

Abstract
Background. Severe influenza is characterized by cytokine storm and multiorgan failure with edema. The aim of this study was to define the impact of the cytokine storm on the pathogenesis of vascular hyperpermeability in severe influenza.
Methods. Weanling mice were infected with influenza A WSN/33(H1N1) virus. The levels of proinflammatory cytokines, tumor necrosis factor (TNF) α, interleukin (IL) 6, IL-1β, and trypsin were analyzed in the lung, brain, heart, and cultured human umbilical vein endothelial cells. The effects of transcriptional inhibitors on cytokine and trypsin expressions and viral replication were determined.
Results. Influenza A virus infection resulted in significant increases in TNF-α, IL-6, IL-1β, viral hemagglutininprocessing protease trypsin levels, and viral replication with vascular hyperpermeability in lung and brain in the first 6 days of infection. Trypsin upregulation was suppressed by transcriptional inhibition of cytokines in vivo and by anti-cytokine antibodies in endothelial cells. Calcium mobilization and loss of tight junction constituent, zonula occludens-1, associated with cytokine- and trypsin-induced endothelial hyperpermeability were inhibited by a protease-activated receptor-2 antagonist and a trypsin inhibitor.
Conclusions. The influenza virus-cytokine-protease cycle is one of the key mechanisms of vascular hyperpermeability in severe influenza.
Influenza A virus is the most common infectious pathogen in humans and causes significant morbidity and mortality, particularly in infants and the elderly population [1, 2]. Multiorgan failure with severe edema is observed in the advanced stage of influenza pneumonia and influenza-associated encephalopathy [3, 4]. However, the relationship between factors that induce vascular hyperpermeability and multiorgan failure in severe influenza remains unclear.
Significant increases in levels of proinflammatory cytokines such as tumor necrosis factor (TNF) α, interleukin (IL) 6, and IL-1β (ie, cytokine storm) affect host survival both positively and negatively [5–7]. The inflammatory response affects cell adhesion, permeability, apoptosis, and mitochondrial reactive oxygen species, potentially resulting in vascular dysfunction and multiorgan failure [8]. In addition, influenza A virus infection upregulates several cellular proteases, including ectopic trypsin [9] and matrix metalloprotease (MMP) 9 [10]. Ectopic trypsin, like tryptase Clara [11], mediates the post-translational proteolytic cleavage of viral envelope hemagglutinin [12], which is crucial for viral entry and replication [13–17] and subsequent tissue damage in various organs [9, 16, 17]. Influenza A virus infection significantly upregulates trypsin in endothelial cells and in hippocampal neurons [9]. Because trypsin efficiently converts pro-MMP-9 to active MMP-9 [18], induction of both proteases synergistically degrades basement membrane proteins, potentially destroying tight junctions and the blood-brain barrier, followed by multiorgan failure [19, 20].
The aim of the present study was to define the pathogenic impact of cytokine storm in influenza A virus infection and the molecular mechanisms by which proinflammatory cytokines cause vascular dysfunction in animal models and in human vein endothelial cells. The results pointed to the role of the influenza virus-cytokine-protease cycle as one of the main mechanisms of vascular dysfunction in severe influenza.
Materials and Methods
Animals and virus. Specified pathogen-free 3-week-old weanling C57BL/6CrSlc female mice were obtained from Japan SLC. Under ketamine anesthesia, 250 or 500 plaque-forming units (PFU) of influenza A/WSN/33(H1N1) [21, 22] in 15 µL of saline or saline alone as the vehicle was instilled intranasally in mice. Mice (n=10) also received inhibitors against nuclear factor-kappa B (NF-κB), such as pyrrolidine dithiocarbamate (10 mg/kg) and N-acetyl-L-cysteine (10 mg/kg) [23, 24], and inhibitor against activator protein 1, nordihydroguaiaretic acid (2.5 mg/kg) [25], intraperitoneally. These inhibitors were administrated once daily for 4 days immediately after viral infection (day 0). Virus titers were determined in Madin-Darby canine kidney cells [11]. All animals were treated in accordance with the guidelines of the animal care committee of the University of Tokushima.
Cell culture. Human umbilical vein endothelial cells (Lonza) were grown using the protocol supplied by the manufacturer. The cells were infected by influenza A virus WSN at amultiplicity of infection of 0.5 or treated with recombinant human IL-6, TNF-α, and IL-1β (10 ng/mL of each) (PeproTec) in the presence or absence of antibodies against these cytokines (Abcam).
Evaluation of vascular permeability. Vascular permeability was analyzed by the Evan's blue extravasation method [26]. One hour after intraperitoneal injection of 400 µL of 2% (w/ v) Evan's blue dye in saline, the whole body was perfused with saline through the cardiac ventricle. The leakage of dye was detected macroscopically and by fluorescence microscope.
Enzyme-linked immunosorbent assay (ELISA). The levels of IL-6, TNF-α, and IL-1β in tissue homogenates and plasma were measured using cytokine ELISA kits (BD Biosciences).
