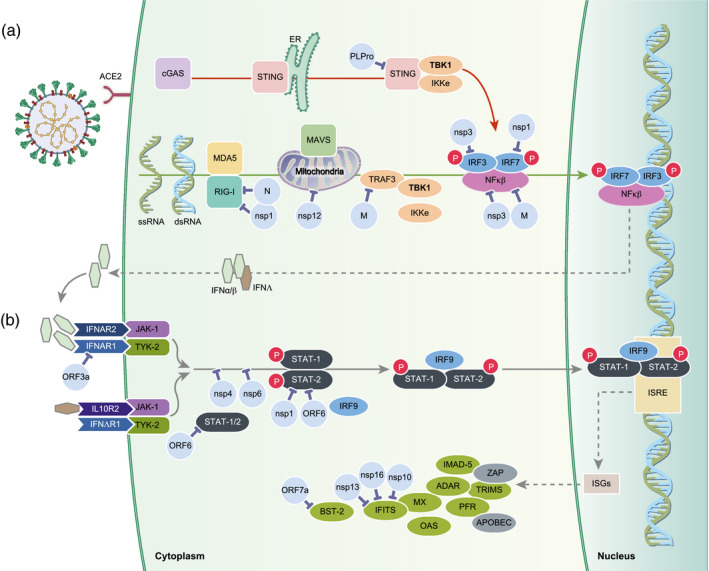
SARS‐CoV‐2 subversion of interferon (IFN) pathways. SARS‐CoV‐2 infects permissible cells via the angiotensin‐converting enzyme 2 (ACE2). Following infection (a) the virion or viral RNA is sensed by either the cGas/STING pathway where stimulator of interferon genes (STING) engages TBK1, or via retinoid inducible gene I (RIG‐I) and melanoma differentiation‐associated gene 5 (MDA‐5). These pathways lead to activation of IFN‐regulatory factor (IRF3) and/or nuclear facror kappa B (NF‐kB) inducing type I/III IFN that is recognized by IFN receptors (b) and subsequent induction of the IFN‐stimulated genes (ISGs) and proteins, many of which have potent anti‐viral activities. Based on the knowledge of other coronaviruses, especially SARS‐CoV, and emerging data from SARS‐CoV‐2, many of the non‐structural, structural and accessory protein subvert and inhibit numerous steps in these pathways, thereby inhibiting IFN production allowing increased viral replication.
Coronaviruses have evolved several strategies to escape such innate immune recognition, allowing widespread replication. Such evasion includes evolution of low genomic CpG, RNA shielding, masking of potential key antigenic epitopes as well as inhibition of steps in the IFN type I/III pathways. Generally, the zinc finger anti‐viral protein (ZAP) specifically binds to and degrades CpG motifs in genomes of RNA viruses. In comparison with other viruses, SARS‐CoV‐2 has evolved the most extreme cytosine–phosphate–guanosine (CpG) deficiency of all betacoronaviruses (Table 2) [26]], thereby evading ZAP action. This suggests that SARS‐CoV‐2 may have evolved under selective pressure in either a new host or tissues expressing high levels of ZAP [26]. Another strategy to protect mRNA used by the host and many viruses is the processing of capping the 5′ end. For both host and virus RNA, capping limits degradation and importantly blocks recognition by cytosolic PPRs. Like many RNA viruses, SARS‐CoV‐2 has exploited several mechanisms to protect the 5′ ends by a cap structure of RNA generated during replication. While some viruses snatch the caps from host RNA, SARS‐CoV‐2, like other coronaviruses, uses its own capping machinery composed of nsp10, nsp13 and the dedicated enzyme nsp16 to generate 2′‐o‐methyltransferase caps (Supporting information, Fig. S1 [41]). SARS‐CoV‐2 yields RNA caps indistinguishable from cellular mRNAs caps, thereby evading detection by MDA‐5 and IFIT activity that target RNA for degradation (Fig. 2). The importance of such capping and viral replication is supported by studies of SARS‐CoV in mice lacking 2′‐O‐MTase activity underscoring that MDA‐5 and the IFN‐induced proteins with tetratricopeptide repeats (IFIT) family are critical for IFN signalling [45] While counterintuitive, SARS‐CoV uses its endoribonuclease (nsp15) to cleave its own viral RNA in the cytosol that would otherwise act as PAMPs, thus evading MDA‐5, protein kinase R (PKR) and OAS/RNAse L [44, 66] However, another strategy used by SARS‐CoV‐2 to protect the viral RNA and proteins generated during replication (Supporting information, Fig. S1) is the use of replicase–transcriptase complex (RTC) or replication organelle, formed of double membrane vesicles [33]. The RTCs link with the endoplasmic reticulum (ER)‐Golgi intermediate compartment (ERGIC) and Golgi apparatus shielding the virus during maturation (Supporting information, Fig. S1). Another immune evasion strategy utilized by coronaviruses is the use of glycans and probably other post‐translational modifications to mask immunogenic viral protein epitopes (Fig. 1c,d). The envelope of SARS‐CoV‐2 is studded with glycoprotein spikes comprised of homotrimers spike proteins of 8–12 nm length that are heavily decorated with glycans. Each spike protein is comprised of two subunits (S1 and S2) that each bear 22 glycan groups [49]. Cell entry of the highly glycosylated S protein of SARS‐CoV is promoted by DC‐SIGN, possibly augmenting virus uptake or aiding capture and transmission of SARS‐CoV by DCs and macrophages [6, 7, 8]. Similar to the spike protein, the other structural, non‐structural and accessory proteins are also modified by glycosylation, palmitoylation, phosphorylation, SUMOylation and ADP‐ribosylation [67]. Conversely, some viral proteins, for example nsp3, possess de‐ubiquitinating (DUB) and deISGylation activity, thereby interfering with host functions targeting those that are critical for signalling transduction of innate immunity [34]. Insertion of the spike protein into cell membranes during replication is a key step for virus budding. While this takes place in the RTC (Supporting information, Fig. S1), receptor‐bound spike proteins interact with TMPRSS2 expressed on the uninfected cell surface and mediates fusion between infected and uninfected cells promoting the formation of syncytia, allowing the virus to spread to adjacent uninfected cells while evading detection by the immune response [68].
