

Abstract
Motivation: Owing to its importance in both basic research (such as molecular evolution and protein attribute prediction) and practical application (such as timely modeling the 3D structures of proteins targeted for drug development), protein remote homology detection has attracted a great deal of interest. It is intriguing to note that the profile-based approach is promising and holds high potential in this regard. To further improve protein remote homology detection, a key step is how to find an optimal means to extract the evolutionary information into the profiles.
Results: Here, we propose a novel approach, the so-called profile-based protein representation, to extract the evolutionary information via the frequency profiles. The latter can be calculated from the multiple sequence alignments generated by PSI-BLAST. Three top performing sequence-based kernels (SVM-Ngram, SVM-pairwise and SVM-LA) were combined with the profile-based protein representation. Various tests were conducted on a SCOP benchmark dataset that contains 54 families and 23 superfamilies. The results showed that the new approach is promising, and can obviously improve the performance of the three kernels. Furthermore, our approach can also provide useful insights for studying the features of proteins in various families. It has not escaped our notice that the current approach can be easily combined with the existing sequence-based methods so as to improve their performance as well.
Availability and implementation: For users’ convenience, the source code of generating the profile-based proteins and the multiple kernel learning was also provided at
http://bioinformatics.hitsz.edu.cn/main/∼binliu/remote/
Contact: bliu@insun.hit.edu.cn or bliu@gordonlifescience.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
By March 2013, 89 003 experimentally determined protein structures were deposited in the Protein Data Bank (Berman et al., 2007). However, this number is only about one-sixth of 539 616, the number of protein sequences held in the UniProtKB/Swiss-Prot database (Wu et al., 2006). To timely use such vast amount of structure-unknown protein sequences for basic research and drug development, it is highly desired to determine their 3D structures and functions by means of homology approaches (Chou, 2004). Unfortunately, protein remote homology detection is still a challenging problem in bioinformatics.
The early methods in dealing with this problem were based on the pairwise sequence comparison approaches, such as BLAST (Altschul et al., 1990) and Smith–Waterman local alignment algorithm (Smith and Waterman, 1981). However, in many cases, this kind of sequence alignment method failed to detect remote homologies due to the low sequence similarities. Later methods to challenge this problem were based on the generative models to induce a probability distribution over the protein family, and then to generate the unknown proteins as new members of the family from the stochastic model. For example, the hidden Markov model (HMM) (Karplus et al., 1998) can be trained iteratively in a semi-supervised manner by using both positively labeled and unlabeled samples of a particular family to generate the positive set (Qian and Goldstein, 2004).
Recently, the discriminative methods, such as support vector machine (SVM) (Vapnik, 1998), were used to address this problem by focusing on the differences between protein families. The key of the SVM methods is the kernel function by which to compute the inner product between two samples in the feature space. The most straightforward approach to generate the kernels was based on the features extracted from protein sequences. SVM-Ngram (Dong et al., 2006), SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004) were three of the most successful sequence-based kernels. SVM-Ngram (Dong et al., 2006) was based on the feature space that contains all short subsequence of length N. In SVM-pairwise (Liao and Noble, 2003), a protein sequence was represented as a vector of pairwise similarities to all protein sequences in the training set, and then inner product between these vector-space representations was taken as the kernel. SVM-LA (Saigo et al., 2004) measured the similarity between a pair of proteins by taking all the optimal local alignment scores with gaps between all possible subsequences into account. Besides these kernels, several other sequence-based kernels were also proposed, such as Mismatch (Leslie et al., 2004) and SVM-BALSA (Webb-Robertson et al., 2005). The profile-based kernels could further improve the performance by using the evolutional information extracted from the profiles. For example, Top-n-grams (Liu et al., 2008) extracted the profile-based patterns by considering the most frequent elements in the profiles; profile kernel (Kuang et al., 2005) extracted the short substrings according to the profile-based ungapped alignment scores; some profile-based methods improved the predictive performance by developing more sensitive profiles. HHsearch method (Söding, 2005) was based on a novel profile using the HMM. In COMPASS (Sadreyev et al., 2009), numerical profiles were generated to construct optimal profile–profile alignments and to estimate the statistical significance of the corresponding alignment scores.
In the meantime, some other features and techniques have been applied to this field to further improve the predictive performance. For instance, the kernel combination methodology (VBKC) (Damoulas and Girolami, 2008) used a single multi-class kernel machine to combine various kernels based on different feature spaces; SVM-physicochemical distance transformation (PDT) (Liu et al., 2012) combined the amino acid physicochemical properties and the profile features via PDT to incorporate the local sequence-order information of the entire protein sequences. Also, based on the similarities between protein sequences and natural languages, the natural language processing techniques were applied to this field. It was shown that the performance of building-block-based methods could be improved by using the latent semantic analysis (LSA) (Dong et al., 2006). Moreover, PROTEMBED (Melvin et al., 2011) detected protein remote homology by embedding protein sequences into a low-dimensional semantic space.
As we can see from the aforementioned introduction, most of the top-performing methods were developed based on the features extracted from profiles. This is consistent with the fact that a profile is much richer than an individual sequence in encoding information. Also, biology is a natural science with historic dimension. All biological species have developed beginning from a limited number of ancestral species. It is true for protein sequence as well (Chou, 2004). Their evolution involves changes of single residues, insertions and deletions of several residues, gene doubling and gene fusion (Chou, 1995). With these changes accumulated for a long period, many similarities between initial and resultant amino acid sequences are gradually eliminated, but the corresponding proteins may still share many common features, such as having basically the same biological function (Loewenstein et al., 2009), folding topology, subcellular location and other attributes (Chou, 2013).
Accordingly, the key to improve the performance of these methods is to find a suitable approach to extract the evolutionary information from the profiles. In view of this, the current study was initiated in an attempt to propose a profile-based protein representation by extracting the evolutionary information from the frequency profiles.
2 MATERIALS AND METHODS
As shown by a series of publications (Chen et al., 2013; Liu et al., 2009; Xiao et al., 2013; Xu et al., 2013) and summarized in a comprehensive review (Chou, 2011), to develop a useful statistical prediction method or model for a biological system, one needs to engage the following procedures: (i) construct or select a valid benchmark dataset to train and test the predictor; (ii) formulate the samples with an effective mathematical expression that can truly reflect their intrinsic correlation with the target to be predicted; (iii) introduce or develop a powerful algorithm (or engine) to operate the prediction; (iv) properly perform cross-validation tests to objectively evaluate the anticipated accuracy of the predictor; and (v) provide the downloadable source code or a web-server for the prediction method. Below, let us describe how to deal these procedures.
