

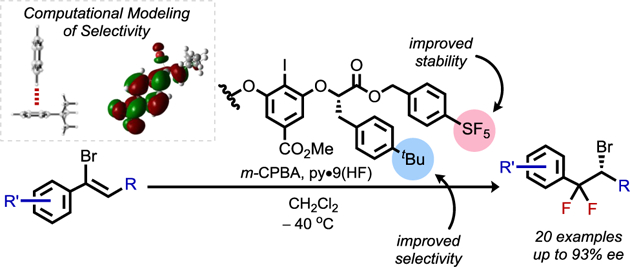
Abstract
We report an iodoarene-catalyzed enantioselective synthesis of β,β-difluoroalkyl halide building blocks. The transformation involves an oxidative rearrangement of α-bromostyrenes, utilizing HF–pyridine as the fluoride source and m-CPBA as the stoichiometric oxidant. A catalyst decomposition pathway was identified, which, in tandem with catalyst structure–activity relationship studies, facilitated the development of an improved catalyst providing higher enantioselectivity with lower catalyst loadings. The versatility of the difluoroalkyl bromide products was demonstrated via highly enantiospecific substitution reactions with suitably reactive nucleophiles. The origins of enantioselectivity were investigated using computed interaction energies of simplified catalyst and substrate structures, providing evidence for both CH–π and π–π transition state interactions as critical features.
Graphical Abstract
The disparity between the prevalence of fluorine in anthropogenic and naturally occurring compounds is striking.1 Few natural products contain carbon–fluorine linkages, whereas medicinal, agricultural, and materials chemistry are replete with examples where fluorination is critical for optimal functional properties.2 This difference manifests synthetically as an absence of naturally occurring chiral-pool sources of enantioenriched alkyl fluorides, necessitating the continuing development of chemical strategies for their selective synthesis.3 In this regard, fluorinated building blocks amenable to diversification are of particular interest.
We envisioned that compounds with the general structure 1 could be of high synthetic value given the unique properties of geminally difluorinated compounds (Figure 1A). The difluoromethylene motif has the highest dipole moment of the fluoromethane series and exerts a bond-angle-widening effect commensurate with sp2 centers, implicating geminal difluorides as both ketone and ether bioisosteres.4 These properties, together with the well-documented effects of fluorination on metabolic stability, lipophilicity, and potency of drug compounds, indicate that compounds such as 1 could be useful building blocks for the preparation of enantioenriched molecules containing geminal difluorides. Though deoxyfluorination and related transformations of carbonyl derivatives provide established approaches to gem-difluorination, they are generally not applicable to the synthesis of compounds bearing α-stereocenters because of epimerization and other nonproductive decomposition pathways.5
Figure 1.
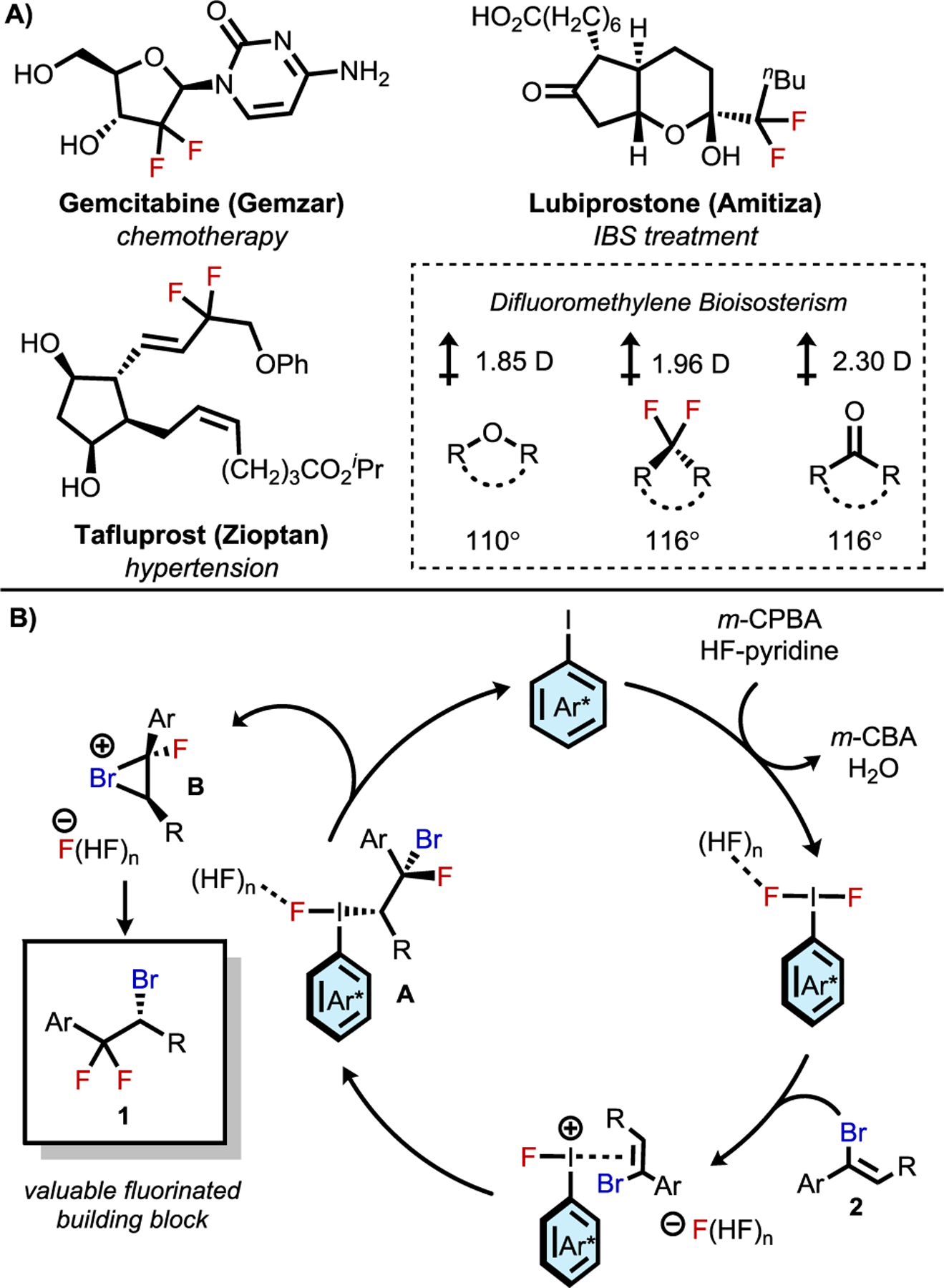
A) Examples of difluroromethylene-containing medicinal compounds and properties of difluoromethylenes as bioisosteres. B) Catalytic cycle for oxidative bromonium formation leading to enantioenriched alkyl bromides.