Western blotting and gelatin zymography. Tissues were homogenized with 3 volumes of Tris-HCl, pH 6.8, containing 2% sodium dodecyl sulphate and 0.5 M NaCl, and centrifuged at 12,000 g for 30 min. Human endothelial cells were lysed in radioimmune precipitation buffer (Nacalai Tesque) at 4°C. These extracts (30 µg protein) were electrophoresed and transferred to polyvinylidene difluoride membranes. Rabbit antizonula occludens-1, anti-occludin antibodies (Zymed), and anti-actin antibody (Chemicon) were used. Immunoreactive bands were detected using chemiluminescence (Amersham Biosciences). For gelatin zymography, the extracts (50 µg protein) were subjected to electrophoresis on 10% gelatin zymogram gels (Invitrogen) as reported previously [9].
Immunohistochemical staining. Immunohistochemical staining was conducted as described elsewhere [9]. Lung and brain sections were reacted overnight with polyclonal antibodies against human influenza A, B virus (Takara) at 4°C, washed, and then reacted for 1 h at room temperature with a biotinylated second antibody. The sections were counterstained with Mayer's hematoxylin.
Permeability assay. Human endothelial cells grown to confluence on 12-well tissue culture plates with FALCON Cell Culture Inserts (1.0 µm), were exposed to the cytokines for 12 h in the presence or absence of 50 µM of aprotinin (Nacalai Tesque). Changes in the monolayer permeability were analyzed and quantified as clearance of fluorescein isothiocyanate-dextran from the upper chamber to lower chamber as reported previously [27].
Reverse-transcription polymerase chain reaction (RT-PCR) and real-time PCR. Total RNA was isolated from human endothelial cells using an RNeasy Mini kit (Qiagen) and reverse transcribed using Oligo primers and SuperScript III RT (Gibco BRL) for complementary DNA synthesis. The following primer pairs were used to amplify human trypsin (hPRSS): hPRSS (forward primer [F], 5'-ATCCAGGTGAGACTGGGAGAGCACA- 3', nucleotide (nt) 222–246, and reverse primer [R], 5'- GTAGACCTTGGTGTAGACTCCAGGC-3', nt 692–716) and those of viral NS1 as reported elsewhere [28]. RT-PCR and quantification of gene expression by real time-PCR were performed using Fast Start SYBR Green Master (Roche Diagnostics) on an ABI Prism 7300 system [28].
Measurement of intracellular calcium levels. Human endothelial cells were cultured on glass chamber slides until confluence. After washing twice with calcium- and magnesiumfree phosphate-buffered saline (PBS—), the cells were incubated with cytokines (10 ng/mL for each cytokine) for 10 h with or without pretreatment for 30 min with 20 µM protease-activated receptor (PAR) 2 antagonist peptide, FSY-NH2 [29], or 50 µM aprotinin at 37°C. The cells were also activated with 1 µg/mL trypsin or 10 µM PAR-2 agonist peptide [30] for 30 min. The cells were also treated for 5 h with 10 nM calcium ionophore A23187 (Calbiochem) or 2 mM CaCl2. For imaging, 10 µM Fluo-3/AM (Invitrogen) was introduced into the cells by incubation for 30 min. The cells were then washed twice with PBS— and incubated with 5 mM glucose in PBS— at 37°C. Intracellular calcium ([Ca2+]i) levels were analyzed using a confocal laser scanning microscope (model CM1900; Leica).
Statistical analysis. Results are presented as mean value ± standard error of the mean (from 3–5 independent experiments). Differences between groups were examined for statistical significance by the paired t test or 1-way analysis of variance. The Wilcoxon test for comparisons of Kaplan-Meier survival curves was used. A P value <.05 was considered to be statistically significant.
Results
Upregulation by influenza A virus infection of cytokines and ectopic trypsin and their suppression by inhibitors of NF-κB and activator protein 1. Mice were infected with influenza A virus WSN to study the pathogenic effects of cytokine storm on vascular dysfunction. The levels of TNF-α and IL-6 in the lungs, the site of initial virus infection, were increased persistently for 6 days, and levels of IL-1β peaked at days 4–6 after infection (Figure 1A). Because these cytokine responses are associated with activation of the transcription factors NF-κB and activator protein 1 [7, 31–33], we treated mice once daily for 4 days with anti-oxidant inhibitors: pyrrolidine dithiocarbamate and N-acetyl-L-cysteine against NF-κB activation, and nordihydroguaiaretic acid against activator protein 1 activation. Pyrrolidine dithiocarbamate and nordihydroguaiaretic acid significantly suppressed the upregulation of TNF-α and IL-1β (P < .001), and N-acetyl-L-cysteine suppressed TNF-α (P < .001) and IL-6 (P < .01) at day 4 after infection (Figure 1A).
Figure 1.

