

Abstract
Background
To better predict response to immune checkpoint therapy and toxicity in healthy tissues, insight in the in vivo behavior of immune checkpoint targeting monoclonal antibodies is essential. Therefore, we aimed to study in vivo pharmacokinetics and whole-body distribution of zirconium-89 (89Zr) labeled programmed cell death protein-1 (PD-1) targeting pembrolizumab with positron-emission tomography (PET) in humanized mice.
Methods
Humanized (huNOG) and non-humanized NOG mice were xenografted with human A375M melanoma cells. PET imaging was performed on day 7 post 89Zr-pembrolizumab (10 µg, 2.5 MBq) administration, followed by ex vivo biodistribution studies. Other huNOG mice bearing A375M tumors received a co-injection of excess (90 µg) unlabeled pembrolizumab or 89Zr-IgG4 control (10 µg, 2.5 MBq). Tumor and spleen tissue were studied with autoradiography and immunohistochemically including PD-1.
Results
PET imaging and biodistribution studies showed high 89Zr-pembrolizumab uptake in tissues containing human immune cells, including spleen, lymph nodes and bone marrow. Tumor uptake of 89Zr-pembrolizumab was lower than uptake in lymphoid tissues, but higher than uptake in other organs. High uptake in lymphoid tissues could be reduced by excess unlabeled pembrolizumab. Tracer activity in blood pool was increased by addition of unlabeled pembrolizumab, but tumor uptake was not affected. Autoradiography supported PET findings and immunohistochemical staining on spleen and lymph node tissue showed PD-1 positive cells, whereas tumor tissue was PD-1 negative.
Conclusion
89Zr-pembrolizumab whole-body biodistribution showed high PD-1-mediated uptake in lymphoid tissues, such as spleen, lymph nodes and bone marrow, and modest tumor uptake. Our data may enable evaluation of 89Zr-pembrolizumab whole-body distribution in patients.
Keywords: immunotherapy, T-lymphocytes, tumor biomarkers
Background
Immune checkpoint inhibitors targeting the programmed cell death protein-1 (PD-1/programmed death ligand-1 (PD-L1) pathway are showing impressive antitumor effects. However, not all patients respond and serious immune-related toxicity has been reported.1 This has raised interest in better understanding the behavior of these drugs in the human body. PD-L1 and PD-1 are expressed by a broad range of immune cells, including T-cells, B-cells, natural killer (NK) cells, monocytes and dendritic cells. PD-L1 can be highly expressed by tumor cells, whereas PD-1 expression is most prominent in T-cells and lower in other immune cells.2 Biodistribution of PD-1 and PD-L1 targeting drugs will likely be influenced by the dynamic expression patterns of these targets.
Molecular imaging has proven to be an useful tool for studying drug biodistribution.3 4 In table 1, we summarized preclinical imaging studies that investigated biodistribution of radiolabeled molecules targeting PD-1 and PD-L1.5–28 Most studies that we reviewed focused on tracer distribution in the tumor and its microenvironment, without considering PD-1 and PD-L1 expression in healthy immune tissues. Studies that do report on tracer uptake in lymphoid tissues are scarce and results are often limited to the spleen. Furthermore, most tracers targeting human PD-1/PD-L1 are not cross-reactive with murine proteins and relevant mouse models reconstituted with (parts of) a human immune system are rarely used. A limited number of studies used NOD scid gamma (NSG) mice engrafted with human peripheral blood mononuclear cells (hNSG model).23–25 27 The hNSG model has a high level of functional T-cells, however, it is also characterized by aberrant distribution of immune cells to murine immune tissues and other cell lineages remain underdeveloped.29 Humanized mice that are engrafted with human CD34 + hematopoietic stem cells (HSCs) establish an immune-competent model with a broader set of developed human immune cells present and might therefore be a better surrogate for the human immune environment.
Table 1.
Preclinical imaging studies targeting PD-L1 and PD-1, using radiolabeled monoclonalantibody or small proteins
| Type of Imaging |
Tracer | Origin and reactivity | Cross reactivity | Animal model | Tumor model | Tracer dose | Imaging / biodistribution time point | Tumor uptake | Uptake lymphoid tissue | Ref |
| Anti-PD-L1 - antibodies | ||||||||||
| SPECT/CT | 111In-PD-L1.3.1 antibody | Murine anti-human | No | Balb/c nude mice 6 to 8 weeks old Immune deficient |
Human breast cancer cell lines | 1.5 µg (15.5 MBq) and 1.0 µg (10.0 MBq) | Imaging and ex vivo biodistribution at 24, 72 and 168 hours pi | 32.8 (±6.8) %ID/g and 6.2 (±1.0) %ID/g at 168 hours pi for MDA-MB-231 and MCF-7 tumors respectively PD-L1 detection at different expression levels in SK-Br-3, SUM149 and BT474 tumors |
No | (5) |
| SPECT | 111In-DTPA-PD-L1 antibody | Hamster anti-mouse | No |
neu-N transgenic mice 8 to 12 weeks old Immune competent |
NT2.5 (mouse mammary tumor) | 7.4 MBq for imaging and 8.4 µg (0.93 MBq) for biodistribution | Imaging on 1, 24, and 72 days pi and ex vivo biodistribution at 1, 24, 72, and 144 hours pi | Tumor uptake of 21.1 (±11.2) %ID/g at 144 hours pi | Yes, spleen (63.5%±25.4 %ID/g) and thymus (16.8%±16.2 %ID/g) at 144 hours pi Spleen uptake was blocked by coinjection of unlabeled antibody |
(6) |
| SPECT | 111In-PD-L1 antibody | Humanized anti-human | Cross-reactive with mouse | NSG mice 6 to 8 weeks old Immune deficient |
Human cell lines | 100 µg (14.8 MBq) for imaging and 8.5 µg (1.48 MBq) for biodistribution | Imaging and ex vivo biodistribution at 24, 48, 72, 96 and 120 hours pi | 8.9 (±0.26) %ID/g at 72 hours pi for MDA-MB-231 tumors and 7.46 (±0.12) at 144 hours pi for H2444 tumors Detection of PD-L1 at different expression levels |
Yes, spleen (23.5±8.2) at 48 hours pi Spleen uptake was blocked by co-injection of unlabeled antibody |
(7) |
| PET | 64Cu-PD-L1 antibody | Humanized anti-human | Cross-reactive with mouse | NSG mice 6 to 8 weeks old Immune deficient |
Human cell lines | 16.7 MBq (40 µg) for imaging and 1.48 MBq (10 µg) for biodistribution | Imaging on 2, 24 and 48 hours pi and ex vivo biodistribution at 24 and 48 hours pi | 40.6 (±6.9) %ID/g, 17.2 (±2.1) %ID/g and 9.4 (±2.3) %ID/g at 48 hours pi for PD-L1 positive CHO, MDA-MB-231 and SUM149 tumors respectively | High spleen uptake (~45 %ID/g) at 24 hours pi after blocking with unlabeled antibody | (8) |
| Balb/c mice 4 to 6 weeks old Immune competent |
4T1 (mouse mammary carcinoma) | 17.0 (±4.3) %ID/g at 48 hours pi for 4T1 tumors | No high uptake observed in spleen (±12 %ID/g) and BAT | |||||||
| SPECT | 111In-DTPA-PD-L1 antibody | Rat anti-mouse | No | C57BL/6 mice 6 to 8 weeks old Immune competent |
