

Abstract
Trisubstituted allenes with a 3‐(1′‐alkenylidene)‐pyrrolidin‐2‐one motif were successfully deracemized (13 examples, 86–98 % ee) employing visible light (λ=420 nm) and a chiral triplet sensitizer as the catalyst (2.5 mol %). The photocatalyst likely operates by selective recognition of one allene enantiomer via hydrogen bonds and by a triplet‐sensitized racemization process. Even a tetrasubstituted allene (45 % ee) and a seven‐membered 3‐(1′‐alkenylidene)‐azepan‐2‐one (62 % ee) could be enantiomerically enriched under the chosen conditions. It was shown that the axial chirality of the allenes can be converted into point chirality by a Diels–Alder (94–97 % ee) or a bromination reaction (91 % ee). Ring opening of the five‐membered pyrrolidin‐2‐one was achieved without significantly compromising the integrity of the chirality axis (92 % ee).
Keywords: allenes, chiral resolution, enantioselectivity, hydrogen bonds, photochemistry, sensitizers
One way street: A photochemical deracemization provided—via sensitization by a chiral photocatalyst—access to a set of enantiomerically enriched allenes. Subsequent chirality transfer or ring opening enabled the synthesis of valuable chiral intermediates with >90 % ee.
Deracemization reactions are defined as processes “in which a racemate is made nonracemic by increasing the quantity of one enantiomer at the expense of the other.”1 Several methods to achieve a deracemization in solution have been described but all of them eventually rely either on the intermediacy of an achiral intermediate which is enantioselectively re‐converted to the substrate or on the use of a stoichiometric chiral reagent.2, 3 If a deracemization is performed catalytically, the entropy loss associated with a deracemization needs to be energetically compensated. Although photochemistry offers in principle a solution to this challenge,4, 5 highly enantioselective catalytic deracemization reactions have remained elusive until very recently. In 2018, it was discovered that a class of allenes, 3‐(1′‐alkenylidene)‐piperidin‐2‐ones, can be deracemized in high yields (>56 %) and with high enantioselectivity (89–97 % ee) by a chiral triplet sensitizer that operates with visible light.6 Chiral cyclopropanes and sulfoxides were subsequently shown to be also amenable to a sensitized deracemization (up to 55 % ee).7 A more recently disclosed photochemical deracemization approach is based on an electron transfer‐mediated process employing a combination of three catalysts. The method was successfully applied to chiral imidazolidin‐2‐ones (92–99 % yield, 62–92 % ee).8
Although more detailed mechanistic work is ongoing, a simplified scheme for the sensitized deracemization of chiral compounds is helpful to illustrate the mode of action (Scheme 1). The chiral photocatalyst distinguishes between the two enantiomeric forms of a compound by non‐covalent binding. The enantiomer that binds to the catalyst is excited via sensitization and subsequently undergoes a racemization via an achiral intermediate. Since, in an ideal scenario, only one enantiomer is processed by the catalyst, the non‐binding enantiomer gets increasingly enriched in solution and is obtained ideally in enantiopure form. In a more realistic scenario, the binding situation is not as perfect as described and other parameters may influence the selectivity.6 The chiral photocatalysts used in our work (see below) rely on a non‐covalent two‐point hydrogen bonding interaction for enantiomer recognition. They operate by triplet energy transfer from a xanthone or thioxanthone chromophore.6, 7, 9
Scheme 1.
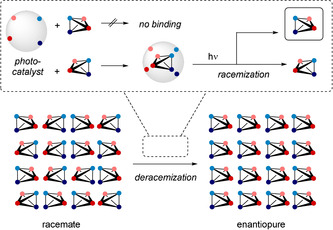
Simplified scheme for a photochemical deracemization achieved by a single chiral photocatalyst. The tetrahedrons represent chiral molecules in both enantiomeric forms.
In the context of allene deracemization, our previous work had focused on a single class of allenes and consecutive reactions had not been studied. Given the importance of allenes in organic synthesis10 it seemed desirable to extend the scope of the deracemization to other allenes and to explore their reactivity. Specifically, the lactam recognition motif of the chiral triplet sensitizer raised the question whether other lactams would be equally suited for binding and what degree of enantioselectivity could be achieved. In the work now presented we focused mainly on 3‐(1′‐alkenylidene)‐pyrrolidin‐2‐ones and their reactivity. A highly enantioselective deracemization was established and three valuable transformations have been found, which gave access to the respective products in high enantioselectivity (>90 % ee). Results of our preliminary experiments are summarized in this Communication.