2.1 SCOP benchmark
Suppose 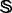 is a remote homology system investigated in the current study that contains 4352 protein sequences, which were taken from (Liao and Noble, 2003) at http://noble.gs.washington.edu/proj/svm-pairwise/. These proteins were selected from SCOP version 1.53 and extracted from the Astral database (Brenner et al., 2000). None of the 4352 proteins has sequence pairwise similarity to any other with higher than
is a remote homology system investigated in the current study that contains 4352 protein sequences, which were taken from (Liao and Noble, 2003) at http://noble.gs.washington.edu/proj/svm-pairwise/. These proteins were selected from SCOP version 1.53 and extracted from the Astral database (Brenner et al., 2000). None of the 4352 proteins has sequence pairwise similarity to any other with higher than 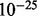 in the E-value [for more about the E-value and its implication in reducing homology bias and redundancy, see (Brenner et al., 2000)]. These proteins were also used by others (Dong et al., 2006; Lingner and Meinicke, 2006; Saigo et al., 2004) to study remote homology detection. The 4352 proteins in
in the E-value [for more about the E-value and its implication in reducing homology bias and redundancy, see (Brenner et al., 2000)]. These proteins were also used by others (Dong et al., 2006; Lingner and Meinicke, 2006; Saigo et al., 2004) to study remote homology detection. The 4352 proteins in 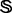 can be classified into 853 superfamilies and 1356 families; i.e.
can be classified into 853 superfamilies and 1356 families; i.e.
| (1) |
where 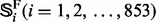 is the
is the 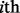 superfamily,
superfamily, 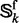
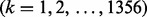 is the
is the 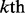 family and the symbol
family and the symbol  represents the ‘union’ in the set theory. For readers’ convenience, the codes of the 4352 proteins and their sequence as well as the attributes of their families and superfamilies are given in Supplementary Material S1.
represents the ‘union’ in the set theory. For readers’ convenience, the codes of the 4352 proteins and their sequence as well as the attributes of their families and superfamilies are given in Supplementary Material S1.
Because some families and superfamilies in 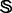 do not contain significant number of protein sequences, and also because the negative dataset for each protein family can be any proteins except those belonging to its own superfamily, it is not so straightforward but a little more complicated and subtle for how to select protein samples from
do not contain significant number of protein sequences, and also because the negative dataset for each protein family can be any proteins except those belonging to its own superfamily, it is not so straightforward but a little more complicated and subtle for how to select protein samples from 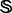 to define the training and testing datasets. To provide a clear description, let us consider a different manner to address this. As demonstrated by many previous studies on a series of important biological topics, such as enzyme-catalyzed reactions (Zhou and Deng, 1984), inhibition of human immunodeficiency virus-1 reverse transcriptase (Althaus et al., 1993), drug metabolism systems (Chou, 2010) and applying wenxiang diagram or graph (Chou et al., 2011) to study protein–protein interactions (Zhou, 2011; Zhou and Huang, 2013), using graphical approaches to study complicated problems can provide an intuitive picture and useful insights for in-depth studying and analyzing various complicated relations in these systems (Lin and Lapointe, 2013). In view of this, let us also use graphic approach to describe the feature and relation of the families and superfamilies in
to define the training and testing datasets. To provide a clear description, let us consider a different manner to address this. As demonstrated by many previous studies on a series of important biological topics, such as enzyme-catalyzed reactions (Zhou and Deng, 1984), inhibition of human immunodeficiency virus-1 reverse transcriptase (Althaus et al., 1993), drug metabolism systems (Chou, 2010) and applying wenxiang diagram or graph (Chou et al., 2011) to study protein–protein interactions (Zhou, 2011; Zhou and Huang, 2013), using graphical approaches to study complicated problems can provide an intuitive picture and useful insights for in-depth studying and analyzing various complicated relations in these systems (Lin and Lapointe, 2013). In view of this, let us also use graphic approach to describe the feature and relation of the families and superfamilies in 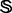 , as shown in Figure 1, where the open circles denote the families or superfamilies that have significant number of protein sequences and the gray circles denote those that do not.
, as shown in Figure 1, where the open circles denote the families or superfamilies that have significant number of protein sequences and the gray circles denote those that do not.
Fig. 1.

A tree (or 3-row) graph to show the remote homology system on the SCOP benchmark. Only the open circles are in the target of the 23 superfamilies and 54 families, while the circles in gray are outside of the target. See the text for further explanation
Of the 1356 families in 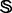 (cf. Equation 1), 54 contain significant number of proteins (see the third row of Fig. 1) and form a target family set
(cf. Equation 1), 54 contain significant number of proteins (see the third row of Fig. 1) and form a target family set 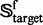 ; i.e.
; i.e.
| (2) |
Of the 853 superfamilies in 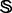 , 23 contain at least one target family (see the open circles in the second row of Fig. 1) and form a target superfamily set
, 23 contain at least one target family (see the open circles in the second row of Fig. 1) and form a target superfamily set 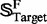 ; i.e.
; i.e.
| (3) |
Thus, we have
| (4) |
meaning that 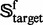 is the subset of
is the subset of 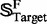 , and
, and 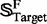 is the subset of
is the subset of 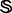 ; each of the three contains 857, 1508 and 4352 proteins, respectively.
; each of the three contains 857, 1508 and 4352 proteins, respectively.
Now, for each of the 54 families in the target family set 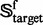 , we can define a training dataset and testing dataset given by
, we can define a training dataset and testing dataset given by
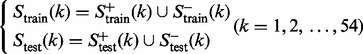 |
(5) |
where the positive training dataset 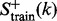 contains at least 10 of its superfamily members, none of which belongs to the
contains at least 10 of its superfamily members, none of which belongs to the 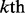 family, and the positive testing dataset
family, and the positive testing dataset 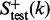 contains at least five protein domains within the family. The proteins in the negative training and testing datasets,
contains at least five protein domains within the family. The proteins in the negative training and testing datasets, 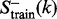 and
and 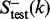 , were picked from
, were picked from 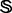 by excluding the superfamily of the
by excluding the superfamily of the 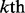 family and randomly split between the two in the same ratio as the positive ones. The 54 training and testing datasets thus obtained are given in the Supplementary Materials S2 and Supplementary Data, respectively.
family and randomly split between the two in the same ratio as the positive ones. The 54 training and testing datasets thus obtained are given in the Supplementary Materials S2 and Supplementary Data, respectively.
2.2 Protein frequency profile
The frequency profile 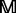 for protein
for protein 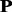 with L amino acids can be represented by
with L amino acids can be represented by
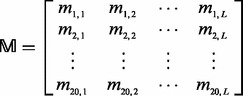 |
(6) |
where 20 is the total number of standard amino acids; mi,j (0 ≤ mi,j ≤ 1) is the target frequency, which reflects the probability of amino acid i (i = 1,2, … ,20) occurring at the sequence position j (j = 1,2, … ,L) in protein 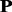 during evolutionary processes. For each column in
during evolutionary processes. For each column in 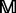 , the elements add up to 1.