B16F10 (murine melanoma) | 15–16 MBq (60 µg) for imaging and 0.37 MBq (0.13 mg/kg) | Imaging on 1, 24 and 72 hours pi and biodistribution at 1, 24, 72 and 96 hours pi | 6.6 (±3.1) %ID/g at 24 hours pi for B16F10 tumors | Yes, spleen (47%±9.5 %ID/g) at 24 hours pi and BAT Spleen uptake was blocked by coinjection of unlabeled antibody |
(9) |
| PET | 89Zr-anti-PD-L1 antibody | Rat anti-mouse | No | C57BL/6 mice 6 to 8 weeks old Immune competent |
MEER (murine tonsil epithelium) or B16F10 (murine melanoma) | 3.7 MBq (50 µg) | Imaging and ex vivo biodistribution at 48 and 96 hours pi | Higher uptake in irradiated (20.1%±2.6 %ID/g) vs non-irradiated (11.1%±1.9 %ID/g) MEER tumors Higher uptake in irradiated (28.0%±4.9 %ID/g) vs non-irradiated (14.4%±1.4 %ID/g) B16F10 tumors |
Yes, spleen (60% to 120%ID/g) and thymus (25% to 35%ID/g) Spleen uptake was blocked by pre-injection of unlabeled antibody |
(10) |
| PET | 89Zr-C4 (recombinant IgG1 antibody) | Engineered anti-human | Cross-reactive mouse | Nu/nu mice 3 to 5 weeks old Immune deficient |
H1975 and A549 (human NSCLC), PC3 (human prostatic small cell carcinoma) | 11.1 MBq for imaging and 1.85 MBq for ex vivo biodistribution | Imaging and ex vivo biodistribution at 8, 24, 48, 72, 120 hours pi | ~9 %ID/g~5 %ID/g and ~7 %ID/g at 48 hours pi for H1975, A459 and PC3 tumors respectively ~13% ID/g at 48 hours pi for B16F16 tumors ~5 %ID/g tumor uptake at 48 hours pi in PDX model Detection of pharmacological-induced changes in PD-L1 expression |
Yes, spleen uptake of ~7 %ID/g and ~6 %ID/g at 48 hours pi in nu/nu and C57BL/6 mice respectively Increased uptake in the spleens of nu/nu mice treated with paclitaxel and spleens of C57BL/6 mice treated with doxorubicin |
(11) |
| C57BL/6 mice 3 to 5 weeks old Immune deficient |
B16F10 (mouse melanoma) | |||||||||
| Not reported | PDX model of EGFR mutant (L858R) NSCLC | |||||||||
| SPECT/CT | 111In-anti-mPD-L1 | Rat anti-murine | No | Balb/c and C57BL/6 6 to 8 weeks old Immune competent |
Murine cell lines | 19.7 (±1.2) MBq (30 µg) | Imaging and ex vivo biodistribution at 72 hours pi | ~14.53 (±5.49) %ID/g,~16.29 (±5.57) %ID/g,~11.06 (±6.54) %ID/g,~14.94 (±4.01) %ID/g,~6.16 (±2.94) %ID/g, for Renca, 4T1, CT26, B16F1 and LLC1 respectively | Yes, spleen varying from 13.09 %ID/g to 40.30 %ID/g. Thymus varying from 6.09 %ID/g to 10.26 %ID/g |
(12) |
| 111In-anti-hPD-L1 | Murine anti-human | No | Non-humanized and humanized NSG mice | MDA-MB-231 (human breast carcinoma) | 11.9±1.6 MBq (1 µg) 111In-anti-hPD-L1 Or 11.5±0.4 MBq (2.8 µg) 111In-control mIgG1 LPS treatment 1 day before tracer injection |
Imaging and ex vivo biodistribution at 72 hours pi | ~40 %ID/g for non-humanized mice and ~60 %ID/g for humanized mice at 72 hours pi ~35 %ID/g for humanized mice after LPS treatment at 72 hours pi ~8 %ID/g for 111In-control mIgG1 in both non-humanized and humanized mice at 72 hours pi |
Yes, spleen uptake ~20% ID/g for non-humanize mice and ~25% ID/g for humanized mice ~80 %ID/g for humanized mice after LPS treatment at 72 hours pi ~10 %ID/g for 111In-control mIgG1 (both groups |
||
| 111In-anti-mPD-L1 | Rat anti-murine | No | Balb/c and C57BL/6 6 to 8 weeks old Immune competent |
Murine cell lines | Irradiation followed on day one by injection of 23.8±1.7 MBq (30 µg) | Imaging and ex vivo biodistribution at 24 hours pi | Higher uptake in irradiated (26.3%±2.0 %ID/g) vs non-irradiated (17.1%±3.1 %ID/g) CT26 tumors Higher uptake in irradiated (15.7%±1.8 %ID/g) vs non-irradiated (12.3%±1.7% ID/g) LLC1 tumors No difference uptake in irradiated (14.9%±6.8 %ID/g) vs non-irradiated (16.7%±3.5% ID/g) for B16F1 tumors |
Spleen uptake ~14% to 17%ID/g for all models Higher uptake in lymph nodes of irradiated tumor models vs non-irradiated tumor models |
||
| Anti-PD-L1 - small molecules | ||||||||||
| PET | 64Cu-WL12 (PD-L1 binding peptide) | Engineered anti-human | No | NSG mice 6 to 8 weeks old Immune deficient |
High PD-L1-expressing CHO cell line | 5.6 MBq for imaging and 1.5 MBq for ex vivo biodistribution | Imaging and ex vivo biodistribution at 10 min, 0.5, 1 and 2 hour pi | 14.9 (±0.8) at 1 hour pi in hPD-L1-expressing CHO tumors | No | (13) |
| PET | 18F-AlF-NOTA-ZPD-L1_ (anti PD-L1 small molecule, affibody) | Engineered anti-human affibody | No | SCID beige mice 6 to 8 weeks old Immune deficient |
LOX-IMVI (human melanoma) and SUDHL6 (human B-cell lymphoma) | 0.2 to 0.6 MBq |
Dynamic PET scan during 90 min | 2.56 (±0.33) %ID/g at 90 min pi for LOX tumors | No | (14) |
| SPECT | 99mTC-anti-PD-L1 nanobodies | Engineered anti-mouse nanobodies | Cross-reactive human | C57BL/6 mice (WT) vs CD8 depleted PD-L1 KO mice 6 weeks old Immune competent |
TC-1 (mouse lung epithelial), WT TC-1 PD-L1+ vs CRISPR/Cas9-modified TC-1 PD-L1 KO |
45 to 155 MBq (10 µg) nanobody | Imaging 1 hour pi and ex vivo biodistribution 80 min pi | 1.7 (±0.1) %ID/g for WT and 1.1 (±0.3) %ID/g for KO at 80 min pi | Yes, spleen 11.4 (±1.4) %ID/g for WT and 1.6±0.2% ID/g for KO at 80 min pi Lymph node uptake 3.5 (±0.8) %ID/g for WT and 0.4 (±0.1) %ID/g for KO at 80 min pi |
(15) |
| PET | 64Cu-PD-1 ectodomain targeting PD-L1 | Engineered anti-human | Not specified | NSG mice Immune deficient |
CT26 (mouse colon cancer) hPD-L1 (+) or hPD-L1(-) | 8.5 MB (25 µg) | Imaging at 1, 2, 4, and 24 hours. Ex vivo biodistribution at 1 and 24 hours | ~3 %ID/g for PD-L1 (+) and ~1.8 %ID/g for PD-L1 (-) at 24 hours pi |
Yes, spleen ~5 %ID/g at 24 hours pi | (16) |
| PET | 18F-BMS-986192 (anti-PD-L1 small molecule) | Engineered anti-human | Affinity for human & cynomolgus PD-L1, no binding to murine PD-L1) | Immune deficient mice | Human L2987 (PD-L1+) and HT-29 (PD-L1-) | 5.6 MBq, block to 3 mg/kg | Dynamic PET scan during 120 min | 2.41 (±0.29) %ID/g for PD-L1 +and 0.82 (±0.11) %ID/g for PD-L1-, 0.79 (±0.12) %ID/g after blocking in PD-L+ | Yes, spleen uptake (no clear numbers) | (17) |
| Cynomolgus monkeys | – | 55.5 MBq | Dynamic PET scan during 150 min | – | Yes, spleen:muscle 12:1, after blocking spleen:muscle1.24:1 | |||||
| PET | 64Cu-PD-1 ectodomains (DOTA-/NOTA-HAC, aglycosylated DOTA-/NOTA-HACA) | Engineered anti-human | Not specified | NSG mice 6 to 8 weeks old Immune deficient |
CT26 (mouse colon cancer) hPD-L1(+) or hPD-L1(-) | 0.7–3.7 MBq (10 to 15 µg) | Imaging and ex vivo biodistribution at 1 hour pi | 1.8 (±0.2) %ID/g for PD-L1(+) and 0.9 (±0.7) %ID/g PD-L1(-) for 64Cu-NOTA-HAC-PD1 at 1 hour pi 4.2 (±0.8) %ID/g for PD-L1(+) and 3.5 (±1.7) %ID/g for PD-L1(-) for 64Cu-NOTA-HAC-PD1 at 1 hour pi 2.7 (±1.1) %ID/g for PD-L1(+) and 0.8 (±0.4) %ID/g for PD-L1(-) for 64Cu-NOTA-HACA-PD1 at 1 hour pi |
Yes, spleen 4.0 (±3.1) %ID/g, 5.5 (±1.4) %ID/g and 1.4 (±0.4) %ID/g for 64Cu-DOTA-HAC-PD1, 64Cu-NOTA-HAC-PD1, and 64Cu-NOTA-HACA-PD1 respectively | (18) |
| 68Ga- PD-1 ectodomains (DOTA-/NOTA-HAC, aglycosylated DOTA-/NOTA-HACA) | Engineered anti-human | Not specified | NSG mice 6 to 8 weeks old Immune deficient |