Upregulation of cytokines and trypsin, increase in viral RNA after influenza A virus infection in mice, and effects of nuclear factor-kappa B (NF−κB) and activator protein 1 inhibitors on the upregulation and survival of infected mice. A, Mice were infected with 250 plaque-forming units (PFU) of influenza A WSN/33(H1N1) virus (WSN) with and without treatment with pyrrolidine dithiocarbamate (PDTC), N-acetyl-L-cysteine (NAC), and nordihydroguaiaretic acid (NDGA). Levels of tumor necrosis factor (TNF) α, interleukin (IL) 6, and IL-1β in lung homogenates (n=3) were analyzed before (Control) and at day 2 (WSN-D2), day 4 (WSN-D4), and day 6 (WSN-D6) after infection. Cytokine levels in lungs of animals treated once daily for 4 days with PDTC (PDTC-D4), NAC (NAC-D4), and NDGA (NDGA-D4) were also measured. Data are mean value ± standard error of the mean (SEM). **P < .01, #P < .001, versus WSN-D4. B, Trypsin activities analyzed by gelatin zymography of infected mice for 0–6 days. Animals were treated with PDTC, NAC, and NDGA once daily for 4 days. Each lane represents the same experimental conditions as in A. C, Mice were infected with WSN and also treated with PDTC, NAC, and NDGA. Quantitative analysis of viral NS1 RNA copies normalized by b-actin at day 4 after infection was conducted by real-time polymerase chain reaction (n=3). Data are mean ± SEM. #P < .001 versus without drug treatment. D, Mice of each group (n=10) were infected with WSN at 250 PFU and 500 PFU. Animals infected with WSN at 250 PFU were treated with PDTC, NAC, and NDGA once daily for 4 days, and the survival rates of the different groups were compared. *P < .05, #P < .001 versus without drug treatment.
Gelatin zymography showed upregulation of ectopic trypsin in mice lung, brain, and heart during infection for 6 days (Figure 1B). Trypsin induction was inhibited by treatment with pyrrolidine dithiocarbamate, N-acetyl-L-cysteine, and nordihydroguaiaretic acid, probably via blockade of NF-κB and activator protein 1 binding in the promoter region of the gene (S. R. Talukder, unpublished data). Viral RNA replication in various organs at day 4 after infection was suppressed by >1 order of magnitude by pyrrolidine dithiocarbamate, N-acetyl- L-cysteine, and nordihydroguaiaretic acid (Figure 1C). Suppression of viral multiplication and induction of cytokines and trypsin by treatment with pyrrolidine dithiocarbamate, N-acetyl- L-cysteine, and nordihydroguaiaretic acid significantly improved the survival of mice at day 14 after infection (ie, the late stage of infection) (Figure 1D).
Viral protein accumulation and increase in vascular permeability in lung and brain. The kinetics of viral replication monitored by viral NS1 gene showed that the level of viral RNA was the highest at day 4 after infection and decreased at day 6 in these organs (Figure 2A). To determine the pathogenesis of tissue injury, the viral protein accumulation in the lung and brain at day 4 after infection was analyzed by immunohistochemical staining (Figure 2B). Viral protein was detected in alveoli and terminal bronchioles in the lung and was also detected in the brain, particularly in the hippocampus, neocortex, brainstem, and brain capillaries.
Figure 2.
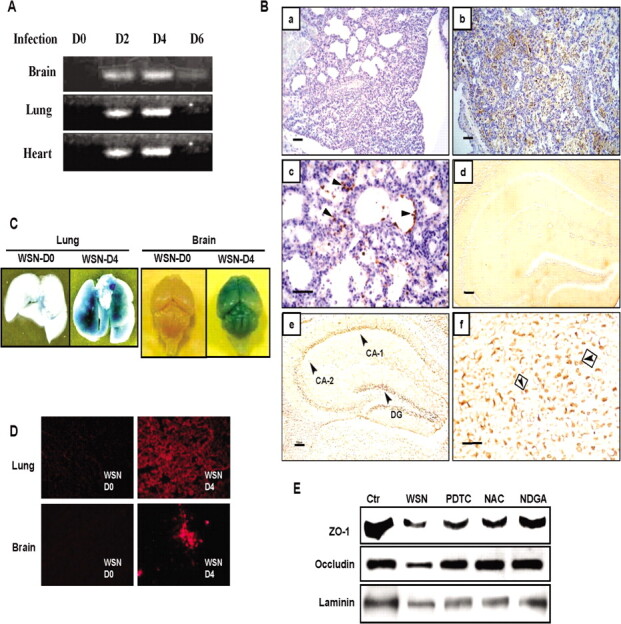
Kinetics of viral proliferation, viral protein accumulation, increase in vascular permeability, and loss of tight-junction proteins in various organs after influenza A WSN/33(H1N1) virus (WSN) infection. A, Detection of viral NS1 gene by reverse-transcription polymerase chain reaction (RTPCR) in the lung, heart, and brain of mice during 0-6 days after infection. B, Immunohistochemical detection of viral antigens in mouse lung and brain at day 4 after infection. a, Hematoxylin and eosin staining of the lung (original magnificationm, ×200). b, Immunoreactive deposits in the lung (original magnification, ×200). c, Viral antigen (arrowheads) in epithelial cells of respiratory bronchioles and infiltrated leukocytes in alveoli (original magnification, ×400). d, No immunoreactive deposits in the brain before infection (original magnification, ×200). e, Virus antigen in the cornu ammonis (CA) 1 and CA-2 and in the stratum granulosum of the dentate gyrus (DG) of the hippocampus (original magnification,×200). f, Virus antigen (arrowheads) in the enlarged image of CA-1 (original magnification, ×400). Scale bars are 100 µm.C, Vascular permeability in the lung and brain analyzed by Evan's blue dye extravasation before (WSN-D0) and after infection at day 4 (WSN-D4). D, Fluorescent micrographs of Evan's blue leakage from capillaries in the brain and lung before and after infection at day 4. E, Loss of tight-junction proteins, zonula occludens (ZO) 1 and occludin, and laminin in the brain analyzed by Western immunoblotting at day 4 after infection and its restoration by pyrrolidine dithiocarbamate (PDTC), N-acetyl-L-cysteine (NAC), and nordihydroguaiaretic acid (NDGA) treatments. The levels before infection are shown as control (Ctr).