To access 1 and related building blocks, we turned to the chemistry of chiral iodoarenes, which have been applied recently by our group and others in catalytic, enantioselective fluorofunctionalizations of diverse olefinic substrates.6–8 We envisioned such catalysts could enable the enantioselective synthesis of β,β-difluorinated alkyl bromides from vinyl bromides of type 2 through a pathway involving initial, enantiodetermining fluoroiodination of the alkene. Subsequent intramolecular, stereospecific displacement of intermediate alkyliodane A to generate bromonium ion B and regioselective ring opening by fluoride would then afford the desired product (Figure 1B). We were encouraged by a report of a related oxidative hydrolysis of vinyl halides, affording racemic α-haloketones using stoichiometric Koser’s reagent.8h
Vinyl bromide 2a was evaluated as a model substrate for the proposed fluorinative bromonium rearrangement (Figure 2A). The α-benzyl-substituted Ishihara-type8c catalyst 3a that was identified previously as optimal in other alkene fluorofunctionalization reactions7a afforded desired product 1a in high conversion and promising levels of enantioselectivity. Systematic evaluation of catalysts bearing varied substitution on the α-benzyl groups (R1) led to the identification of para-tert-butyl-substituted derivative 3b as optimal. We hypothesized initially that the beneficial effect of tert-butyl substituents might have a steric origin, and this notion was seemingly supported by a good correlation between Charton parameters for the alkyl substituents and enantioselectivity (Figure 2B, black diamonds, R2alkyl = 0.91).9,10 However, the correlation fails if electron-withdrawing para-substituents are included (R2all = 0.22). We will return to an analysis of the intriguing catalyst substituent effects below.
Figure 2.
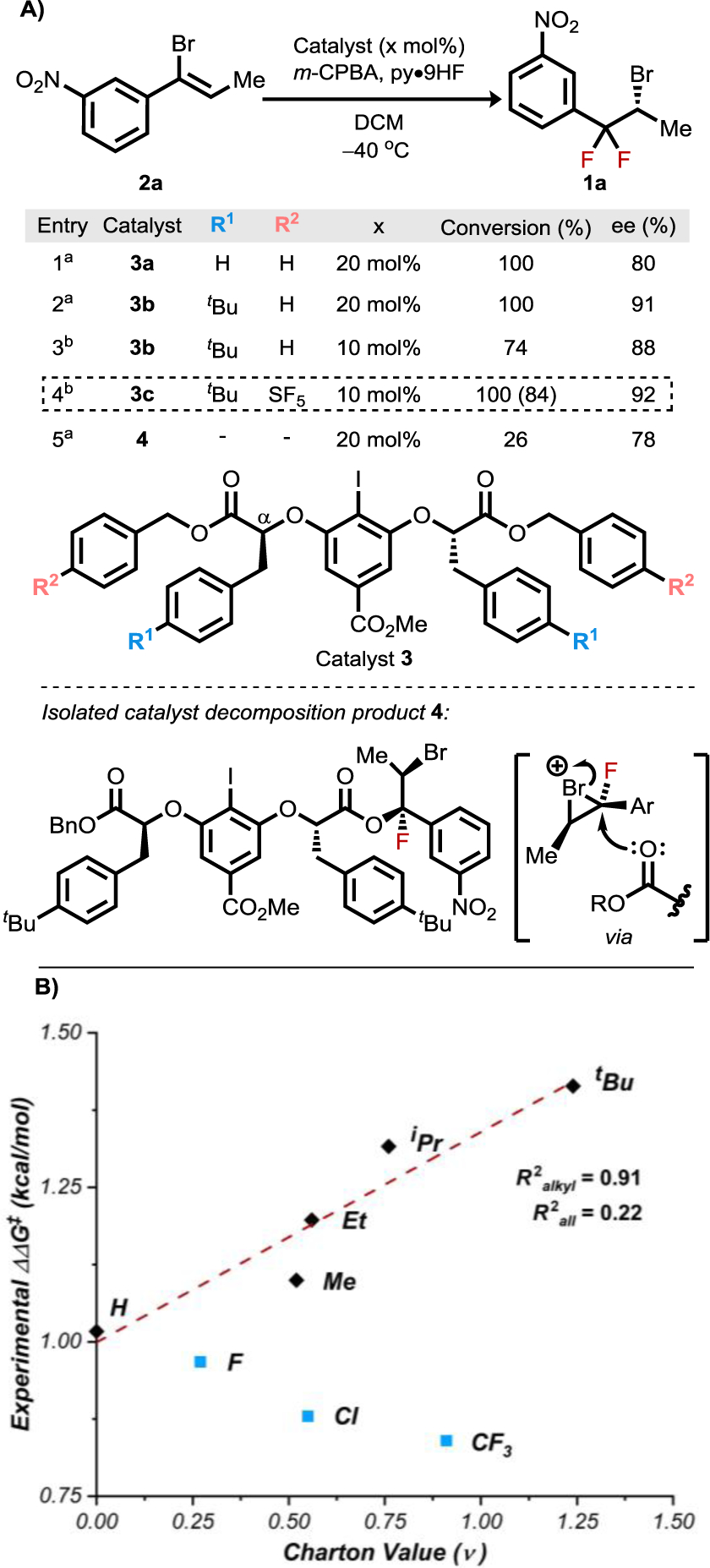
A) Reaction optimization and isolation of catalyst decomposition product 4. Conditions: 0.05 mmol scale, 11 equiv. py•9HF, 1.42 equiv. m-CPBA, 0.08M. a16 h. b48 h. Isolated yield in parentheses. B) Charton plot of catalyst substituents (R1) vs. experimental enantioselectivity (ΔΔG‡ = −RTln(e.r.)).
Lowering the catalyst loading of 3b from 20 mol% to 10 mol% led to a substantial decrease in reaction conversion with a concomitant decrease in enantioselectivity, the latter being unexpected given the lack of an observable background reaction in the absence of an iodoarene catalyst. The decrease in conversion was not offset by extending reaction times, indicating that catalyst deactivation was occurring. Indeed, efforts to recover the catalyst led to the identification of catalyst decomposition product 4, in which one benzyloxy substituent was replaced with a monofluorinated derivative of the vinyl bromide substrate. Reactions carried out with 4 as the catalyst produced the same difluorinated product 1a as parent catalyst 3b, but with substantially lower levels of conversion and diminished enantioselectivity.
We reasoned that the formation of 4 results from attack of a carbonyl group of catalyst 3b on the bromonium ion intermediate (Figure 2A).11 Catalysts bearing electron-withdrawing substituents on the benzyl ester (R2) were therefore prepared and evaluated with the goal of attenuating the nucleophilicity of the ester carbonyl and thereby limiting the decomposition pathway. Indeed, after examining several candidates, the p-SF5 derivative (3c) was identified as optimal for both conversion and enantioselectivity. Use of 3c at 10 mol% loading in the model transformation resulted in generation of 1a in 84% isolated yield and 92% ee with no decomposition products analogous to 4 observed (Figure S7).
Having identified 3c as an effective catalyst for the enantioselective difluorinative rearrangement with model substrate 2a, we evaluated the scope of this transformation with respect to the substitution pattern on the aromatic and aliphatic portions of the substrate (Figure 3). Styrenyl bromides bearing electron-withdrawing meta- and para-substituents were effective substrates (products 1a–1q), affording products in 52–84% yield and 70–93% ee. Substrates bearing free alcohol (1s) or primary aliphatic bromide (1t) substituents also reacted smoothly, affording the desired products with 90% ee and 76% ee, respectively. However, substrates possessing more electron-rich aromatic substituents or ortho-substituted arenes were either too unstable to isolate in sufficient quantities or decomposed under the reaction conditions, precluding their evaluation in this reaction.
Figure 3.