CT26 (mouse colon cancer) hPD-L1(+) or hPD-L1(-) | 0.7 to 3.7 MBq (10 to 15 µg) | Imaging and ex vivo biodistribution at 1 hour pi | 3.8 (±1.6) %ID/g for PD-L1(+) and 1.7 (±1.3) %ID/g for PD-L1(-) for 68Ga-NOTA-HACA-PD1 at 1 hour pi 2.8 (±1.5) %ID/g for PD-L1(+) and 0.8 (±0.1) %ID/g for PD-L1(-) for 68Ga-DOTA-HACA-PD1 at 1 hour pi |
Yes, spleen 3.5 (±0.6) %ID/g and 0.2 (±0.2) %ID/g for 68Ga-NOTA-HACA-PD1 and 68Ga-DOTA-HACA-PD1 respectively |
||
| PET | 64Cu-FN3hPD-L1 | Small molecule anti-human | No | CT26/hPD-L1 | 3.7 (±0.4) MBq (8 to 10 µg) | Imaging at 0.5, 1, 4, 18, and 24 hours pi followed by ex vivo biodistribution | 5.6 (±0.9) %ID/g at 24 hours pi for CT26/hPD-L1 tumors | No | (19) | |
| MDA-MB-231 (human breast cancer) | 3.6 (±0.5) %ID/g at 24 hours pi for MDA-MB-231 tumors | |||||||||
| PET | 68Ga-WL12 (PD-L1 binding peptide) | Engineered anti-human | No | NSG mice 6 to 8 weeks old Immune deficient |
Human cell lines | ±7.4 MBq for imaging and ±0.9 MBq for ex vivo biodistribution | Imaging and ex vivo biodistribution at 15, 60, and 120 min pi | 11.56 (±3.18) %ID/g, 4.97 (±0.8) %ID/g and 1.9 (±0.1) %ID/g for hPD-L1, MDA-MB-231 and SUM149 tumors respectively at 60 min pi |
No | (20) |
| PET |
64Cu-WL12 (PD-L1 binding peptide) |
Engineered anti-human | No | NSG mice 5 to 6 weeks old Immune deficient |
Human cell lines: H226, HCC827, CHO-hPD-L1+, CHO-hPDL1-, MDAMB231 | ±7.4 MBq for imaging and ±0.74 MBq for ex vivo biodistribution | Imaging and ex vivo biodistribution at 120 min pi Treatment with atezolizumab 24 hours prior to tracer injection (20 mg/kg) |
~5.5 %ID/g,~8 %ID/g,~18 %ID/g,~5 %ID/g,~8 %ID/g for H226, HCC827, CHO-PDL1+, CHO-PDL1- and MDAMB231 respectively at 120 min pi Treatment reduced uptake in all cell lines? Tumors models? |
Yes, spleen ~4 %ID/g, after treatment ~3.5 %ID/g | (21) |
| Anti-PD1 - antibodies | ||||||||||
| PET | 64Cu-PD-1 antibody | Hamster anti-mouse | No | Treg+transgenic mice (Foxp3+.LuciDTR) Immune competent |
B16F10 (mouse melanoma) | 7.4 (±0.4) MBq (10–12 µg) Blocking with fivefold molar excess |
Imaging and ex vivo biodistribution at 1 hour, 24 hours, and 48 hours pi | 7.4 (±0.71) %ID/g for non-block vs 4.51 (±0.26) %ID/g for blocking 48 hours pi | Yes, spleen 23.04 (±4.97) %ID/g for non-block vs 14.39±0.53) %ID/g for blocking 48 hours pi | (22) |
| PET | 89Zr-pembrolizumab | Humanized anti-human | Not specified | NSG and humanized NSG mice (hNSG) | A375 (human melanoma) | 3.2 (±0.4) MBq (15 to 16 µg) | Imaging at 1, 4, 18, 24, 48, 72, 96, 120 and 144 hours pi, ex vivo biodistribution at 144 hours pi | 1.8 (±0.4) %ID/g for NSG and 3.2 (±0.7) %ID/g for hNSG at 144 hours pi | Yes, spleen ~19 %ID/g for NSG and ~28 %ID/g for hNSG at 144 hours pi |
(23) |
| 64Cu-pembrolizumab | 7.4 (±0.4) MBq (20 to 25 µg) | Imaging at 1, 4, 18, 24 and 48 hours pi, ex vivo biodistribution at 48 hours pi | 5.7 (±0.6) %ID/g for NSG, 9.4 (±2.5) %ID/g for hNSG and 5.9 (±2.1) %ID/g for hNSG block at 48 hours pi | Yes, spleen ~6.5 %ID/g for NSG,~10.5 for hNSG, and ~7% ID/g for hNSG block at 48 hours pi |
||||||
| PET | 89Zr-pembrolizumab | Humanized anti-human | No | ICR (CD-1) mice and Hsd Sprague-Dawley rats, 5 weeks old Immune competent |
No tumor model | Mice: 5 to 10 MBq (7 to 14 µg) Rats: 50 MBq (14 µg) |
Imaging at 3, 6, 12, 24, 48, 72, and 168 hours pi, ex vivo biodistribution at 168 hours pi | No tumor model | Yes, spleen ~2.5 %ID/g for mice and ~1% ID/g for rats 168 hours pi | (24) |
| NSG mice and humanized NSG mice engrafted with human PBMCs (hu-PBL-SCID), 5–8 weeks old | No tumor model; PBMC engraftment | No tumor model | Yes, spleen ~8 %ID/g for NSG and ~4.5 %ID/g for hu-PBL-SCID) at 168 hours pi | |||||||
| PET | 89Zr-Df-nivolumab | Humanized anti-human | No | NSG mice and humanized NSG mice engrafted with human PBMCs (hu-PBL-SCID 3–5 weeks old |
A549 (human lung cancer) | 5 to 10 MBq (7 to 14 µg) | Imaging at 3, 6, 12, 24, 48, 72, and 168 hours pi, and ex vivo biodistribution at 168 hours pi. | 3.88 (±0.38) %ID/g for NSG and 9.85 (±2.73) %ID/g for hu-PBL-SCID at 168 hours pi 2.85 (±0.39) %ID/g for hu-PBL IgG control at 168 hours pi |
Yes, 7.48 (±0.47) %ID/g for NSG and 4.32 (±0.40) %ID/g for hu-PBL-SCID) at 168 hours pi 3.05 (±0.79) %ID/g for hu-PBL-SCID IgG control at 168 hours pi |
(25) |
| PET | 89Zr-nivolumab | Humanized anti-human | Affinity for cynomolgus monkey | Healthy non-human primates | – | 54.5 (±11.0) MBq (237 µg) | Imaging at 24 hours, 96 hours, 144 hours and 192 hours | – | Yes, spleen at 192 hours SUV=17.63 Blocking 1 mg/kg at 192 hours SUV=2.5, 3 mg/kg SUV=2.62 |
(26) |
| PET | 64Cu-pembrolizumab | Humanized anti-human | No | Humanized NSG mice | 293T (human embryonic kidney cell line) expressing hPD-L1 | 7.4 (±0.4) MBq (20 to 25 µg) | Dynamic PET scans on 1, 2, and 4 hour pi during 3 min, at 18 and 24 hours pi during 5 min, at 24 hours pi during 10 min and at and 48 hours pi during 15 min Ex vivo biodistribution at 1, 12, 24, and 48 hours pi |
14.8 (±1.2) %ID/g for 293T tumors at 48 hours pi 0.44 (±0.01) %ID/g for A375 tumors at 48 hours pi |
Yes, spleen (17.5%±1.6 %ID/g) at 48 hours pi | (27) |
| A375 (human melanoma) | ||||||||||
| Anti-PDL1 + anti-PD1 antibodies | ||||||||||
| PET | 64Cu-PD-1 and 64Cu-PD-L1 antibody | Murine anti-mouse | No | C57BL/6N mice PD-1-deficient mice PD-L1-deficient mice Immune competent |
B16F10 (mouse melanoma) | 1.13 (±0.31) MBq (1.5 µg) 64Cu-PD-1 and 6.38 (±0.35) MBq (20 µg) 64Cu-PD-L1 | Dynamic PET scan during 45–55 and 15–20 min at 24 hours pi for 64Cu-PD-1 and 64Cu-PD-L1 respectively Ex vivo biodistribution at 48 hours pi |
±14 %IA/cm3 in B16F10 tumor at 24 hours pi in vivo for 64Cu-anti-PD-1 and 64Cu-anti-PD-L1 ±12 %IA/cm3 in B16F10 tumor at 24 hours pi ex vivo for 64Cu-anti-PD-L1 |
Yes, spleen (±20 %IA/cm3) and lymph nodes (20%–30%IA/cm3) for 64Cu-PD-1, spleen (15 %IA/cm3), lymph nodes (7.5%–15%IA/cm3) and BAT (±12 %IA/cm3) for 64Cu-PD-L1 Detection of PD-1 +TILs after immunoradiothera-py PD-L1 upregulation (mainly in lung) by IFN-γ treatment visualized |
(28) |
WT; wild-type; AlF, aluminum fluoride; BAT, brown adipose tissue; DOTA, 1,4,7,10-tetraazacyclododecane- N, N', N″, N'″-tetraacetic acid; DTPA, diethylenetriaminepentaacetic acid; EGFR, epidermal growth factor receptor; %ID/g, percentage of injected dose per gram; IFN-γ, interferon-gamma; KO, knock-out; LPS, lipopolysaccharide; NOTA, 1,4,7-triazacyclononane-N, N', N''-triacetic acid; NSCLC, non-small cell lung cancer; NSG, NOD SCID gamma; PBMC, peripheral blood mononuclear cell; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PDX, patient-derived xenograft; PET, positron emission tomography; pi, post-injection; SPECT, single photon emission CT; TILs, tumor-infiltrating lymphocytes.