Racemic allenes rac‐1 (Scheme 2) were prepared from N‐para‐methoxybenzyl(PMB)‐pyrrolidin‐2‐one by a four‐step sequence, including α‐iodination, conversion to the triphenylphosphonium salt, Wittig reaction and final deprotection (see SI for further details). The compounds are stable at room temperature but show some decomposition under acidic conditions. The deracemization was attempted with thioxanthone 2 that exhibits a significant absorption in the visible part of the electromagnetic spectrum. In line with previous data on the triplet energy (E T) of allenes we estimate the triplet energies of compounds 1 to be in the range of 250–270 kJ mol−1.6, 11 In preliminary experiments, we verified the configurational lability of the enantioenriched allene 1 a (separation by chiral HPLC) in the presence of parent thioxanthone with a reported E T=268 kJ mol−1 (77 K, EtOH).12 The triplet energy of thioxanthone 2 had been determined as E T=263 kJ mol−1 (77 K, PhCF3)6 which indicated that it should be suitable for the planned deracemization. The reactions were preferably performed in acetonitrile at −40 °C and were terminated after eight hours (for optimization see the SI).
Scheme 2.
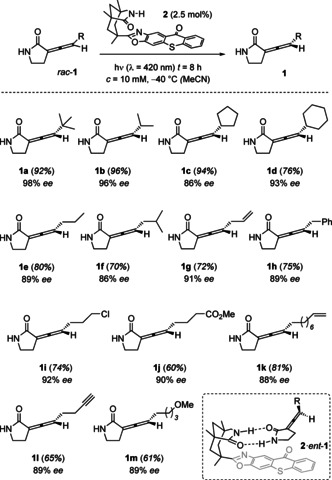
Photochemical deracemization of various 3‐(1′‐alkenylidene)‐pyrrolidin‐2‐ones rac‐1 upon visible light irradiation in the presence of chiral sensitizer 2 and binding of allene enantiomer ent‐1 to catalyst 2 (inset).
Given that we had seen a lower enantioselectivity for five‐membered lactams in reactions which require coordination to a 1,5,7‐trimethyl‐3‐azabicyclo[3.3.1]nonan‐2‐one core,13 it was gratifying to note that the enantioselectivity achieved in the deracemization reaction of 3‐(1′‐alkenylidene)‐pyrrolidin‐2‐ones was high at a catalyst loading of only 2.5 mol %. For the trisubstituted allenes (1 a–1 m), the enantiopurity varied between 86 % to 98 % ee. The assignment of the absolute configuration was deduced from a consecutive reaction (vide infra) and matched our expectation. In analogy to the general picture (Scheme 1), complex formation between enantiomer ent‐1 and the sensitizer by hydrogen bonding14 is preferred (2⋅ent‐1) which leads to an enrichment of the observed enantiomer 1. A key aspect is the fact that the energy transfer to the allene is highly efficient within complex 2⋅ent‐1 while an intermolecular energy transfer is slower.5, 6 The functional group tolerance of the reaction was demonstrated for alkenyl, phenyl, chloro, methoxycarbonyl, alkynyl, and methoxy (1 g–1 m) although some yields suffered due to the instability of the allenes during purification. No decomposition was observed during the irradiation.
In order to explore the limits of the deracemization, the tetrasubstituted allene rac‐3 was submitted to the optimized irradiation conditions (Scheme 3). In this instance the size difference between methyl and ethyl is responsible for the different binding properties and it is remarkable that sensitizer 2 does allow for a significant distinction which in turn results in 45 % ee. Likewise, even a seven‐membered lactam, azepan‐2‐one rac‐4, with a higher flexibility within the ring could be partially deracemized and delivered a major enantiomer with the putative structure 4.
Scheme 3.

Photochemical deracemization reaction of tetrasubstituted allene rac‐3 and of 3‐(3′,3′‐dimethyl‐1′‐butenylidene)‐azepan‐2‐one (rac‐4) upon visible light irradiation in the presence of chiral sensitizer 2.
Subsequent reactions of allenes 1 were mainly performed with the most easily accessible product 1 a which was obtained in high enantiopurity (98 % ee). A concerted cycloaddition15 seemed most rewarding regarding the desired transfer of axial into point chirality.16, 17 After some optimization we found the Diels–Alder reaction with cyclopentadiene to be feasible if performed under Lewis acid catalysis at low temperature (Scheme 4). Two diastereoisomers 5 a and 5 a′ were obtained and could be fully separated. The cycloaddition occurred exclusively at the internal α,β‐unsaturated but not at the β,γ‐unsaturated double bond. After N‐substitution of the major diastereoisomer with para‐nitrobenzenesulfonyl chloride a crystalline product was obtained which was suitable for X‐ray crystallography.18 In line with NOESY (nuclear Overhauser enhancement spectroscopy) data, the crystal structure supported the assignment of the relative configuration to the exo product. Anomalous diffraction established the absolute configuration of product 5 a which in turn confirmed the assumed absolute configuration of allenes 1. Indeed, diene addition is known to occur from the more accessible face of a trisubstituted allene.19 In the specific case of allenes 1 a and 1 b there is an exo preference for the approach of the diene as seen by the observed diastereomeric ratio (d.r.). The chirality transfer in the Diels–Alder reaction is close to perfect both for allenes 1 a and 1 b. Products 5 invite further functionalization at several positions and represent potentially useful building blocks.