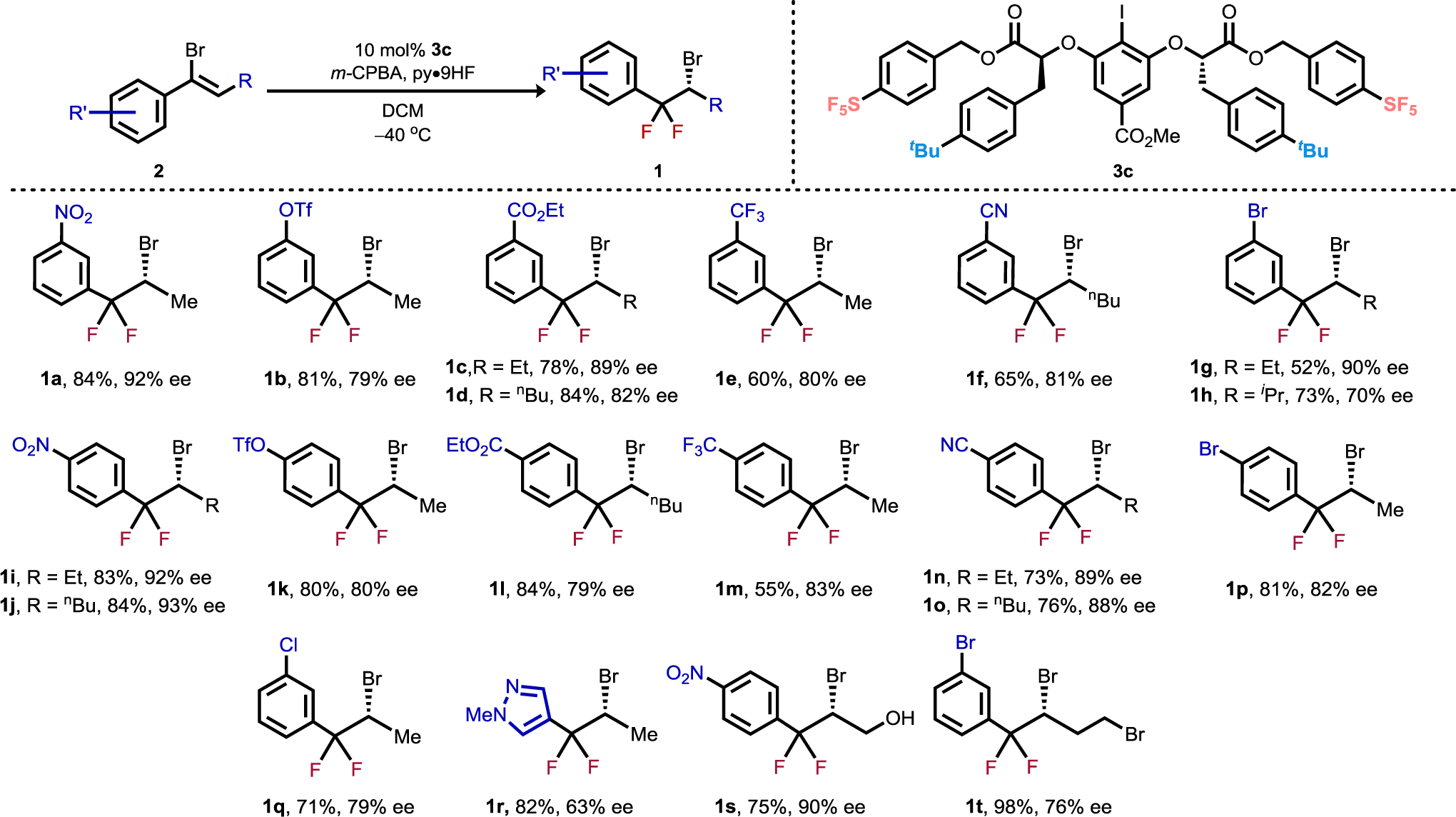
Scope studies. Enantiomeric excess determined by chiral HPLC or GC. Conditions: 0.2 mmol scale, 11 equiv. py•9HF, 1.2 equiv. m-CPBA, 0.08 M, 48 h, isolated yields. Absolute configuration of 1i established through X-ray crystal structure determination of a derivative (S4), remaining products assigned by analogy.
 |
(1) |
 |
(2) |
 |
(3) |
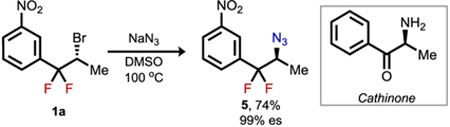
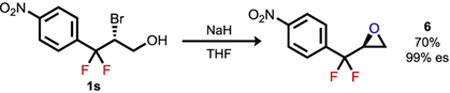
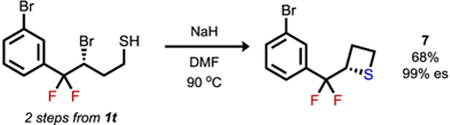
To assess the synthetic utility of the fluorinated alkyl bromide products, a series of derivatization reactions were explored (eqs 1–3). Both intermolecular (eq 1) and intramolecular substitution (eqs 2 and 3, respectively) occurred smoothly with nitrogen, oxygen, and sulfur nucleophiles to afford the corresponding products with complete enantiospecificity. Such ready access to azide 5 and its derivatives provides an appealing entry to gem-difluorinated bioisosteric analogues of the ketone-containing cathinone subclass of phenethylamines.12 Invertive nucleophilic substitution of the bromide in 1 required more forcing conditions than substitutions with typical secondary aliphatic alkyl bromides.13 However, the deactivating effect of gem-difluorination in SN2 pathways does not preclude stereospecific substitution by strong nucleophiles, presumably because of an even greater deactivation toward SN1-like pathways.14,15
As noted above, catalyst enantioselectivity is subject to strong substituent effects that do not correlate with steric parameters. Given that the substituted arenes of the catalyst are insulated electronically and remote from the reactive iodine center, we hypothesized that attractive secondary noncovalent interactions (NCIs) influenced by the electronic properties of the substituents may play a critical role. This idea was also supported by earlier computational studies conducted in collaboration with Houk and Xue of related styrene difluorination reactions, wherein several key attractive NCIs were invoked to underlie enantioinduction.16
We anticipated any attractive NCIs might be analyzed effectively using linear free energy relationships (LFERs) between experimental enantioselectivities and catalyst parameters. Such an approach is not predicated on knowledge of the enantiodetermining step because ΔΔG‡ (calculated from e.r.) provides an intrinsic readout of the energy difference in the competing major and minor transition states in that step. While the LFER approach does not provide a three-dimensional transition state structure as might be attainable with density functional theory (DFT) calculations, it leverages the full body of experimental data to evaluate the factors governing stereocontrol.