To gain more insight in the in vivo behavior of a human PD-1 targeting monoclonal antibody (mAb), not cross-reactive with murine PD-1, we aimed to study the biodistribution of zirconium-89 (89Zr) radiolabeled pembrolizumab in melanoma-bearing humanized NOG mice (huNOG) engrafted with HSCs using positron-emission tomography (PET) imaging. To enable consecutive clinical translation of this approach, we developed and validated a good manufacturing practices (GMP) compliant production process for 89Zr-pembrolizumab. Finally, we put our data in perspective by summarizing results from current in vivo preclinical studies with PD-1 and PD-L1 targeting radiolabeled molecules.
Methods
Cell lines
The human melanoma cell line A375M was purchased from the American Type Culture Collection. Cell lines were confirmed to be negative for microbial contamination and were authenticated on August 6, 2018, by BaseClear using short tandem repeat profiling. A375M cells were routinely cultured in Roswell Park Memorial Institute 1640 medium (Invitrogen) containing 10% fetal calf serum (Bodinco BV), under humidified conditions at 37°C with 5% CO2. Cells were passaged 1:10, twice a week. For in vivo experiments, cells in the exponential growth phase were used.
Development of 89Zr-pembrolizumab and 89Zr-IgG4
First, the buffer of pembrolizumab (25 mg/mL, Merck) was exchanged for NaCl 0.9% (Braun) using a Vivaspin-2 concentrator (30 kDa) with a polyethersulfon filter (Sartorius). Next, pembrolizumab was conjugated with the tetrafluorphenol-N-succinyldesferal-Fe(III) ester (TFP-N-sucDf; ABX) as described earlier, in a 1:2 TFP-N-sucDf:mAb ratio.30 Conjugated product was purified from unbound chelator using Vivaspin-2 concentrators and stored at −80 °C. On the day of tracer injection, N-sucDf-pembrolizumab was radiolabeled with 89Zr, delivered as 89Zr-oxalate dissolved in oxalic acid (PerkinElmer), as described previously.30 For in vivo studies, pembrolizumab was radiolabeled at a specific activity of 250 MBq/mg. IgG4 control molecule (Sigma-Aldrich) was conjugated with TFP-N-sucDf at a 1:3 molar ratio, followed by radiolabeling with 89Zr at similar specific activity of 250 MBq/mg.
Quality control of 89Zr-pembrolizumab
Size exclusion high-performance liquid chromatography (SE-HPLC) was used to determine the final number of TFP-N-sucDf ligands per antibody (chelation ratio). SE-HPLC analysis was also performed to assess potential aggregation and fragmentation for both N-sucDf-pembrolizumab and 89Zr-pembrolizumab. An HPLC system (Waters) equipped with an isocratic pump (Waters), a dual wavelength absorbance detector (Waters), in-line radioactivity detector (Berthold) and a TSK-GEL G3000SWXL column (Tosoh Biosciences) was used with phosphate buffered saline (PBS, sodium chloride 140.0 mmol/L, sodium hydrogen phosphate 0.9 mmol/L, sodium dihydrogen phosphate 1.3 mmol/L; pH 7.4) as mobile phase (flow 0.7 mL/min). Radiochemical purity of 89Zr-pembrolizumab was measured by trichloroacetic acid precipitation assay.31 Immunoreactivity of 89Zr-pembrolizumab was analyzed by a competition binding assay with unlabeled pembrolizumab. Nunc-immuno break apart 96-wells plates (Thermo Scientific) were coated overnight at 4°C with 100 µL of 1 µg/mL PD-1 extracellular domain (R&D Systems) in PBS, set to pH 9.6 with Na2CO3 2M. Plates were washed with 0.1% Tween 80 in PBS and blocked for 1 hour at room temperature (RT) with 150 µL 1% human serum albumin (Albuman, Sanquin) in PBS. Multiple 1:1 mixtures of 89Zr-pembrolizumab with unlabeled pembrolizumab were prepared, using a fixed concentration of 89Zr-pembrolizumab (7000 ng/mL) and varying concentrations of unlabeled pembrolizumab (from 3.75 ng/mL to 12.5×106 ng/mL). Of each mixture, 100 µL was added to the 96-wells plate and incubated for 2 hours at RT. After washing twice with washing buffer, radioactivity in each well was counted using a gamma counter (Wizard2 2480–0019, SW 2.1, PerkinElmer). Counts were plotted against the concentration of competing unlabeled pembrolizumab. The half maximal inhibitory concentration (IC50) was calculated using GraphPad Prism 7 (GraphPad software). Immunoreactivity was expressed as the IC50 value divided by the 89Zr-pembrolizumab concentration to calculate the immune reactive fraction (IRF).
Animal studies
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Groningen. Studies were performed in humanized NOG mice (NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac, Taconic) and non-humanized NOG mice (Taconic) were used for control experiments. HuNOG mice are sublethally irradiated 3 weeks after birth and subsequently reconstituted with human CD34+ hematopoietic stem cells derived from fetal cord blood to express a functional human immune system including B-cells, T-cells, NK-cells, dendritic cells and monocytes. HuNOG and NOG mice were subcutaneously xenografted with 5×106 A375M human melanoma cells in 300 µL of a 1:1 mixture of PBS and Matrigel (BD Biosciences) on the right flank. Tumor growth was assessed by caliper measurements. When tumor volumes reached 100 to 200 mm3 (after 2 weeks), 2.5 MBq 89Zr-pembrolizumab (10 µg) was administered via retro-orbital injection. Mice were anesthetized using isoflurane/medical air inhalation (5% induction, 2.5% maintenance).
The first group of huNOG mice received 10 µg 89Zr-pembrolizumab (n=5). In addition, a second group of huNOG mice xenografted with the same tumor model received a co-injection of 10 µg 89Zr-pembrolizumab and 90 µg unlabeled pembrolizumab (n=4). To a third group of huNOG mice, 2.5 MBq 89Zr-IgG4 control (10 µg) was administered (n=4). Control NOG mice received 10 µg 89Zr-pembrolizumab (n=4).