, the elements add up to 1.
The target frequency is calculated from the multiple sequence alignments generated by running PSI-BLAST (Altschul et al., 1997) against the NCBI’s NR with default parameters except that the number of iterations was not set at 1 but was set at 10 in the current study. The target frequency of amino acid i in sequence position j is calculated as:
| (7) |
where fij is the observed frequency of amino acid i in column j; β is a free parameter set to a constant value of 10, which is initially used by PSI-BLAST; α is the number of different amino acids in column j − 1; and gij is the pseudo-count for amino acid i in protein sequence position j, which can be calculated as:
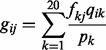 |
(8) |
where pk is the background frequency of amino acid k, and qik is the score of amino acid i being aligned to amino acid k in BLOSUM62 substitution matrix, which is the default score matrix of PSI-BLAST (Altschul et al., 1997).
2.3 Profile-based protein representation
Although the methods by using amino acid sequence information achieve certain degree of success, only using sequence information cannot accurately detect protein remote homology. Recent studies demonstrated that the profile-based methods would show better performance because a profile is richer than an individual sequence as far as the encoding information is concerned. However, a profile is a 2D matrix, whereas a protein sequence is an amino acid string. Therefore, the 2D evolutionary profile information cannot be directly incorporated into the sequence-based methods for prediction. To deal with this problem, we propose an approach to convert the frequency profiles into a series of profile-based proteins. Thus, the existing sequence-based methods can be directly performed on these proteins for the prediction. The target frequencies in the frequency profiles reflect the probabilities of the corresponding amino acids appearing in the specific sequence positions. The higher the frequency is, the more likely the corresponding amino acid occurs. It is reasonable to use the nth most frequent amino acids in the frequency profiles to represent the protein sequences. Below is the description on how to convert frequency profiles into profile-based proteins.
Given the frequency profile 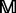 for protein
for protein 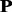 (Equation 6), we can sort the amino acids in each column according to a descending order. The frequency profile thus obtained by the sorting operation is called the sorted frequency profile and denoted by
(Equation 6), we can sort the amino acids in each column according to a descending order. The frequency profile thus obtained by the sorting operation is called the sorted frequency profile and denoted by 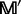 . For each row in
. For each row in 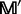 , the amino acids are combined to produce the profile-based protein. By following this approach, the frequency profile
, the amino acids are combined to produce the profile-based protein. By following this approach, the frequency profile 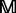 is converted into 20 profile-based proteins p1, p2, … , p20 (Supplementary Fig. S1 in Supplementary Material S4), which contain the evolutionary information in the frequency profile. These 20 proteins have different importance. During evolutionary process, protein
is converted into 20 profile-based proteins p1, p2, … , p20 (Supplementary Fig. S1 in Supplementary Material S4), which contain the evolutionary information in the frequency profile. These 20 proteins have different importance. During evolutionary process, protein 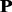 is preferred to transform into p1, but not preferred to transform into p20. For reader’s convenience, the source code for generating the profile-based proteins is accessible by clicking the link at http://bioinformatics.hitsz.edu.cn/main/∼binliu/remote/.
is preferred to transform into p1, but not preferred to transform into p20. For reader’s convenience, the source code for generating the profile-based proteins is accessible by clicking the link at http://bioinformatics.hitsz.edu.cn/main/∼binliu/remote/.
2.4 Sequence-based kernels
Three state-of-the-art sequence-based kernels [SVM-Ngram (Dong et al., 2006), SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004)] were used to validate whether the proposed approach could improve their performance.
In SVM-Ngram (Dong et al., 2006) method, the Ngrams were the set of all possible subsequences of amino acids of a fixed length. A protein sequence was mapped to a feature vector by the occurrence frequency of each Ngram. The value of N was set at 3 as suggested by the authors (Dong et al., 2006), and therefore the dimension of the vector is 8000. At the heart of the SVM is a kernel function that acts as a similarity score between pairs of vectors. The kernel was normalized so that each vector had length 1 in the feature space:
| (9) |
where X and Y are two proteins in the dataset. This normalized step was also used by SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004). The normalized kernel thus obtained was then transformed into a radial basis kernel.
In the SVM-pairwise (Liao and Noble, 2003) method, the feature vector was a list of pairwise sequence similarity scores, computed with respect to all of the sequences in the training set. The radial basis function was used as the kernel. The rest steps were the same as the ones used in SVM-Ngram (Dong et al., 2006).
In the SVM-LA (Saigo et al., 2004), the kernel was calculated by summing up scores obtained from the local alignments with gaps between the two sequences, computed by Smith–Waterman dynamic programming algorithm. Such kernel might not be a positive definite kernel and the authors (Saigo et al., 2004) provided two solutions for this problem. Owing to its performance and simplicity, we implemented one of the two methods, namely, the LA-ekm kernel. The parameters of LA-ekm kernel took the optimal values (β = 0.5, d = −11, e = −4).
2.5 Multiple kernel learning
The kernel described in the previous section can be used by kernel methods to train the SVM classifier. Each kernel contains different discriminative information, and therefore combining the kernels automatically is a promising way to improve the performance. In machine learning field, this approach is called multiple kernel learning (MKL) (Cortes et al., 2010; Varma and Babu, 2009), which has attracted a lot of attention recently. The MKL technique aimed to combine different kernels to improve the performance, and showed the state-of-the-art results on image classification field (Varma and Babu, 2009). In this article, we focused on the weighted linear combination of kernels. The weight of each kernel can be optimized based on different criterion, which can be categorized by two groups. One group is the one-stage kernel learning methods, which optimize the weight and the SVM objective function simultaneously (Varma and Babu, 2009). These methods suffer from the high training complexity. The other group is two-stage kernel learning methods, which optimize the weight by using a criterion first and then train the SVM classifier using the kernel combined by the learned weight of each kernel. Compared with one-stage learning methods, the two-stage kernel learning methods showed better performance with reduced training cost. Therefore, in this study, we adopted the two-stage kernel learning method. Specifically, the kernel target alignment (KTA) objective function was used to optimize the weight of each kernel, which showed theoretical guarantees and can improve the performance in practice (Cortes et al., 2010; Varma and Babu, 2009).