Scheme 4.
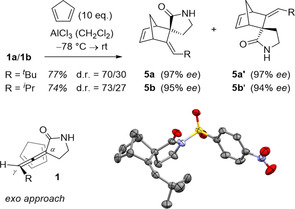
Diels–Alder reaction of allenes 1 a and 1 b with cyclopentadiene: Transfer of axial chirality to point chirality in products 5 (top); exo approach of the diene to substrates 1 (bottom left); structure of the N‐para‐nitrobenzenesulfonyl derivative of compound 5 a as determined by anomalous X‐ray diffraction18 (bottom right).
When searching for possible ways which allow for an electrophilic attack on allenes, we discovered a useful bromination protocol.20 Treatment with bromine in CH2Cl2 resulted in the formation of a dibrominated product to which we assign structure 7. The compound was formed with high enantioselectivity indicating an efficient, previously unknown chirality transfer.21 The relative configuration of compound 7 is suggested by NOESY data and it is assumed that initial bromine attack occurs at the α,β‐unsaturated double bond opposite to the large tert‐butyl group. Intermediate bromonium ion 6 22 would subsequently be opened by bromide attack which, if not concerted, benefits from the neighboring effect of the existing C−Br bond.
Scheme 5.
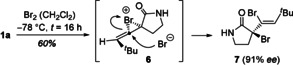
Chirality transfer in the addition of bromine to allene 1 a and formation of product 7, presumably via bromonium ion intermediate 8.
A final set of experiments was conducted to investigate the ring opening of the pyrrolidinone ring and the stereochemical integrity of the allene during this process. In the first step, a protecting group was installed to avoid complications from the acidic proton at the nitrogen atom. The most versatile protecting group for this purpose appeared to be the tert‐butoxycarbonyl (Boc) group. However, classical conditions for its introduction had been reported to cause partial isomerization in other α,β‐unsaturated cyclic amides, presumably due to reversible 1,4‐addition by the ancillary base 4‐(N,N‐dimethylamino)pyridine.23 Therefore, a careful optimization of the reaction conditions was needed to retain the chirality axis. Eventually, it was found that compound 1 a was most reliably protected by complete deprotonation of the lactam and subsequent treatment with di‐tert‐butyl dicarbonate (Boc2O). The N‐Boc protected lactam 8 was expected to be susceptible to a ring opening reaction by nucleophiles.24 As an example, we employed phenyl magnesium bromide as a potential reagent which indeed led to a smooth conversion. The desired trisubstituted allene was obtained in 78 % and with 92 % ee. The erosion of enantiomeric purity in the process is notable but fortunately at a level that should still allow its use in stereoselective synthesis.
Scheme 6.
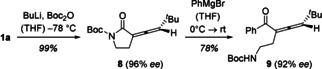
Ring opening of the pyrrolidin‐2‐one ring by a Grignard reagent (PhMgBr) after protection of the nitrogen atom of 1 a with a tert‐butyloxycarbonyl (Boc) group: Synthesis of acyclic allene 9 via intermediate 8.
In summary, a new, versatile class of allenes has become accessible in enantiomerically enriched form by a visible light‐mediated deracemization reaction. Since either enantiomer of the deracemization catalyst is available25 not only allenes 1 but also their enantiomers ent‐1 can be selectively obtained. The success of the reactions underlines the high efficiency of deracemization processes with substrates which display a lactam binding motif. Applications of the allenes have been successfully explored by addition to the internal allene double bond with concomitant axial‐to‐point chirality transfer. An alicyclic allene could be obtained with high enantioselectivity after lactam ring opening. Applications of chiral allenes in more complex synthetic sequences are currently being pursued.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (Ba 1372/24‐1) is gratefully acknowledged. O. Ackermann, F. Pecho, and J. Kudermann are acknowledged for their help with HPLC and GLC analyses. Open access funding enabled and organized by Projekt DEAL.