PET imaging and ex vivo biodistribution
On day 7 post tracer injection (pi), PET scanning was performed. We selected this day based on optimal tumor-to-blood ratio and technical aspects, including feasible tracer specific activity and animal welfare. Mice were placed in a Focus 220 rodent scanner (CTI Siemens) on heating matrasses. Acquisition time was 60 min. A transmission scan of 515 s was performed using a 57Co point source to correct for tissue attenuation. After scanning, mice were sacrificed for ex vivo biodistribution. Bone marrow was collected from the femur bone by centrifugal-based separation. All other organs were dissected and counted in a gamma-counter (Wizard2 2480–0019, SW 2.1, PerkinElmer). Tracer uptake in each organ was expressed as percentage of the injected dose per gram tissue weight, calculated by the following formula: %ID/g = (activity in tissue (MBq)/total injected activity (MBq))/tissue weight (g)×100. To compare ex vivo and in vivo uptake, ex vivo uptake was also calculated as mean radioactivity per gram tissue, adjusted for total body weight (SUVmean ex vivo), calculated with the following formula: SUVmean ex vivo = (activity in tissue (MBq)/total injected activity (MBq))×mouse weight (g). Calculations are corrected for decay and background.
PET data was reconstructed and in vivo quantification was performed using PMOD software (V.4.0, PMOD technologies LCC). Three-dimensional regions of interest were drawn around the tumor. For other organs and tissues, a size-fixed sphere was drawn in representative tissue parts. PET data was presented as mean standardized uptake value (SUVmean in vivo), calculated by the following formula: SUVmean (g/mL) = (activity concentration (Bq/mL)/applied dose (Bq))×weight (kg)×1000.
Autoradiography
Tumor and spleen from ex vivo biodistribution studies were formalin-fixed and paraffin embedded (FFPE). FFPE tissue blocks where cut into slices of 4 µM. These slices were exposed to a phosphor imaging screen (PerkinElmer) for 72 hours and then scanned using a Cyclone phosphor imager (PerkinElmer).
Immunohistochemistry
Subsequent slices of the same tumor, spleen and mesenteric lymph node tissue were stained for H&E, CD3, CD8 and PD-1. FFPE tumor, spleen and lymph node tissue were cut into 4 µm slices using a microtome (Microm Hm 355 s, Thermo Scientific) and mounted on glass slides. Tissue sections were deparaffinized and rehydrated using xylene and ethanol. Heat-induced antigen retrieval was performed in citrate buffer (pH=6) at 100°C for 15 min. Endogenous peroxidase was blocked by 30 min incubation with 0.3% H2O2 in PBS. For CD3 staining, slides were incubated with rabbit anti-human CD3-antibody (Spring bioscience; clone SP162) in a 1:100 dilution in PBS/1% bovine serum albumin (BSA) at RT for 15 min. For CD8 staining, slides were incubated with rabbit anti-human CD8-antibody (Abcam; clone SP16) in a 1:50 dilution in PBS/1% BSA at 4°C overnight. For PD-1 staining, slides were incubated with rabbit anti-human PD-1-antibody (Abcam, clone EPR4877(2)) in a 1:500 dilution in PBS/1% BSA at RT for 30 min. Human tonsil or lymph nodes tissues sections served ad positive control and were incubated with either CD3, CD8 or PD-1 antibody. As a negative control human tonsil or lymph nodes sections were incubated with rabbit IgG monoclonal antibody (Abcam, clone EPR25A) or PBS/1% BSA.
For CD3, CD8 and PD-1 staining, incubation with secondary antibody (anti-rabbit EnVision+, Dako) was performed for 30 min, followed by application of diaminobenzidine chromogen for 10 min. Hematoxylin counterstaining was applied and tissue sections were dehydrated using ethanol and imbedded using mounting medium (Eukitt). H&E staining served to analyze tissue viability and morphology. Digital scans were acquired by a Nanozoomer 2.0-HT multi slide scanner (Hamamatsu).
89Zr-pembrolizumab manufacturing according to GMP
To enable clinical application, GMP-compliant 89Zr-pembrolizumab was developed. First, N-sucDf-pembrolizumab intermediate product was produced on a larger scale (60 mg batch, divided in 2.5 mg aliquots) and subsequently radiolabeled with 89Zr, followed by purification, dilution and sterile filtration (online supplemental figure S1). Release specifications were defined, as shown in online supplemental table S1. All analytical methods for quality control (QC) were validated. According to protocol validation of both N-sucDf-pembrolizumab and 89Zr-pembrolizumab, manufacturing consisted of three independent validation runs, including complete release QC. Stability of N-sucDf-pembrolizumab stored at −80 °C was studied up to 6 months and stability of 89Zr-pembrolizumab was determined up to 168 hours at 2°C to 8°C stored in a sterile, type 1 glass injection vial. In addition, in use stability was demonstrated at RT in a polypropylene syringe for up to 4 hours (online supplemental table S2).
jitc-2020-000938supp001.pdf (1.8MB, pdf)
Statistical analysis
Data are presented as median±IQR. A Mann-Whitney U test, followed by a Bonferroni correction was performed to compare groups (GraphPad, Prism 7). P values ≤0.05 were considered significant. If not indicated otherwise, results were not statistically significant.
Results
89Zr-pembrolizumab development for in vivo studies
We optimized the conjugation processes of pembrolizumab with the TFP-N-sucDf chelator and its subsequent radiolabeling with 89Zr. For in vivo studies, N-sucDf-pembrolizumab was produced with >60% yield and average 1.7 chelators per antibody (online supplemental figure S2, table S1). N-sucDf-pembrolizumab was subsequently radiolabeled with 89Zr at a specific activity of 250 MBq/mg, with radiochemical purity of >95% after purification. Both N-sucDf-pembrolizumab and 89Zr-pembrolizumab were stable, as shown in online supplemental table S1, S2 and figure S2. Immunoreactivity was not impaired by conjugation or radiolabeling.
89Zr-pembrolizumab imaging and biodistribution in humanized mice
PET imaging revealed 89Zr-pembrolizumab uptake in tumor, but also in healthy tissues, including liver, spleen and lymph nodes, of A375M tumor-bearing huNOG mice (figure 1A, B). Consistent with these results, ex vivo biodistribution at day 7 pi showed highest 89Zr-pembrolizumab uptake in spleen (SUVmean 30.5, IQR 15.8 to 67.7), mesenteric lymph nodes (SUVmean 20.4, IQR 8.0 to 25.2), bone marrow (SUVmean 14.5, IQR 6.1 to 32.8), thymus (SUVmean 1.3, IQR 1.1 to 2.1), liver (SUVmean, IQR 6.0, IQR 3.4 to 9.9) and tumor (SUVmean 5.1, IQR 3.3 to 8.9) (figure 1C, online supplemental table S3).
Figure 1.

In vivo PET imaging and ex vivo biodistribution of 89Zr-pembrolizumab in immunocompetent humanized NOG mice. Mice were xenografted with A375M tumor cells and received tracer injection at day 0. For blocking studies huNOG mice received a 10-fold excess of unlabeled pembrolizumab (huNOG excess). As a control for non-specific uptake huNOG mice were injected with 89Zr-IgG4. PET imaging performed on day 7 post injection (pi). (A) In vivo PET examples (maximum intensity projections) at day 7 pi showing uptake in tumor (T), axillary lymph nodes (LN), liver (L) and spleen (S). (B) In vivo uptake of 89Zr-pembrolizumab in spleen, lymph nodes (axillary), liver and tumor, at day 7 pi. Uptake is expressed as SUVmean. (C) Ex vivo biodistribution of 89Zr-pembrolizumab in humanized NOG mice. Uptake is expressed as mean radioactivity per gram tissue, adjusted for total body weight (SUVmean ex vivo). Data expressed as median±IQR *p≤0.05. BAT, brown adipose tissue; huNOG, humanized NOG mice; MLN, mesenteric lymph nodes; PET, positron emission tomography.
Tumor uptake of 89Zr-pembrolizumab was variable and slightly higher than tumor uptake observed for 89Zr-IgG4 control, however not significant due to small groups of mice (SUVmean 5.1, IQR 3.3 to 8.9 vs SUVmean 3.5, IQR 2.7 to 4.4) (figure 1C). This may be explained by low PD-1 expression found in all tumors by immunohistochemical (IHC) analysis (figure 2). 89Zr-pembrolizumab tumor-to-blood ratio also did not differ from 89Zr-IgG4 control (figure 1D).
Figure 2.

IHC analysis of spleen, mesenteric lymph node and tumor tissue humanized NOG mice. Formalin-fixed and paraffin embedded tissue blocks where cut into slices of 4 µM and stained for PD-1, CD3 and CD8 (40x). H&E staining served to analyze tissue viability and morphology (40x). Scalebar: 50 µm. IHC, immunohistochemical; PD-1, programmed cell death protein-1.