A modest linear correlation (R2 = 0.76) was obtained between the experimental enantioselectivities and the classical Hammett σp/m values of the catalyst arene substituents examined in the original optimization process (defined as the training set). The σm parameter has previously been correlated to the strength of NCIs with arenes,17 but the relationship is indirect given that σ values are derived from experimental pKa values of substituted benzoic acids. Examining a wide range of DFT-calculated molecular properties (including natural bond orbital charges, IR frequencies/intensities, and NMR chemical shifts) did not produce good correlations with experimental enantioselectivity unless incorporated into complex multivariate statistical models. Recognizing that such multivariate correlations can result from overfitting the limited data set,18 we sought to identify other computational descriptors that could capture the relevant NCIs more directly.19
This endeavor was facilitated by the use of symmetry-adapted perturbation theory (SAPT),20 wherein interaction energies were evaluated between R1-substituted benzenes serving as truncated versions of the catalysts and probe molecules serving as representative partners for different NCIs (Figure 4A). These calculated interaction energies provided significantly improved correlations to the experimental enantioselectivity data.18 A particularly strong univariate correlation (with R2 = 0.93, training set) was obtained using the CH–π interaction energy between the truncated catalyst arene and an orthogonally disposed benzene probe (Figure 4B). This correlation enabled the successful prediction and experimental validation of two new catalysts, defining both more (1-adamantyl-substituted) and less (3,4,5-trifluoro-substituted) enantioselective catalysts than those in the training set. Significant correlations were also obtained using certain cation–π interaction energies (R2 ≤ 0.84, training set), but such correlations displayed systematic curvatures indicating that the beneficial effect on enantioselectivity of electron-donating substituents was being captured less accurately. Moreover, the cation–π correlations also failed to predict the performance of 1-adamantyl- and 3,4,5-trifluoro-substituted catalysts as accurately as the CH–π correlation. Thus, the SAPT analysis validated the conclusion that the substituent effect on catalyst enantioselectivity is not steric in nature, and reveals instead that it is best ascribed to a selective, attractive CH–π interaction in the enantiodetermining transition state(s).21
Figure 4.
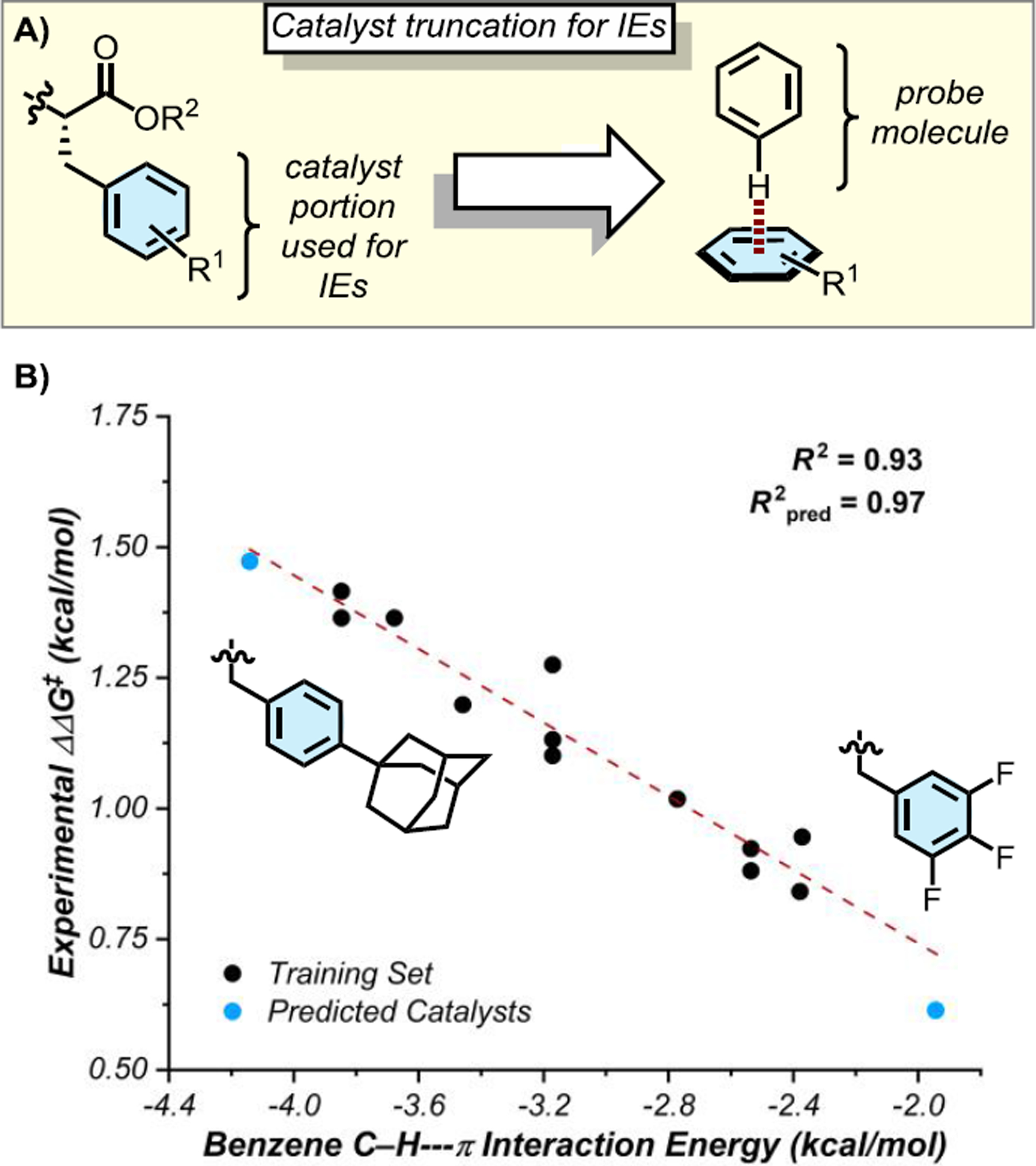
A) Catalyst truncation for computed arene–arene interaction energies (IEs). B) Correlation of experimental enantioselectivity (ΔΔG‡ = –RTln(e.r.)) with computed interaction energies between substituted catalyst arenes and a benzene probe. The line of best fit for the training set was used to extrapolate catalyst performance.
To further interrogate the role of attractive NCIs as important features of this enantioselective process, both DFT properties and SAPT-derived interaction energies were computed for the vinyl bromide substrates 2a–2t and compared to experimental enantioselectivities.15 From a training set of thirteen substrates (defined chronologically by the first substrates evaluated), a univariate correlation was obtained using the LUMO energies of the substrates (R2 = 0.90, Figure 5A). External validation of this set with seven additional entries revealed a poorer overall correlation with the LUMO energy (R2 = 0.68) as a result of three outliers. Nevertheless, this simple model could be used to predict the performance of pyrazole-containing substrate 2r, despite structural dissimilarities compared to the training set. Multivariable linear regression analysis was also conducted on the entire data set in attempts to reconcile outliers in the LUMO correlation; however, expansion to two-parameter models did not afford any significant improvement to the observed correlation with enantioselectivity.22,23
Figure 5.