SARS‐CoV‐2 subversion of innate immune responses
In addition to strategies to evade PPR recognition, SARS‐CoV‐2 has also evolved strategies to inhibit steps in the pathway leading to type I/III IFN production. This may be especially relevant in the lungs, where type IFN III (lambda) is considered to be more effective in controlling viral infections and critically affected in COVID‐19. Knowledge arising from the study of other coronaviruses, especially SARS‐CoV and MERS, has shown that many of the non‐structural, structural and accessory proteins interfere with elements of the IFN pathway (Table 2, Fig. 2), essential for the development of effective immunity. IFN antagonism has been attributed to several of the structural, non‐structural and accessory proteins that interfere with the STING–TRAF3–TBK1 complex, thereby blocking STING/TBK1/IKKε‐induced type I IFN production, signal transducer and activator of transcription (STAT)‐1/2 translocation to the nucleus, IRF3, NF‐κB signalling as well as interfering with the actions of the ISG products, including IFITs (Table 2). As examples, nsp1, 4 and 6 and ORF6 interfere with STAT‐1/2 signalling while nsp 10, 13 and 16 cap the viral RNA (Table 2), preventing recognition by RIG‐I, MDA‐5 and IFITs. Nsp3 also acts by DUB proteins, thereby preventing their activity such as RIG‐I and other steps in the IFN pathways for which ubiquitination is essential. CoV PLPro (nsp 3) also interrupts the stimulator of IFN genes STING–TRAF3–TBK1 complex, thereby blocking STING/TBK1/IKKε‐type I IFN production [32, 34]. As well as subversion of the IFN pathway, SARS‐CoV ORF7a (also present in SARS‐CoV‐2) blocks the activity of tetherin, also known as bone marrow stromal antigen 2 (BST‐2) [58]. BST2 acts by tethering budding viruses to the cell membrane, thus preventing its release from the cells. ORF7a removes this inhibition aiding the release of mature virions.
In summary, emerging evidence from SARS‐CoV‐2, and comparison with other SARS‐CoV and MERS, reveals many strategies used to evade the innate immune response and subvert the IFN pathway. While this facilitates widespread viral replication, increasing the viral load also promotes the viral cytopathic effects leading to tissue damage described below, and probably leads to exacerbation and hyperinflammation of the innate immune response once triggered.
Triggering innate immunity
Despite immune evasion and subverting innate immune responses during early infection, SARS‐CoV‐2 effectively initiates immune signalling pathways. This is probably due to the increased viral load that exponentially produces viral RNA and viral proteins [pathogen‐associated molecular patterns (PAMPS)], and also induces cell damage that release damage‐associated molecular patterns (DAMPS), both of which trigger innate immune pathways.
Like SARS‐CoV and NL63, SARS‐CoV‐2 uses the angiotensin (Ang)‐converting enzyme‐2 (ACE2) as a cell receptor (Table 1), expressed on epithelia in renal, cardiovascular and gastrointestinal tract tissues, testes and on pneumocytes and vascular endothelia [66]. ACE2 regulates the renin–angiotensin system (RAS) by balancing the conversion of angiotensin I to angiotensins 1–9 and angiotensin II to angiotensins 1–7. Binding of SARS‐CoV‐2 to ACE2 leads to endosome formation, reducing ACE2 expression on the cell surface (Figs. 3 and 4) and pushing the RAS system to a proinflammatory mode, triggering production of reactive oxygen species (ROS), fibrosis, collagen deposition and a proinflammatory environment, including IL‐6 and IL‐8 production, by macrophages and recruitment of neutrophils (Fig. 4). Thus, binding and entry of SARS‐CoV‐2 via the ACE2 is likely to be the first step in a line of augmented and detrimental immune responses in COVID‐19 that involves complement activation, innate immune activation via PAMPS and DAMPS, inflammasome activation and pyroptosis, natural killer (NK) cell activation, hyperactivation of macrophages, neutrophils and innate T cells and induction of a cytokine storm, as discussed below.
Fig. 3.

SARS‐CoV‐2 activates innate immune pathways. SARS‐CoV‐2 infects permissible cells via the angiotensin‐converting enzyme 2 (ACE2) and is taken by in the endosome where the virus is recognized by Toll‐like receptors 7/9 triggering the myeloid differentiation primary response 88 (MyD88) pathway, or Toll‐like receptor (TLR)‐3 via the TIR‐domain‐containing adapter‐inducing interferon‐β (TRIF) pathway (a). Pathogen‐associated molecular patterns (PAMPS) and damage‐associated molecular patterns (DAMPS) are also recognized by TLR‐4 (b) or receptor for advanced glycation end (RAGE) (d) triggering high mobility group box 1 (HMGB1)‐induced damage and NOD pyrin domain‐containing 3 (NLRP3) inflammasome activation. During viral replication ORF3a and E proteins form viroporins that augment reactive oxygen species (ROS) production and inflammasome activation.
Fig. 4.

SARS‐CoV‐2 is a vascular and coagulation disease. (a) Binding of SARS‐CoV‐2 to angiotensin‐converting enzyme 2 (ACE2) blocks ACE2‐induced formation of anti‐oxidant angiotensin, facilitating oxygen free‐radical formation. Infection in some people also triggers pyroptosis, complement activation (b) and hyperinflammation with influx of macrophages, natural killer (NK) cells and neutrophils (c). This self‐augmenting cycle triggers further cell damage and damage‐associated molecular patterns (DAMPS) and pathogen‐associated molecular patterns (PAMPS) release, as well as reactive oxygen species (ROS) production. (d) Activation of neutrophils induces neutrophil extracellular traps (NET) aided by the N protein and generated in response to ROS‐induced endothelial cell damage. Disruption of the vascular barrier and endothelial cell exposure to proinflammatory cytokine and ROS increases expression of P‐selectin, von Willebrand factor (vWF) and fibrinogen that attract platelets triggering expression of tissue factor. Together, this sequence activates the complement system, one of many pathways that crucially activates the coagulation cascade leading to thrombi formation.