Given m training samples x1, x2, … , xm and their corresponding labels y1, y2, … , ym, the ideal kernel matrix can be formulated as Ky = yTy, where y is the vector of labels [y1, y2, … , ym]. For the given n kernels K1, K2, … Kn, the aim is to learn the weight of each kernel. To avoid the kernel scaling problem, we center kernel Kk and the corresponding ideal kernel Ky in feature space by the following equation:
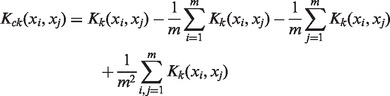 |
(10) |
where Kck is normalized by:
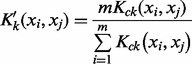 |
(11) |
Following the above steps, each kernel is normalized, and then these n kernels are linearly combined by the following equation:
| (12) |
where wk (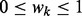 ,
, 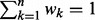 ) is the weight of kernel
) is the weight of kernel 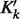 . The weight is learned by KTA objective function, which maximizes the alignment between
. The weight is learned by KTA objective function, which maximizes the alignment between 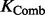 and the centered ideal kernel
and the centered ideal kernel 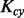 .
.
| (13) |
This leads to a quadratic program problem and can be solved quite efficiently. For implementation details, please refer to (Cortes et al., 2010).
In this study, three kernels (Kp, Kp1 and Kp2) for each selected sequence-based method were linearly combined by using the above KTA approach to further improve the performance. For reader’s convenience, the source code of the MKL is accessible by clicking the link at http://bioinformatics.hitsz.edu.cn/main/∼binliu/remote/.
2.6 SVM
SVM is a class of supervised learning algorithms first introduced by Vapnik (1998). SVM-based machine learning algorithm has been successfully used to investigate various problems in molecular biology, such as identifying DNA recombination spots (Chen et al., 2013), membrane protein types (Cai et al., 2003) and heat-shock protein functions (Feng et al., 2013), among many others. In this study, the publicly available Gist SVM package (http://www.chibi.ubc.ca/gist/) was used.
2.7 Evaluation methodology
Because the test sets have many more negative than positive samples, simply measuring error-rates will not give a good evaluation of performance. For the cases in which the positive and negative samples are not evenly distributed, the best way to evaluate the trade-off between the specificity and sensitivity is to use a receiver operating characteristic (ROC) score (Gribskov and Robinson, 1996). An ROC score is the normalized area under a curve that plots true positives against false positives for different classification thresholds. A score of 1 means perfect separation of positive samples from negative ones, whereas a score of 0 means that none of the sequences selected by the algorithm is positive. Another performance measure is ROC50 score, which is the area under the ROC curve up to the first 50 false positives.
3 RESULTS AND DISCUSSION
3.1 Profile-based protein representation can improve the performance of methods based on sequence composition
The frequency profile of a protein 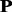 can be converted into 20 profile-based proteins (p1, p2, … , p20) by using the proposed approach (see Section 2 for details). These 20 proteins have different importance. p1 is the most important protein, as it is the combination of the top frequent amino acids in frequency profile, whereas p20 is the profile-based protein to which protein
can be converted into 20 profile-based proteins (p1, p2, … , p20) by using the proposed approach (see Section 2 for details). These 20 proteins have different importance. p1 is the most important protein, as it is the combination of the top frequent amino acids in frequency profile, whereas p20 is the profile-based protein to which protein 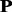 is the most unlikely to convert because it is the combination of the amino acids with lowest frequencies in frequency profile. If all the 20 profile-based proteins are used in the prediction, the computational cost is relatively high. In this study, only the top n most important profile-based proteins (p1, … , pn) were used in the prediction. To select the value of n, the following experiment was conducted. The frequencies of 20 standard amino acids in each column of a frequency profiles add up to 1. Therefore, the average frequency is 0.05 (1/20 = 0.05). If an amino acid with frequency >0.05, it is likely to occur during evolutionary process; otherwise, it is not likely to occur. The percentage of the amino acids with frequencies >0.05 in each profile-based protein on the SCOP benchmark was calculated, and the results are shown in Figure 2. As we can see from the figure, such amino acids are abundant in profile-based proteins p1, p2 and p3 (99.99%, 99.60% and 98.13%, respectively), but for the other 17 profile-based proteins, the percentage decreases significantly (from 89.28 to 0%). Therefore, in this study, only the top three profile-based proteins were used in the prediction. These profile-based proteins were combined with three state-of-the-art methods based on sequence composition, including SVM-Ngram (Dong et al., 2006), SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004), and the results are shown in Supplementary Table S1 of Supplementary Material S4. For each of the three methods, the best performance was achieved for the top important protein p1. Compared with the methods performed on the raw protein sequence
is the most unlikely to convert because it is the combination of the amino acids with lowest frequencies in frequency profile. If all the 20 profile-based proteins are used in the prediction, the computational cost is relatively high. In this study, only the top n most important profile-based proteins (p1, … , pn) were used in the prediction. To select the value of n, the following experiment was conducted. The frequencies of 20 standard amino acids in each column of a frequency profiles add up to 1. Therefore, the average frequency is 0.05 (1/20 = 0.05). If an amino acid with frequency >0.05, it is likely to occur during evolutionary process; otherwise, it is not likely to occur. The percentage of the amino acids with frequencies >0.05 in each profile-based protein on the SCOP benchmark was calculated, and the results are shown in Figure 2. As we can see from the figure, such amino acids are abundant in profile-based proteins p1, p2 and p3 (99.99%, 99.60% and 98.13%, respectively), but for the other 17 profile-based proteins, the percentage decreases significantly (from 89.28 to 0%). Therefore, in this study, only the top three profile-based proteins were used in the prediction. These profile-based proteins were combined with three state-of-the-art methods based on sequence composition, including SVM-Ngram (Dong et al., 2006), SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004), and the results are shown in Supplementary Table S1 of Supplementary Material S4. For each of the three methods, the best performance was achieved for the top important protein p1. Compared with the methods performed on the raw protein sequence 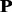 , the performance of the proposed methods can be improved by 3.7∼7.5% and 9.6∼13.7% in terms of average ROC and ROC50 scores, respectively, indicating that the proposed profile-based protein representation is useful for protein remote homology detection. The performance of the methods performed on p2 is similar as that of the methods performed on the raw protein
, the performance of the proposed methods can be improved by 3.7∼7.5% and 9.6∼13.7% in terms of average ROC and ROC50 scores, respectively, indicating that the proposed profile-based protein representation is useful for protein remote homology detection. The performance of the methods performed on p2 is similar as that of the methods performed on the raw protein 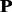 . The predictive results of the methods performed on p3 were the lowest. These results are consistent with the different importance of the three profile-based proteins p1, p2 and p3.
. The predictive results of the methods performed on p3 were the lowest. These results are consistent with the different importance of the three profile-based proteins p1, p2 and p3.
Fig. 2.

