

Abstract
Objectives
This study aimed to assess the antiviral properties of a unique lectin (NICTABA) produced by the tobacco plant, Nicotiana tabacum.
Methods
Cellular assays were used to investigate the antiviral activity of NICTABA and Urtica dioica agglutinin (UDA). Surface plasmon resonance (SPR) studies were performed to study the sugar specificity and the interactions of both lectins with the envelope glycoproteins of HIV-1.
Results
The N-acetyl-d-glucosamine (GlcNAc)-binding lectins exhibited broad-spectrum activity against several families of enveloped viruses including influenza A/B, Dengue virus type 2, herpes simplex virus types 1 and 2 and HIV-1/2. The IC50 of NICTABA for various HIV-1 strains, clinical isolates and HIV-2 assessed in PBMCs ranged from 5 to 30 nM. Furthermore, NICTABA inhibited syncytium formation between persistently HIV-1-infected T cells and uninfected CD4+ T lymphocytes and prevented DC-SIGN-mediated HIV-1 transmission to CD4+ target T lymphocytes. However, unlike many other antiviral carbohydrate-binding agents (CBAs) described so far, NICTABA did not block HIV-1 capture to DC-SIGN+ cells and it did not interfere with the binding of the human monoclonal antibody 2G12 to gp120. SPR studies with HIV-1 envelope glycoproteins showed that the affinity of NICTABA for gp120 and gp41 was in the low nanomolar range. The specific binding of NICTABA to gp120 could be prevented in the presence of a GlcNAc trimer, but not in the presence of mannose trimers. NICTABA displayed no antiviral activity against non-enveloped viruses.
Conclusions
Since CBAs possess a high genetic barrier for the development of viral resistance and NICTABA shows a broad antiviral activity profile, this CBA may qualify as a potential antiviral candidate with a pleiotropic mode of action aimed at targeting the entry of enveloped viruses.
Keywords: glycosylation, broad spectrum, antiviral peptides
Introduction
Viruses such as HIV, herpes simplex virus (HSV), influenza virus and hepatitis C virus (HCV) contain highly glycosylated proteins in their envelope. The HIV envelope protein gp120 is among the most heavily glycosylated proteins, with almost 50% of its molecular weight being due to the presence of N-linked glycans.1,2 These glycans play an important role in the conformation of the envelope during synthesis and in viral transmission and subsequent infection.3–7 They also function as a shield to reduce the accessibility of the immunogenic protein core in order to prevent an efficient immune response.8–11 Given the important structural and functional role of the viral envelope glycans, various carbohydrate-binding agents (CBAs) have been investigated for their potential to interfere with virus infection. In fact, CBAs have been proposed as a novel interesting class of antiviral drugs since they have been shown to have the capacity to inhibit both the infection of cells by cell-free virus particles and the interaction between HIV-infected leucocytes and uninfected CD4+ cells, as well as the capture of virus by DC-SIGN-expressing dendritic cells (DCs) and macrophage mannose receptor-expressing macrophages12,13 and the transmission of the captured virus to uninfected CD4+ T lymphocytes.14
Because these agents block both the entry and the transmission of the virus, they would qualify as potent antiviral compounds with a pleiotropic mode of action. Several CBAs have therefore been studied in greater detail for their antiviral properties.12,15–24 It has been shown that, apart from their ability to prevent virus infection, capture and transmission, they are also able to force the virus to delete N-glycosylation sites in its envelope after prolonged drug exposure. Indeed, after the prolonged exposure of HIV-1 gp120 to escalating doses of a broad variety of CBAs [i.e. HHA, GNA, CV-N, 2G12, AH, MVN, PRM-S, GRFT or Urtica dioica agglutinin (UDA)], up to eight or nine N-glycosylation sites were deleted in the envelope of such mutant virus strains.25–29 This implies a high genetic barrier for the CBA treatment of viral infections since often more than three to five N-glycan deletions in the HIV gp120 are required to afford significant phenotypic resistance to CBAs. In addition, the uncovering of previously hidden immunogenic epitopes on the surface of the virion by the glycan deletions may trigger an immune response against these epitopes. It has indeed been shown that mutant HIV-1 strains that have several N-glycan deletions in their envelope are more prone to neutralizing antibodies directed against gp120.11,14,30 For this reason, the CBAs display a unique combination of antiviral mechanisms. A specific subgroup of CBAs are the lectins. Although lectins of natural origin have garnered much interest due to their specific interaction with carbohydrates and their strong antiviral properties, many hurdles have hampered the translation of their preclinical success. First, lectins as a therapeutic strategy must overcome the issues posed by their protein nature, more specifically their sensitivity to proteolytic degradation, their large size, their short t1/2 and their potential immunogenicity. Second, the large-scale production and purification of these natural products is very costly and technically challenging. However, computational methods (e.g. EpiSweep) are currently available that aid in the design of immunotolerant therapeutic proteins with preserved structural properties.31 Furthermore, low molecular weight synthetic compounds with lectin-like properties could be an alternative solution to these problems. Pradimicin A, pradimicin S and benanomicin A have also shown antiviral activities and represent the first prototype drugs of non-peptidic low molecular weight CBAs.32–34 These compounds may be good candidate CBAs for further preclinical research. Although peptidic lectins are currently not being evaluated in clinical trials, they should nevertheless be considered as valuable preclinical screening tools to find novel drug candidates that mimic their activity.
UDA is a chitin-binding protein from the hevein family. It adopts a monomeric form in solution and is isolated from the rhizome of the stinging nettle (U. dioica) as a complex mixture of isolectins.35 With its 8.5 kDa molecular weight, it is one of the smallest lectins reported and has specific N-acetyl-d-glucosamine (GlcNAc)-binding activity.36 Two hevein domains of 43 amino acids are connected by a short linker of four amino acids in the UDA molecule. Through binding studies with GlcNAc oligomers, it has been determined that there are presumably three sugar-binding areas in both hevein domains, meaning that a trimer of GlcNAc is the actual substrate of the binding site. Although the presence of a second binding site on the protein has been postulated by several groups, the affinity of this site for glycans would be more than 10-fold lower than the affinity of the first binding site.37,38 More recent studies have revealed that UDA isolectin I can form dimers mediated by two Zn2+ ions bound to the sugar-binding site.39 UDA is known to be an immunomodulatory agent with insecticidal and fungistatic properties.40 Earlier, this agent was shown to possess pronounced activity against viruses such as HIV,41 HCV42 and human cytomegalovirus (CMV),41 with a low agglutination activity against human red blood cells.25,41
We focus here on the tobacco agglutinin or NICTABA, another GlcNAc-specific CBA, isolated from the plant Nicotiana tabacum.43 This lectin is a cytoplasmic/nuclear chito-oligosaccharide-binding protein whose expression is induced in tobacco leaves by jasmonates or stress situations such as insect attacks. It forms a homodimer consisting of two subunits with a dimer molecular weight of ∼38 kDa. The lectin shows a strong affinity for GlcNAc oligomers as well as for high-mannose-type N-glycans.44 In this study, the broad antiviral properties of UDA and NICTABA are compared and their mechanism of interaction with the HIV envelope investigated.
Materials and methods
Compounds
The chitin-binding lectin from the stinging nettle (UDA) was purified using a combination of cation exchange chromatography and affinity chromatography on a chitin column, as previously described.35 The tobacco (N. tabacum cv Samsun NN) lectin NICTABA was purified from tobacco leaves treated with methyl jasmonate using affinity chromatography on chitin followed by anion exchange chromatography as described by Chen et al.43 The mannose-specific lectin from Hippeastrum hybrid (HHA) was purified from Hippeastrum hybrid bulbs using affinity chromatography on mannose-Sepharose, as previously described.45 The 2G12 monoclonal antibody (MAb) was generously provided by Polymun Scientific (Vienna, Austria).
Cell line cultures and primary leucocytes
Human T lymphocytic C8166, HUT-78, SupT1 and CEM cells were obtained from ATCC (Manassas, VA, USA). MT-4 cells were provided by Dr L. Montagnier (at that time at the Institut Pasteur, Paris, France). The Raji DC-SIGN-expressing (Raji DC-SIGN+) Raji/DC-SIGN cells and the TZM-bl cells46 were kindly provided by Dr L. Burleigh (Institut Pasteur, Paris, France) and by Dr G. Van Ham (ITG, Antwerp, Belgium), respectively. The CrFK cells were obtained from Dr H. Egberink (University of Utrecht, the Netherlands). Buffy coat preparations from healthy donors were obtained from the Blood Bank (Red Cross) in Leuven, Belgium. PBMCs were activated with phytohaemagglutinin (PHA) at 2 μg/mL (Sigma, Bornem, Belgium) for 3 days at 37°C before further use in antiviral assays as PHA-activated PBMCs. All cell lines mentioned were cultivated in RPMI-1640 medium supplemented with 10% FBS (BioWhittaker Europe, Verviers, Belgium), 2 mM l-glutamine and 0.075 M NaHCO3.