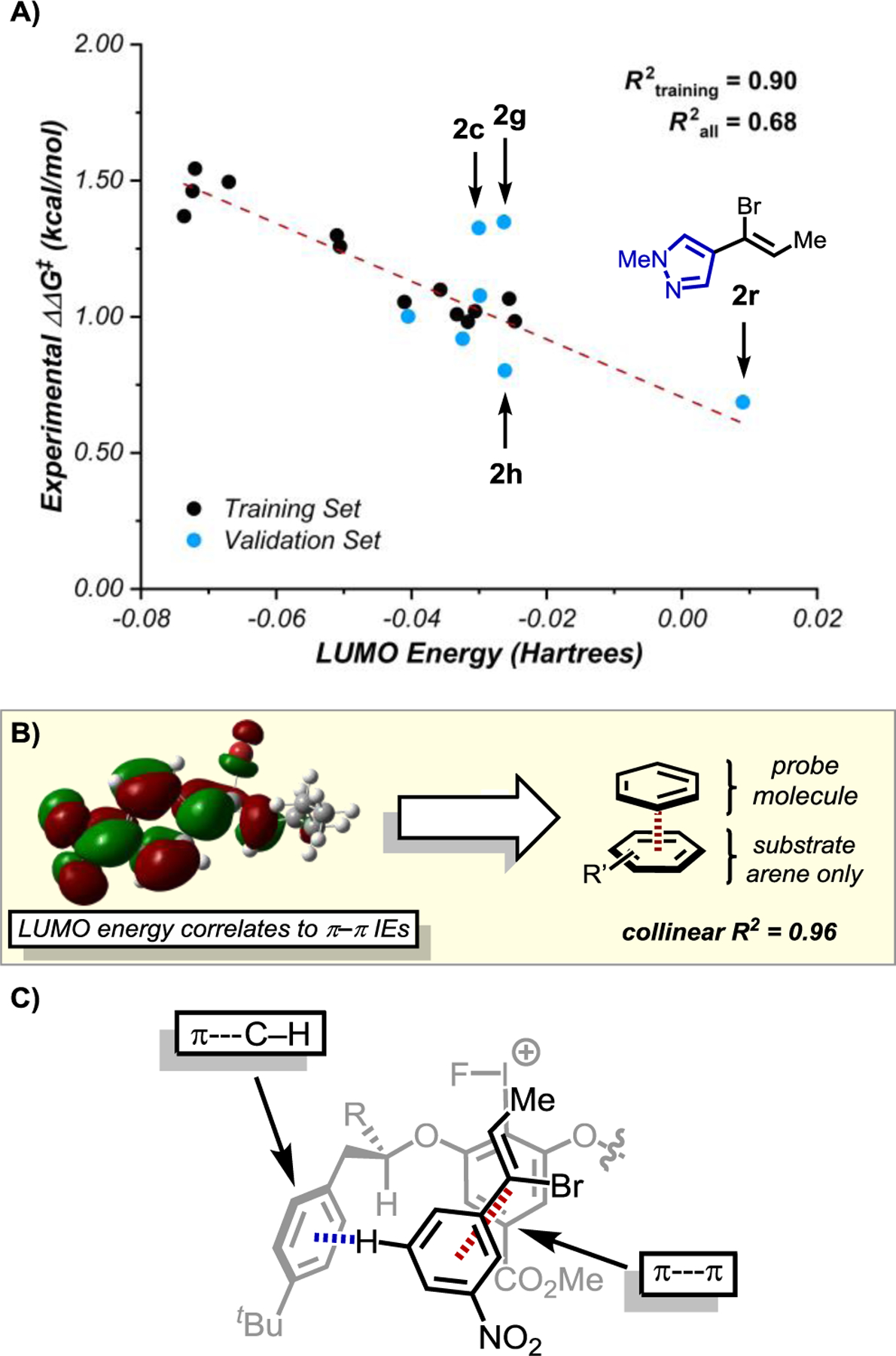
A) Univariate correlation of experimental enantioselectivity (ΔΔG‡ = –RTln(e.r.)) to the calculated LUMO energy of each vinyl bromide substrate. The line of best fit for the training set (red) was extended to the full range of values in the data set without recalculating the fit to include the validation set. B) LUMO energies likely reflect the ability of substrates to participate in π–π interactions. C) Representation of how catalyst 3c and substrate 2a may interact in the enantiodetermining transition state based on SAPT studies (R = CO2CH2C6H4(p-SF5), one catalyst sidearm omitted for clarity).
To interpret the correlation of calculated substrate LUMO energies to enantioselectivity, we assessed the LUMO energies against a wide set of other DFT-derived parameters and SAPT interaction energies. The best correlation was with calculated energies of a directly stacked π–π interaction between the substrate arene and a benzene probe (R2 = 0.96, Figure 5B).17b We therefore hypothesize that the LUMO correlation reflects the degree to which the substrates can engage in selective π-interactions in the enantiodetermining transition states Taken together with the catalyst correlation and the previous computational study,16 these observations are consistent with a network of attractive NCIs being responsible for enantioinduction.
In conclusion, we have developed a synthetic method that leverages the chemistry of chiral iodoarene difluoride intermediates to generate enantioenriched, gem-difluorinated, secondary aliphatic bromides with high enantioselectivity. Systematic optimization of the catalyst structure and identification of a catalyst decomposition pathway led to a protocol that was both more enantioselective and more efficient. A single SAPT-derived interaction energy was sufficient to correlate enantioselectivities to catalyst substituent effects. Efforts to capture substrate effects on enantioselectivity with calculated parameters proved more challenging, although a correlation with substrate electrophilicity was established and interpreted as the ability for the substrate to participate in selective π-interactions. Together, these studies add to a growing body of evidence for the important structural features underpinning the activity, stability, and enantioselectivity of this increasingly important class of iodoarene catalysts.24
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (GM043214 to E.N.J. and GM121383 to M.S.S.), NIH postdoctoral fellowships to M.D.L. (F32GM125187) and J.A.R. (F32GM128351), and an NSF predoctoral fellowship to J.M.O. (DGE1745303). We thank Shao-Liang Zheng (Harvard University) for determination of X-Ray crystallographic structures. Dr. Adam Trotta (Harvard University), Dr. Tobias Gensch (TU Berlin), and Dr. Nathaniel Kenton (Ohio State) are thanked for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and characterization data (PDF)
Computational procedures and data (PDF)
Crystallographic data for derivatized product S4 (CIF)
REFERENCES
- 1.(a) O’Hagan D; Harper B, Fluorine-Containing Natural Products D. Journal of Fluorine Chemistry 1999, 100, 127–133. 10.1016/S0022-1139(99)00201-8. [DOI] [Google Scholar]; (b) Chan KK; O’Hagan D The rare fluorinated natural products and biotechnological prospects for fluorine enzymology. Methods Enzymol. 2012, 516, 219–235. 10.1016/B978-0-12-394291-3.00003-4. [DOI] [PubMed] [Google Scholar]; (c) Walker MC; Thuronyi BW; Charkoudian LK; Lowry B; Khosla C; Chang MCY Expanding the Fluorine Chemistry of Living Systems Using Engineered Polyketide Synthase Pathways. Science 2013, 341, 1089–1094. 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Selected reviews:Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; (b) Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (c) Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114, 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (d) Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 3.(a) Selected reviews:Lectard S; Hamashima Y; Sodeoka M Recent Advances in Catalytic Enantioselective Fluorination Reactions. Adv. Synth. Catal 2010, 352, 2708–2732. 10.1002/adsc.201000624. [DOI] [Google Scholar]; (b) Yang X; Wu T; Phipps RJ; Toste FD Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev 2015, 115, 826–870. 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) O’Hagan D; Rzepa HS; Schüler M; Slawin AMZ The Vicinal Difluoro Motif: The Synthesis and Conformation of Erythro- and Threo- Diastereoisomers of 1,2-Difluorodiphenylethanes, 2,3-Difluorosuccinic Acids and Their Derivatives. Beilstein J. Org. Chem 2006, 2, 19 10.1186/1860-5397-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) O’Hagan D; Wang Y; Skibinski M; Slawin AMZ Influence of the Difluoromethylene Group (CF2) on the Conformation and Properties of Selected Organic Compounds. Pure Appl. Chem 2012, 84, 1587–1595. 10.1351/PAC-CON-11-09-26. [DOI] [Google Scholar]; (c) Corr MJ; Cormanich RA; von Hahmann CN; Bühl M; Cordes DB; Slawin AMZ; O’Hagan D Fluorine in Fragrances: Exploring the Difluoromethylene (CF2) Group as a Conformational Constraint in Macrocyclic Musk Lactones. Org. Biomol. Chem 2015, 14, 211–219. 10.1039/C5OB02023A. [DOI] [PubMed] [Google Scholar]; (d) Callejo R; Corr MJ; Yang M; Wang M; Cordes DB; Slawin AMZ; O’Hagan D Fluorinated Musk Fragrances: The CF2 Group as a Conformational Bias Influencing the Odour of Civetone and (R)-Muscone. Chem. – Eur. J 2016, 22, 8137–8151. 10.1002/chem.201600519. [DOI] [PubMed] [Google Scholar]