Illustration to show the feature of frequency profile. Percentage of amino acids with frequencies > 0.05 in the 20 profile-based proteins derived from SCOP benchmark
3.2 Comparison with closely related methods
Besides the current method, there are some other methods for predicting protein remote homologies based on profiles, such as SVM-Top-n-gram-combine-LSA (Liu et al., 2008), SVM-PDT-Profile (Liu et al., 2012), Profile (Kuang et al., 2005), BioSVM-2L (Muda et al., 2011) and HHSearch (Söding, 2005). SVM-Top-n-gram-combine-LSA (Liu et al., 2008) extracted the building blocks of proteins from the frequency profiles, which could be treated as the ‘words’ of protein language. The LSA (Dong et al., 2006) was applied to further improve the performance of this method. SVM-PDT-Profile (Liu et al., 2012) combined the amino acid physicochemical properties in the Amino Acid Index (AAIndex) (Kawashima et al., 2008) with the frequency profiles for the prediction. The feature vector of Profile method (Kuang et al., 2005) was constructed by the short subsequences whose PSSM-based ungapped alignment score was above a predefined threshold. BioSVM-2L constructed two-layer SVM classifiers with profile-based kernels (Muda et al., 2011). All the above three methods were based on SVM, and the difference among them was in the extracted features. HHSearch (Söding, 2005) was one of the best protein remote homology detection methods, which used a novel profile-based HMM. The results obtained by these four methods on the SCOP benchmark are listed in Supplementary Table S1 of Supplementary Material S4, from which we can see that the current method outperforms SVM-Top-n-gram-combine-LSA (Liu et al., 2008), SVM-PDT-Profile (Liu et al., 2012) and BioSVM-2L (Muda et al., 2011) and is highly comparable with Profile (Kuang et al., 2005) and HHSearch (Söding, 2005), indicating that the profile-based protein representation is a promising approach to extract the evolutionary information from frequency profiles for protein remote homology detection.
3.3 Combining different methods via MKL
As mentioned above, the approaches based on the top two profile-based proteins p1, p2 and the raw protein 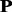 are among the top performing methods. It is interesting to investigate whether these methods can be combined to further improve the performance. In this study, the MKL framework was used to combine these methods. The KTA method was used to automatically optimize the weight of each kernel on the training set, and then these kernels are combined with weights into a single kernel for the SVM-based prediction. The results are shown in Supplementary Table S2 of Supplementary Material S4 as well as Supplementary Materials S5–S7. The MKL approach can improve the performance of SVM-Ngram (Dong et al., 2006), but only has minor impact on the SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004). To uncover the reason, the weight of each kernel was analyzed. For each kernel, the average weight on all the 54 protein families is shown in Supplementary Table S2 of Supplementary Material S4. For these three methods, the p1-based kernel was weighted most heavily. For SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004), the weight values of their corresponding
are among the top performing methods. It is interesting to investigate whether these methods can be combined to further improve the performance. In this study, the MKL framework was used to combine these methods. The KTA method was used to automatically optimize the weight of each kernel on the training set, and then these kernels are combined with weights into a single kernel for the SVM-based prediction. The results are shown in Supplementary Table S2 of Supplementary Material S4 as well as Supplementary Materials S5–S7. The MKL approach can improve the performance of SVM-Ngram (Dong et al., 2006), but only has minor impact on the SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004). To uncover the reason, the weight of each kernel was analyzed. For each kernel, the average weight on all the 54 protein families is shown in Supplementary Table S2 of Supplementary Material S4. For these three methods, the p1-based kernel was weighted most heavily. For SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004), the weight values of their corresponding 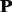 and p2 kernels are <0.1, indicating these kernels only have minor impact on the final results, and hence the performance improvement is modest. VBKC (Damoulas and Girolami, 2008) is another method based on the MKL, which combined four string kernels: SVM-pairwise (Liao and Noble, 2003), SVM-LA (Saigo et al., 2004), SVM-MM (Leslie et al., 2004) and SVM-Mono (Lingner and Meinicke, 2006). Our proposed SVM-pairwise-KTA and SVM-LA-KTA outperform VBKC (Damoulas and Girolami, 2008) by 1.2 ∼ 2.2% and 29.9 ∼ 31.3% according to the average ROC and ROC50 scores, respectively. The obvious performance improvement is mainly due to the proposed profile-based protein representation and MKL approach.
and p2 kernels are <0.1, indicating these kernels only have minor impact on the final results, and hence the performance improvement is modest. VBKC (Damoulas and Girolami, 2008) is another method based on the MKL, which combined four string kernels: SVM-pairwise (Liao and Noble, 2003), SVM-LA (Saigo et al., 2004), SVM-MM (Leslie et al., 2004) and SVM-Mono (Lingner and Meinicke, 2006). Our proposed SVM-pairwise-KTA and SVM-LA-KTA outperform VBKC (Damoulas and Girolami, 2008) by 1.2 ∼ 2.2% and 29.9 ∼ 31.3% according to the average ROC and ROC50 scores, respectively. The obvious performance improvement is mainly due to the proposed profile-based protein representation and MKL approach.
3.4 Correlations between discriminative power of Ngrams and protein families
The SVM-Ngram (Dong et al., 2006) method is based on the explicit feature space representation, which provides the possibility to measure the correlations between Ngrams and protein families. The sequence-specific weight learnt from the SVM training process can be used to calculate the discriminant weight for each Ngram, which indicates the importance of the corresponding Ngram. By following Lingner and Meinicke’s approach (Lingner and Meinicke, 2008), given the weight vector of a set of M sequences obtained from the kernel-based training process α = [α1, α2, α3, … , αM], the discriminant weight vector w in the feature space can be calculated by the following equation:
| (14) |
where F is the matrix of sequence representatives. The magnitude of the element in w represents the discriminative power of the corresponding feature.