Complement
SARS‐CoV‐2 is heavily decorated with glycans (Fig. 1) that are recognized by DC‐SIGN and other lectins that facilitate viral uptake by dendritic cells (DCs). Glycans also activate the lectin complement pathway following binding of mannose‐binding protein (MBP) to SARS‐CoV‐2 viral proteins expressed on infected cells (Figs. 1 and 2). Pathology studies and transcriptional profiles of tissues from COVID‐19 cases reveal robust activation of the complement system with the deposition of MBL, C4d, C3 and C5b‐9, forming the membrane attack complex (MAC) in alveolar and epithelial cells [68, 69] (Fig. 4). In addition, C4d and C5b‐9 deposits in lung and skin microvasculature co‐localized with spike glycoproteins indicates systemic complement activation, supporting the role of complement in tissue damage [68]. Importantly, activation of the lectin, as well as the classical pathway following antibody binding to viral proteins, probably contributes to cell damage (Fig. 4) [70] by either direct complement‐mediated lysis or via antibody‐dependent cell‐mediated cytotoxicity. Of relevance to the coagulation dysfunction, thrombosis and vascular damage observed following SARS‐CoV‐2 infection is that complement components induce secretion of von Willebrand factor [71], but also promotes monocyte and neutrophil recruitment as well as stimulating NET formation [72] that, in turn, perpetuates complement activation (Fig. 4). Complement may thus contribute to widespread tissue damage in SARS‐CoV‐2‐infected cases. The pathogenic role of complement in disease is supported by findings in mice. For example, mice deficient in C3 had similar viral load as wild‐type mice, but lacked the overt pathology with fewer neutrophils and macrophages in the lung [73]. Thus, while complement activation is not required for control of virus infection it probably plays a key role in the tissue damage.
PAMPS and DAMPS mediate innate immune signalling
Infected pneumocytes and other permissible cells undergo cell damage and cell death releasing virally associated molecules, so‐called PAMPS. In addition, intracellular components released due to damage, so‐called DAMPS, include ATP, oxidized lipids, heat shock proteins and other components associated with regulated cell death programmes including apoptosis, autophagy, necroptosis and pyroptosis (Figs. 3 and 4). Thus, both DAMPS and PAMPS contribute to innate immune activation in COVID‐19.
RNA viruses trigger several Toll‐like receptors (TLRs), including TLR‐7/8 and TLR‐3, and elegant molecular in‐silico docking studies show that the spike protein of SARS‐CoV‐2 can bind to TLR‐1, TLR‐4 and TLR‐6 [74] (Fig. 3), whereas in vitro the SARS‐CoV spike protein triggers NF‐κB activation and IL‐8 production via TLR‐2 signalling in human peripheral blood mononuclear cells (PBMCs) [75]. In mice in which specific points in the TLR pathway were deleted; that is, TLR‐3−/−, TLR‐4−/− and TRIF‐related adaptor molecule (TRAM)−/− animals, were more susceptible to SARS‐CoV infection, although the clinical severity of disease was dramatically reduced. This was in direct contrast to deficiency in TIR‐domain‐containing adapter‐inducing IFN‐β (TRIF, the TLR adaptor protein (Fig. 3) in which TRIF−/− mice developed severe disease, exacerbated influx of macrophages and neutrophils and lung pathology indicative of COVID‐19 pathology. Thus, a balanced response to infection via the TLR‐3 pathway is essential to trigger a protective response to SARS‐CoV [76]. This study also supports the idea that in addition, PAMPS, immune pathways triggered by DAMPS such as oxidized phospholipids, high mobility group box 1 (HMGB1), histones, heat shock proteins and adenosine triphosphate released by damaged cells may contribute to COVID‐19 outcome (Figs. 3 and 4). In addition to RIG‐I, MDA‐5 and MAVS, RNA viruses are also sensed by the STING that is activated by cGAMP when enveloped RNA viruses interact with the host membranes [64]. Downstream, STING engages TBK1 that actives IRF3 and/or NF‐κB inducing type 1 IFN and/or proinflammatory cytokines. That hyperactivation of STING contributes to severe COVID‐19, as has been hypothesized by Berthelot and Lioté [77]. These authors present several lines of evidence, the strongest being that gain‐of‐function mutations of STING associated with hyperactivation of type I IFN induces the disease SAVI (STING‐associated vasculopathy with onset in infancy). Affected children with SAVI present with pulmonary inflammation, vasculitis and endothelial‐cell dysfunction that mimics many aspects of COVID‐19 [78]. Furthermore, STING polymorphisms are associated with ageing‐related diseases such as obesity and cardiovascular disease, possibility explaining the impact of co‐morbidities and development of severe COVID‐19 [78]. Also, in bats, in which SARS‐CoV‐2 may have arisen, STING activation, and thus consequently IFN‐β, is blunted [79], probably aiding viral replication and spread, as observed in early SARS‐CoV‐2 infection in humans. That DAMPS released due to viral cytotoxicity may contribute to severe COVID‐19, which is best exemplified by HMBG1 released by damaged and dying cells, as well as activated innate immune cells, especially in sepsis [80]. Depending on its conformation, HMGB1 triggers TLR‐2, TLR‐4 and TLR‐9, the receptor for advanced glycation end‐products (RAGE) and triggering receptor expressed in myeloid cells 1 (TREM‐1) (Fig. 3). In mice, intratracheal administration of HMGB1 activates mitogen‐activated protein kinase (MAPK) and NF‐κB, inducing proinflammatory cytokines, activating the endothelium and recruiting neutrophils in the lung: key pathological features of severe COVID‐19 [80, 81]. HMGB1, and especially the platelet‐derived source, may play a crucial role in SARS‐CoV‐2 vascular damage as HMGB1−/− mice display delayed coagulation, reduced thrombus formation and platelet aggregation [82]. Furthermore, blocking HMGB1 is beneficial in experimental lung injury and sepsis, suggesting that therapies targeting HMGB1 might also be beneficial in severe COVID‐19 [83,84].
Inflammasome activation and pyroptosis
Studies of peripheral blood and post‐mortem tissues from severe COVID‐19 cases reveal high levels of IL‐1β and IL‐6 and increased numbers of CD14+IL‐1β monocytes, suggesting activation of the NOD‐like receptor family, pyrin domain‐containing 3 (NLRP3) inflammasome pathway [85]. Activation of the NLRP3 inflammasome, essential for effective anti‐viral immune responses, is elicited by several factors associated with SARS‐CoV infection, including RAS disbalance, engagement of PPR, TNFR and IFNAR, mitochondrial ROS production and complement components including MAC, as well as SARS‐CoV viral proteins such as ORF3a, N and E [86] (Fig. 3, Table 2). As a consequence, NLRP3 interaction with adaptor apoptosis speck‐like protein (ASC) recruits and activates procaspase‐1, processing pro‐IL‐1β and pro‐IL‐18 to the activated forms (Fig. 3). This drives the propyroptotic factor gasdermin D (GSDMD) formation of pores in the cell membrane; that is, pyroptosis that facilitates the release of proinflammatory cytokines. The pores also aid the release of cellular DAMPS such as HMGB1 and viral PAMPS that further exacerbate inflammation, suggesting that targeting the NLRP3 pathway might be beneficial in severe COVID‐19 cases.