89Zr-pembrolizumab in huNOG mice showed higher uptake in lymphoid tissues compared with 89Zr-IgG4 control: spleen (SUVmean 13.9, IQR 7.1 to 21.4, NS, p=0.254), mesenteric lymph nodes (SUVmean 2.3, IQR 1.4 to 4.4, NS, p=0.114), salivary gland (SUVmean 2.1, IQR 1.2 to 2.9, NS, p=0.635), bone marrow (SUVmean 8.8, IQR 7.6 to 10.0, NS, p=1.714) and thymus (SUVmean 0.5, IQR 0.4 to 1.1, p=0.1714), indicating that 89Zr-pembrolizumab uptake in these tissues is, at least partly, PD-1-mediated. 89Zr-pembrolizumab tissue-to-blood (T:B) and tissue-to-muscle (T:M) ratios in lymphoid organs confirmed high uptake in these tissues (figure 1D, E). Additionally, relatively high 89Zr-IgG4 uptake was found in spleen, bone marrow and liver compared with other organs, suggesting 89Zr-pembrolizumab uptake in these tissues is also due to Fcγ receptor (FcγR)-binding of the antibody’s Fc-tail. High 89Zr-IgG4 uptake was less evident in lymph nodes and thymus.
89Zr-pembrolizumab spleen uptake in huNOG mice was blocked by the addition of a 10-fold excess unlabeled pembrolizumab (SUVmean 30.5, IQR 15.8 to 67.7 versus SUVmean 5.1, IQR 4.3 to 7.0, p=0.032) (figure 1B, C). Uptake in other lymphoid organs and liver was also reduced by addition of unlabeled mAb dose, whereas uptake in non-lymphoid tissues was unaffected (online supplemental table S3). Tracer activity in blood pool was increased by addition of unlabeled mAb (SUVmean 0.1, IQR 0.0 to 1.8 to SUVmean 2.2, IQR 1.4 to 7.4), but uptake in tumor did not change.
Autoradiography confirmed PET imaging results on a macroscopic level, showing high uptake in spleens of huNOG mice compared with spleens of mice that received an additional unlabeled pembrolizumab dose (figure 3). Furthermore, comparable tumor uptake was found for different dose groups. IHC analysis on spleen and lymph node tissue of huNOG mice revealed that PD-1, CD3 and CD8 positive cells were present. CD3 and CD8 cells were also present in tumor tissue of huNOG mice (figure 2), however, PD-1 staining of these tumors was negative.
Figure 3.
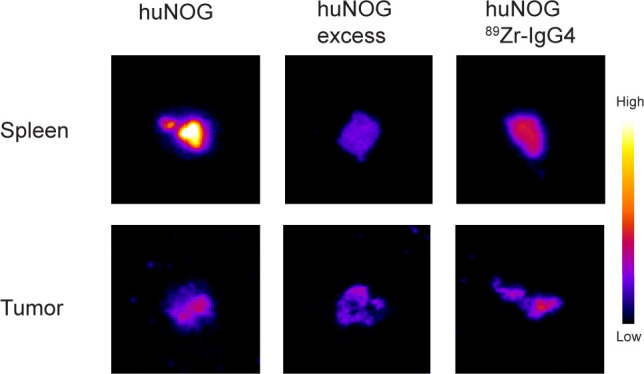
Autoradiography of spleen and tumor tissue humanized NOG mice (huNOG). Formalin-fixed and paraffin embedded tissue blocks where cut into slices of 4 µM. These slices were exposed to a phosphor imaging screen for 72 hours and were then scanned using a Cyclone phosphor imager.
89Zr-pembrolizumab biodistribution in NOG control mice clearly showed a different pattern than in huNOG mice, with high uptake in liver (SUVmean 16.9, IQR 5.1 to 26.2) and spleen (SUVmean 49.6, IQR 16.6 to 135.6), whereas 89Zr-pembrolizumab tumor uptake in NOG mice was similar to huNOG mice (SUVmean 9.3, IQR 4.5 to 15.7 vs SUVmean 5.1, IQR 3.3 to 8.9) (online supplemental figure S3). High 89Zr-pembrolizumab spleen uptake in this model may be unexpected, since limited T-cells are present in NOG mice (online supplemental figure S3). However, high spleen uptake in severely immunocompromised mice has been described previously and is potentially Fcγ receptor-mediated.23 24 32 Moreover, spleen weights in NOG mice were lower than in huNOG mice (NOG: 0.017 g±0.015 g; huNOG: 0.037 g±0.016 g, p=0.036), which resulted in higher tracer uptake expressed as %ID per gram spleen tissue for NOG mice. A low spleen weight may result from high radiosensitivity of NOG splenocytes, which can lead to toxicity.33
Critical steps in 89Zr-pembrolizumab manufacturing
The production processes for N-sucDf-pembrolizumab intermediate product and 89Zr-pembrolizumab for in vivo studies were modified to comply with GMP requirements. In the conjugation reaction, pH is increased from 4.5 to 8.5, performed in small titration steps, as described earlier by Verel et al.30 During this pH transition, precipitation occurred at 6.5 to 7.0, which was re-dissolved at pH >7.5. No precipitation was observed when pH was changed abruptly, for example, by buffer exchange, to pH 8.5 during conjugation and to pH 4.5 for removal of Fe(III). This indicates potential instability of pembrolizumab at pH 6.5 to 7.0. Formation of aggregates may be explained by the fact that pembrolizumab is an IgG4 type mAb, which forms non-classical disulfide bonds. In contrast, IgG1 type antibodies can only form classical disulfide bonds. There are many other determinants of antibody stability besides disulfide bond formation, however, this phenomenon was not seen previously with the radiolabeling of IgG1 type antibodies.31 33 34
Immunoreactivity was not affected when pembrolizumab showed precipitation during pH transition, demonstrated by comparable IRF for precipitated N-sucDf-pembrolizumab and for non-precipitated N-sucDf-pembrolizumab (online supplemental figure S4). However, it is unknown whether the pembrolizumab structure is modified by the formation of precipitates. Therefore, the method for pH transition by buffer exchange was incorporated in the conjugation protocol for pembrolizumab. Production of clinical grade 89Zr-pembrolizumab was performed as previously described by Verel et al.30
89Zr-pembrolizumab GMP validation
Three consecutive batches of conjugated and radiolabeled pembrolizumab were produced at clinical scale and complied with all release specifications (online supplemental tables S1 and S2), indicating that our process for manufacturing clinical grade 89Zr-pembrolizumab is consistent and robust. 89Zr-pembrolizumab was obtained with a specific activity of 37 MBq/mg and mean IRF of 1.35±0.6 (n=3). Stability studies revealed that N-sucDf-pembrolizumab remained compliant to release specifications up to 6 months storage at −80°C, therefore N-sucDf-pembrolizumab shelf-life was set at 6 months. Stability studies are ongoing and shelf-life may be extended if future time points remain within specifications. Data obtained during process development and validation were used to compile the investigational medicinal product dossier (IMPD), which includes all information regarding quality control, production and validation of 89Zr-pembrolizumab. Based on this IMPD, 89Zr-pembrolizumab has been approved by competent authorities for use in clinical studies.
Discussion
This study reveals 89Zr-pembrolizumab whole-body distribution in tumor-bearing huNOG mice established with a broad set of developed immune cells. Tumor uptake of 89Zr-pembrolizumab was markedly lower than uptake in lymphoid tissues such as spleen, lymph nodes and bone marrow, but higher than uptake in other organs. Importantly, high uptake in lymphoid tissues could be reduced with a 10-fold excess of unlabeled pembrolizumab. This contrasts with 89Zr-pembrolizumab tumor uptake, which was not reduced by the addition of unlabeled pembrolizumab.
Our study nicely shows the in vivo behavior of 89Zr-pembrolizumab, which, apart from IgG pharmacokinetics determined by its molecular weight and Fc tail, is predominantly driven by its affinity for PD-1 (Kd:~30 pM). The PD-1 cell surface receptor is primarily expressed on activated T-cells and pro B-lymphocytes, which are abundantly present in our huNOG mouse model. Lymphocytes are highly concentrated in organs that are key players of the immune system: lymph nodes, spleen, thymus, bone marrow as well as tonsils, adenoid and Peyer’s patches. From our PET imaging and ex vivo biodistribution data, we learned that 89Zr-pembrolizumab distributed mainly to lymphoid organs, where PD-1 expressing immune cells are present.