M. Plaza, C. Jandl, T. Bach, Angew. Chem. Int. Ed. 2020, 59, 12785.
Contributor Information
Dr. Manuel Plaza, http://www.oc1.ch.tum.de/home_en/
Prof. Dr. Thorsten Bach, Email: thorsten.bach@ch.tum.de.
References
- 1. Pirkle W. H., Reno D. S., J. Am. Chem. Soc. 1987, 109, 7189–7190. [Google Scholar]
- 2.Reviews:
- 2a. Palmans A. R. A., Mol. Syst. Des. Eng. 2017, 2, 34–46; [Google Scholar]
- 2b. Rachwalski M., Vermue N., Rutjes F. P. J. T., Chem. Soc. Rev. 2013, 42, 9268–9282; [DOI] [PubMed] [Google Scholar]
- 2c. Servi S., Tessaro D., Pedrocchi-Fantoni G., Coord. Chem. Rev. 2008, 252, 715–726; [Google Scholar]
- 2d. Gruber C., Lavandera I., Faber K., Kroutil W., Adv. Synth. Catal. 2006, 348, 1789–1805. [Google Scholar]
- 3.For examples in allene chemistry, see:
- 3a. Naruse Y., Watanabe H., Ishiyama Y., Yoshida T., J. Org. Chem. 1997, 62, 3862–3866; [Google Scholar]
- 3b. Varghese J. P., Zouev I., Aufauvre L., Knochel P., Marek I., Eur. J. Org. Chem. 2002, 4151–4158; [Google Scholar]
- 3c. Ye J., Ma S., Org. Chem. Front. 2014, 1, 1210–1224. [Google Scholar]
- 4.For some early attempts, see:
- 4a. Murov S. L., Cole R. S., Hammond G. S., J. Am. Chem. Soc. 1968, 90, 2957–2958; [Google Scholar]
- 4b. Ouannès C., Beugelmans R., Roussi G., J. Am. Chem. Soc. 1973, 95, 8472–8474; [Google Scholar]
- 4c. Drucker C. S., Toscano V. G., Weiss R. G., J. Am. Chem. Soc. 1973, 95, 6482–6484; [Google Scholar]
- 4d. Balavoine G., Juge S., Kagan H. B., Tetrahedron Lett. 1973, 14, 4159–4162. [Google Scholar]
- 5.Recent reviews:
- 5a. Shi Q., Ye J., Angew. Chem. Int. Ed. 2020, 59, 4998–5001; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5030–5033; [Google Scholar]
- 5b. Yang C., Inoue Y., Nature 2018, 564, 197–199. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Hölzl-Hobmeier A., Bauer A., Silva A. V., Huber S. M., Bannwarth C., Bach T., Nature 2018, 564, 240–243. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Tröster A., Bauer A., Jandl C., Bach T., Angew. Chem. Int. Ed. 2019, 58, 3538–3541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3576–3579; [Google Scholar]
- 7b. Wimberger L., Kratz T., Bach T., Synthesis 2019, 51, 4417–4424. [Google Scholar]
- 8. Shin N. Y., Ryss J. M., Zhang X., Miller S. J., Knowles R. R., Science 2019, 366, 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For a recent review on reactions mediated by triplet energy transfer, see: Zhou Q.-Q., Zou Y.-Q., Lu L.-Q., Xiao W.-J., Angew. Chem. Int. Ed. 2019, 58, 1586–1604; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1600–1619. [Google Scholar]
- 10.For the synthesis of enantiomerically enriched allenes and their use in organic synthesis, see:
- 10a. Krause N., Hashmi A. S. K., Modern Allene Chemistry, Wiley-VCH, Weinheim, 2004; [Google Scholar]
- 10b. Yu S., Ma S., Angew. Chem. Int. Ed. 2012, 51, 3074–3112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3128–3167; [Google Scholar]
- 10c. Chu W., Zhang Y., Wang J., Catal. Sci. Technol. 2017, 7, 4570–4579; [Google Scholar]
- 10d. Huang X., Ma S., Acc. Chem. Res. 2019, 52, 1301–1312. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Gotthardt H., Hammond G. S., Chem. Ber. 1975, 108, 657–663; [Google Scholar]
- 11b. Kamphuis J., Bos H. J. T., Visser R. J., Huizer B. H., Varma C. A. G. O., J. Chem. Soc. Perkin Trans. 2 1986, 1867–1874; [Google Scholar]
- 11c. Tsuno T., Hoshino H., Sugiyama K., Tetrahedron Lett. 1997, 38, 1581–1584; [Google Scholar]
- 11d. Alcaide B., Almendros P., Aragoncillo C., Chem. Soc. Rev. 2010, 39, 783–816. [DOI] [PubMed] [Google Scholar]
- 12. Iyer A., Clay A., Jockusch S., Sivaguru J., J. Phys. Org. Chem. 2017, 30, e3738. [Google Scholar]
- 13.