Hyperinflammation and severe COVID‐19
The delayed IFN response, increased viral load and virus dissemination, coupled with the release of DAMPS and PAMPs, lead to activation of several innate immune pathways. Following infection, pneumocytes, epithelial and alveolar cells and infiltrating monocyte–macrophages and neutrophils probably produce the first wave of tumour necrosis factor (TNF)‐α, IL‐6, IFN‐γ‐induced protein 10 (IP‐10), monocyte chemoattractant protein‐1 (MCP‐1), macrophage inflammatory protein (MIP)‐1α and regulated on activation, normal T‐expressed and secreted (RANTES) production [87, 88]. Hyperinflammation is probably promoted by co‐morbidities due to increased ACE2 expression, concurrent bacterial infections and ageing as well as a direct effect of SARS‐CoV‐2 replication, as virus–host interactome studies reveal that SARS‐CoV‐2 nsp10 regulates the NF‐κB repressor factor NKRF, facilitating IL‐8 production [89]. This is followed by a second wave of cell recruitment, including NK cells that produce IFN‐γ and further recruitment of (alternatively activated) monocytes/macrophages and neutrophils (Fig. 4), as observed in bronchial lavages, post‐mortem tissues and peripheral blood studies [88, 89]. NK cells are key players in disease outcome of infection, critically balancing the direct response to the virus by eliminating infected cells while also augmenting tissue damage (Fig. 4). Probably aided by IFN‐γ induction by NK cells, hyperinflammation in severe COVID‐19 is also characterized by recruitment of immature and mature human monocyte‐derived DCs that harbour SARS‐CoV infection; however, infection is abortive and mature virions are not released. During infection, DCs express only low levels of cytokines probably due to innate immune subversion strategies. The sustained activation of infiltrating monocytes and monocyte‐derived macrophages [90] observed in severe COVID‐19 cases is probably driven by a number of factors, including oxidative stress, anti‐SARS‐CoV‐2 antibody–antigen complexes, NLRP3 inflammasome activation and complement activation that converge to sustain an aberrant hyperinflammatory response, or cytokine storm [91]. Following SARS‐CoV‐2 infection, one of the first innate immune cells to infiltrate into the tissues are neutrophils, probably recruited by chemokine (C‐X‐C motif) ligand (CXCL)2 and CXCL8 generated by infected cells [92]. While neutrophils do not clear viral particles, they phagocytose apoptotic bodies containing virus and debris, releasing proteolytic enzymes, anti‐microbial peptides, matrix metalloproteinases and high levels of ROS to inactivate viruses. A key function of neutrophils relevant to the pathology of SARS‐CoV‐2 is the production of neutrophil extracellular traps (NETs) generated in response to endothelial damage, ROS production, IL‐1β production and virus replication (Fig. 4, reviewed in [93]). The formation of NETs by neutrophils are aided by activated platelets associated with damaged endothelial cells that further activate the complement, fuelling the coagulation cascade and thrombi formation. While the NETs act to prevent further spread of the virus, they trigger platelet activation and bind erythrocytes thereby promoting (micro)thrombi formation (Fig. 4).
SARS‐CoV‐2 is a vascular and coagulation disease
While respiratory damage and complications are the major clinical signs of severe COVID‐19 many tissues and organs are affected often prior to, or independently of lung pathology, for example Kawasaki‐like vascular disease in children [94]. Clinical, post‐mortem studies and experimental animal models of SARS‐CoV reveal infection of endothelial cells and the widespread damage of endothelial cells, vascular dysfunction and thrombosis [94, 95] that are emerging as a common pathological feature of SARS‐CoV‐2 infection. The link between SARS‐CoV‐2 infection, vascular damage and thrombosis is evidenced by high levels of D‐dimers in 20–40% critically ill patients probably produced in an attempt to dissolve thrombotic clots. The endothelial cell damage is supported by the finding that endothelial cells express ACE2 and are thus permissible to SARS‐CoV‐2 infection [94]. Thus, infection not only leads to reduced ACE2 in endothelial cells, but also direct viral cytopathic damage and increased vascular permeability (Fig. 4), although more recent data challenge this view, suggesting that pericytes and not endothelial cells are permissible to infection and viral‐induced damage [95, 96]. Damage of endothelial cells and pericytes leads to vascular permeability in severe COVID‐19 that is probably amplified by activation of complement components widely expressed in post‐mortem tissues of COVID‐19 cases [68, 69]. Disruption of the vascular barrier and endothelial cell exposure to IL‐1β, TNF‐α and ROS increase expression of P‐selectin, von Willebrand factor (vWF) and fibrinogen, and attracting platelets that trigger expression of tissue factor (Fig. 4). Together, this sequence triggers the coagulation cascade and explains the finding of increased D‐dimer and fibrin, abnormal clotting times in severe COVID‐19 cases and widespread disseminated thrombi in post‐mortem tissues.
Disease severity, co‐morbidities and innate immunity
SARS‐CoV‐2 exploits many strategies to subvert innate immune responses allowing the virus to replicate and disseminate within the host. The extent to which the virus replicates within the host, and the efficacy of the host innate immune response to eradicate the infection and trigger effective adaptive immune responses, but not hyper‐responsiveness of innate immunity, strongly determines the disease outcome (Table 3). The severity of infection has been linked to age, smoking, co‐morbidities such as cancer, immune suppression, autoimmune diseases, inflammatory disease, neurodegenerative diseases, obesity, gender and race [97, 98, 99, 100, 101, 102, 103, 104, 105, 106]. For example, in a large cohort of 72 314 cases the case fatality ratio for more than 80 years was 14·8 versus 2·3% in the total cohort [97]. This is probably higher due to inflamm‐ageing, an aberrant innate immune response such as lower production of IFN‐β [98], increased oxidative stress [99] and sensence of macrophages that become less effective in their reparative functions with age [100]. Similarly, viral load, obesity, gender, race, blood groups and co‐morbidities have all been reported to influence the response to SARS‐CoV‐2 infection (Table 4) [101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112], although few studies have fully examined the extent to which subversion and activation of innate immune components contribute to susceptibility in these cases.
Table 3.