89Zr-pembrolizumab showed relatively low and variable tumor uptake, however, this uptake could be visualized with PET imaging 7 days pi and was higher than in non-lymphoid tissues. We hypothesized there may be PD-1-mediated 89Zr-pembrolizumab tumor uptake, but we also found tumor uptake for 89Zr-IgG4, suggesting part of the 89Zr-pembrolizumab tumor uptake is FcγR-mediated. In our mouse model, few PD-1 positive immune cells may have traveled to the tumor, thereby potentially limiting 89Zr-pembrolizumab tumor uptake. Interestingly, the addition of unlabeled pembrolizumab did not influence tumor uptake. This is likely caused by substantial increase of 89Zr-pembrolizumab in blood pool as a direct consequence of adding excess unlabeled pembrolizumab, warranting a continuous pembrolizumab supply to the tumor.
Ex vivo immunohistochemical analysis revealed CD3 and CD8 positive lymphocytes were present in tumor, but limited PD-1-expression was found. Immune checkpoint protein expression status in tumor-infiltrating lymphocytes is highly dynamic.35 36 This so-called ‘immune phenotype’ depends on several factors, including tumor type, location and mutational burden. Our results indicate that, whereas PD-1 expression may demonstrate large variation, 89Zr-pembrolizumab PET imaging is able to capture PD-1 dynamics in both tumor and healthy tissues.
Compared with earlier preclinical studies with radiolabeled pembrolizumab in the hNSG model, we found higher 89Zr-pembrolizumab uptake in spleen and other lymphoid tissues.23 24 This likely reflects the presence of multiple hematopoietic cell lineages, including B-cells, T-cells, NK-cells, dendritic cells and monocytes, and thus higher PD-1 expression, in our huNOG model compared with the hNSG model. Molecular imaging studies with radiolabeled antibodies generally show distribution to the spleen. It also known that Fc/FcγR-mediated immunobiology of the experimental mouse model plays a key role in the in vivo biodistribution and tumor targeting.33 In our mouse model, we also observed 89Zr-IgG4 uptake in lymphoid tissues, indicating 89Zr-pembrolizumab uptake in these organs may have an FcγR-mediated component. For most radiolabeled antibodies without an immune target, spleen uptake in patients is ~5 %ID/kg.37 This supports the idea that, independent of their target, antibodies often show distribution to the spleen. However, spleen uptake may be higher if PD-1 or PD-L1 is present.
Pembrolizumab has an IgG4κ backbone with a stabilizing SER228PRO sequence alteration in the Fc-region to prevent the formation of half molecules. The IgG4 backbone of pembrolizumab may slightly differ from the IgG4 control molecule that we used for our experiments, however, FcγR-binding affinity and kinetics of pembrolizumab appears to be very similar to IgG4.38 We, therefore, consider the used IgG4 control molecule to provide a useful indication of the extent of FcγR-mediated uptake. In this respect, FcγR-mediated uptake may be present in the spleen but potentially also in liver and tumor, since these tissues demonstrate relatively high uptake of 89Zr-IgG4.
PD-1 is predominantly expressed on activated T-cells while its ligand PD-L1 is expressed by a broader range of immune cells as well as tumor cells. It is therefore to be expected that biodistribution of antibody tracers targeting PD-L1 may deviate from the biodistribution results that we described here for 89Zr-pembrolizumab. In table 1, we presented an overview of preclinical imaging and biodistribution studies using anti-PD-1 and anti-PD-L1 tracers. Data turned out to be highly variable, mostly focused on tumor and not on the immune system, and therefore not just comparable. From our results, we increasingly realize that it is extremely important for interpretation of these type of data to know the characteristics of the antibody (origin, cross-reactivity, Fc-backbone, target, target-affinity and dose), the animal model (mouse strain, age, immune status and tumor cell line) and time points, variables we detailed in the table.
As for preclinical studies, data on the distribution of PD-1 and PD-L1 targeting antibodies to lymphoid organs in patients is still limited. A clinical imaging study in 13 patients demonstrated modest 89Zr-nivolumab spleen uptake of SUVmean 5.8±0.7, whereas uptake of this radiolabeled antibody targeting PD-1 in other lymphoid tissues was not addressed.39 89Zr-atezolizumab (anti-PD-L1 antibody) imaging in 22 patients revealed spleen uptake with an SUVmean of 15. 89Zr-atezolizumab also distributed to other lymphoid tissues and sites of inflammation, whereas uptake in non-lymphoid organs was low. The high spleen uptake could at least partly be explained by presence of PD-L1 in endothelial littoral cells of the spleen.40 To perceive what can be expected for 89Zr-pembrolizumab PET imaging in patients, how results may be interpreted and potentially translated to predicting response, knowledge on which immune cells express PD-1 and where these cells are located in the human body is of utmost importance.
With our study, we validated the use of 89Zr-pembrolizumab PET imaging to evaluate PD-1-mediated uptake in tumor and immune tissues in a setting that allowed for comparing tracer uptake and whole tumor tissue analysis. To enable evaluation of 89Zr-pembrolizumab biodistribution in humans, we developed clinical grade 89Zr-pembrolizumab. Clinical 89Zr-pembrolizumab PET imaging in patients with melanoma and NSCLC before treatment with pembrolizumab is currently performed at our center (ClinicalTrials.gov Identifier NCT02760225), and may elucidate if tracer tumor uptake correlates to response and if uptake in healthy PD-1 expressing tissues correlates to toxicity.
Conclusion
We demonstrated the in vivo biodistribution of 89Zr-pembrolizumab in humanized mice, and found uptake in tumor with the highest uptake in the lymphoid system, reflecting the presence of PD-1. Insight in the in vivo behavior and biodistribution of immune checkpoint targeting monoclonal antibodies might aid in better understanding immune checkpoint inhibition therapy and could potentially help explaining variation in response as well as potential toxicity due to uptake in healthy (immune) tissues.
Footnotes
Twitter: @VriesElisabeth
Contributors: ELvdV was involved in project design and conceptualization, was involved in tracer development and GMP validation, wrote the IMPD, performed animal studies, performed ex vivo analyses, data analysis and wrote the manuscript; DG was involved in study conceptualization, data analysis, performed ex vivo analyses and wrote the manuscript; LPdJ was involved in tracer development and GMP validation, performed animal studies, performed ex vivo analyses and edited the manuscript; AJS was involved in GMP validation, wrote the IMPD and edited the manuscript; EGEdV was involved in project design and conceptualization, supervised the study and edited the manuscript; MNLdH was involved in project design and conceptualization, supervised the study and edited the manuscript. All authors read and approved the final manuscript.
Funding: The research leading to these results received funding from the Innovative Medicines Initiatives 2 Joint Undertaking under grant agreement No 116106 (TRISTAN). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program and EFPIA.