In most protein families, kernel p1 is weighted more heavily than kernel 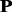 and kernel p1. Two such protein families (SCOP ID: 2.1.1.4 and 3.2.1.5) were selected from the SCOP benchmark for further study, and the results are shown in Supplementary Tables S3 and Supplementary Data of Supplementary Material S4, respectively. For each kernel, the top 10 most discriminative Ngram features calculated by Equation 14 are shown in the tables too. For protein family 2.1.1.4, kernel
and kernel p1. Two such protein families (SCOP ID: 2.1.1.4 and 3.2.1.5) were selected from the SCOP benchmark for further study, and the results are shown in Supplementary Tables S3 and Supplementary Data of Supplementary Material S4, respectively. For each kernel, the top 10 most discriminative Ngram features calculated by Equation 14 are shown in the tables too. For protein family 2.1.1.4, kernel 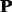 and kernel p1 share some common most discriminative Ngrams, such as ‘mtm’, ‘yty’, ‘mtf’ and ‘wwf’, indicating these Ngrams remain stable during evolutionary process and therefore these Ngrams would be the important sequence patterns for maintaining the structure and function of this protein family (Supplementary Table S3 of Supplementary Material S4). However, there are a few common most discriminative Ngrams between kernel
and kernel p1 share some common most discriminative Ngrams, such as ‘mtm’, ‘yty’, ‘mtf’ and ‘wwf’, indicating these Ngrams remain stable during evolutionary process and therefore these Ngrams would be the important sequence patterns for maintaining the structure and function of this protein family (Supplementary Table S3 of Supplementary Material S4). However, there are a few common most discriminative Ngrams between kernel 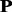 and kernel p1 in protein family 3.2.1.5. The top 10 most discriminative Ngrams of kernel p1 are all different from those in kernel
and kernel p1 in protein family 3.2.1.5. The top 10 most discriminative Ngrams of kernel p1 are all different from those in kernel 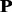 (Supplementary Table S4 of Supplementary Material S4). These Ngrams would contribute to the higher discriminative power of kernel p1 for this protein family.
(Supplementary Table S4 of Supplementary Material S4). These Ngrams would contribute to the higher discriminative power of kernel p1 for this protein family.
Although in most cases, kernel p1 was weighted most heavily, some exceptions were observed. For example, for protein family 7.3.6.1, kernel p2 is the most discriminative kernel with weight value of nearly 1, while the other two kernels only have little contribution to the MKL (Supplementary Table S5 of Supplementary Material S4). The top 10 most discriminative Ngrams for each kernel were investigated, and the results are shown in Supplementary Table S5 of Supplementary Material S4, from which some interesting patterns can be observed. The Ngrams containing amino acids ‘n’ and ‘f’ tended to show strong discriminative power in both kernel 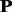 and kernel p1, whereas amino acid ‘a’ was abundant in the top discriminative Ngrams in kernel p2, indicating the Ngrams with amino acid ‘a’ could better describe the prosperities of protein family 7.3.6.1 in the evolutionary process.
and kernel p1, whereas amino acid ‘a’ was abundant in the top discriminative Ngrams in kernel p2, indicating the Ngrams with amino acid ‘a’ could better describe the prosperities of protein family 7.3.6.1 in the evolutionary process.
3.5 Application of the proposed remote homology detection methods for studying the 3D structure of Nck5a
In addition to provide useful insights for evolution study, protein remote homology detection is useful for drug development as well. As is well known, many drug-targeted proteins are still without X-ray or nuclear magnetic resonance structure. Pharmaceutical scientists have to resort to the homology modeling technique or structural bioinformatics tools (Chou, 2004) to timely develop their 3D structures, so as to be able to conduct molecular docking study (Chou et al., 2003; Wang et al., 2009), one of the key steps in structure-based drug design. However, a reliable template, or a structure-known protein homologous to the target protein, is the necessary prerequisite in this regard (Chou, 2004). Unfortunately, many target proteins did not have significant sequence similarity with any structure-known proteins, and hence it was hard to find a proper template to develop their 3D structures. Actually, many of them did have structure-known homologous proteins, but the problem was how to detect them. For example, the sequence similarity between Nck5a and CyclinA was <20% (Chou et al., 1999) and hence their homologous relationship could not be detected by the simple sequence alignment technique (Mohabatkar, 2010). Now let us see what will happen if the current remote homology detection technique is applied.
To realize this, a dataset was constructed based on SCOP, from which 11 proteins were selected as the positive samples in the cyclin family (SCOP ID: a.74.1.1), while 3605 negative samples were selected from the SCOP version 1.67 by excluding all the proteins within the cyclin-like superfamily. None of these proteins shares >95% sequence similarity. Trained with such 11 positive proteins and 3605 negative proteins, the proposed best performing method SVM-LA (p1) was used to predict Nck5a. It was found that Nck5a is homologous to CyclinA, fully consistent with the experimental results obtained by the site-directed mutagenesis studies (Tang et al., 1997). Actually, Chou et al. (1999) did use CyclinA as a template to construct the 3D structure of activation domain of Nck5a, one of the important parts of tau protein kinase II, an important therapeutic target against Alzheimer’s disease. Furthermore, based on the computed structure thus obtained, the molecular truncation experiments (Zhang et al., 2002) were conducted with an outcome that confirmed and validated the structure computed by using such a remote homologous protein as a template. Therefore, it is anticipated that the proposed method for detecting remote homology proteins will certainly enhance the power of homology modeling, and hence have impacts on drug development as well.
4 CONCLUSION
Discriminative methods based on SVM are the most effective and accurate methods for protein remote homology detection. The performance of the SVM-based methods depends on the kernel function, which measures the similarity between the samples in any pair. Varieties of kernels based on sequence composition have been proposed. However, these methods often fail to accurately predict the proteins sharing low sequence similarity. Recently, methods using the evolutionary information extracted from profiles achieved great success, such as Profile (Kuang et al., 2005), SW-PSSM (Rangwala and Karypis, 2005), SVM-Top-Ngram (Liu et al., 2008) and SVM-ACC (Liu et al., 2011). A key step to improve the performance of these methods is in how to find a suitable approach to incorporate the evolutionary information extracted from the profiles for prediction. In this article, we proposed a method that can convert the frequency profile into a series of profile-based proteins. Three state-of-the-art sequence-based kernels, i.e. SVM-Ngram (Dong et al., 2006), SVM-pairwise (Liao and Noble, 2003) and SVM-LA (Saigo et al., 2004), were selected for demonstration on a well-known benchmark. It was shown that the methods based on the profile-based proteins p1 and p2 achieved the best performance, outperforming the original three string kernels by 3.7 ∼ 7.5% and 9.6 ∼ 13.7%, respectively, according to the average ROC and ROC50 scores. These results are fully consistent with our previous findings that the top two most frequent amino acids show stronger discriminative power than the other low frequent amino acids in the frequency profiles (Liu et al., 2008), further confirming that the proposed profile-based protein representation is a promising approach in extracting the evolutionary information from frequency profiles for protein remote homology detection.
It has not escaped our notice that the current approach can be easily combined with sequence-based methods, and hence, with the development of the sequence-based kernels, the currently proposed method can be further improved accordingly. It is instructive to point out that since the concept of pseudo amino acid composition, or Chou’s PseAAC (Lin and Lapointe, 2013), was introduced in 2001 (Chou, 2001), it has been successfully used to predict various attributes of proteins (e.g. Chen and Li, 2013; Chou, 2005; Georgiou et al., 2009; Huang and Yuan, 2013; Khosravian et al., 2013; Mohabatkar, 2010; Mohabatkar et al., 2011, 2013; Mohammad Beigi et al., 2011; Nanni et al., 2012; Sahu and Panda, 2010; Zhang et al., 2008; Zhou et al., 2007; Liu et al., 2013). Accordingly, the potential would be high to develop a powerful method for protein remote homology detection by combing PseAAC with profile-based protein representation. In the original PseAAC, it only uses three indices, including the hydrophobicity index, hydrophilicity index and side-chain mass index. Because protein remote homology detection is a more difficult problem, proteins in the dataset only share low sequence similarity. Only these three indices would not be enough to capture the different properties of various proteins. Therefore, our further research will focus on incorporating new amino acid indices into PseAAC and applying it to protein remote homology detection.