Impact of innate immune suppression on disease outcome
| Asymptomatic or mild | Moderate | Severe | |
|---|---|---|---|
| Type I/III IFN | Increased and prolonged production | Moderate suppression | High and prolonged suppression |
| Viral replication | Limited | Mild or chronic | High and sustained |
| Immune response | Strong and rapid induction of adaptive immunity, viral clearance | NK cell and complement mediated clearance of infected cells, reduced adaptive immune responses | Hyperinflammation, cytokine storm, for example, IL‐6, IL‐8, TNF‐α, delayed or ineffective adaptive immune response, innate T cell activity |
| Pathology | None, subclinical | Tissue damage due to inflammatory response | Viral‐induced cytotoxicity, ADCC, complement‐mediated damage, pyroptosis, necroptosis, neutrophil‐driven NETosis |
| Vascular damage and thrombosis | No | Not likely or mild | Highly likely and contributes to clinical disease and tissue damage, hence the finding of high levels of D‐dimer produced to counteract thrombi formation |
ADCC = antibody‐mediated cell cytotoxicity; IFN = interferon; IL = interleukin; TNF = tumour necrosis factor; NK = natural killer.
Table 4.
Factors and co‐morbidities, aberrant innate immune responses and COVID‐19
| Factor or co‐morbidity | Impact on COVID‐19 | Proposed impact on innate immune responses | Refs. |
|---|---|---|---|
| Age | Increased CFR | Increased oxidative stress, decreased IFN responses | [97, 98, 99, 101] |
| > 80–14·8% vf 2·3% | Elevated proinflammatory cytokines | ||
| 70–79 to 8·0% | |||
| Blood groups | Higher risk in blood group A and protective effect in blood group O in a cohort of 1610 cases | Neutralizing antibodies against protein‐linked N‐glycans on SARS‐CoV‐2, or stabilization of vWF | [102, 103] |
| Cardiovascular disease | Increased CFR | Infection of cardiomyocytes, Increased myocarditis, impact of drugs on RAS. Increase levels of vWF | [97, 104] |
| 10·5% vf 2·3% | |||
| Cancer | 4·7%, 5·6% | unknown | [97, 105] |
| Diabetes mellitus | 7·3% | Reduced ACE2 levels in diabetes already predispose to a proinflammatory environment. Increased IL‐6 levels. Increased levels of vWF | [97, 106] |
| Gender | Increased CFR for males across all ages | Differential expression levels of ACE2, hormonal regulation of immune reposes, IL‐6 higher in men | [107, 108] |
| Ethnicity | Higher risk in some ethnic groups not due to socio‐economic conditions | Difference in TLR expression, levels of IL‐6 and TNF‐α | [109, 110] |
| Reduced levels of VitD | |||
| Obesity | BMI > 25 or 30 increased risk of severe pneumonia by 86% and 140% | Dysregulated NK cells, increased numbers of myeloid cells in adipose tissues and expression of ACE2 by adipocytes | [111, 112] |
ACE 2 = angiotensin‐converting enzyme 2; BMI = body mass index; CFR = case fatality ratio; NK cells = natural killer cells; TLR = Toll‐like receptors; vWF = von Willebrand factor.
Future perspectives
Understanding the innate immune factors that exacerbate the vascular complications will be crucial to control severe disease following SARS‐CoV‐2 infection. Rapidly emerging studies reveal the extent to which therapeutic approaches for other viral infections and inflammatory diseases can be repurposed to target innate immunity to treat COVID‐19 patients [113, 114]. Similarly, novel approaches have been put forward to target the susceptible ageing population or those with co‐morbidities. One approach under investigation is to re‐establish the youthful function of macrophages and repair mechanisms using metformin, a drug used in type II diabetes that has been shown to attenuate hallmarks of ageing [115]. In a retrospective study of 25 326 subjects tested for COVID‐19, while diabetes was reported to be an independent risk factor for COVID‐19‐related mortality [116], the risk in subjects taking metformin was significantly reduced (odds ratio = 0·33; 95% confidence interval = 0·13–0·84), suggesting that metformin might be protective in high‐risk populations, especially as metformin has also been reported to suppress neutrophil‐induced NETosis in vitro and reduce circulating NETosis biomarkers in vivo [117]. Thus, metformin and other drugs such as niacin [118],that rejuvenate the innate immune system, may be useful in COVID‐19.
Disclosures
D. B. and S. A. have received compensation for consultancies, presentations and advisory board activities, but no companies were involved in the decision to write and submit this manuscript. L. F. B. has nothing to disclose. S. A. has received consultancy from Novartis and Roche. D. B. has received compensation for activities related to Canbex therapeutics, InMune Biol, Lundbeck, Japan Tobacco, Merck and Novartis.
Author contributions
S. A. wrote the paper, L. F. B. produced Figs. 1and 2 and the Supplementary figure and made the first draft of the paper. D. B. made the second draft. All authors read and approved the final draft.
Supporting information
Fig. S1. Replication Cycle of SARS‐CoV‐2. Based on knowledge emerging from the SARS‐CoV‐2 infection and other coronaviruses, the cycle comprises of viral binding to the host cell via ACE‐2 (1) and virion uptake into the endosome. (2) The positive stranded RNA allows direct translation of the genome (which is capped by nsp – see text for details) generating the replicase polyprotein and subsequent viral proteins some of which are necessary to form the double membrane vesicles (DMV) (3). The replicase polyprotein enzyme synthesises the negative strands to transcribe the small subgenomic positive RNAs (4). These are used to produce the other viral proteins (N, S, M and E and accessory proteins) and the positive RNA strands for the new virions. The nucleocapsid protein binds to the RNA. The S, M and E proteins are incorporated in the lipid envelope in the ER. The new virons assemble in the ER‐Golgi intermediate compartment (ERGIC) and exocytosed in the Golgi complex (6). Finally, mature SARS‐CoV‐2 virions are r eleased from the host cell (7).
Acknowledgements
This study received no funding. The authors thank Alison Schroeer (Schroeer Scientific Illustration) for advice and assistance concerning the figure production and Dr Roger D. Seheult, (MedCram) for invaluable insights relating to pulmonary medicine and COVID‐19.