Vascular hyperpermeability is one of the main complications of organ injury in severe influenza. Vascular permeability was analyzed by infiltration of Evans Blue dye in the lung and brain after infection (Figure 2C and 2D). In contrast to no dye infiltration in uninfected animals, infected mice showed a progressive increase in vascular permeability in the lung and brain at day 4 after infection. Fluorescence microscopy showed leakage of dye from the blood vessels in these organs (Figure 2D).
To elucidate the mechanisms underlying vascular dysfunction in the brain, changes in the levels of tight-junction proteins, intracellular zonula occludens-1 and transmembrane occludin, and the matrix protein laminin were analyzed byWestern blotting. Marked reductions in the expression levels of tight-junction constituents were detected at day 4 after infection, which were partly rescued by pyrrolidine dithiocarbamate, N-acetyl- L-cysteine, or nordihydroguaiaretic acid (Figure 2E). No other tight-junction protein, claudin-5, or matrix fibronectin and type IV collagen was affected (data not shown).
Effects of influenza A virus infection and cytokine treatment on human endothelial cells in culture. To clarify the linkage between upregulated cytokines and trypsin and vascular hyperpermeability after viral infection, the relationships among these findings were examined in human endothelial cells. Viral infection significantly increased TNF-α and IL-6 levels (2.7- fold and 7.1-fold, respectively) but not IL-1β levels in the culture media in a time-dependent manner over a 24-h period (Table 1).
Table 1.

Proinflammatory Cytokine Levels in the Culture Media of Human Endothelial Cells after Influenza A WSN/33(H1N1) Virus Infection
Influenza A virus infection upregulated human trypsin/ hPRSS gene by approximately 2-fold in the cells after infection for 6–12 h (Figure 3A). To analyze the linkage between cytokines and trypsin in the cells, changes in the expression of hPRSS gene were analyzed after exposure to 10 ng/mL TNF-α, IL-6, and IL-1β instead of viral infection for 6 h (Figure 3B). All tested cytokines tended to upregulate hPRSS expression levels, especially TNF-α (P < .01) and IL-1β (P < .05), although less effectively than did viral infection, and the upregulation was inhibited by simultaneous treatment of the respective neutralizing antibodies (100 ng/mL) with these cytokines (P < .05 for TNF-α; P < .01 for IL-1β).
Figure 3.

Increase in human trypsin (hPRSS) expression in endothelial cells after influenza A WSN/33(H1N1) virus (WSN) infection and cytokine treatment and its suppression by anti-cytokine antibodies. A, hPRSS messenger RNA (mRNA) levels in the cells measured by reverse-transcription polymerase chain reaction (RT-PCR) after viral infection for 0–12 h and the percentage change in the expression. B, Increase in hPRSS mRNA levels in the cells after treatment with cytokine (tumor necrosis factor [TNF] α, interleukin [IL] 6, and IL-1β) for 6 h and its suppression by anti-cytokine antibodies. Data are mean value ± standard error of the mean. ##P < .01, #P < .05 versus the control. **P < .01, *P < .05, versus treatment with each antibod
Relationship between cytokines and trypsin upregulation in the loss of tight-junction proteins in human endothelial cells. Treatment of the cells for 12 h with TNF-α, IL-6, and IL-1β markedly suppressed tight-junction protein zonula occludens- 1 levels and occluding levels slightly, and loss of these proteins were abrogated by simultaneous treatment of the cells with 50 µM of the nonpermeable trypsin inhibitor aprotinin (Figure 4A). Furthermore, cytokine treatment disrupted the continuous and linear arrangement of zonula occludens-1 among the cells, and aprotinin inhibited the disruption (Figure 4B). Accordingly, treatment with cytokines, especially IL-1β and TNF-α, tended to increase endothelial cell monolayer permeability and this effect was blocked by 50 µM of aprotinin (P < .05) (Figure 4C). These findings suggest that cytokines upregulate trypsin in vascular endothelial cells and that secreted trypsin plays an important mechanistic role in the loss of zonula occludens-1 and increased permeability.