- 5.(a) Singh RP; Shreeve JM Recent Advances in Nucleophilic Fluorination Reactions of Organic Compounds Using Deoxofluor and DAST. Synthesis 2002, 2002 (17), 2561–2578. 10.1055/s-2002-35626; for an alternative approach see [DOI] [Google Scholar]; (b) Geri JB; Wade Wolfe MM; Szymczak NK The Difluoromethyl Group as a Masked Nucleophile: A Lewis Acid/Base Approach. J. Am. Chem. Soc 2018, 140 (30), 9404–9408. 10.1021/jacs.8b06093; see also ref 7c and references therein. [DOI] [PubMed] [Google Scholar]
- 6.(a) For a recent review, see:Parra A Chem. Rev 2019, 119, 12033–12088. 10.1021/acs.chemrev.9b00338. Seminal reports on iodoarene difluoride chemistry: [DOI] [PubMed] [Google Scholar]; (b) Carpenter W Aryliodosodifluorides. J. Org. Chem 1966, 31, 2688–2689. 10.1021/jo01346a512. [DOI] [Google Scholar]; (c) Gregorcic A; Zupan M Fluorination with Substituted (Difluoroiodo)Arenes. BCSJ 1977, 50, 517–520. 10.1246/bcsj.50.517. [DOI] [Google Scholar]; (d) Gregorčič A; Zupan M Fluorination of Norbornadiene and 1,4-Dihydro-1,4-Methanonaphthalene with Substituted (Difluoroiodo)Benzenes. J. Chem. Soc., Perkin Trans. 1 1977, 12, 1446–1449. 10.1039/P19770001446. [DOI] [Google Scholar]; (e) Patrick TB; Scheibel JJ; Hall WE; Lee YH Substituent Effects in the Fluorination-Rearrangement of 1,1-Diarylethenes with Aryliodine(III) Difluorides. J. Org. Chem 1980, 45, 4492–4494. 10.1021/jo01310a043. [DOI] [Google Scholar]; (f) Hara S; Nakahigashi J; Ishi-i K; Sawaguchi M; Sakai H; Fukuhara T; Yoneda N Difluorination of Alkenes with Iodotoluene Difluoride. Synlett 1998, 5, 495–496. 10.1055/s-1998-1714. [DOI] [Google Scholar]; (g) Hara S; Nakahigashi J; Ishi-i K; Fukuhara T; Yoneda N Fluorinative Ring-Contraction of Cyclic Alkenes with p-Iodotoluene Difluoride. Tetrahedron Letters 1998, 39, 2589–2592. 10.1016/S0040-4039(98)00276-7. [DOI] [Google Scholar]
- 7.(a) Iodoarene-catalyzed alkene fluorofunctionalization reactions, asymmetric and racemic:Banik SM; Medley JW; Jacobsen EN Catalytic, Diastereoselective 1,2-Difluorination of Alkenes. J. Am. Chem. Soc 2016, 138, 5000–5003. 10.1021/jacs.6b02391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Molnár IG; Gilmour R Catalytic Difluorination of Olefins. J. Am. Chem. Soc 2016, 138, 5004–5007. 10.1021/jacs.6b01183. [DOI] [PubMed] [Google Scholar]; (c) Banik SM; Medley JW; Jacobsen EN Catalytic, Asymmetric Difluorination of Alkenes to Generate Difluoromethylated Stereocenters. Science 2016, 353, 51–54. 10.1126/science.aaf8078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Woerly EM; Banik SM; Jacobsen EN Enantioselective, Catalytic Fluorolactonization Reactions with a Nucleophilic Fluoride Source. J. Am. Chem. Soc 2016, 138, 13858–13861. 10.1021/jacs.6b09499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sarie JC; Thiehoff C; Mudd RJ; Daniliuc CG; Kehr G; Gilmour R Deconstructing the Catalytic, Vicinal Difluorination of Alkenes: HF-Free Synthesis and Structural Study of p-TolIF2. J. Org. Chem 2017, 82, 11792–11798. 10.1021/acs.joc.7b01671. [DOI] [PubMed] [Google Scholar]; (f) Banik SM; Mennie KM; Jacobsen EN Catalytic 1,3-Difunctionalization via Oxidative C–C Bond Activation. J. Am. Chem. Soc 2017, 139, 9152–9155. 10.1021/jacs.7b05160. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Scheidt F; Thiehoff C; Yilmaz G; Meyer S; Daniliuc CG; Kehr G; Gilmour R Fluorocyclisation via I(I)/I(III) Catalysis: A Concise Route to Fluorinated Oxazolines. Beilstein J. Org. Chem 2018, 14, 1021–1027. 10.3762/bjoc.14.88. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Mennie KM; Banik SM; Reichert EC; Jacobsen EN Catalytic Diastereo- and Enantioselective Fluoroamination of Alkenes. J. Am. Chem. Soc 2018, 140, 4797–4802. 10.1021/jacs.8b02143. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Scheidt F; Schafer M; Sarie J; Daniliuc C; Molloy J; Gilmour R Enantioselective, Catalytic Vicinal Difluorination of Alkenes. Angew. Chem., Int. Ed 2018, 57, 16431–16435. https://doi/10.1002/anie.201810328. [DOI] [PubMed] [Google Scholar]; (j) Haj MK; Banik SM; Jacobsen EN Catalytic, Enantioselective 1,2-Difluorination of Cinnamamides. Org. Lett 2019, 21, 4919–4923. 10.1021/acs.orglett.9b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Enantioselective iodoarene-catalyzed oxy- and aminofunctionalization:Dohi T; Maruyama A; Takenaga N; Senami K; Minamitsuji Y; Fujioka H; Caemmerer SB; Kita Y A Chiral Hypervalent Iodine(III) Reagent for Enantioselective Dearomatization of Phenols. Angew. Chem. Int. Ed 2008, 47, 3787–3790. 