References
- 1. Yeager CL, Ashmun RA, Williams RK et al Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992; 357:420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bertram S, Dijkman R, Habjan M et al TMPRSS2 activates the human coronavirus 229E for cathepsin‐independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 2013; 87:6150–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonavia A, Arbour N, Yong VW, Talbot PJ. Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J Virol 1997; 71:800–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collins AR. HLA class I antigen serves as a receptor for human coronavirus OC43. Immunol Invest 1993; 22:95–103. [DOI] [PubMed] [Google Scholar]
- 5. Vlasak R, Luytjes W, Spaan W, Palese P. Human and bovine coronaviruses recognize sialic acid‐containing receptors similar to those of influenza C viruses. Proc Natl Acad Sci USA 1988; 85:4526–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marzi A, Gramberg T, Simmons G et al DC‐SIGN and DC‐SIGNR interact with the glycoprotein of marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J Virol 2004; 78:12090–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeffers SA, Tusell SM, Gillim‐Ross L et al CD209L (L‐SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci USA 2004; 101:15748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hofmann H, Pyrc K, Van Der Hoek L, Geier M, Berkhout B, Pöhlmann S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci USA 2005; 102:7988–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan CM, Lau SKP, Woo PCY et al Identification of major histocompatibility complex class I C molecule as an attachment factor that facilitates coronavirus HKU1 spike‐mediated infection. J Virol 2009; 83:1026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two‐step, furin‐mediated activation of the spike protein. Proc Natl Acad Sci USA 2014; 111:15214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann M, Kleine‐Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS‐CoV‐2 is essential for infection of human lung cells. Mol Cell 2020; 78:779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen G, Wu D, Guo W et al Clinical and immunologic features in severe and moderate coronavirus disease 2019. J Clin Invest 2020; 130:2620–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee S, Meyler P, Mozel M, Tauh T, Merchant R. Asymptomatic carriage and transmission of SARS‐CoV‐2: what do we know? Can J Anaesth 2020; 67:1424–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rokkas T. Gastrointestinal involvement in COVID‐19: a systematic review and meta‐analysis. Ann Gastroenterol 2020; 33:355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hendren NS, Drazner MH, Bozkurt B, Cooper LT. Description and proposed management of the acute COVID‐19 cardiovascular syndrome. Circulation 2020; 141:1903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ellul MA, Benjamin L, Singh B et al Neurological associations of COVID‐19. Lancet Neurol 2020; 19:767–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshimoto FK. The proteins of severe acute respiratory syndrome coronavirus‐2 (SARS CoV‐2 or n‐COV19), the cause of COVID‐19. Protein J 2020; 39:198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thevarajan I, Nguyen THO, Koutsakos M et al Breadth of concomitant immune responses prior to patient recovery: a case report of non‐severe COVID‐19. Nat Med 2020; 26:453–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grifoni A, Weiskopf D, Ramirez SI et al Targets of T cell responses to SARS‐CoV‐2 coronavirus in humans with COVID‐19 disease and unexposed individuals. Cell 2020; 181:1489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Biasi S, Meschiari M, Gibellini L et al Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID‐19 pneumonia. Nat Commun 2020; 11:3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu R, Zhao X, Li J et al Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020; 395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grant OC, Montgomery D, Ito K, Woods RJ. 3D Models of glycosylated SARS‐CoV‐2 spike protein suggest challenges and opportunities for vaccine development. bioRxiv 2020. 10.1101/2020.04.07.030445. [DOI] [Google Scholar]
- 23. Walls AC, Tortorici MA, Frenz B et al Glycan shield and epitope masking of a coronavirus spike protein observed by cryo‐electron microscopy. Nat Struct Mol Biol 2016; 23:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daly JL, Simonetti B, Antón‐Plágaro C et al Neuropilin‐1 is a host factor for SARS‐CoV‐2 infection. bioRxiv 2020. 10.1101/2020.07.17.209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu DX, Fung TS, Chong KK‐L, Shukla A, Hilgenfeld R. Accessory proteins of SARS‐CoV and other coronaviruses. Antiviral Res 2014; 109:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xia X. Extreme genomic CpG deficiency in SARS‐CoV‐2 and evasion of host antiviral defense. Mol Biol Evol 2020; 37:2699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jauregui AR, Savalia D, Lowry VK, Farrell CM, Wathelet MG. Identification of residues of SARS‐CoV nsp1 that differentially affect inhibition of gene expression and antiviral signaling. PLOS ONE 2013; 8:e62416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol 2007; 81:11620–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kamitani W, Narayanan K, Huang C et al Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci USA 2006; 103:12885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graham RL, Sims AC, Brockway SM, Baric RS, Denison MR. The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J Virol 2005; 79:13399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cornillez‐Ty CT, Liao L, Yates JR 3rd, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol 2009; 83:10314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Báez‐Santos YM, St. John SE, Mesecar AD. The SARS‐coronavirus papain‐like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 2015; 115:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double‐membrane vesicles. MBio 2013; 4:e00524–e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z. SARS coronavirus papain‐like protease inhibits the type I interferon signaling pathway through interaction with the STING–TRAF3–TBK1 complex. Protein Cell 2014; 5:369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin C‐W, Lin K‐H, Hsieh T‐H, Shiu S‐Y, Li J‐Y. Severe acute respiratory syndrome coronavirus 3C‐like protease‐induced apoptosis. FEMS Immunol Med Microbiol 2006; 46:375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kirchdoerfer RN, Ward AB. Structure of the SARS‐CoV nsp12 polymerase bound to nsp7 and nsp8 co‐factors. Nat Commun 2019; 10:2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sutton G, Fry E, Carter L et al The nsp9 replicase protein of SARS‐coronavirus, structure and functional insights. Structure 2004; 12:341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Egloff M‐P, Ferron F, Campanacci V et al The severe acute respiratory syndrome‐coronavirus replicative protein nsp9 is a single‐stranded RNA‐binding subunit unique in the RNA virus world. Proc Natl Acad Sci USA 2004; 101:3792–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma Y, Wu L, Shaw N et al Structural basis and functional analysis of the SARS coronavirus nsp14‐nsp10 complex. Proc Natl Acad Sci USA 2015; 112:9436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Q, Wang L, Dong C et al The interaction of the SARS coronavirus non‐structural protein 10 with the cellular oxido‐reductase system causes an extensive cytopathic effect. J Clin Virol 2005; 34:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Decroly E, Debarnot C, Ferron F et al Crystal structure and functional