Figure 4.

Loss of tight-junctions by cytokine treatment and its rescue by trypsin inhibitor. A, Western blotting analysis of tight-junction proteins, zonula occludens (ZO) 1 and occludin after treatment of the cells with cytokines for 12 h in the absence and presence of 50 µM aprotinin. Actin as an internal control (Ctr). B, Representative example (from 3 separate experiments) of immunofluorescence showing decreased ZO-1 with cytokine treatment and its restoration by aprotinin. C, Increased permeability of the cells treated with cytokines and its rescue by aprotinin (n=3). Data are mean value ± standard error of the mean. *P < .05 between the values with and without aprotinin.
Figure 6.

The hypothesis of influenza virus-cytokine-protease cycle, which may affect the pathogenesis of vascular hyperpermeability and tissue destruction in severe influenza. AP-1, activator protein 1; BBB, blood-brain barrier; PAR-2, protease-activated receptor 2; ZO-1 zonula, occludens-1.
Cytokines increase in the intracellular Ca2+ levels via PAR- 2 in human endothelial cells. Inflammatory mediators cause an increase in [Ca2+]i through G protein-coupled receptors, leading to cytoskeletal reorganization in the microvascular endothelium and consequent increase in permeability and tissue edema [34]. Trypsin receptor PAR-2 is a G protein-coupled receptor activated by trypsin and tryptase and plays an important role in increasing [Ca2+]i [35]. To investigate the mechanisms underlying vascular hyperpermeability in severe influenza, we treated human endothelial cells with cytokines, trypsin, and PAR-2 agonist peptide in PBS— and thenmeasured [Ca2+]i (Figure 5). Marked [Ca2+]i mobilization was found after treatment of the cells with trypsin, PAR-2 agonist peptide, and CaCl2 for 10 h, whereas calcium ionophore A23187 decreased [Ca2+]i. Treatment with TNF-α, IL-1β, and IL-6 also increased [Ca2+]i, and the mobilization was suppressed by pretreatment of the cells with PAR-2 antagonist, FSY-NH2, or aprotinin for 30 min. These results suggest that [Ca2+]i mobilization by proinflammatory cytokines through activation of trypsin and its receptor PAR-2 is one of the main mechanisms underlying increased endothelial cell permeability.
Figure 5.
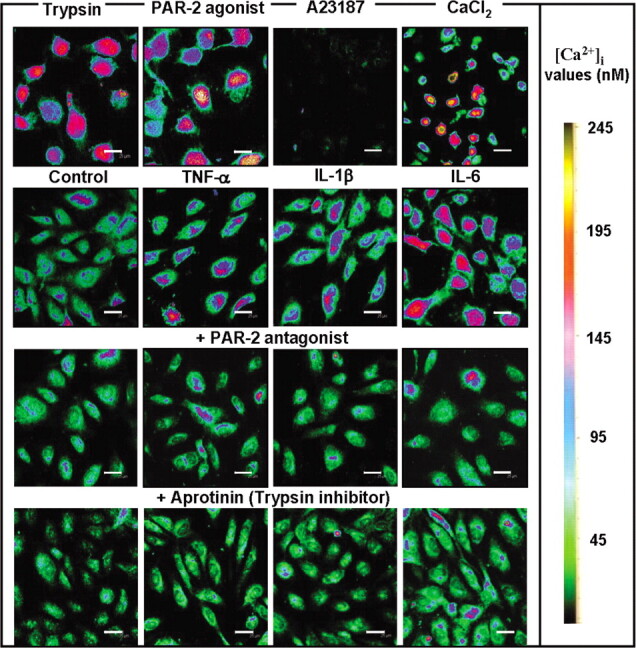
Effects of cytokines trypsin, and protease-activated receptor (PAR) 2 agonist on [Ca2+]i and rescue of the increase in [Ca2+]i by PAR-2 antagonist and aprotinin in endothelial cells. The cells were treated for 10 h with 1 µg/mL trypsin, 10 µM PAR-2 agonist, 10 mM calcium ionophore A23187, and 2 mM CaCl2. The cells were stimulated without (control) or with 10 ng/mL tumor necrosis factor (TNF) α, interleukin (IL) 1β, and IL-6. For inhibition studies on [Ca2+]i, the cells were pretreated for 30 min with 20 µM PAR-2 antagonist FSY-NH2 or 50 µM aprotinin and then treated with these cytokines. Fluorescence images of the cells were analyzed by a confocal microscope. [Ca2+]i values (nM) are displayed using a color scale. Scale bars are 25 µm.