10.1002/anie.200800464. [DOI] [PubMed] [Google Scholar]; (b) Altermann SM; Richardson RD; Page TK; Schmidt RK; Holland E; Mohammed U; Paradine SM; French AN; Richter C; Bahar AM; Witulski B; Wirth T Catalytic Enantioselective α-Oxysulfonylation of Ketones Mediated by Iodoarenes. Eur. J. Org. Chem 2008, 31, 5315–5328. 10.1002/ejoc.200800741. [DOI] [Google Scholar]; (c) Uyanik M; Yasui T; Ishihara K Enantioselective Kita Oxidative Spirolactonization Catalyzed by In Situ Generated Chiral Hypervalent Iodine(III) Species. Angew. Chem. Int. Ed 2010, 49, 2175–2177. 10.1002/anie.200907352. [DOI] [PubMed] [Google Scholar]; (d) Uyanik M; Yasui T; Ishihara K Chiral Hypervalent Iodine-Catalyzed Enantioselective Oxidative Kita Spirolactonization of 1-Naphthol Derivatives and One-Pot Diastereo-Selective Oxidation to Epoxyspirolactones. Tetrahedron 2010, 66, 5841–5851. 10.1016/j.tet.2010.04.060. [DOI] [Google Scholar]; (e) Dohi T; Takenaga N; Nakae T; Toyoda Y; Yamasaki M; Shiro M; Fujioka H; Maruyama A; Kita Y Asymmetric Dearomatizing Spirolactonization of Naphthols Catalyzed by Spirobiindane-Based Chiral Hypervalent Iodine Species. J. Am. Chem. Soc 2013, 135, 4558–4566. 10.1021/ja401074u. [DOI] [PubMed] [Google Scholar]; (f) Uyanik M; Yasui T; Ishihara K Hydrogen Bonding and Alcohol Effects in Asymmetric Hypervalent Iodine Catalysis: Enantioselective Oxidative Dearomatization of Phenols. Angew. Chem. Int. Ed 2013, 52, 9215–9218. 10.1002/anie.201303559. [DOI] [PubMed] [Google Scholar]; (g) Haubenreisser S; Wöste TH; Martínez C; Ishihara K; Muñiz K Structurally Defined Molecular Hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed 2016, 55, 413–417. 10.1002/anie.201507180. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Jobin-Des Lauriers A; Legault CY Iodine(III)-Mediated Oxidative Hydrolysis of Haloalkenes: Access to α-Halo Ketones by a Release-and-Catch Mechanism. Org. Lett 2016, 18, 108–111. 10.1021/acs.orglett.5b03345. [DOI] [PubMed] [Google Scholar]; (i) Muñiz K; Barreiro L; Romero RM; Martínez C Catalytic Asymmetric Diamination of Styrenes. J. Am. Chem. Soc 2017, 139, 4354–4357. 10.1021/jacs.7b01443. [DOI] [PubMed] [Google Scholar]; (j) Dohi T; Sasa H; Miyazaki K; Fujitake M; Takenaga N; Kita Y Chiral Atropisomeric 8,8′-Diiodobinaphthalene for Asymmetric Dearomatizing Spirolactonizations in Hypervalent Iodine Oxidations. J. Org. Chem 2017, 82 (22), 11954–11960. 10.1021/acs.joc.7b02037. [DOI] [PubMed] [Google Scholar]; (k) Flores A; Cots E; Berges J; Muniz K Enantioselective Iodine(I/III) Catalysis in Organic Synthesis. Adv. Syn. Catal 2019, 361, 2–25. 10.1002/adsc.201800521. [DOI] [Google Scholar]
- 9.(a) Taft RW Polar and Steric Substituent Constants for Aliphatic and O-Benzoate Groups from Rates of Esterification and Hydrolysis of Esters1. J. Am. Chem. Soc 1952, 74 (12), 3120–3128. 10.1021/ja01132a049; [DOI] [Google Scholar]; (b) Charton M Steric Effects. I. Esterification and Acid-Catalyzed Hydrolysis of Esters. J. Am. Chem. Soc 1975, 97 (6), 1552–1556. 10.1021/ja00839a047. [DOI] [Google Scholar]
- 10.(a) Bisel P; Al-Momani L; Müller M The Tert-Butyl Group in Chemistry and Biology. Org. Biomol. Chem 2008, 6 (15), 2655–2665. 10.1039/B800083B. [DOI] [PubMed] [Google Scholar]; (b) Zhang W; Loebach JL; Wilson SR; Jacobsen EN Enantioselective Epoxidation of Unfunctionalized Olefins Catalyzed by Salen Manganese Complexes. J. Am. Chem. Soc 1990, 112 (7), 2801–2803. 10.1021/ja00163a052. [DOI] [Google Scholar]; (c) Evans DA; Woerpel KA; Hinman MM; Faul MM Bis(Oxazolines) as Chiral Ligands in Metal-Catalyzed Asymmetric Reactions. Catalytic, Asymmetric Cyclopropanation of Olefins. J. Am. Chem. Soc 1991, 113 (2), 726–728. 10.1021/ja00002a080. [DOI] [Google Scholar]; (d) Sigman MS; Vachal P; Jacobsen EN A General Catalyst for the Asymmetric Strecker Reaction. Angew. Chem. Int. Ed 2000, 39 (7), 1279–1281. . [DOI] [PubMed] [Google Scholar]; (e) Austin JF; MacMillan DWC Enantioselective Organocatalytic Indole Alkylations. Design of a New and Highly Effective Chiral Amine for Iminium Catalysis. J. Am. Chem. Soc 2002, 124 (7), 1172–1173. 10.1021/ja017255c [DOI] [PubMed] [Google Scholar]; (f) Banert K; Heck M; Ihle A; Kronawitt J; Pester T; Shoker T Steric Hindrance Underestimated: It Is a Long, Long Way to Tri-Tert-Alkylamines. J. Org. Chem 2018, 83 (9), 5138–5148. 10.1021/acs.joc.8b00496. [DOI] [PubMed] [Google Scholar]
- 11.The mass balance of catalyst represented by recovered 3b and 4 is not complete. However, the basic alumina quench utilized in the workup precludes isolation of any carboxylic acid-containing catalyst decomposition products. To probe the possible relevance of such species, we prepared a dealkylated mono-acid catalyst derived from 3a, which provided 1a with 72% conversion and 69% ee. Reactions with this catalyst also resulted in the formation of 4, suggesting multiple catalyst-derived species may interact with the intermediate bromonium ion. When 4 is employed as a catalyst, the corresponding bis(transesterified) catalyst decomposition product is also observed in small quantities.