analysis of the SARS‐Coronavirus RNA cap 2′‐O‐methyltransferase nsp10/nsp16 complex. Rey FA, editor. PLOS Pathog 2011; 7:e1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ivanov KA, Thiel V, Dobbe JC, van der Meer Y, Snijder EJ, Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J Virol 2004; 78:5619–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Minskaia E, Hertzig T, Gorbalenya AE et al Discovery of an RNA virus 3′–>5′ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc Natl Acad Sci USA 2006; 103:5108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deng X, Hackbart M, Mettelman RC et al Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc Natl Acad Sci USA 2017; 114:4251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Menachery VD, Yount BL, Josset L et al Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′‐O‐methyltransferase activity. J Virol 2014; 88:4251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Menachery VD, Debbink K, Baric RS. Coronavirus non‐structural protein 16: evasion, attenuation, and possible treatments. Virus Res 2014; 194:191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walls AC, Park Y‐J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS‐CoV‐2 spike glycoprotein. Cell 2020; 81:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Imai Y, Kuba K, Penninger JM. The discovery of angiotensin‐converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol 2008; 93:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe Y, Allen JD, Wrapp D, McLellan JS, Crispin M. Site‐specific glycan analysis of the SARS‐CoV‐2 spike. Science 2020; 369:330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Padhan K, Tanwar C, Hussain A et al Severe acute respiratory syndrome coronavirus ORF3a protein interacts with caveolin. J Gen Virol 2007; 88:3067–77. [DOI] [PubMed] [Google Scholar]
- 51. Kopecky‐Bromberg SA, Martínez‐Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 2007; 81:548–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, Jameel S. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand‐independent downregulation of the type 1 interferon receptor. PLOS ONE 2009; 4:e8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nieto‐Torres JL, DeDiego ML, Verdiá‐Báguena C et al Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLOS Pathog 2014; 10:e1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Álvarez E, DeDiego ML, Nieto‐Torres JL, Jiménez‐Guardeño JM, Marcos‐Villar L, Enjuanes L. The envelope protein of severe acute respiratory syndrome coronavirus interacts with the non‐structural protein 3 and is ubiquitinated. Virology 2010; 402:281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fang X, Gao J, Zheng H et al The membrane protein of SARS‐CoV suppresses NF‐kappaB activation. J Med Virol 2007; 79:1431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Siu K‐L, Kok K‐H, Ng M‐HJ et al Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem 2009; 284:16202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chan C‐M, Ma C‐W, Chan W‐Y, Chan HYE. The SARS–coronavirus membrane protein induces apoptosis through modulating the Akt survival pathway. Arch Biochem Biophys 2007; 459:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Taylor JK, Coleman CM, Postel S et al Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J Virol 2015; 89:11820–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuan X, Wu J, Shan Y et al SARS coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology 2006; 346:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schaecher SR, Mackenzie JM, Pekosz A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS‐CoV) is expressed in virus‐infected cells and incorporated into SARS‐CoV particles. J Virol 2007; 81:718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Y, Zhang J, Chen Y, et al The ORF8 protein of SARS‐CoV‐2 mediates immune evasion through potently downregulating MHC‐I. bioRxiv. 2020. Available at: https://www.biorxiv.org/content/10.1101/2020.05.24.111823v1. [Google Scholar]
- 62. Hu Y, Li W, Gao T et al The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25‐mediated RIG‐I ubiquitination. J Virol 2017; 91:e02143–e2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shi C‐S, Qi H‐Y, Boularan C et al SARS‐coronavirus open reading frame‐9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol 2014; 193:3080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ma Z, Damania B. The cGAS‐STING defense pathway and its counteraction by viruses. Cell Host Microbe 2016; 19:150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bai J, Liu F. The cGAS–cGAMP–STING pathway: a molecular link between immunity and metabolism. Diabetes 2019; 68:1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Posthuma CC, te Velthuis AJW, Snijder EJ. Nidovirus RNA polymerases: complex enzymes handling exceptional RNA genomes. Virus Res 2017; 234:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fung TS, Liu DX. Post‐translational modifications of coronavirus proteins: roles and function. Future Virol 2018; 13:405–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xia S, Liu M, Wang C et al Inhibition of SARS‐CoV‐2 (previously 2019‐nCoV) infection by a highly potent pan‐coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 2020; 30:343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shang J, Wan Y, Luo C et al Cell entry mechanisms of SARS‐CoV‐2. Proc Natl Acad Sci 2020; 117:11727–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Magro C, Mulvey JJ, Berlin D et al Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID‐19 infection: a report of five cases. Transl Res 2020; 220:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ramlall V, Thangaraj P, Meydan C, et al Identification of immune complement function as a determinant of adverse SARS‐CoV‐2 infection outcome. medRxiv 2020. 10.1101/2020.05.05.20092452. [DOI] [Google Scholar]
- 72. Ip WKE, Chan KH, Law HKW et al Mannose‐binding lectin in severe acute respiratory syndrome coronavirus infection. J Infect Dis 2005; 191:1697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b‐9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP‐140 to the cell surface. J Biol Chem 1989; 264:9053–60. [PubMed] [Google Scholar]
- 74. de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol 2019; 16:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gralinski LE, Sheahan TP, Morrison TE et al Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. MBio 2018; 9:e01753–e1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS‐CoV‐2 spike glycoprotein with ACE‐2 receptor homologs and human TLRs. J Med Virol 2020; 92:2105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dosch SF, Mahajan SD, Collins AR. SARS coronavirus spike protein‐induced innate immune response occurs via activation of the NF‐κB pathway in human monocyte macrophages in vitro . Virus Res 2009; 142:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Totura AL, Whitmore A, Agnihothram S et al Toll‐like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio 2015; 6:e00638‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Berthelot J‐M, Lioté F. COVID‐19 as a STING disorder with delayed over‐secretion of interferon‐beta. EBioMed 2020; 56:102801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu Y, Jesus AA, Marrero B et al Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014; 7:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xie J, Li Y, Shen X et al Dampened STING‐dependent interferon activation in bats. Cell Host Microbe 2018; 23:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen G, Chen D, Li J et al Pathogenic role of HMGB1 in SARS? Med Hypoth 2004; 63:691–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schaefer L. Complexity of danger: the diverse nature of damage‐associated molecular patterns. J Biol Chem 2014; 289:35237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vogel S, Bodenstein R, Chen Q et al Platelet‐derived HMGB1 is a critical mediator of thrombosis. J Clin Invest 2015; 125:4638–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Andersson U, Ottestad W, Tracey KJ. Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID‐19? Mol Med 2020; 26:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nieto‐Torres JL, Verdiá‐Báguena C, Jimenez‐Guardeño JM et al Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015; 485:330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Xiong Y, Liu Y, Cao L et al Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID‐19 patients. Emerg Microbes Infect 2020; 9:761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bost P, Giladi A, Liu Y et al Host–viral infection maps reveal signatures of severe COVID‐19 patients. Cell 2020; 181:1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li J, Guo M, Tian X et al Virus–host interactome and proteomic survey of PBMCs from COVID‐19 patients reveal potential virulence factors influencing SARS‐CoV‐2 pathogenesis. Med (NY) 2020. 10.1016/j.medj.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Law HKW, Cheung CY, Ng HY et al Chemokine up‐regulation in SARS‐coronavirus‐infected, monocyte‐derived human dendritic cells. Blood 2005; 106:2366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The cytokine storm in COVID‐19: an overview of the involvement of the chemokine/chemokine‐receptor system. Cytokine Growth Factor Rev 2020; 53:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Blanco‐Melo D, Nilsson‐Payant BE, Liu W‐C et al Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell 2020; 181:1036–1045.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schönrich G, Raftery MJ, Samstag Y. Devilishly radical NETwork in COVID‐19: oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv Biol Regul 2020; 77:100741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Varga Z, Flammer AJ, Steiger P et al Endothelial cell infection and endotheliitis in COVID‐19. Lancet 2020; 395:1417–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. He L, Mäe MA, Sun Y, et al Pericyte‐specific vascular expression of SARS‐CoV‐2 receptor ACE2 – implications for microvascular inflammation and hypercoagulopathy in COVID‐19 patients. bioRxiv 2020. 10.1101/2020.05.11.088500. [DOI] [Google Scholar]
- 96. Cardot‐Leccia N, Hubiche T, Dellamonica J, Burel‐Vandenbos F, Passeron T. Pericyte alteration sheds light on micro‐vasculopathy in COVID‐19 infection. Intens Care Med 2020; 46:1777–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China. JAMA 2020; 323:1239. [DOI] [PubMed] [Google Scholar]
- 98. Molony RD, Malawista A, Montgomery RR. Reduced dynamic range of antiviral innate immune responses in aging. Exp Gerontol 2018; 107:130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cannizzo ES, Clement CC, Sahu R, Follo C, Santambrogio L. Oxidative stress, inflamm‐aging and immunosenescence. J Proteomics 2011; 74:2313–23. [DOI] [PubMed] [Google Scholar]
- 100. Ellinghaus D, Degenhardt F, Bujanda L et al Genomewide association study of severe COVID‐19 with respiratory failure. N Engl J Med 2020. 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM. Metabolic alterations in aging macrophages: ingredients for inflammaging? Trends Immunol 2019; 40:113–27. 10.1016/j.it.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 102. Breiman A, Ruvën‐Clouet N, Le Pendu J. Harnessing the natural anti‐glycan immune response to limit the transmission of enveloped viruses such as SARS‐CoV‐2. PLOS Pathog 2020; 16:e1008556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Murray GP, Post SR, Post GR. ABO blood group is a determinant of von Willebrand factor protein levels in human pulmonary endothelial cells. J Clin Pathol 2020; 73:347–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Guzik TJ, Mohiddin SA, Dimarco A et al COVID‐19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res 2020; 116:1666–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sidaway P. COVID‐19 and cancer: what we know so far. Nat Rev Clin Oncol 2020; 17:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pal R, Bhadada SK. COVID‐19 and diabetes mellitus: an unholy interaction of two pandemics. Diabetes Metab Syndr Clin Res Rev 2020; 14:513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gebhard C, Regitz‐Zagrosek V, Neuhauser HK, Morgan R, Klein SL. Impact of sex and gender on COVID‐19 outcomes in Europe. Biol Sex Differ 2020; 11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bonafè M, Prattichizzo F, Giuliani A, Storci G, Sabbatinelli J, Olivieri F. Inflamm‐aging: why older men are the most susceptible to SARS‐CoV‐2 complicated outcomes. Cytokine Growth Factor Rev 2020; 53:33–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Niedzwiedz CL, O’Donnell CA, Jani BD et al Ethnic and socioeconomic differences in SARS‐CoV‐2 infection: prospective cohort study using UK Biobank. BMC Med 2020; 18:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lyn‐Cook BD, Xie C, Oates J et al Increased expression of Toll‐like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol 2014; 61:38–43. [DOI] [PubMed] [Google Scholar]
- 111. Cai Q, Chen F, Wang T et al Obesity and COVID‐19 severity in a designated hospital in Shenzhen, China. Diabetes Care 2020; 43:1392–8. [DOI] [PubMed] [Google Scholar]
- 112. O’Shea D, Hogan AE. Dysregulation of natural killer cells in obesity. Cancers 2019; 11:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Schijns V, Lavelle EC. Prevention and treatment of COVID‐19 by control of innate immunity. Eur J Immunol 2020; 50:932–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Encinar JA, Menendez JA. Potential drugs targeting early innate immune evasion of SARS‐coronavirus 2 via 2'‐O‐methylation of viral RNA. Viruses 2020; 12:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab 2020; 32:15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Crouse A, Grimes T, Li P, Might M, Ovalle F, Shalev A. Metformin use is associated with reduced mortality in a diverse population with COVID‐19 and diabetes. medRxiv 2020. 10.1101/2020.07.29.20164020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Menegazzo L, Scattolini V, Cappellari R. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo . Acta Diabetol 2018; 55:593–601. [DOI] [PubMed] [Google Scholar]
- 118. Rawji KS, Young AMH, Ghosh T et al Niacin‐mediated rejuvenation of macrophage/microglia enhances remyelination of the aging central nervous system. Acta Neuropathol 2020; 139:893–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Replication Cycle of SARS‐CoV‐2. Based on knowledge emerging from the SARS‐CoV‐2 infection and other coronaviruses, the cycle comprises of viral binding to the host cell via ACE‐2 (1) and virion uptake into the endosome. (2) The positive stranded RNA allows direct translation of the genome (which is capped by nsp – see text for details) generating the replicase polyprotein and subsequent viral proteins some of which are necessary to form the double membrane vesicles (DMV) (3). The replicase polyprotein enzyme synthesises the negative strands to transcribe the small subgenomic positive RNAs (4). These are used to produce the other viral proteins (N, S, M and E and accessory proteins) and the positive RNA strands for the new virions. The nucleocapsid protein binds to the RNA. The S, M and E proteins are incorporated in the lipid envelope in the ER. The new virons assemble in the ER‐Golgi intermediate compartment (ERGIC) and exocytosed in the Golgi complex (6). Finally, mature SARS‐CoV‐2 virions are r eleased from the host cell (7).