Discussion
The present study reports several new observations: (1) proinflammatory cytokines, TNF-α, IL-1β, and IL-6, when upregulated by influenza A virus infection, induce trypsin expression in various organs and human endothelial cells; (2) the upregulated trypsin induces [Ca2+]i mobilization via activation of the PAR-2, followed by loss of zonula occludens-1 and vascular hyperpermeability; (3) inhibitors of NF-κB and activator protein 1 effectively suppress the upregulation of proinflammatory cytokines and trypsin and improve the survival rates of infected mice. Based on these results, we propose the influenza virus- cytokine-protease cycle hypothesis as one of the mechanisms of vascular dysfunction in multiorgan failure with cytokine storm in severe influenza and influenza-associated encephalopathy (Figure 6).
The significance of proinflammatory hypercytokinemia, or cytokine storm, in the pathogenesis of influenza A virus infection remains unclear. The positive effects include that cytokines promote lymphocyte activation and infiltration at the sites of infection and exert direct antiviral effects. However, the negative effects of excess cytokines include the fact that the hyperinflammatory process evoked by viral infection [8, 36] may become harmful through intracellular activation of NF-κB, activator protein 1, and the Janus kinase-signal transducers and activators of transcription signaling pathways [31–33, 37, 38]. The in vivo experiments presented here showed that NF-κB and activator protein 1 inhibitors markedly suppress the expression of cytokines and trypsin, viral replication, and endothelial dysfunction and result in a significant increase in the survival of infected mice. Furthermore, cytokines interact with mitochondria to increase the production of reactive oxygen species, resulting in the production and activation of vasodilatory mediators, such as nitric oxide and bradykinin, and subsequent endothelial dysfunction and edema in various organs [8] (Figure 6).
The molecular mechanisms underlying tight-junction disruption in endothelial cells and vascular hyperpermeability following the cytokine storm remain unclear. TNF-α upregulation alters the cellular redox state, reduces the expression of 4 complex I subunits by increasing mitochondrial O2− production and depleting adenosine triphosphate (ATP) synthesis, decreases oxygen consumption (resulting in mitochondrial damage) [8, 39], and increases [Ca2+]i [40]. ATP depletion dissociates zonula occludens-1 from the actin cytoskeleton and thereby increases junctional permeability [41]. The present results allow us to propose a new mechanism of junctional permeability regulation: upregulated trypsin by influenza A virus and/or proinflammatory cytokines induces increase in [Ca2+]i and loss of zonula occludens-1 in endothelial cells via PAR-2 signaling. In contrast to the marked upregulation of cytokines in the lungs (Figure 1A), upregulation of cytokines in the brain was mild (data not shown), which suggests that the bloodbrain barrier destruction is the result of systemic effects of cytokines produced in the lung in severe influenza. Anti-cytokine antibodies and trypsin inhibitors may effectively suppress junctional permeability.
Endothelial dysfunction induced by the influenza virus-cytokine-protease cycle in the early stage of severe influenza may also affect various circulating factors, coagulation factors, and complement systems, as well as vascular interacting cells, such as neutrophils, macrophages, and lymphocytes. Multiorgan failure is the final outcome of metabolic and mitochondrial fuel disorder, immunosuppression, endocrine disorder, and tissue injury followed by endothelial dysfunction in many organs. Another key pathway of acute lung injury in the highly pathogenic avian influenza virus H5N1 and acute respiratory syndrome- corona virus infection reported recently involves oxidative stress and the formation of oxidized phospholipids, which induce lung injury via Toll-like receptor 4 signaling pathway [42]. In addition to these data, upregulated trypsin and proinflammatory cytokines may also affect tissue destruction and immunosuppression in the late stage of influenza A virus infection. Further studies are required on the role of the influenza virus-cytokine-protease cycle in the pathogenesis of multiorgan failure, particularly in the late stage of viral infection.
Acknowledgments
We are grateful to Mayumi Shiota for expert assistance.
Footnotes
Potential conflicts of interest: none reported.
Financial support: Grant in Aid (21249061) and the Special Coordination Funds for Promoting Science and Technology of Ministry of Education Culture, Sports, Science and Technology of Japan. Present affiliation: School of Nursing, Nantong University, Nantong 226001, China.