- 12.Meyer MR; Wilhelm J; Peters FT; Maurer HH Beta-Keto Amphetamines: Studies on the Metabolism of the Designer Drug Mephedrone and Toxicological Detection of Mephedrone, Butylone, and Methylone in Urine Using Gas Chromatography–Mass Spectrometry. Anal. Bioanal. Chem 2010, 397, 1225–1233. 10.1007/s00216-010-3636-5. [DOI] [PubMed] [Google Scholar]
- 13.(a) For example, nucleophilic substitutions of secondary aliphatic bromides with azide typically proceed at ambient temperature:Ito M; Koyakumaru K; Ohta T; Takaya H A Simple and Convenient Synthesis of Alkyl Azides under Mild Conditions. Synthesis 1995, 1995 (4), 376–378. 10.1055/s-1995-3928; previous reports have noted an influence of vicinal fluorination: [DOI] [Google Scholar]; (b) Benaïssa T; Hamman S; Beguin CG Synthesis of β-Fluoroazides: A Route to Primary β-Fluoro Amines. Journal of Fluorine Chemistry 1988, 38 (2), 163–173. 10.1016/S0022-1139(00)83025-0. [DOI] [Google Scholar]
- 14.(a) Electronic influence on the rate of SN2:Wu C-H; Galabov B; Wu JI-C; Ilieva S; Schleyer P. von R.; Allen WD Do π-Conjugative Effects Facilitate SN2 Reactions? J. Am. Chem. Soc 2014, 136, 3118–3126. 10.1021/ja4111946. [DOI] [PubMed] [Google Scholar]; (b) Robiette R; Trieu-Van T; Aggarwal VK; Harvey JN Activation of the SN2 Reaction by Adjacent π Systems: The Critical Role of Electrostatic Interactions and of Dissociative Character. J. Am. Chem. Soc 2016, 138, 734–737. 10.1021/jacs.5b11402. [DOI] [PubMed] [Google Scholar]
- 15.(a) Reports of nucleophilic substitution vicinal to difluoromethylenes:Zhang D; Li P; Lin Z; Huang H An Efficient and Convenient Protocol for the Synthesis of 1,1-Difluoro-6-Nitro-2,3-Dihydro-1H-Indene Derivatives. Synthesis 2014, 46, 613–620. 10.1055/s-0033-1340595. [DOI] [Google Scholar]; (b) Xia G; Benmohamed R; Morimoto RI; Kirsch DR; Silverman RB Deuteration and Fluorination of 1,3-Bis(2-Phenylethyl)Pyrimidine-2,4,6(1H,3H,5H)-Trione to Improve Its Pharmacokinetic Properties. Bioorg. Med. Chem. Lett 2014, 24, 5098–5101. 10.1016/j.bmcl.2014.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brown K; Dixey M; Weymouth-Wilson A; Linclau B The Synthesis of Gemcitabine. Carbohydrate Research 2014, 387, 59–73. 10.1016/j.carres.2014.01.024. [DOI] [PubMed] [Google Scholar]
- 16.Zhou B; Haj MK; Jacobsen EN; Houk KN; Xue X-S Mechanism and Origins of Chemo- and Stereoselectivities of Aryl Iodide-Catalyzed Asymmetric Difluorinations of β-Substituted Styrenes. J. Am. Chem. Soc 2018, 140, 15206–15218. 10.1021/jacs.8b05935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Mecozzi S; West AP; Dougherty DA Cation−π Interactions in Simple Aromatics: Electrostatics Provide a Predictive Tool. J. Am. Chem. Soc 1996, 118 (9), 2307–2308. 10.1021/ja9539608. [DOI] [Google Scholar]; (b) Wheeler SE; Houk KN Substituent Effects in the Benzene Dimer Are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene. J. Am. Chem. Soc 2008, 130, 10854–10855. 10.1021/ja802849j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigman MS; Harper KC; Bess EN; Milo A The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond. Acc. Chem. Res 2016, 49, 1292–1301. 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]
- 19.(a) Orlandi M; Coelho JAS; Hilton MJ; Toste FD; Sigman MS Parametrization of Non-Covalent Interactions for Transition State Interrogation Applied to Asymmetric Catalysis. J. Am. Chem. Soc 2017, 139 (20), 6803–6806. 10.1021/jacs.7b02311. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miró J; Gensch T; Ellwart M; Han S-J; Lin H-H; Sigman MS; Toste FD Enantioselective Allenoate-Claisen Rearrangement Using Chiral Phosphate Catalysts. J. Am. Chem. Soc 2020, 142, 6390–6399. 10.1021/jacs.0c01637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Jeziorski B; Moszynski R; Szalewicz K Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van Der Waals Complexes. Chem. Rev 1994, 94, 1887–1930. 10.1021/cr00031a008. [DOI] [Google Scholar]; (b) Hohenstein EG; Sherrill CD Wavefunction Methods for Non-Covalent Interactions. WIREs. Comput. Mol. Sci 2012, 2, 304–326. 10.1002/wcms.84. [DOI] [Google Scholar]; (c) Sherrill CD Energy Component Analysis of π Interactions. Acc. Chem. Res 2013, 46, 1020–1028. 10.1021/ar3001124. [DOI] [PubMed] [Google Scholar]; (d) Parker TM; Burns LA; Parrish RM; Ryno AG; Sherrill CD Levels of Symmetry Adapted Perturbation Theory (SAPT). I. Efficiency and Performance for Interaction Energies. J. Chem. Phys 2014, 140, 094106 10.1063/1.4867135. [DOI] [PubMed] [Google Scholar]; (e) Gonthier JF; Sherrill CD Density-Fitted Open-Shell Symmetry-Adapted Perturbation Theory and Application to π-Stacking in Benzene Dimer Cation and Ionized DNA Base Pair Steps. J. Chem. Phys 2016, 145, 134106 10.1063/1.4963385. [DOI] [PubMed] [Google Scholar]
- 21.Interaction energies with many other charged and neutral probe molecules representing a wide range of potential NCIs were also evaluated. The computed SAPT energies are reflective of the subtle differences in substituent effects manifested by each class of NCI, supporting the premise that they can in fact be differentiated. See sections S3 and S4 of the computational SI for details.
- 22.More traditional parameters such as σp/m and σ+ failed to account for the variation in enantioselectivity as a function of substrate structure (R2 values with ΔΔG‡ of 0.45 and 0.41, respectively).
- 23.Introduction of a second parameter into the LUMO model corrects for outlier 2h, bearing a branched alkyl group, but does not reconcile the remaining outliers. See Figure S14 in the computational SI for details.
- 24.(a) Hirt UH; Spingler B; Wirth T New Chiral Hypervalent Iodine Compounds in Asymmetric Synthesis. J. Org. Chem 1998, 63 (22), 7674–7679. 10.1021/jo980475x. [DOI] [Google Scholar]; (b) Zhdankin VV; Protasiewicz JD Development of New Hypervalent Iodine Reagents with Improved Properties and Reactivity by Redirecting Secondary Bonds at Iodine Center. Coordination Chemistry Reviews 2014, 275, 54–62. 10.1016/j.ccr.2014.04.007. [DOI] [Google Scholar]; (c) Berthiol F Reagent and Catalyst Design for Asymmetric Hypervalent Iodine Oxidations. Synthesis 2015, 47 (5), 587–603. 10.1055/s-0034-1379892. [DOI] [Google Scholar]; (d) Brown M; Kumar R; Rehbein J; Wirth T Enantioselective Oxidative Rearrangements with Chiral Hypervalent Iodine Reagents. Chemistry – A European Journal 2016, 22 (12), 4030–4035. 10.1002/chem.201504844. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Fujita M Mechanistic Aspects of Alkene Oxidation Using Chiral Hypervalent Iodine Reagents. Tetrahedron Letters 2017, 58 (47), 4409–4419. 10.1016/j.tetlet.2017.10.019. [DOI] [Google Scholar]; (f) Sreenithya A; Patel C; Hadad CM; Sunoj RB Hypercoordinate Iodine Catalysts in Enantioselective Transformation: The Role of Catalyst Folding in Stereoselectivity. ACS Catal. 2017, 7 (6), 4189–4196. 10.1021/acscatal.7b00975. [DOI] [Google Scholar]; (g) Uyanik M; Sasakura N; Mizuno M; Ishihara K Enantioselective Synthesis of Masked Benzoquinones Using Designer Chiral Hypervalent Organoiodine(III) Catalysis. ACS Catal. 2017, 7 (1), 872–876. 10.1021/acscatal.6b03380. [DOI] [Google Scholar]; (h) Ghosh S; Pradhan S; Chatterjee I A Survey of Chiral Hypervalent Iodine Reagents in Asymmetric Synthesis. Beilstein J. Org. Chem 2018, 14 (1), 1244–1262. 10.3762/bjoc.14.107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Parra A Chiral Hypervalent Iodines: Active Players in Asymmetric Synthesis. Chem. Rev 2019, 119 (24), 12033–12088. 10.1021/acs.chemrev.9b00338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.