Competing interests: EGEdV reports grants from IMI TRISTAN (GA no.116106), during the conduct of the study; consulting and advisory role for NSABP, Daiichi Sankyo, Pfizer, Sanofi, Merck, Synthon Biopharmaceuticals; grants from Amgen, Genentech, Roche, Chugai Pharma, CytomX Therapeutics, Nordic Nanovector, G1 Therapeutics, AstraZeneca, Radius Health, Bayer, all made available to the institution, outside the submitted work.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available upon reasonable request. The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Postow MA, Sidlow R, Hellmann MD. Immune-Related adverse events associated with immune checkpoint blockade. N Engl J Med 2018;378:158–68. 10.1056/NEJMra1703481 [DOI] [PubMed] [Google Scholar]
- 2.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immunol 2015;15:45–56. 10.1038/nri3790 [DOI] [PubMed] [Google Scholar]
- 3.van der Veen EL, Bensch F, Glaudemans AWJM, et al. Molecular imaging to enlighten cancer immunotherapies and underlying involved processes. Cancer Treat Rev 2018;70:232–44. 10.1016/j.ctrv.2018.09.007 [DOI] [PubMed] [Google Scholar]
- 4.Lamberts LE, Williams SP, Terwisscha van Scheltinga AGT, et al. Antibody positron emission tomography imaging in anticancer drug development. J Clin Oncol 2015;33:1491–504. 10.1200/JCO.2014.57.8278 [DOI] [PubMed] [Google Scholar]
- 5.Heskamp S, Hobo W, Molkenboer-Kuenen JDM, et al. Noninvasive imaging of tumor PD-L1 expression using radiolabeled anti-PD-L1 antibodies. Cancer Res 2015;75:2928–36. 10.1158/0008-5472.CAN-14-3477 [DOI] [PubMed] [Google Scholar]
- 6.Josefsson A, Nedrow JR, Park S, et al. Imaging, biodistribution, and dosimetry of radionuclide-labeled PD-L1 antibody in an immunocompetent mouse model of breast cancer. Cancer Res 2016;76:472–9. 10.1158/0008-5472.CAN-15-2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee S, Lesniak WG, Gabrielson M, et al. A humanized antibody for imaging immune checkpoint ligand PD-L1 expression in tumors. Oncotarget 2016;7:10215–27. 10.18632/oncotarget.7143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lesniak WG, Chatterjee S, Gabrielson M, et al. PD-L1 Detection in Tumors Using [(64)Cu]Atezolizumab with PET. Bioconjug Chem 2016;27:2103–10. 10.1021/acs.bioconjchem.6b00348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nedrow JR, Josefsson A, Park S, et al. Imaging of programmed cell death ligand 1: impact of protein concentration on distribution of anti-PD-L1 SPECT agents in an immunocompetent murine model of melanoma. J Nucl Med 2017;58:1560–6. 10.2967/jnumed.117.193268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kikuchi M, Clump DA, Srivastava RM, et al. Preclinical immunoPET/CT imaging using Zr-89-labeled anti-PD-L1 monoclonal antibody for assessing radiation-induced PD-L1 upregulation in head and neck cancer and melanoma. Oncoimmunology 2017;6:e1329071. 10.1080/2162402X.2017.1329071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Truillet C, Oh HLJ, Yeo SP, et al. Imaging PD-L1 expression with ImmunoPET. Bioconjug Chem 2018;29:96–103. 10.1021/acs.bioconjchem.7b00631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heskamp S, Wierstra PJ, Molkenboer-Kuenen JDM, et al. Pd-L1 microSPECT/CT imaging for longitudinal monitoring of PD-L1 expression in syngeneic and humanized mouse models for cancer. Cancer Immunol Res 2019;7:150–61. 10.1158/2326-6066.CIR-18-0280 [DOI] [PubMed] [Google Scholar]
- 13.Chatterjee S, Lesniak WG, Miller MS, et al. Rapid PD-L1 detection in tumors with PET using a highly specific peptide. Biochem Biophys Res Commun 2017;483:258–63. 10.1016/j.bbrc.2016.12.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez Trotter DE, Meng X, McQuade P, et al. In vivo imaging of the programmed death ligand 1 by 18F positron emission tomography. J Nucl Med 2017;25:1852–7. [DOI] [PubMed] [Google Scholar]
- 15.Broos K, Keyaerts M, Lecocq Q, et al. Non-Invasive assessment of murine PD-L1 levels in syngeneic tumor models by nuclear imaging with nanobody tracers. Oncotarget 2017;8:41932–46. 10.18632/oncotarget.16708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maute RL, Gordon SR, Mayer AT, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc Natl Acad Sci U S A 2015;112:E6506–14. 10.1073/pnas.1519623112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donnelly DJ, Smith RA, Morin P, et al. Synthesis and Biologic Evaluation of a Novel 18F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J Nucl Med 2018;59:529–35. 10.2967/jnumed.117.199596 [DOI] [PubMed] [Google Scholar]
- 18.Mayer AT, Natarajan A, Gordon SR, et al. Practical immuno-PET radiotracer design considerations for human immune checkpoint imaging. J Nucl Med 2017;58:538–46. 10.2967/jnumed.116.177659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natarajan A, Patel CB, Ramakrishnan S, et al. A novel engineered small protein for positron emission tomography imaging of human programmed death ligand-1: validation in mouse models and human cancer tissues. Clin Cancer Res 2019;25:1774–85. 10.1158/1078-0432.CCR-18-1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Silva RA, Kumar D, Lisok A, et al. Peptide-Based 68Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol Pharm 2018;15:3946–52. 10.1021/acs.molpharmaceut.8b00399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar D, Lisok A, Dahmane E, et al. Peptide-Based PET quantifies target engagement of PD-L1 therapeutics. J Clin Invest 2019;129:616–30. 10.1172/JCI122216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natarajan A, Mayer AT, Xu L, et al. Novel radiotracer for immunoPET imaging of PD-1 checkpoint expression on tumor infiltrating lymphocytes. Bioconjug Chem 2015;26:2062–9. 10.1021/acs.bioconjchem.5b00318 [DOI] [PubMed] [Google Scholar]
- 23.Natarajan A, Mayer AT, Reeves RE, et al. Development of novel immunoPET tracers to image human PD-1 checkpoint expression on tumor-infiltrating lymphocytes in a humanized mouse model. Mol Imaging Biol 2017;19:903–14. 10.1007/s11307-017-1060-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.England CG, Ehlerding EB, Hernandez R, et al. Preclinical pharmacokinetics and biodistribution studies of 89Zr-labeled pembrolizumab. J Nucl Med 2017;58:162–8. 10.2967/jnumed.116.177857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.England CG, Jiang D, Ehlerding EB, et al. 89Zr-labeled nivolumab for imaging of T-cell infiltration in a humanized murine model of lung cancer. Eur J Nucl Med Mol Imaging 2018;45:110–20. 10.1007/s00259-017-3803-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole EL, Kim J, Donnelly DJ, et al. Radiosynthesis and preclinical PET evaluation of 89Zr-nivolumab (BMS-936558) in healthy non-human primates. Bioorg Med Chem 2017;25:5407–14. 10.1016/j.bmc.2017.07.066 [DOI] [PubMed] [Google Scholar]
- 27.Natarajan A, Patel CB, Habte F, et al. Dosimetry Prediction for Clinical Translation of 64Cu-Pembrolizumab ImmunoPET Targeting Human PD-1 Expression. Sci Rep 2018;8:633. 10.1038/s41598-017-19123-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hettich M, Braun F, Bartholomä MD, et al. High-Resolution PET imaging with therapeutic antibody-based PD-1/PD-L1 checkpoint tracers. Theranostics 2016;6:1629–40. 10.7150/thno.15253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De La Rochere P, Guil-Luna S, Decaudin D, et al. Humanized mice for the study of immuno-oncology. Trends Immunol 2018;39:748–63. 10.1016/j.it.2018.07.001 [DOI] [PubMed] [Google Scholar]
- 30.Verel I, Visser GWM, Boellaard R, et al. 89Zr immuno-PET: comprehensive procedures for the production of 89Zr-labeled monoclonal antibodies. J Nucl Med 2003;44:1271–81. [PubMed] [Google Scholar]
- 31.Nagengast WB, de Vries EG, Hospers GA, et al. In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J Nucl Med 2007;48:1313–9. 10.2967/jnumed.107.041301 [DOI] [PubMed] [Google Scholar]
- 32.Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 2012;4:17–23. 10.4161/mabs.4.1.18347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma SK, Chow A, Monette S, et al. Fc-Mediated anomalous biodistribution of therapeutic antibodies in immunodeficient mouse models. Cancer Res 2018;78:1820–32. 10.1158/0008-5472.CAN-17-1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dijkers ECF, Kosterink JGW, Rademaker AP, et al. Development and characterization of clinical-grade 89Zr-trastuzumab for HER2/neu immunoPET imaging. J Nucl Med 2009;50:974–81. 10.2967/jnumed.108.060392 [DOI] [PubMed] [Google Scholar]
- 35.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133–50. 10.1038/s41568-019-0116-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simon S, Labarriere N. Pd-1 expression on tumor-specific T cells: friend or foe for immunotherapy? Oncoimmunology 2017;7:e1364828. 10.1080/2162402X.2017.1364828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bensch F, Smeenk MM, van Es SC, et al. Comparative biodistribution analysis across four different 89Zr-monoclonal antibody tracers-The first step towards an imaging warehouse. Theranostics 2018;8:4295–304. 10.7150/thno.26370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang T, Song X, Xu L, et al. The binding of an anti-PD-1 antibody to FcγRΙ has a profound impact on its biological functions. Cancer Immunol Immunother 2018;67:1079–90. 10.1007/s00262-018-2160-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niemeijer AN, Leung D, Huisman MC, et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat Commun 2018;9:4664. 10.1038/s41467-018-07131-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bensch F, van der Veen EL, Lub-de Hooge MN, et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat Med 2018;24:1852–8. 10.1038/s41591-018-0255-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-000938supp001.pdf (1.8MB, pdf)