References
- 1.Lipatov AS, Govorkova EA, Webby RJ, et al. Influenza: emergence and control. J Virol. 2004;78:8951–8959. doi: 10.1128/JVI.78.17.8951-8959.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HM, Brandt CD, Arrobio JO, Murphy B, Chanock RM, Parrott RH. Influenza A and B virus infection in infants and young children during the years 1957–1976. Am J Epidemiol. 1979;109:464–79. doi: 10.1093/oxfordjournals.aje.a112704. [DOI] [PubMed] [Google Scholar]
- 3.Dolorme L, Middleton PJ. Influenza A virus associated with acute encephalopathy. Am J Dis Child. 1979;133:822–824. doi: 10.1001/archpedi.1979.02130080062011. [DOI] [PubMed] [Google Scholar]
- 4.Fujimoto S, Kobayashi M, Uemura O, et al. PCR on cerebrospinal fluid to show influenza-associated acute encephalopathy or encephalitis. Lancet. 1998;352:873–875. doi: 10.1016/S0140-6736(98)12449-2. [DOI] [PubMed] [Google Scholar]
- 5.Kawada J, Kimura H, Ito Y, et al. Systemic cytokine responses in patients with influenza-associated encephalopathy. J Infect Dis. 2003;188:690–698. doi: 10.1086/377101. [DOI] [PubMed] [Google Scholar]
- 6.Cheung CY, Poon LL, Lau AS, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360:1801–1802. doi: 10.1016/s0140-6736(02)11772-7. [DOI] [PubMed] [Google Scholar]
- 7.Julkunen I, Sareneva T, Pirhonen J, Ronni T, Melén K, Matikainen S. Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev. 2001;12:171–180. doi: 10.1016/s1359-6101(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 8.Spraque AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le QT, Kawachi M, Yamada H, Shiota M, Okumura Y, Kido H. Identification of trypsin I as a candidate for influenza A virus and Sendai virus envelope glycoprotein processing protease in rat brain. Biol Chem. 2006;387:467–475. doi: 10.1515/BC.2006.062. [DOI] [PubMed] [Google Scholar]
- 10.Ichiyama T, Morishima T, Kajimoto M, Matsushige T, Matsubara T, Furukawa S. Matrix metalloprotease-9 and tissue inhibitors of metalloproteinases I in influenza-associated encephalopathy. Pediatr Infect Dis J. 2007;26:542–544. doi: 10.1097/INF.0b013e31803994a0. [DOI] [PubMed] [Google Scholar]
- 11.Kido H, Yokogoshi Y, Sakai K, et al. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelia Clara cells. A possible activator of the viral fusion glycoprotein. J Biol Chem. 1992;267:13573–13579. [PubMed] [Google Scholar]
- 12.Klenk HD, Rott R, Orlich M, Blödom J. Activation of influenza A viruses by trypsin treatment. Virology. 1975;68:426–439. doi: 10.1016/0042-6822(75)90284-6. [DOI] [PubMed] [Google Scholar]
- 13.Homma M, Ohuchi M. Trypsin action on the growth of Sendai virus in tissue culture cells: structural difference of Sendai viruses grown in eggs and tissue culture cells. J Virol. 1973;12:1457–1465. doi: 10.1128/jvi.12.6.1457-1465.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheid A, Choppin P. Identification of biological activity of paramyxovirus glycoprotein: activation of cell fusion, hemolysis and infectivity by proteolytic cleavage of an inactive precursor protein of Sendai virus. Virology. 1974;57:475–490. doi: 10.1016/0042-6822(74)90187-1. [DOI] [PubMed] [Google Scholar]
- 15.Klenk HD, Rott R. The molecular of influenza virus pathogenicity. Adv Virus Res. 1988;34:247–281. doi: 10.1016/S0065-3527(08)60520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kido H, Okumura Y, Yamada H, Le QT, Yano M. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr Pharm Des. 2007;13:405–414. doi: 10.2174/138161207780162971. [DOI] [PubMed] [Google Scholar]
- 17.Kido H, Okumura Y, Takahashi E, et al. Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J Mol Genet Med. 2008;3:167–175. [PMC free article] [PubMed] [Google Scholar]
- 18.Duncan ME, Richardson JP, Murray GI, Melvin WT, Fothergill JE. Human matrix metalloproteinase—9: activation by limited trypsin treatment and generation of monoclonal antibodies specific for the activated form. Eur J Biochem. 1998;258:37–43. doi: 10.1046/j.1432-1327.1998.2580037.x. [DOI] [PubMed] [Google Scholar]
- 19.Agrawal S, Anderson P, Durbeej M, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med. 2006;203:1007–1019. doi: 10.1084/jem.20051342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanumegowda UM, Copple BL, Shibuya M, Malle E, Ganey PE, Roth RA. Basement membrane and matrix metalloproteinases in monocrotaline- induced liver injury. Toxicol Sci. 2003;76:237–246. doi: 10.1093/toxsci/kfg222. [DOI] [PubMed] [Google Scholar]
- 21.Mori I, Kimura Y. Neuropathogenesis of influenza virus infection in mice. Microbes Infect. 2001;3:475–479. doi: 10.1016/s1286-4579(01)01403-4. [DOI] [PubMed] [Google Scholar]