Funding: National Natural Science Foundation of China [numbers 61300112, 61370165, 61173075, 61272383 and 61370010), the Natural Science Foundation of Guangdong Province [numbers S2012040007390 and S2013010014475], the Scientific Research Innovation Foundation in Harbin Institute of Technology (project number HIT.NSRIF.2013103), the Shanghai Key Laboratory of Intelligent Information Processing, China [grant number IIPL-2012-002].
Conflict of Interest: none declared
Supplementary Material
REFERENCES
- Althaus IW, et al. Steady-state kinetic studies with the non-nucleoside HIV-1 reverse transcriptase inhibitor U-87201E. J. Biol. Chem. 1993;268:6119–6124. [PubMed] [Google Scholar]
- Altschul SF, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H, et al. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids. Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner SE, et al. The ASTRAL compendium for sequence and structure analysis. Nucleic Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai YD, et al. Support vector machines for predicting membrane protein types by using functional domain composition. Biophys. J. 2003;84:3257–3263. doi: 10.1016/S0006-3495(03)70050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, et al. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gks1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YK, Li KB. Predicting membrane protein types by incorporating protein topology, domains, signal peptides, and physicochemical properties into the general form of Chou's pseudo amino acid composition. Journal of Theoretical Biology. 2013;318:1–12. doi: 10.1016/j.jtbi.2012.10.033. [DOI] [PubMed] [Google Scholar]
- Chou KC. The convergence-divergence duality in lectin domains of the selectin family and its implications. FEBS Lett. 1995;363:123–126. doi: 10.1016/0014-5793(95)00240-a. [DOI] [PubMed] [Google Scholar]
- Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. Proteins. 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- Chou KC. Review: structural bioinformatics and its impact to biomedical science. Curr. Med. Chem. 2004;11:2105–2134. doi: 10.2174/0929867043364667. [DOI] [PubMed] [Google Scholar]
- Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005;21:10–19. doi: 10.1093/bioinformatics/bth466. [DOI] [PubMed] [Google Scholar]
- Chou KC. Graphic rule for drug metabolism systems. Curr. Drug Metab. 2010;11:369–378. doi: 10.2174/138920010791514261. [DOI] [PubMed] [Google Scholar]
- Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th anniversary year review) J. Theor. Biol. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC. Some remarks on predicting multi-label attributes in molecular biosystems. Mol. Biosyst. 2013;9:1092–1100. doi: 10.1039/c3mb25555g. [DOI] [PubMed] [Google Scholar]
- Chou KC, et al. A model of the complex between cyclin-dependent kinase 5 (Cdk5) and the activation domain of neuronal Cdk5 activator. Biochem. Biophys. Res. Commun. 1999;259:420–428. doi: 10.1006/bbrc.1999.0792. [DOI] [PubMed] [Google Scholar]
- Chou KC, et al. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC, et al. Wenxiang: a web-server for drawing wenxiang diagrams. Natural Science. 2011;3:862–865. doi: 10.4236/ns.2011.310111. [DOI] [Google Scholar]
- Cortes C, et al. Proceedings of the 27th International Conference on Machine Learning. 2010. Two-stage learning kernel algorithms; pp. 239–246. [Google Scholar]
- Damoulas T, Girolami MA. Probabilistic multi-class multi-kernel learning: on protein fold recognition and remote homology detection. Bioinformatics. 2008;24:1264–1270. doi: 10.1093/bioinformatics/btn112. [DOI] [PubMed] [Google Scholar]
- Dong QW, et al. Application of latent semantic analysis to protein remote homology detection. Bioinformatics. 2006;22:285–290. doi: 10.1093/bioinformatics/bti801. [DOI] [PubMed] [Google Scholar]
- Feng PM, et al. iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013;442:118–125. doi: 10.1016/j.ab.2013.05.024. [DOI] [PubMed] [Google Scholar]
- Georgiou DN, et al. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou's pseudo amino acid composition. J. Theor. Biol. 2009;257:17–26. doi: 10.1016/j.jtbi.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Gribskov M, Robinson NL. Use of receiver operating characteristic (Roc) analysis to evaluate sequence matching. Comput. Chem. 1996;20:25–33. doi: 10.1016/s0097-8485(96)80004-0. [DOI] [PubMed] [Google Scholar]
- Huang C, Yuan JQ. A multilabel model based on Chou's pseudo-amino acid composition for identifying membrane proteins with both single and multiple functional types. J. Membr. Biol. 2013;246:327–334. doi: 10.1007/s00232-013-9536-9. [DOI] [PubMed] [Google Scholar]
- Karplus K, et al. Hidden markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Kawashima S, et al. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 2008;36:D202–D205. doi: 10.1093/nar/gkm998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravian M, et al. Predicting antibacterial peptides by the concept of Chou's pseudo-amino acid composition and machine learning methods. Protein Pept. Lett. 2013;20:180–186. doi: 10.2174/092986613804725307. [DOI] [PubMed] [Google Scholar]
- Kuang R, et al. Profile-based string kernels for remote homology detection and motif extraction. J. Bioinform. Comput. Biol. 2005;3:527–550. doi: 10.1142/s021972000500120x. [DOI] [PubMed] [Google Scholar]
- Leslie CS, et al. Mismatch string kernels for discriminative protein classification. Bioinformatics. 2004;20:467–476. doi: 10.1093/bioinformatics/btg431. [DOI] [PubMed] [Google Scholar]
- Liao L, Noble WS. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J. Comput Biol. 2003;10:857–868. doi: 10.1089/106652703322756113. [DOI] [PubMed] [Google Scholar]
- Lin SX, Lapointe J. Theoretical and experimental biology in one. J. Biomed. Sci. Eng. 2013;6:435–442. doi: 10.4236/jbise.2013.64054. [DOI] [Google Scholar]
- Lingner T, Meinicke P. Remote homology detection based on oligomer distances. Bioinformatics. 2006;22:2224–2231. doi: 10.1093/bioinformatics/btl376. [DOI] [PubMed] [Google Scholar]