HIV strains
HIV-1 (IIIB) was provided by Dr R. C. Gallo and Dr M. Popovic (Institute of Human Virology, University of Maryland, Baltimore, MD, USA). HIV-2 (ROD) was provided by Professor L. Montagnier (at that time at the Institut Pasteur, Paris, France). The primary clinical isolates representing different HIV-1 clades and HIV-2 isolates were all kindly provided by Dr J. Lathey from BBI Biotech Research Laboratories, Inc., Gaithersburg, MD, USA, and their co-receptor use (R5 or X4) was determined in our laboratory.47
Antiviral assays
The antiviral assays were based on the inhibition of virus-induced cytopathicity in confluent cell cultures, and the cytostatic assays on the inhibition of cell proliferation in exponentially growing cell cultures according to previously described methodology.48,49 The antiviral assays, other than the anti-HIV assays, were based on the inhibition of virus-induced cytopathicity in HEL [(HSV-1) (KOS), HSV-2 (G), Vero (reovirus-1), HeLa (coxsackie virus B4, parainfluenza-3 virus and respiratory syncytial virus), CrFK (feline corona virus and herpes virus) or MDCK [influenza A (H1N1, H3N2) and influenza B] cell cultures. Confluent cell cultures (or nearly confluent for MDCK cells) in microtitre 96-well plates were inoculated with 100 times the median cell culture infective dose (100 CCID50) of virus, and the cell cultures were incubated in the presence of varying concentrations of the compounds. The viral CPE was recorded as soon as it reached completion in the control virus-infected cell cultures that were not treated with the compounds. The minimal cytotoxic concentration (MCC) of the compounds was defined as the concentration of the compound that caused a microscopically visible alteration in cell morphology. For the antiviral assay with Dengue virus (DENV), the Raji/DC-SIGN+ cells were infected with DENV serotype 2 in the absence or presence of the CBAs and analysed by flow cytometry 4 days post-infection, as previously described.50
Anti-HIV replication assays
The methodology of the anti-HIV assays was as follows: human CEM (∼3 × 105 cells/cm3) cells were infected with 100 CCID50 of HIV-1 (IIIB) and seeded in the 200 μL wells of a microtitre plate containing appropriate dilutions of the lectins. After 4 days of incubation at 37°C, HIV-induced CEM giant cell formation was examined microscopically. The antiretroviral assays in MT-4 cells and T cell blasts have previously been described in detail.51 Briefly, MT-4 cells (1 × 106 cells/mL) were pre-incubated for 30 min at 37°C with the test compounds in a 96-well plate. Next, NL4.3 virus was added according to the CCID50 of the viral stock. The CPE was scored microscopically 5 days post-infection, and the 50% effective concentration (EC50) values were determined using the MTS/phenazine ethosulfate method.51
The PHA-stimulated blasts were seeded at 0.5 × 106 cells per well into a 48-well plate containing varying concentrations of compound in medium containing IL-2 (25 U/mL; R&D Systems Europe, Abingdon, UK). After 30 min of pre-incubation with the test compounds, 1000 pg/mL of virus stock (HIV-1, HIV-2 or clinical isolates) was added. On Days 3 and 6 post-infection, 2 ng/mL of IL-2 was once more added to the cells. The cell supernatant was collected on Day 10 and HIV-1 core antigen (Ag) in the culture supernatant was analysed using a p24 Ag ELISA kit (PerkinElmer, Zaventem, Belgium) according to the manufacturer's guidelines. The INNOTEST from Innogenetics (Temse, Belgium) was used to detect HIV-2 p27 Ag.
TZM-bl cells were seeded at a density of 17 × 104 cells/well in a 96-well plate and were pre-incubated with varying concentrations of the compounds at 37°C. After the pre-incubation step, a stock of BaL virus was added at a final concentration of 500 pg/mL and incubated for an additional 48 h. Luminescence was measured after 10 min of incubation with Steadylite Plus reagent (PerkinElmer). To determine the loss of cell viability, luminescence from the sample-treated wells was compared with that of the cell controls. The EC50 is reported as the concentration of the sample that reduced the relative luminescence units by 50% compared with the virus control.
Effect of NICTABA or UDA on giant cell formation in co-cultures of SupT1 cells with persistently HIV-1-infected HUT-78/HIV-1 cells (giant cell assay)
Persistently HIV-1-infected HUT-78 cell cultures were generated as previously described.24 For the co-cultivation assays, HUT-78/HIV-1 cells were thoroughly washed to remove free virus from the culture medium, and 5 × 104 cells (50 μL) were transferred to 96-well plates. A comparable amount of SupT1 cells (50 μL), along with an appropriate concentration of test compound (100 μL), was then added to each well. After 2 days, the EC50 values were determined microscopically based on the appearance of syncytia in the cell co-cultures.
Effect of short exposure of HIV-1 to test compounds on virus capture by Raji/DC-SIGN cells (HIV-1 capture assay)
High amounts of HIV-1 particles (100 μL; 2.2 × 106 pg/mL p24) were exposed to serial dilutions of the compounds (400 μL) for 30 min as previously described.13 The drug-exposed virus suspensions (500 μL) were then mixed with Raji/DC-SIGN+ cell suspensions (500 μL; 106 cells) for 60 min at 37°C, after which the cells were thoroughly washed twice with 40 mL of culture medium as described earlier. This procedure resulted in a final dilution of the concentrations of the initial compound by at least 160 000-fold. The Raji/DC-SIGN+ cell cultures were then analysed for p24 Ag content.
Exposure of DC-SIGN+ cells to HIV-1 and subsequent co-cultivation with CD4+ T cells (HIV-1 transmission assay)
The B lymphocyte Raji DC-SIGN+ cells were suspended in cell culture medium at 6 × 106 cells/400 μL as described by Balzarini et al.13 Briefly, 0.4 mL cell suspensions were exposed to 600 μL of WT HIV-1 (IIIB or HE) (2.2 × 106 pg/mL p24) for 60 min, after which the cell/virus suspension was diluted 40-fold in culture medium. After centrifugation at 1250 rpm for 10 min, the pellet obtained was resuspended in a large volume of medium. Following a second centrifugation step, the supernatant was once more removed and the remaining 0.1 mL cell suspension was diluted 10-fold in cell culture medium. Under these experimental (washing) conditions, a maximum of 8 pg HIV-1 p24 could have remained in the 1 mL supernatant (or 0.4 pg in 50 μL). A 50 μL cell suspension was withdrawn for determination of p24 Ag by a specific HIV-1 p24 ELISA, and 50 μL of the Raji/DC-SIGN+ cell suspension was added to 96-well microplates in which 100 μL compound dilutions were present. Then, 50 μL of C8166 cells (107/mL) were added to each well. These mixed cell cultures were incubated at 37°C in a CO2-controlled humidified incubator and were microscopically scored for syncytium formation at ∼36–48 h post-virus exposure/co-cultivation. It should be noted that the maximum number of free viral particles that could have remained in the culture medium after the extended washing steps (<1 pg of HIV-1 p24) could not result in HIV-1-induced giant cell formation in CD4+ T cell cultures within the time period of the analysis (36–48 h).
2G12 MAb binding assay
CD4+ MT-4 cells were infected with HIV-1 NL4.3 and analysed when the CPE started to occur (3–4 days post-infection). Shortly after washing with PBS supplemented with 2% FCS (PBS/FCS2%), the cells were pre-incubated with various concentrations of NICTABA, HHA or UDA for 30 min, extensively washed and incubated with 2G12 MAb for 30 min. After this, the cells were washed and further incubated with RaH-IgG-FITC (Dako) for 30 min. As a control for non-specific background staining, the cells were stained in parallel with RaH-IgG-FITC only. After washing, the cells were fixed with a 1% aqueous formaldehyde solution and analysed with a FACSCalibur flow cytometer using CellQuest software (BD Biosciences, San Jose, CA, USA). To calculate the MAb binding, the mean fluorescence intensity (MFI) of each sample was expressed as a percentage of the MFI of the control cells (after subtracting the MFI of the background staining).
Surface plasmon resonance (SPR)
Kinetic experiments
For the HIV envelope binding experiments, the recombinant glycoprotein gp120 from the HIV-1 IIIB strain (ImmunoDiagnostics Inc., Woburn, MA, USA; produced in cell cultures of Chinese hamster ovaries) and the recombinant glycoprotein gp41 from the HIV-1 HxB2 strain (Acris Antibodies GmbH, Herford, Germany; produced in Pichia pastoris cells) were covalently coupled to the carboxymethylated dextran matrix-covered CM5 sensor chips at flow channels two and four, respectively (GE Healthcare, Uppsala, Sweden), in 10 mM sodium acetate, pH 4.0 using the standard amine coupling chemistry, affording a final density of 148 response units (RUs) for gp120 (1.25 fmol) and 265 RUs for gp41 (6.5 fmol). The flow cells in channels one and three were used as controls for non-specific binding and changes in refractive index.
All interaction studies were performed at 25°C on a Biacore T100 instrument (GE Healthcare, Uppsala, Sweden). The compounds were diluted in HBS-P (10 mM HEPES, 150 mM NaCl and 0.05% surfactant P20; pH 7.4) supplemented with 10 mM Ca2+, using 2-fold dilution steps. UDA and NICTABA were used at concentrations ranging from 1.0 to 63 nM. Samples were injected over 3 min at a flow rate of 30 μL/min, followed by a dissociation phase of 5 min. The sensor chip was regenerated with 50 mM NaOH. Several blanks were included throughout the experiment as a second reference.
To determine the kinetic parameters, the curves were fitted using the 1: 1 binding model (Biacore T100 Evaluation software 2.0.1).
Inhibition of the binding by sugars
The sensor surface that was prepared as described for the kinetic experiments was used to analyse the binding of UDA and NICTABA in the presence or absence of a GlNAc trimer [GlcNAc β-(1,4)-GlcNAc/β-(1,4) GlcNAc] (Dextra Laboratories Ltd, Reading, UK), a mannose/α-(1,2)-mannose/α-(1,2)-mannose trisaccharide (Carbohydrate Synthesis, Oxford, UK) or a mannose/α-(1,3)-mannose/α-(1,6)-mannose trisaccharide (Dextra Laboratories Ltd, Reading, UK). The concentration of UDA was kept constant at 100 nM throughout the whole experiment. NICTABA was used at a concentration of 80 nM. The concentrations of the trisaccharides were 200 or 100 μM. The lectins, trisaccharides or pre-incubated mixtures of lectin and trisaccharide were injected across the sensor surface as described for the kinetic experiments.
Results
Broad antiviral activity profile of NICTABA and UDA in cell cultures
NICTABA and UDA have been evaluated for their inhibitory activity against a wide variety of enveloped DNA and RNA viruses in cell culture using the α-(1,3)-α-(1,6)-mannose-specific HHA as a control (Table 1). Whereas HHA was inhibitory only against influenza A and B viruses, DENV serotype 2 (DENV-2) and HIV (Table 2), the GlcNAc-specific CBAs displayed a much broader antiviral spectrum. They not only inhibited the same viruses as HHA, but also showed inhibitory activity against HSV type 1 (HSV-1) and HSV-2, varicella zoster virus (VZV) and parainfluenza virus. NICTABA often showed an activity superior to UDA. Interestingly, respiratory syncytial virus and HSV-1 were only suppressed by NICTABA but not by UDA (Table 1). None of the three CBAs displayed antiviral activity to non-enveloped viruses such as coxsackie virus and reovirus. The CC50 value of NICTABA in HEL cells was >50 μg/mL (>1.5 μM).
Table 1.
Antiviral activity of NICTABA, UDA and HHA against a broad spectrum of viruses evaluated in HEL, MDCK, HeLa, Raji/DC-SIGN, CrFK or Vero cell cultures
| Viruses | EC50a (nM) |
||
|---|---|---|---|
| NICTABAb | UDAb | HHAb | |
| Enveloped viruses | |||
| HSV-1 (KOS) (HEL) | 263 ± 113 | >11 800 | >2000 |
| HSV-2 (G) (HEL) | 53 ± 11 | 1294 ± 82 | >2000 |
| HSV-1 TK− KOS ACV (HEL) | 171 ± 47.0 | 9647 ± 3764 | >2000 |
| VZV (HEL) | 129 ± 1.32 | ND | ND |
| influenza A H1N1 subtype (MDCK) | 32 ± 11 | 506 ± 153 | 540 ± 360 |
| influenza A H3N2 subtype (MDCK) | 18 ± 5.3 | 188 ± 47 | 2 ± 0.2 |
| influenza B (MDCK) | 11 ± 0.0 | 224 ± 176 | 6 ± 2 |
| parainfluenza-3 virus (HeLa) | 50 ± 26 | 235 ± 0.0 | >2000 |
| respiratory syncytial virus (HeLa) | 105 ± 0.0 | >11 800 | >2000 |
| DENV type 2 (Raji/DC-SIGN+) | 526 ± 2.3 | 1176 ± 423 | 12 ± 0.2 |
| feline herpes virus (CrFK) | 24 ± 0.3 | 647 ± 470 | 220 ± 86 |
| feline corona virus (CrFK) | 153 ± 1.7 | 1024 ± 106 | 520 ± 12 |
| Non-enveloped viruses | |||
| coxsackie virus B4 (HeLa) | >2600 | >5900 | >2000 |
| reovirus-1 (Vero) | >2600 | >11 800 | >2000 |
ND, not determined.
The mean IC50 ± SEM from three or four independent experiments are shown.
MCC for NICTABA (μg/mL): HEL >100, HeLa >100 and Vero >100.
CC50 (median cytotoxic concentration) for NICTABA (μg/mL): MDCK >50, CrFK >50 and Raji/DC-SIGN+ >50.
aIC50 (50% inhibitory concentration) is the concentration that inhibits virus-induced CPE by 50%.
bMolecular weights: NICTABA, 38 kDa; UDA, 8.5 kDa; and HHA, 50 kDa.
Table 2.
Anti-HIV activity (EC50 in μM) of NICTABA, UDA and HHA against laboratory-adapted strains and clinical isolates of HIV in cell cultures and PHA-activated PBMCs
| HIV-1 strains (tropism; subtype) | NICTABA | UDA | HHA |
|---|---|---|---|
| Laboratory-adapted strains | |||
| MT-4/NL4.3 (X4; B) | 0.17 ± 0.08 | 0.28 ± 0.08a | 0.004 ± 0.001a |
| TZM-bl/BaL (R5; B) | 0.23 ± 0.05 | 0.5 ± 0.1 | 0.010 ± 0.001 |
| CEM/IIIB (X4; B) | 0.023 ± 0.003 | 0.23 ± 0.00 | 0.0060 ± 0.0005 |
| PBMCs/NL4.3 (X4; B) | 0.018 ± 0.003 | 0.23 ± 0.06 | 0.005 ± 0.002 |
| PBMCs/BaL (R5; B) | 0.014 ± 0.005 | 0.38 ± 0.14 | 0.01 ± 0.004 |
| PBMCs/HE (X4/R5; B) | 0.015 ± 0.004 | 1.80 ± 0.02 | 0.240 ± 0.039 |
| PBMCs/ROD (HIV-2) | 0.005 ± 0.003 | 0.06 ± 0.03 | ND |
| Clinical isolates | |||
| PBMCs/UG273 (R5; A) | 0.008 ± 0.002 | 1.6 ± 0.7 | ND |
| PBMCs/DJ259 (R5; C) | 0.020 ± 0.004 | 0.1 ± 0.2 | 0.10 ± 0.02 |
| PBMCs/ID12 (R5; A/E) | 0.010 ± 0.003 | 0.4 ± 0.2 | ND |
| PBMCs/ETH2220 (R5; C) | 0.03 ± 0.01 | 0.8 ± 0.3 | ND |
| PBMCs/US2 (R5; B) | 0.03 ± 0.03 | ND | ND |
ND, not determined.
Mean EC50 values ± SEM from at least 3–5 independent experiments are shown.
CC50 for NICTABA (μg/mL): MT-4 >20, TZM-bl >20, CEM >20 and PBMCs 14.4.
aData obtained from Huskens et al.17
Broad-spectrum anti-HIV activity profile of NICTABA and UDA
Our first set of experiments demonstrated a broad activity spectrum of the CBAs against enveloped viruses, which are heavily glycosylated. Since HIV is one of the most glycosylated viruses, we evaluated the antiviral activity of CBAs and their mode of action against different HIV strains.
In this part of the study, we measured the activity of NICTABA and UDA against cell-line-adapted HIV-1 strains (NL4.3, BaL, IIIB) in different cell models including genital epithelial TZM-bl cells, as well as in susceptible CD4+ T cell lines such as MT-4 and CEM cells. As a reference, we compared the activity of the GlcNAc-binding lectins with the mannose-specific CBA HHA. As shown in Table 2, both NICTABA and UDA was able to inhibit infection by HIV strains with an EC50 in the lower micromolar range (EC50 0.023–0.28 μM).
Second, the anti-HIV activity of NICTABA and UDA was tested against clade B HIV-1 strains and HIV-2 in PHA-activated PBMCs (or T cell blasts). We found that both CBAs inhibited the infection of CXCR4-using (X4) NL4.3, CCR5-using (R5) BaL and dual-tropic HE HIV-1 strains in the lower nanomolar range (IC50 NICTABA 14–18 nM). Moreover, NICTABA and UDA prevented the HIV-2 infection of PBMCs with mean EC50 values of 5 and 60 nM, respectively. Overall, the mannose-specific lectin HHA showed better antiviral activity against HIV-1 laboratory strains than both GlcNAc-binding CBAs, but NICTABA demonstrated less variability in its virus-suppressive potential and was 10- to 100-fold more potent than UDA (Table 2).
Third, primary HIV-1 clinical isolates representing different HIV-1 clades (A, B, C and A/E) were tested for their susceptibility to neutralization by NICTABA and UDA in PBMCs. Both CBAs were found to be protective, but the neutralization activity of NICTABA was higher than that of UDA, with EC50 values varying from 8 to 30 nM. In addition, the antiviral activity of NICTABA was evaluated in monocyte-derived macrophages against the HIV-1 BaL strain and was found to be protective with a mean EC50 of 0.016 ± 0.004 μM (data not shown). The CC50 value for NICTABA obtained in PBMCs was 0.38 μM. We can conclude that NICTABA and UDA exhibited a consistent and broad anti-HIV activity in both CD4+ T lymphoma cell cultures and PBMCs (Table 2). In all cases, the antiviral potency of the GlcNAc-binding agent NICTABA was superior to that of UDA.
NICTABA and UDA inhibit HIV-induced cell–cell syncytium formation
When NICTABA and UDA were examined for their potential to prevent syncytium formation between persistently HIV-1 IIIB-infected cells (HUT-78/IIIB) and uninfected CD4+ SupT1 cells, both CBAs inhibited giant cell formation to a similar extent (IC50 0.32–0.48 μM), but with results inferior to those of the mannose-specific HHA (0.08 μM) (Table 3).
Table 3.
Inhibitory activity of NICTABA, UDA and HHA against syncytium formation between persistently HIV-1-infected cells and uninfected CD4+ T cells
| CBA | EC50a (μM) |
|---|---|
| NICTABA | 0.32 ± 0.09 |
| UDA | 0.48 ± 0.1 |
| HHA | 0.08 ± 0.02 |
Mean EC50 values ± SEM of three independent experiments are shown.
aEC50, the effective concentration of CBA necessary to inhibit syncytium formation between persistently HIV-infected HUT-78/IIIB cells and uninfected CD4+ SupT1 T cells by 50%.
NICTABA and UDA have also been investigated for their potential to prevent HIV-1 capture by DC-SIGN-expressing Raji/DC-SIGN+ cells and to block the transmission of DC-SIGN-captured virus to CD4+ T lymphocyte C8166 cell cultures. Whereas NICTABA could not prevent virus capture by Raji/DC-SIGN+ cells, UDA could. However, HHA was much more efficient than UDA in preventing virus capture (Table 4). On the other hand, the transmission of HIV-1-captured virus by Raji/DC-SIGN+ cells to the uninfected CD4+ T cells was dose-dependently inhibited by both NICTABA and UDA (Table 4).
Table 4.
Inhibition of HIV-1 capture and transmission by DC-SIGN+ cells to human CD4+ T lymphocytes
| CBA | EC50a (μM) |
|
|---|---|---|
| capture | transmissionb | |
| NICTABA | >1.3 | 0.14 |
| UDA | 0.44 | 0.61 |
| HHA | 0.01 | 0.06 |
aEC50, the effective concentration of CBA necessary to inhibit HIV-1 capture by Raji DC-SIGN and the transmission of DC-SIGN-captured virus to uninfected CD4+ C8166 T cells by 50%.
bIn co-cultivation cultures with CD4+ C8166 T cells, samples were taken 44 h post-co-cultivation.
Effect of NICTABA, UDA and HHA on the binding of the carbohydrate-binding MAb 2G12 to HIV-1 infected MT-4 cells
NICTABA at the highest concentration tested (0.52 μM; 20 μg/mL) had no effect on the binding of the carbohydrate-binding MAb 2G12 to HIV-1-infected MT-4 cells, whereas HHA (0.4 μM; 20 μg/mL) and UDA (2.4 μM; 20 μg/mL) efficiently inhibited this process (Figure 1). Thus NICTABA, in contrast to all the other CBAs examined so far, including the MAb 2G12, does not interact with the 2G12 MAb binding epitope on gp120.
Figure 1.
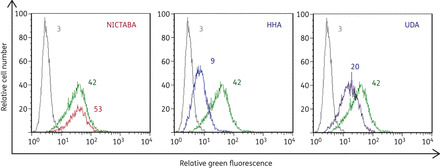
UDA and HHA, but not NICTABA, inhibit the binding of the 2G12 MAb to HIV-1-NL4.3-infected MT-4 cells. MT-4 cells were infected with HIV-1 strain NL4.3. After 3–4 days, the cells were incubated with the 2G12 MAb (20 μg/mL) + RaH-IgG-FITC in the absence of NICTABA, HHA or UDA (green curves) or the presence of NICTABA (red curves), HHA (blue curves) or UDA (purple curves). The light grey curves show the background fluorescence. The average MFI values of three independent experiments are indicated above each histogram and reflect the degree of binding of the 2G12 MAb to the HIV-1-infected cells. One representative experiment out of three is shown.
In addition, NICTABA, but also the other CBAs HHA and UDA at similar concentrations, had no effect on the binding of the MAb (clone 9205) recognizing the V3 loop of gp120 to HIV-1-infected MT-4 cells (data not shown).
Kinetic interactions of NICTABA and UDA with the HIV envelope glycoproteins gp120 and gp41
The binding of NICTABA and UDA to the virus envelope was kinetically characterized by SPR analysis. In this experiment, gp120 and gp41 were covalently immobilized on a CM5 sensor chip. The CBAs UDA and NICTABA were used as analytes in a concentration range of 1.8–63 nM for NICTABA and 2.0–63 nM (gp120) or 2.0–31 nM (gp41) for UDA. The analyte solutions were injected across the sensor chip for 3 min, after which the analyte was allowed to dissociate from the surface for 5 min. The residual analyte was then removed from the surface using a pulse of 50 mM NaOH. The binding curves of NICTABA and UDA to gp120, together with the fitted curves, are shown in Figure 2(a).
Figure 2.
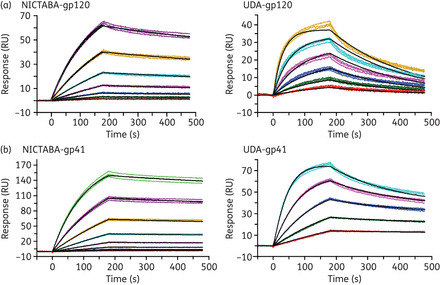
Kinetic analysis of interactions between the CBAs and the immobilized envelope proteins of HIV. Binding of NICTABA (left panels) or UDA (right panels) to HIV-1 IIIB gp120 (a) or HIV-1 HxB2 gp41 (b) immobilized on a CM5 sensor chip. Serial 2-fold analyte dilutions were injected over the surface of the HIV-1 envelope-protein-bound sensor chip. These dilutions covered a concentration range from 1.8 to 63 nM for NICTABA and from 2.0 to 63 nM (gp120) or 2.0 to 31 nM (gp41) for UDA. Experiments were carried out in triplicate (coloured lines). The curves were fitted using the 1: 1 binding model (black curves) to determine the kinetic parameters.
The kinetic parameters that were determined for the binding of NICTABA and UDA to gp120 and gp41 are summarized in Table 5. Both CBAs showed KD (affinity) values for HIV-1 gp120 in the lower nanomolar range (KD 4–10 nM). Whereas the on-rates (association rate constant, ka) to gp120 were fairly comparable for UDA and NICTABA, the dissociation rate constant (kd) for NICTABA was 6-fold lower than for UDA.
Table 5.
Kinetic parameters of the interaction between NICTABA or UDA and immobilized HIV-1 envelope proteins gp120 and gp41 determined by SPR
| k a (1/Ms)a | k d (1/s)a | K D (M)a,b | |
|---|---|---|---|
| HIV-1 gp120 | |||
| NICTABA | 1.8 × 105 ± 9.3 × 101 | 7.0 × 10–4 ± 9.5 × 10–7 | 3.8 × 10–9 |
| UDA | 4.8 × 105 ± 5.7 × 102 | 4.2 × 10–3 ± 3.5 × 10–6 | 8.7 × 10–9 |
| HIV-1 gp41 | |||
| NICTABA | 1.7 × 105 ± 6.1 × 102 | 2.6 × 10–4 ± 9.1 × 10–7 | 1.5 × 10–9 |
| UDA | 1.0 × 106 ± 2.9 × 103 | 1.6 × 10–3 ± 3.3 × 10–6 | 1.6 × 10–9 |
Mean values ± SEM from three independent experiments are shown.
a k a, association rate constant; kd, dissociation rate constant; KD, affinity constant.
bThe affinity constant KD is the ratio kd/ka.
Their affinities for gp41 were at least as good as, if not better than, their affinities for gp120 (Figure 2b and Table 5). Comparable apparent KD values (1.5–1.6 nM) were detected for the binding of both CBAs to gp41. For UDA, the KD value was 5.4-fold lower (higher affinity) for gp41 than for gp120 (Table 5).
Overall, the sensorgrams show that both CBAs have a high affinity for the envelope proteins gp120 and gp41, with KD values ranking in the subnanomolar range.
Effect of carbohydrate oligomers on the binding of NICTABA and UDA to HIV-1 gp120
To verify the nature of the sugar specificity of the two lectins, the binding capacity of NICTABA and UDA to immobilized gp120 was investigated in the presence or absence of different glycan structures such as a GlcNAc β-(1,4)/GlcNAc/β-(1,4) GlcNAc, a mannose α-(1,2)-mannose/α-(1,2)-mannose or a mannose/α-(1,3)-mannose/α-(1,6)-mannose oligosaccharide.
Prior to the gp120 binding, the CBAs were incubated in the presence of the respective trisaccharides (trisaccharides at a final concentration of 100 or 200 μM) for a short time. The GlcNAc trimer and the α-(1,2)/α-(1,2) mannose and α-(1,3)/α-(1,6) mannose trimers were also injected in the absence of the CBAs at a concentration of 200 μM as a reference. The final concentrations of NICTABA and UDA in these experiments were 80 and 100 nM, respectively. The results are shown in Figure 3.
Figure 3.
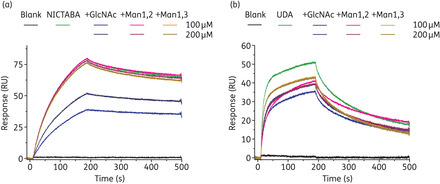
Affinity analysis of NICTABA (a) and UDA (b) for immobilized HIV-1 IIIB gp120 in the presence or absence of various oligosaccharides. The concentrations of the trisaccharides [GlcNAc β-(1,4)-GlcNAc/β-(1,4)-GlcNAc, mannose α-(1,2)-mannose/α-(1,2)-mannose and mannose α-(1,3)-mannose/α-(1,6) mannose] were 200 or 100 μM. The concentration of UDA was kept constant at 100 nM and the concentration of NICTABA at 80 nM throughout the whole experiment. The lectins, trisaccharides or pre-incubated mixtures of lectin and trisaccharide were injected across the sensor surface.
The binding of NICTABA to gp120 was dose-dependently reduced by the addition of the GlcNAc trimer to the NICTABA solution, while the other two mannose trisaccharides only had a minor, if any, effect on the binding of NICTABA to immobilized gp120 (Figure 3a). Instead, the binding of UDA to gp120 could be reduced by adding either one of the trisaccharides, although the highest level of inhibition was observed with the GlcNAc trimer. Clearly, both mannose trimers interfered with the binding of UDA to gp120 although to a lesser extent compared with the effect of GlcNAc trimers on UDA binding (Figure 3b).
Discussion
Many viruses affecting human health use the host cell machinery to assemble glycans on their surface, which are important for viral entry and for escape from the host immune system.8,9,52 Glycans are bound by natural ligands called lectins that have differential binding preferences for them. Targeting viral glycans is a promising antiviral strategy since there are multiple glycosylated sites on the virus capsid.53,54 In addition, the emergence of resistance to such anti-carbohydrate therapies will most likely involve the removal of multiple sugar residues from the viral envelope, thereby more efficiently exposing the surface of the virus to host-derived neutralizing antibodies.25,53,55–58 Moreover, Balzarini et al.25 demonstrated that the kinetics of resistance development are also likely to depend on the type of glycan targeted as it took 3-fold longer before phenotypic resistance became apparent against UDA, a GlcNAc-binding CBA, than against mannose-specific CBAs. Here, we evaluated the activity of the tobacco plant lectin NICTABA, which has exclusive affinity for GlcNAc residues, and compared it with UDA and the mannose-specific HHA.
We found that NICTABA and UDA are both inhibitory against a very broad spectrum of enveloped viruses. Interestingly, this spectrum is much broader than that of HHA. This may be due to the fact that GlcNAc is more abundantly present in complex-type than in high-mannose-type glycans. As HIV is one of the most heavily glycosylated viruses,59,60 we evaluated the activity of NICTABA, UDA and HHA against a variety of HIV strains in different target cells. Indeed, Raska et al.61 reported that the glycosylation pattern is profoundly influenced by the cell type and metabolic activity of the producing cells, resulting in distinct gp120 N-glycan contents and heterogeneity. While such high variability of the HIV envelope compromises the efficacy of the neutralizing antibodies (which only recognize one specific envelope epitope),62 GlcNAc-specific CBAs maintained their antiviral activity with EC50 values ranking between the lower nano- and micromolar ranges. Furthermore, NICTABA and UDA were endowed with much less variation in their antiviral spectrum than HHA. An important factor contributing to these differences may again be the sugar specificity of these CBAs. While HHA predominantly recognizes α-(1,3)- and/or α-(1,6)-mannose residues located at the pentasaccharide core, GlcNAc oligomers are invariably present at the base of the pentasaccharide core in each N-glycosylation site of gp120. In addition, there was a clear tendency for NICTABA to be more potent than UDA in inhibiting virus replication. This may be related to a more pronounced binding of NICTABA to the viral envelope glycans than UDA. Indeed, the apparent affinity of NICTABA for HIV-1 gp120 was 2-fold higher than of UDA, but, importantly, this was mainly due to the contribution of the off-rate, which was ∼6-fold lower for NICTABA than UDA.
The measurements of affinity between gp120 and NICTABA or UDA showed low KD values in the low to sub-nanomolar range. Since the dissociation rates of these CBAs were very low, they can be virtually considered to be irreversible gp120 binders. These tight-binding properties are in agreement with the increased antiviral activities upon pre-incubation of HIV-1 or HIV-1-infected cells with NICTABA and UDA. However, Doores et al.63 found that the glycan composition of recombinant monomeric gp120 comprised mainly complex glycans while the envelope proteins of intact virions almost exclusively carried oligomannose glycans. This difference in composition may affect the binding capacity of CBAs, and therefore our comparative binding studies performed with recombinant monomeric gp120 need to be interpreted with some caution and in close connection with the antiviral cell culture data. It was interesting to notice that the affinity of the CBAs for gp41 (KD ∼ 1.5 nM) was higher than for gp120 (KD 3.8–8.7 nM) while the envelope protein gp120 contained 6-fold more sites for N-linked glycosylation than gp41, which typically contains only four sites for N-glycan attachment.64
Many viruses including HIV,65 respiratory syncytial virus66 and VZV67 induce membrane fusion between cells to form giant multinucleated cells, called syncytia, which allows the virion to kill many cells by just infecting one single cell. NICTABA and UDA were able to inhibit syncytium formation between persistently HIV-1-infected T cells and uninfected CD4+ T cells, suggesting an inhibitory role of the GlcNAc-binding CBAs during the attachment of gp120 to the cellular receptors and in the subsequent fusion steps. This is an important property of the CBAs that is shared with other HIV entry inhibitors [dextran sulphate (DS-5000),68 enfuvirtide69 or the CXCR4 antagonists47], but is absent in drugs that target a site in the replication cycle of HIV located at a timepoint after viral entry.
DC-SIGN is a lectin of our innate immune system predominantly present on immature DCs. It functions in DC recognition and the uptake and processing of pathogens leading to Ag presentation to the T cells. It has been shown that DC-SIGN can recognize and internalize numerous bacteria, protozoa70 and viruses such as HIV,70 DENV,71 human CMV,72 HCV73 and Ebola virus74 to allow for efficient trans infection of the target cells. Preventing interaction with DCs, and in particular DC-SIGN, represents an attractive approach to prevent viral transmission and infection of the host. We showed that UDA, but not NICTABA, efficiently prevents the DC-SIGN-directed capture of HIV-1. Interestingly, both NICTABA and UDA prevented the transmission of DC-SIGN-captured HIV-1 to uninfected CD4+ T lymphocytes in the same concentration range needed for the inhibition of cell-free viral transmission in T lymphoma cell lines (EC50 for NICTABA ∼140 nM and for UDA ∼600 nM). Some CBAs have been shown to be not equally effective in their inhibitory potential for the two modes of transmission.14,47 NICTABA is the first CBA reported so far that does not block DC-SIGN-directed HIV capture. This suggests that NICTABA recognizes different glycan epitopes than does DC-SIGN, while UDA, at least partially, shares certain glycan epitopes present on gp120 or hinders DC-SIGN binding. Several studies have revealed that the carbohydrate-recognition domain of DC-SIGN recognizes α-(1,3)- and α-(1,2)-linked mannose oligomers75 as well as fucosylated glycans.76 That NICTABA prevented HIV-1 transmission, but not viral capture, could be explained by the assumption that its binding to the GlcNAc-containing glycans on gp120 prevents conformational changes and hinders the flexibility of gp120 that is required to properly interact with the cell membrane receptor necessary for fusion.
When taking a closer look at the results of the sugar inhibition experiments, it is clear that the binding of NICTABA and UDA to gp120 in the presence of a GlcNAc β-1,4-trimer was dose-dependently decreased. This is in line with previous reports on the GlcNAc specificity of both lectins. While this appears to be in contrast to the recent finding from a microarray screen that NICTABA also efficiently recognizes high-mannose glycans,77 it should be mentioned that these studies found a high affinity of GlcNAcβ1,4GlcNAc configurations that are extended with mannose residues. This may imply that NICTABA shows a certain affinity for mannose oligomers on the condition that they are part of a complex with GlcNAc oligomers. Generating crystal structures with (GlcNA)2Manx oligomers might be useful to clarify this observation.
In this report, α-(1,2)-mannose and α-(1,3/1,6)-mannose trimers (devoid of GlcNAc residues) hardly influenced NICTABA binding but they did affect UDA binding to gp120, albeit to a lesser degree than in the presence of GlcNAc trisaccharides. This finding suggests that UDA has a high affinity for GlcNAc residues, but, like DC-SIGN, can also recognize α-(1,3)- and α-(1,2)-linked mannose oligomers. Another observation that supports the broader sugar specificity of UDA is that binding of the MAb 2G12 is diminished in the presence of UDA but not in the presence of NICTABA. 2G12 MAb is one of the few broadly neutralizing anti-HIV MAbs that solely recognizes a specific configuration of three α1,2-mannose glycan oligomers on gp120, which lies around the C4–V4 region.78–80
2G12 MAb activity has been shown to vary depending on the nature of the HIV-1 isolates that are evaluated. Indeed, Travers58 has demonstrated that the 2G12 epitope is poorly conserved across the HIV-1 group M due to strong strain-specific glycosylation patterns. In this report, the observed narrow variation in the potency of NICTABA represents an interesting property with respect to the potential use for NICTABA as an antiviral agent. Furthermore, Huskens et al.15 observed that under increased drug pressure from the 2G12 MAb, HIV-1 became resistant rather quickly (six cell culture passages) and only one glycan deletion appeared sufficient for the 2G12 MAb to lose its antiviral activity. This observation was also confirmed in vivo where the potency of 2G12 and two other neutralizing antibodies to gp41 were evaluated in HIV-1-infected individuals.81 Although plasma levels of 2G12 were the highest and exceeded the in vitro required 90% inhibitory doses, viral escape emerged very rapidly and at high titers.81 In contrast, only after more than 90 cell culture subcultivations did a UDA-resistant virus emerge possessing nine mutations at N-linked glycosylation sites.25 Interestingly, more high-mannose glycan-containing glycosylation sites were deleted compared with complex mannose-type glycans of gp120.25 Complex N-linked glycans differ from the high-mannose and hybrid glycans by having added other sugars including GlcNAc residues at both the α-3 and α-6 mannose sites. Cross-resistance was observed with multiple mannose-specific CBAs (HHA, GNA, CV-N, 2G12).25 It would be interesting to see whether NICTABA-resistant virus would have a similar preference for the annihilation of high-mannose type N-glycans in gp120 than for complex N-linked glycans. However, after 70 subcultivations, no phenotypical and genotypical resistance of HIV-1 was observed against the GlcNAc-specific NICTABA (data not shown). Unfortunately, due to a limited selectivity index (SI = 21) in cell culture, we will probably not be able to increase drug pressure and thus generate an HIV-1-resistant NICTABA strain. It has been shown that a mitogenic response can be induced by certain lectins, for instance cyanovirin, which, despite being a very potent inhibitor of viral entry, can bind to the host cell surface and induce T cell activation markers and the production of various pro-inflammatory cytokines.82 In contrast, griffithsin, one of the most potent CBAs identified to date, has also been shown to bind the host cell surface, but this binding did not result in cellular activation.82 Whether NICTABA also binds to the cell surface remains unclear since there are currently no specific antibodies available to resolve this issue.
A better understanding of the molecular interaction between NICTABA and the GlcNAc-sugar residues on gp120 using NMR or crystallography would enable a rational design of more potent synthetic GlcNAc-specific CBAs. Further research to feasibly scale up synthetic GlcNAc-specific CBAs should be explored due to the high genetic barrier for the development of resistance, even more so than for mannose-specific CBAs. Another concern that is often raised for CBAs in general is their potential inability to discriminate between pathogen and cellular glycans. Nevertheless, it has been shown that the three-dimensional configuration of the glycans present on pathogens is important for its specific interaction.14 In fact, DC-SIGN is a very good example of a lectin that can distinguish between pathogen-associated glycoproteins and cellular glycoproteins, thereby enabling a selective elimination of the pathogen. Furthermore, the intravenous administration of UDA and the mannose-specific GNA and HHA to adult mice has been shown to lack toxicity.83 In addition, for UDA, it has also been reported that, at concentrations that were substantially higher than its antiviral activity in cell culture, it did not agglutinate human red blood cells and was poorly mitogenic.25
All things considered, targeting the envelope N-glycans of HIV and other viruses might be a promising strategy. This is also corroborated by the fact that a new generation of potent, broadly neutralizing antibodies has recently been identified from HIV-1-infected individuals; these target novel epitopes on HIV gp120 that are partly or exclusively composed of glycans.84–88 These MAbs have been shown to exhibit great breadth and antiviral potency against various HIV-1 isolates in vitro and have shown promising results in a vaccination study in macaques.85 These recent findings indicate that CBAs may have potential as future antiviral drugs.
In summary, NICTABA is, unlike UDA, a clearly GlcNAc-specific CBA and is endowed with broad-spectrum antiviral activity. It may concomitantly suppress both well-known co-pathogens HIV and HSV, which is beneficial from a microbicidal viewpoint.89 Furthermore, its neutralizing activity was striking, in the sense that NICTABA displayed minimal variation in antiviral activity irrespective of the nature of the HIV viral strain and host cell type. In addition, NICTABA was found to be a potent inhibitor of syncytium formation and of DC-SIGN-directed transmission to CD4+ T lymphocytes. These properties together make NICTABA an appealing and potent candidate for further drug development against enveloped viruses. The therapeutic exploitation of a GlcNAc-specific CBA will require the design of synthetic mimics that share the same broad activity, but lack the potential cellular side effects, of NICTABA.
Funding
This work was supported by grants from the KU Leuven (GOA 10/014 and PF/10/018), the Foundation of Scientific Research (FWO no. G-0485-08, G-0528-12), the Foundation Dormeur, Vaduz and the CHAARM project (No. 242135) of the European Commission.
Transparency declarations
None to declare.
Acknowledgements
We are grateful to Leentje Persoons, Leen Ingels, Frieda De Meyer, Lizette van Berckelaer, Sandra Claes, Rebecca Provinciael, Evelyne Van Kerckhove, Daisy Ceusters, Eric Largy and Eric Fonteyn for excellent technical assistance. We thank Sam Noppen for the critical advice on the generated SPR data.
References
- 1. Leonard CK, Spellman MW, Riddle L, et al. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem 1990; 265: 10373–82. [PubMed] [Google Scholar]
- 2. Scanlan CN, Offer J, Zitzmann N, et al. Exploiting the defensive sugars of HIV-1 for drug and vaccine design. Nature 2007; 446: 1038–45. [DOI] [PubMed] [Google Scholar]
- 3. Rudd PM, Dwek RA. Glycosylation: heterogeneity and the 3D structure of proteins. Crit Rev Biochem Mol Biol 1997; 32: 1–100. [DOI] [PubMed] [Google Scholar]
- 4. Ezekowitz RA. Role of the mannose-binding lectin in innate immunity. J Infect Dis 2003; 187Suppl 2: S335–9. [DOI] [PubMed] [Google Scholar]
- 5. Etzioni A. Adhesion molecules in leukocyte endothelial interaction. Adv Exp Med Biol 1996; 408: 151–7. [DOI] [PubMed] [Google Scholar]
- 6. Meunier JC, Fournillier A, Choukhi A, et al. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J Gen Virol 1999; 80: 887–96. [DOI] [PubMed] [Google Scholar]
- 7. White JM, Delos SE, Brecher M, et al. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 2008; 43: 189–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang M, Gaschen B, Blay W, et al. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 2004; 14: 1229–46. [DOI] [PubMed] [Google Scholar]
- 9. Cook JD, Lee JE. The secret life of viral entry glycoproteins: moonlighting in immune evasion. PLoS Pathog 2013; 9: e1003258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morozov VA, Morozov AV, Semaan M, et al. Single mutations in the transmembrane envelope protein abrogate the immunosuppressive property of HIV-1. Retrovirology 2012; 9: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reitter JN, Means RE, Desrosiers RC. A role for carbohydrates in immune evasion in AIDS. Nat Med 1998; 4: 679–84. [DOI] [PubMed] [Google Scholar]
- 12. Balzarini J, Schols D, Neyts J, et al. α-(1–3)- and α-(1–6)-d-Mannose-specific plant lectins are markedly inhibitory to human immunodeficiency virus and cytomegalovirus infections in vitro. Antimicrob Agents Chemother 1991; 35: 410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balzarini J, Van Herrewege Y, Vermeire K, et al. Carbohydrate-binding agents efficiently prevent dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN)-directed HIV-1 transmission to T lymphocytes. Mol Pharmacol 2007; 71: 3–11. [DOI] [PubMed] [Google Scholar]
- 14. Balzarini J. Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat Rev Microbiol 2007; 5: 583–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huskens D, Van Laethem K, Vermeire K, et al. Resistance of HIV-1 to the broadly HIV-1-neutralizing, anti-carbohydrate antibody 2G12. Virology 2007; 360: 294–304. [DOI] [PubMed] [Google Scholar]
- 16. Ferir G, Huskens D, Palmer KE, et al. Combinations of griffithsin with other carbohydrate-binding agents demonstrate superior activity against HIV type 1, HIV type 2, and selected carbohydrate-binding agent-resistant HIV Type 1 strains. AIDS Res Hum Retroviruses 2012; 28: 1513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huskens D, Ferir G, Vermeire K, et al. Microvirin, a novel α(1,2)-mannose-specific lectin isolated from Microcystis aeruginosa, has anti-HIV-1 activity comparable with that of cyanovirin-N but a much higher safety profile. J Biol Chem 2010; 285: 24845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pollicita M, Schols D, Aquaro S, et al. Carbohydrate-binding agents (CBAs) inhibit HIV-1 infection in human primary monocyte-derived macrophages (MDMs) and efficiently prevent MDM-directed viral capture and subsequent transmission to CD4+ T lymphocytes. Virology 2008; 370: 382–91. [DOI] [PubMed] [Google Scholar]
- 19. Hoorelbeke B, Huskens D, Ferir G, et al. Actinohivin, a broadly neutralizing prokaryotic lectin, inhibits HIV-1 infection by specifically targeting high-mannose-type glycans on the gp120 envelope. Antimicrob Agents Chemother 2010; 54: 3287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boyd MR, Gustafson KR, McMahon JB, et al. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob Agents Chemother 1997; 41: 1521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buffa V, Stieh D, Mamhood N, et al. Cyanovirin-N potently inhibits human immunodeficiency virus type 1 infection in cellular and cervical explant models. J Gen Virol 2009; 90: 234–43. [DOI] [PubMed] [Google Scholar]
- 22. Swanson MD, Winter HC, Goldstein IJ, et al. A lectin isolated from bananas is a potent inhibitor of HIV replication. J Biol Chem 2010; 285: 8646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mori T, O'Keefe BR, Sowder RC, 2nd, et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J Biol Chem 2005; 280: 9345–53. [DOI] [PubMed] [Google Scholar]
- 24. Ferir G, Huskens D, Noppen S, et al. Broad anti-HIV activity of the Oscillatoria agardhii agglutinin homologue lectin family. J Antimicrob Chemother 2014; 69: 2746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balzarini J, Van Laethem K, Hatse S, et al. Carbohydrate-binding agents cause deletions of highly conserved glycosylation sites in HIV GP120: a new therapeutic concept to hit the Achilles heel of HIV. J Biol Chem 2005; 280: 41005–14. [DOI] [PubMed] [Google Scholar]
- 26. Balzarini J, Van Laethem K, Hatse S, et al. Marked depletion of glycosylation sites in HIV-1 gp120 under selection pressure by the mannose-specific plant lectins of Hippeastrum hybrid and Galanthus nivalis. Mol Pharmacol 2005; 67: 1556–65. [DOI] [PubMed] [Google Scholar]
- 27. Balzarini J, Van Laethem K, Hatse S, et al. Profile of resistance of human immunodeficiency virus to mannose-specific plant lectins. J Virol 2004; 78: 10617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pashov A, MacLeod S, Saha R, et al. Concanavalin A binding to HIV envelope protein is less sensitive to mutations in glycosylation sites than monoclonal antibody 2G12. Glycobiology 2005; 15: 994–1001. [DOI] [PubMed] [Google Scholar]
- 29. Alexandre KB, Moore PL, Nonyane M, et al. Mechanisms of HIV-1 subtype C resistance to GRFT, CV-N and SVN. Virology 2013; 446: 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu Q, Mahmood N, Shattock RJ. High-mannose-specific deglycosylation of HIV-1 gp120 induced by resistance to cyanovirin-N and the impact on antibody neutralization. Virology 2007; 368: 145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parker AS, Choi Y, Griswold KE, et al. Structure-guided deimmunization of therapeutic proteins. J Comput Biol 2013; 20: 152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Balzarini J, Francois KO, Van Laethem K, et al. Pradimicin S, a highly soluble nonpeptidic small-size carbohydrate-binding antibiotic, is an anti-HIV drug lead for both microbicidal and systemic use. Antimicrob Agents Chemother 2010; 54: 1425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakagawa Y, Ito Y. Carbohydrate-binding molecules with non-peptidic skeletons. Trends Glycosci Glycotechnol 2012; 24: 1–12. [Google Scholar]
- 34. Hoshino H, Seki J, Takeuchi T. New antifungal antibiotics, benanomicins A and B inhibit infection of T-cell with human immunodeficiency virus (HIV) and syncytium formation by HIV. J Antibiot (Tokyo) 1989; 42: 344–6. [DOI] [PubMed] [Google Scholar]
- 35. Van Damme EJ, Broekaert WF, Peumans WJ. The Urtica dioica agglutinin is a complex mixture of isolectins. Plant Physiol 1988; 86: 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Beintema JJ, Peumans WJ. The primary structure of stinging nettle (Urtica dioica) agglutinin. A two-domain member of the hevein family. FEBS Lett 1992; 299: 131–4. [DOI] [PubMed] [Google Scholar]
- 37. Hom K, Gochin M, Peumans WJ, et al. Ligand-induced perturbations in Urtica dioica agglutinin. FEBS Lett 1995; 361: 157–61. [DOI] [PubMed] [Google Scholar]
- 38. Lee RT, Gabius HJ, Lee YC. Thermodynamic parameters of the interaction of Urtica dioica agglutinin with N-acetylglucosamine and its oligomers. Glycoconj J 1998; 15: 649–55. [DOI] [PubMed] [Google Scholar]
- 39. Harata K, Schubert WD, Muraki M. Structure of Urtica dioica agglutinin isolectin I: dimer formation mediated by two zinc ions bound at the sugar-binding site. Acta Crystallogr D Biol Crystallogr 2001; 57: 1513–7. [DOI] [PubMed] [Google Scholar]
- 40. Chrubasik JE, Roufogalis BD, Wagner H, et al. A comprehensive review on the stinging nettle effect and efficacy profiles. Part II: urticae radix. Phytomedicine 2007; 14: 568–79. [DOI] [PubMed] [Google Scholar]
- 41. Balzarini J, Neyts J, Schols D, et al. The mannose-specific plant lectins from Cymbidium hybrid and Epipactis helleborine and the (N-acetylglucosamine)n-specific plant lectin from Urtica dioica are potent and selective inhibitors of human immunodeficiency virus and cytomegalovirus replication in vitro. Antiviral Res 1992; 18: 191–207. [DOI] [PubMed] [Google Scholar]
- 42. Bertaux C, Daelemans D, Meertens L, et al. Entry of hepatitis C virus and human immunodeficiency virus is selectively inhibited by carbohydrate-binding agents but not by polyanions. Virology 2007; 366: 40–50. [DOI] [PubMed] [Google Scholar]
- 43. Chen Y, Peumans WJ, Hause B, et al. Jasmonic acid methyl ester induces the synthesis of a cytoplasmic/nuclear chito-oligosaccharide binding lectin in tobacco leaves. FASEB J 2002; 16: 905–7. [DOI] [PubMed] [Google Scholar]
- 44. Lannoo N, Peumans WJ, Pamel EV, et al. Localization and in vitro binding studies suggest that the cytoplasmic/nuclear tobacco lectin can interact in situ with high-mannose and complex N-glycans. FEBS Lett 2006; 580: 6329–37. [DOI] [PubMed] [Google Scholar]
- 45. Kaku H, Van Damme EJ, Peumans WJ, et al. Carbohydrate-binding specificity of the daffodil (Narcissus pseudonarcissus) and amaryllis (Hippeastrum hybr.) bulb lectins. Arch Biochem Biophys 1990; 279: 298–304. [DOI] [PubMed] [Google Scholar]
- 46. Montefiori DC. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol 2009; 485: 395–405. [DOI] [PubMed] [Google Scholar]
- 47. Ferir G, Hanchen A, Francois KO, et al. Feglymycin, a unique natural bacterial antibiotic peptide, inhibits HIV entry by targeting the viral envelope protein gp120. Virology 2012; 433: 308–19. [DOI] [PubMed] [Google Scholar]
- 48. Tzioumaki N, Tsoukala E, Manta S, et al. Synthesis, antiviral and cytostatic evaluation of unsaturated exomethylene and keto d-lyxopyranonucleoside analogues. Arch Pharm (Weinheim) 2009; 342: 353–60. [DOI] [PubMed] [Google Scholar]
- 49. Manta S, Tzioumaki N, Tsoukala E, et al. Unsaturated dideoxy fluoro-ketopyranosyl nucleosides as new cytostatic agents: a convenient synthesis of 2,6-dideoxy-3-fluoro-4-keto-β-d-glucopyranosyl analogues of uracil, 5-fluorouracil, thymine, N4-benzoyl cytosine and N6-benzoyl adenine. Eur J Med Chem 2009; 44: 4764–71. [DOI] [PubMed] [Google Scholar]
- 50. Alen MM, De Burghgraeve T, Kaptein SJ, et al. Broad antiviral activity of carbohydrate-binding agents against the four serotypes of dengue virus in monocyte-derived dendritic cells. PLoS One 2011; 6: e21658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vermeire K, Princen K, Hatse S, et al. CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. AIDS 2004; 18: 2115–25. [DOI] [PubMed] [Google Scholar]
- 52. Van Breedam W, Pohlmann S, Favoreel HW, et al. Bitter-sweet symphony: glycan-lectin interactions in virus biology. FEMS Microbiol Rev 2014; 38: 598–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Balzarini J. Carbohydrate-binding agents: a potential future cornerstone for the chemotherapy of enveloped viruses? Antivir Chem Chemother 2007; 18: 1–11. [DOI] [PubMed] [Google Scholar]
- 54. Vigerust DJ, Shepherd VL. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol 2007; 15: 211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Balzarini J, Van Laethem K, Daelemans D, et al. Pradimicin A, a carbohydrate-binding nonpeptidic lead compound for treatment of infections with viruses with highly glycosylated envelopes, such as human immunodeficiency virus. J Virol 2007; 81: 362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Balzarini J, Van Laethem K, Peumans WJ, et al. Mutational pathways, resistance profile, and side effects of cyanovirin relative to human immunodeficiency virus type 1 strains with N-glycan deletions in their gp120 envelopes. J Virol 2006; 80: 8411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Witvrouw M, Fikkert V, Hantson A, et al. Resistance of human immunodeficiency virus type 1 to the high-mannose binding agents cyanovirin N and concanavalin A. J Virol 2005; 79: 7777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Travers SA. Conservation, compensation, and evolution of N-linked glycans in the HIV-1 group M subtypes and circulating recombinant forms. ISRN AIDS 2012; 2012: 823605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bonomelli C, Doores KJ, Dunlop DC, et al. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS One 2011; 6: e23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Myers G, MacInnes K, Korber B. The emergence of simian/human immunodeficiency viruses. AIDS Res Hum Retroviruses 1992; 8: 373–86. [DOI] [PubMed] [Google Scholar]
- 61. Raska M, Takahashi K, Czernekova L, et al. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J Biol Chem 2010; 285: 20860–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Raska M, Czernekova L, Moldoveanu Z, et al. Differential glycosylation of envelope gp120 is associated with differential recognition of HIV-1 by virus-specific antibodies and cell infection. AIDS Res Ther 2014; 11: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Doores KJ, Bonomelli C, Harvey DJ, et al. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc Natl Acad Sci USA 2010; 107: 13800–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Drummer HE, Hill MK, Maerz AL, et al. Allosteric modulation of the HIV-1 gp120-gp41 association site by adjacent gp120 variable region 1 (V1) N-glycans linked to neutralization sensitivity. PLoS Pathog 2013; 9: e1003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sun J, Barbeau B, Sato S, et al. Syncytium formation and HIV-1 replication are both accentuated by purified influenza and virus-associated neuraminidase. J Biol Chem 2002; 277: 9825–33. [DOI] [PubMed] [Google Scholar]
- 66. Pastey MK, Crowe JE, Jr, Graham BS. RhoA interacts with the fusion glycoprotein of respiratory syncytial virus and facilitates virus-induced syncytium formation. J Virol 1999; 73: 7262–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang E, Arvin AM, Oliver SL. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin pathogenesis. PLoS Pathog 2014; 10: e1004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ida H, Kurata A, Eguchi K, et al. Mechanism of inhibitory effect of dextran sulfate and heparin on human T-cell lymphotropic virus type I (HTLV-I)-induced syncytium formation in vitro: role of cell-to-cell contact. Antiviral Res 1994; 23: 143–59. [DOI] [PubMed] [Google Scholar]
- 69. Matthews T, Salgo M, Greenberg M, et al. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov 2004; 3: 215–25. [DOI] [PubMed] [Google Scholar]
- 70. Koppel EA, van Gisbergen KP, Geijtenbeek TB, et al. Distinct functions of DC-SIGN and its homologues L-SIGN (DC-SIGNR) and mSIGNR1 in pathogen recognition and immune regulation. Cell Microbiol 2005; 7: 157–65. [DOI] [PubMed] [Google Scholar]
- 71. Tassaneetrithep B, Burgess TH, Granelli-Piperno A, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med 2003; 197: 823–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Halary F, Amara A, Lortat-Jacob H, et al. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity 2002; 17: 653–64. [DOI] [PubMed] [Google Scholar]
- 73. Lozach PY, Amara A, Bartosch B, et al. C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J Biol Chem 2004; 279: 32035–45. [DOI] [PubMed] [Google Scholar]
- 74. Alvarez CP, Lasala F, Carrillo J, et al. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J Virol 2002; 76: 6841–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feinberg H, Mitchell DA, Drickamer K, et al. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 2001; 294: 2163–6. [DOI] [PubMed] [Google Scholar]
- 76. van Liempt E, Bank CM, Mehta P, et al. Specificity of DC-SIGN for mannose- and fucose-containing glycans. FEBS Lett 2006; 580: 6123–31. [DOI] [PubMed] [Google Scholar]
- 77. Stefanowicz K, Lannoo N, Proost P, et al. Arabidopsis F-box protein containing a Nictaba-related lectin domain interacts with N-acetyllactosamine structures. FEBS Open Bio 2012; 2: 151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Scanlan CN, Pantophlet R, Wormald MR, et al. The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of α1→2 mannose residues on the outer face of gp120. J Virol 2002; 76: 7306–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Trkola A, Purtscher M, Muster T, et al. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol 1996; 70: 1100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Calarese DA, Lee HK, Huang CY, et al. Dissection of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proc Natl Acad Sci USA 2005; 102: 13372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Trkola A, Kuster H, Rusert P, et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med 2005; 11: 615–22. [DOI] [PubMed] [Google Scholar]
- 82. Kouokam JC, Huskens D, Schols D, et al. Investigation of griffithsin's interactions with human cells confirms its outstanding safety and efficacy profile as a microbicide candidate. PLoS One 2011; 6: e22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Balzarini J, Hatse S, Vermeire K, et al. Mannose-specific plant lectins from the Amaryllidaceae family qualify as efficient microbicides for prevention of human immunodeficiency virus infection. Antimicrob Agents Chemother 2004; 48: 3858–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Horiya S, MacPherson IS, Krauss IJ. Recent strategies targeting HIV glycans in vaccine design. Nat Chem Biol 2014; 10: 990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Shingai M, Donau OK, Plishka RJ, et al. Passive transfer of modest titers of potent and broadly neutralizing anti-HIV monoclonal antibodies block SHIV infection in macaques. J Exp Med 2014; 211: 2061–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Burton DR, Poignard P, Stanfield RL, et al. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 2012; 337: 183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kwong PD, Mascola JR. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity 2012; 37: 412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Klein F, Mouquet H, Dosenovic P, et al. Antibodies in HIV-1 vaccine development and therapy. Science 2013; 341: 1199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Koharudin LM, Gronenborn AM. Antiviral lectins as potential HIV microbicides. Curr Opin Virol 2014; 7: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]