- 22.Aronsson F, Lannebo C, Paucar M, Brask J, Kristensson K, Karlsson H. Persistence of viral RNA in the brain of offspring to mice infected with influenza A/WSN/33 virus during pregnancy. J Neurovirol. 2002;8:353–357. doi: 10.1080/13550280290100480. [DOI] [PubMed] [Google Scholar]
- 23.Siegel AL, Bledsoe C, Lavin J, et al. Treatment with inhibitors of the NF-κB pathway improves whole body tension development in the mdx mouse. Neuromuscular Disorders. 2009;19:131–139. doi: 10.1016/j.nmd.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Rebeca GR, Daniel SG, Sandra R, et al. The differential NF-κB modulation by S-adenosyl-L-methionine, N-acetylcysteine and quercetin on the promotion stage of chemical hepatocarcinogenesis. Free Radic Res. 2008;42:331–343. doi: 10.1080/10715760802005169. [DOI] [PubMed] [Google Scholar]
- 25.Kwon H, Park S, Lee S, Lee DK, Yang CH. Determination of binding constant of transcription factor AP-1 and DNA: application of inhibitors. Eur J Biochem. 2001;268:565–572. doi: 10.1046/j.1432-1327.2001.01897.x. [DOI] [PubMed] [Google Scholar]
- 26.Yao D, Kuwajima M, Chen Y, et al. Impaired long-chain fatty acid metabolism in mitochondria causes brain vascular invasion by a nonneurotropic epidemic influenza A virus in the newborn/suckling period: implications for influenza-associated encephalopathy. Mol Cell Biochem. 2007;299:85–92. doi: 10.1007/s11010-005-9046-x. [DOI] [PubMed] [Google Scholar]
- 27.Nakamuta S, Endo H, Higashi Y, et al. Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J Neurovirol. 2008;14:186–195. doi: 10.1080/13550280801993630. [DOI] [PubMed] [Google Scholar]
- 28.Sawabuchi T, Suzuki S, Iwase K, et al. Boost of mucosal secretory immunoglobulin A response by clarithromycin in pediatric influenza. Respirology. 2009;14:1173–1179. doi: 10.1111/j.1440-1843.2009.01639.x. [DOI] [PubMed] [Google Scholar]
- 29.Wilson S, Greer B, Hooper J, et al. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem J. 2005;388:967–972. doi: 10.1042/BJ20041066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blackhart BD, Emilsson K, Nguyen D, et al. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- 31.Nimmerjahn F, Dudziak D, Dirmeier U, et al. Active NF-κB signaling is a prerequisite for influenza virus infection. J Gen Virol. 2004;85:2347–2356. doi: 10.1099/vir.0.79958-0. [DOI] [PubMed] [Google Scholar]
- 32.Mori I, Goshima F, Koshizuka T, et al. Differential activation of the c- Jun N-terminal kinase/stress-activated protein kinase and p38 mitogenactivated protein kinase signal transduction pathways in the mouse brain upon infection with neurovirulent influenza A virus. J Gen Virol. 2003;84:2401–2408. doi: 10.1099/vir.0.19188-0. [DOI] [PubMed] [Google Scholar]
- 33.Santoro MG, Rossi A, Amici C. NF-κB and virus infection: who controls whom. EMBO J. 2003;22:2552–2560. doi: 10.1093/emboj/cdg267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation. 2006;13:693–708. doi: 10.1080/10739680600930347. [DOI] [PubMed] [Google Scholar]
- 35.Cottrell GS, Amadesi S, Schmidlin F, Bunnett N. Protease-activated receptor 2: activation, signalling and function. Biochem Soc Trans. 2003;31:1191–1197. doi: 10.1042/bst0311191. [DOI] [PubMed] [Google Scholar]
- 36.Xu T, Qiao J, Zhao L, et al. Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am J Respir Crit Care Med. 2006;174:1011–1017. doi: 10.1164/rccm.200511-1751OC. [DOI] [PubMed] [Google Scholar]
- 37.Moon SK, Cha BY, Kim CH. ERK1/2 mediates TNF-α-induced matrix metalloprotease-9 expression in human vascular smooth muscle cells via the regulation of NF-κB and AP-1: Involvement of the Ras dependent pathway. J Cell Physiol. 2004;198:417–427. doi: 10.1002/jcp.10435. [DOI] [PubMed] [Google Scholar]
- 38.Granet C, Maslinski W, Miossec P. Increased AP-1 and NF-κB activation and recruitment with the combination of the proinflammatory cytokines IL-1b, tumor necrosis factor alpha and IL-17 in rheumatoid synoviocytes. Arthritis Res Ther. 2004;6:R190–R198. doi: 10.1186/ar1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mariappan N, Elks CM, Fink B, Francis J. TNF-induced mitochondrial damage: a link between mitochondrial complex I activity and left ventricular dysfunction. Free Radic Biol Med. 2009;46:462–470. doi: 10.1016/j.freeradbiomed.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizuguchi M, Yamanouchi H, Ichiyama T, Shiomi M. Acute encephalopathy associated with influenza and other viral infections. Acta Neurol Scand. 2007;115:45–56. doi: 10.1111/j.1600-0404.2007.00809.x. [DOI] [PubMed] [Google Scholar]
- 41.Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. Am J Physiol. 1998;274:F1–F9. doi: 10.1152/ajprenal.1998.274.1.F1. [DOI] [PubMed] [Google Scholar]
- 42.Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]