- Lingner T, Meinicke P. Word correlation matrices for protein sequence analysis and remote homology detection. BMC Bioinformatics. 2008;9:259. doi: 10.1186/1471-2105-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. A discriminative method for protein remote homology detection and fold recognition combining Top-n-grams and latent semantic analysis. BMC Bioinformatics. 2008;9:510. doi: 10.1186/1471-2105-9-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. Prediction of protein binding sites in protein structures using hidden Markov support machine. BMC Bioinformaitcs. 2009;10:381. doi: 10.1186/1471-2105-10-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. Using amino acid physicochemical distance transformation for fast protein remote homology detection. PLoS One. 2012;7:e46633. doi: 10.1371/journal.pone.0046633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. Protein remote homology detection by combining Chou's pseudo amino acid composition and profile-based protein representation. Molecular Informatics. 2013;32:775–782. doi: 10.1002/minf.201300084. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. Protein remote homology detection based on auto-cross covariance transformation. Comput. Biol. Med. 2011;41:640–647. doi: 10.1016/j.compbiomed.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Loewenstein Y, et al. Protein function annotation by homology-based inference. Genome Biol. 2009;10:207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melvin I, et al. Detecting remote evolutionary relationships among proteins by large-scale semantic embedding. PLoS Comput. Biol. 2011;7:e1001047. doi: 10.1371/journal.pcbi.1001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohabatkar H. Prediction of cyclin proteins using Chou's pseudo amino acid composition. Protein Pept. Lett. 2010;17:1207–1214. doi: 10.2174/092986610792231564. [DOI] [PubMed] [Google Scholar]
- Mohabatkar H, et al. Prediction of GABA(A) receptor proteins using the concept of Chou's pseudo-amino acid composition and support vector machine. J. Theor. Biol. 2011;281:18–23. doi: 10.1016/j.jtbi.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Mohabatkar H, et al. Prediction of allergenic proteins by means of the concept of Chou's pseudo amino acid composition and a machine learning approach. Med. Chem. 2013;9:133–137. doi: 10.2174/157340613804488341. [DOI] [PubMed] [Google Scholar]
- Mohammad Beigi M, et al. Prediction of metalloproteinase family based on the concept of Chou's pseudo amino acid composition using a machine learning approach. J. Struct. Funct. Genomics. 2011;12:191–197. doi: 10.1007/s10969-011-9120-4. [DOI] [PubMed] [Google Scholar]
- Muda HM, et al. Remote protein homology detection and fold recognition using two-layer support vector machine classifiers. Comput. Biol. Med. 2011;41:687–699. doi: 10.1016/j.compbiomed.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Nanni L, et al. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of chou's pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012;9:467–475. doi: 10.1109/TCBB.2011.117. [DOI] [PubMed] [Google Scholar]
- Qian B, Goldstein RA. Performance of an iterated T-HMM for homology detection. Bioinformatics. 2004;20:2175–2180. doi: 10.1093/bioinformatics/bth181. [DOI] [PubMed] [Google Scholar]
- Rangwala H, Karypis G. Profile-based direct kernels for remote homology detection and fold recognition. Bioinformatics. 2005;21:4239–4247. doi: 10.1093/bioinformatics/bti687. [DOI] [PubMed] [Google Scholar]
- Sadreyev RI, et al. COMPASS server for homology detection: improved statistical accuracy, speed and functionality. Nucleic Acids Res. 2009;37:W90–W94. doi: 10.1093/nar/gkp360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu SS, Panda G. A novel feature representation method based on Chou's pseudo amino acid composition for protein structural class prediction. Comput. Biol. Chem. 2010;34:320–327. doi: 10.1016/j.compbiolchem.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Saigo H, et al. Protein homology detection using string alignment kernels. Bioinformatics. 2004;20:1682–1689. doi: 10.1093/bioinformatics/bth141. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Söding J. Protein homology detection by HMM–HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Tang D, et al. Cyclin-dependent kinase 5 (Cdk5) activation domain of neuronal Cdk5 activator. Evidence of the existence of cyclin fold in neuronal Cdk5a activator. J. Biol. Chem. 1997;272:12318–12327. doi: 10.1074/jbc.272.19.12318. [DOI] [PubMed] [Google Scholar]
- Vapnik VN. Statistical Learning Theory. New York: Wiley-Interscience; 1998. [Google Scholar]
- Varma M, Babu BR. Proceedings of the 26th International Conference on Machine Learning. 2009. More generality in efficient multiple kernel learning; pp. 1065–1072. [Google Scholar]
- Wang JF, et al. Insights from investigating the interactions of adamantane-based drugs with the M2 proton channel from the H1N1 swine virus. Biochem. Biophys. Res. Commun. 2009;388:413–417. doi: 10.1016/j.bbrc.2009.08.026. [DOI] [PubMed] [Google Scholar]
- Webb-Robertson B-J, et al. SVM-BALSA: remote homology detection based on Bayesian sequence alignment. Comput. Biol. Chem. 2005;29:440–443. doi: 10.1016/j.compbiolchem.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Wu CH, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids. Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, et al. iCDI-PseFpt: identify the channel-drug interaction in cellular networking with PseAAC and molecular fingerprints. J. Theor. Biol. 2013;337C:71–79. doi: 10.1016/j.jtbi.2013.08.013. [DOI] [PubMed] [Google Scholar]
- Xu Y, et al. iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013;1:e171. doi: 10.7717/peerj.171. https://peerj.com/articles/171.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, et al. Identification of the N-terminal functional domains of Cdk5 by molecular truncation and computer modeling. Proteins. 2002;48:447–453. doi: 10.1002/prot.10173. [DOI] [PubMed] [Google Scholar]
- Zhang SW, et al. Using the concept of Chou's pseudo amino acid composition to predict protein subcellular localization: an approach by incorporating evolutionary information and von Neumann entropies. Amino Acids. 2008;34:565–572. doi: 10.1007/s00726-007-0010-9. [DOI] [PubMed] [Google Scholar]
- Zhou GP, Deng MH. An extension of Chou's graphic rules for deriving enzyme kinetic equations to systems involving parallel reaction pathways. Biochem. J. 1984;222:169–176. doi: 10.1042/bj2220169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou GP. The disposition of the LZCC protein residues in wenxiang diagram provides new insights into the protein-protein interaction mechanism. J. Theor. Biol. 2011;284:142–148. doi: 10.1016/j.jtbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XB, et al. Using Chou's amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J. Theor. Biol. 2007;248:546–551. doi: 10.1016/j.jtbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Zhou GP, Huang RB. The pH-Triggered Conversion of the PrP(c) to PrP(sc.) Curr Top Med Chem. 2013;13:1152–1163. doi: 10.2174/15680266113139990003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.