- 13a. Kapitán P., Bach T., Synthesis 2008, 1559–1564; [Google Scholar]
- 13b. Lenhart D., Bauer A., Pöthig A., Bach T., Chem. Eur. J. 2016, 22, 6519–6523; [DOI] [PubMed] [Google Scholar]
- 13c.For a review on hydrogen bonding to this core, see: Burg F., Bach T., J. Org. Chem. 2019, 84, 8815–8836. [DOI] [PubMed] [Google Scholar]
- 14.For a related sensitizer that operates by hydrogen bonding, see: Skubi K. L., Kidd J. B., Jung H., Guzei I. A., Baik M.-H., Yoon T. P., J. Am. Chem. Soc. 2017, 139, 17186–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Examples of Diels–Alder reactions with electron-deficient allenes:
- 15a. Kimura H., Fujiwara T., Katoh T., Nishide K., Kajimoto T., Node M., Chem. Pharm. Bull. 2006, 54, 399–402; [DOI] [PubMed] [Google Scholar]
- 15b. Crouch I. T., Neff R. K., Frantz D. E., J. Am. Chem. Soc. 2013, 135, 4970–4973; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Barber J. S., Yamano M. M., Ramirez M., Darzi E. R., Knapp R. R., Liu F., Houk K. N., Garg N. K., Nat. Chem. 2018, 10, 953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For reviews on general axial-to-point chirality transfer employing allenes, see:
- 16a. Ma S., Chem. Soc. Rev. 2005, 105, 2829–2871; [Google Scholar]
- 16b. Campolo D., Gastaldi S., Roussel C., Betrand M. P., Nechab M., Chem. Soc. Rev. 2013, 42, 8434–8466; [DOI] [PubMed] [Google Scholar]
- 16c. Alonso J. M., Quirós M. T., Muñoz M. P., Org. Chem. Front. 2016, 3, 1186–1204. [Google Scholar]
- 17.For key references to photochemical axial-to-point chirality transfer, see:
- 17a. Ayitou A. J.-L., Jesuraj J. L., Barooah N., Ugrinov A., Sivaguru J., J. Am. Chem. Soc. 2009, 131, 11314–11315; [DOI] [PubMed] [Google Scholar]
- 17b. Kumarasamy E., Ayitou A. J.-L., Vallavoju N., Raghunathan R., Iyer A., Clay A., Kandappa S. K., Sivaguru J., Acc. Chem. Res. 2016, 49, 2713–2724. [DOI] [PubMed] [Google Scholar]
- 18.CCDC 1988993 (5 a) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 19.For examples of a chirality transfer in Diels–Alder reactions of trisubstituted allenes with an electron withdrawing group, see:
- 19a. Yu J., Chen W.-J., Gong L.-Z., Org. Lett. 2010, 12, 4050–5053; [DOI] [PubMed] [Google Scholar]
- 19b. Ma Z.-G., Wei J.-L., Lin J.-B., Wang G.-J., Zhou J., Chen K., Fan C.-A., Zhang S.-Y., Org. Lett. 2019, 21, 2468–2472. [DOI] [PubMed] [Google Scholar]
- 20. Marx V. M., Cameron T. S., Burnell D. J., Tetrahedron Lett. 2009, 50, 7213–7216. [Google Scholar]
- 21. Byrd L. R., Caserio M. C., J. Am. Chem. Soc. 1971, 93, 5758–5764. [Google Scholar]
- 22. Smadja W., Chem. Rev. 1983, 83, 263–320. [Google Scholar]
- 23.
- 23a. Zhao G.-L., Huang J. W., Shi M., Org. Lett. 2003, 5, 4737–4739; [DOI] [PubMed] [Google Scholar]
- 23b. Rassu G., Zambrano V., Tanca R., Sartori A., Battistini L., Zanardi F., Curti C., Casiraghi G., Eur. J. Org. Chem. 2012, 466–470; [Google Scholar]
- 23c. Manoni F., Connon S. J., Angew. Chem. Int. Ed. 2014, 53, 2628–2632; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2666–2670. [Google Scholar]
- 24. Chen C., Kattanguru P., Tomashenko O. A., Karpowicz R., Siemiaszko G., Bhattacharya A., Calasans V., Six Y., Org. Biomol. Chem. 2017, 15, 5364–5372. [DOI] [PubMed] [Google Scholar]
- 25. Alonso R., Bach T., Angew. Chem. Int. Ed. 2014, 53, 4368–4371; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4457–4460. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary