

Abstract
Protein aggregates have negative implications in disease. While reductionist experiments have increased our understanding of aggregation processes, the systemic view in biological context is still limited. To extend this understanding, we used mass spectrometry‐based proteomics to characterize aggregation and disaggregation in human cells after non‐lethal heat shock. Aggregation‐prone proteins were enriched in nuclear proteins, high proportion of intrinsically disordered regions, high molecular mass, high isoelectric point, and hydrophilic amino acids. During recovery, most aggregating proteins disaggregated with a rate proportional to the aggregation propensity: larger loss in solubility was counteracted by faster disaggregation. High amount of intrinsically disordered regions were associated with faster disaggregation. However, other characteristics enriched in aggregating proteins did not correlate with the disaggregation rates. In addition, we analyzed changes in protein thermal stability after heat shock. Soluble remnants of aggregated proteins were more thermally stable compared with control condition. Therefore, our results provide a rich resource of heat stress‐related protein solubility data and can foster further studies related to protein aggregation diseases.
Keywords: aggregation, disaggregation, heat shock, proteomics, thermal proteome profiling
Subject Categories:
An in situ proteome‐wide analysis of protein solubility upon heat shock and during recovery reveals protein aggregation and disaggregation features in human cells.

Introduction
Insoluble protein deposits are a hallmark for many devastating neurodegenerative diseases, such as Alzheimer's, Parkinson's, and Huntington's disease (Valastyan & Lindquist, 2014). Understanding the basic principles of protein (mis)folding, (dis)aggregation, and other features of protein quality control is essential when attempting to tackle and interfere with the causes of those diseases.
Mass spectrometry‐based proteomics has become an effective tool for unbiased analysis of the effects of cellular perturbations on a system‐wide scale (Cox & Mann, 2007). Modern mass spectrometry analysis allows the quantification of thousands of proteins from multiple samples simultaneously by using one of many labeling techniques such as stable isotope labeling by amino acids in cell culture (SILAC), isobaric tags for relative and absolute quantification (iTRAQ), or tandem mass tags (TMT) (Bantscheff et al, 2012). Recently, combination of protein and peptide level labeling, termed hyperplexing (Dephoure & Gygi, 2012), has allowed to quantify proteomes from even tens of samples in one mass spectrometry experiment (Dephoure & Gygi, 2012; Savitski et al, 2018; Aggarwal et al, 2019).
Proteome‐wide mass spectrometry‐based studies have been previously used to characterize aggregation‐prone proteins in different organisms and conditions. These include, for example, aging nematode (David et al, 2010; Reis‐Rodrigues et al, 2012; Walther et al, 2015), mice expressing disease‐causing mutant of huntingtin protein (Hosp et al, 2017), mice cells exposed to different stress conditions (Sui et al, 2020), and yeast under chemical or heat stress (Ibstedt et al, 2014; O'Connell et al, 2014; Wallace et al, 2015; Weids et al, 2016). Although these studies involved quite different organisms and conditions, some similarities could be found. For example, chaperones and other proteostasis components were enriched in aggregates from aging nematode (David et al, 2010) and mice expressing huntingtin with disease‐causing mutation (Hosp et al, 2017). Similarly, chaperones were found in aggregates when yeast cells were exposed to hydrogen peroxide, arsenite, or azetidine‐2‐carboxylic acid (Weids et al, 2016). Another similarity is the aggregation of ribosomal proteins in stress conditions, such as aging in nematode (Reis‐Rodrigues et al, 2012) and heat (Wallace et al, 2015) or arsenite stress (Ibstedt et al, 2014) in yeast. However, ribosomal proteins were also found in aggregates at physiological conditions in yeast (Ibstedt et al, 2014).
Cells have multiple ways to handle protein aggregates (Tyedmers et al, 2010; Miller et al, 2015; Mogk et al, 2018). Irreversibly damaged proteins can be degraded by the ubiquitin‐proteasome system (Balchin et al, 2016). Larger aggregates can be cleared by autophagy or secreted out from the cells (Tyedmers et al, 2010). To maintain protein homeostasis, protein degradation can be balanced by upregulated protein synthesis. In addition, protein disaggregation and re‐folding allow to rescue functional proteins from the aggregates (Doyle et al, 2013; Mogk et al, 2018; Nillegoda et al, 2018).
Disaggregation of aggregated proteins was initially observed and characterized in yeast (Parsell et al, 1994). A proteome‐wide study showed that the disaggregation of heat‐induced aggregates is the main strategy for yeast to deal with the aggregates (Wallace et al, 2015). However, disaggregation in yeast is conducted by a Hsp100 disaggregase (Sanchez & Lindquist, 1990; Parsell et al, 1994) that has no homologue in the human genome (Mosser et al, 2004; Shorter, 2011; Doyle et al, 2013; Nillegoda & Bukau, 2015). In metazoans (including humans), the disaggregase activity of Hsp100 is likely replaced by a Hsp70 chaperone system (Rampelt et al, 2012; Doyle et al, 2013; Nillegoda & Bukau, 2015; Nillegoda et al, 2015; Mogk et al, 2018) which opens the question of how human cells handle aggregates of endogenous proteins.
Here, we studied heat‐induced aggregation and disaggregation of endogenous human proteins in situ. We developed a hyperplexed quantitative mass spectrometry assay to measure protein solubility after transient non‐lethal heat shock and during recovery. The aggregating proteins were enriched in nuclear proteins, intrinsically disordered regions, high molecular mass, high isoelectric point, and hydrophilic amino acids. After characterizing the features of aggregation‐prone proteins, we analyzed the dynamics of disaggregation patterns. We found that the majority of aggregating proteins were rescued from the aggregates. The disaggregation rates correlated with the initial loss of solubility in heat shock and with the proportion of disordered regions in the proteins. In addition to aggregating proteins, we analyzed proteins that remained soluble after heat shock by monitoring their changes in thermal stability. Strikingly, non‐lethal heat shock triggered thermal stabilization of aggregation‐prone proteins invoking immediate protection against aggregation. We also detected changes in thermal stability for a large number of proteins, including proteins related to stress signaling, DNA binding, and protein quality control complexes.
Results
Monitoring protein solubility after heat shock and during recovery
Dynamic SILAC labeling (Ong et al, 2002; Doherty et al, 2009) was used to distinguish between pre‐existing proteins and newly synthesized proteins (Fig 1A). K562 human leukemia cells were cultured in light SILAC medium. The medium was switched to heavy SILAC 90 min before heat treatment. We assume that during this period prior to heat treatment, the intracellular pool of arginine and lysine from light SILAC medium would be consumed. That would allow a more accurate quantification of pre‐existing proteins since the signal from newly synthesized proteins can be filtered out.
Figure 1. Experimental design for quantitative proteome‐wide solubility measurements after heat shock.
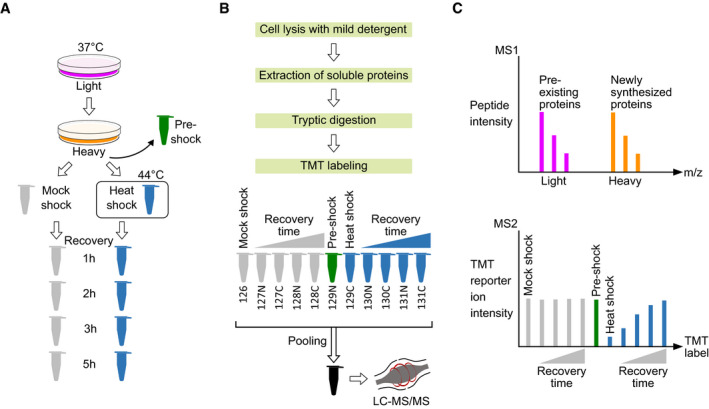
- Dynamic SILAC, heat treatment and recovery. K562 human cells were grown at 37°C in light SILAC medium. 90 min before heat treatment, the medium was changed to heavy SILAC containing stable heavy carbon and nitrogen isotopes in arginine and lysine amino acids. These heavy amino acids are incorporated into newly synthesized proteins while the light versions remain in pre‐existing proteins. A control sample was collected (pre‐shock) prior to partitioning cells for heat treatment. The cells were treated either with heat shock (44°C) or mock shock (37°C) for 10 min. After heat treatment, cells were allowed to recover at 37°C and samples were collected at different time points.
- Sample processing. Samples were lysed with mild detergent (0.8% NP‐40), soluble proteins were extracted and digested to tryptic peptides. Peptides were labeled with TMT labels and pooled. After offline reversed phase fractionation, samples were analyzed with mass spectrometer.
- Quantification of soluble protein fraction. MS1 scan allows to separate peptides from newly synthesized proteins (heavy) and pre‐existing proteins (light). MS2 scan allows for peptide (and later protein) identification and, based on TMT reporter ion intensities, quantification from different samples. For MS2 scan, a hypothetical example is shown for aggregating and disaggregating protein from pre‐existing (light) fraction.
Prior to heat treatment, cells were partitioned into two aliquots which were exposed to either 44°C (heat shock) or 37°C (mock shock) for 10 min (Fig 1A). A control sample was collected before partitioning the cells (pre‐shock). The heat shock temperature was chosen so that it did not compromise cell viability (Fig EV1).
Figure EV1. Cell viability after heat shocks with different temperatures and during recovery.
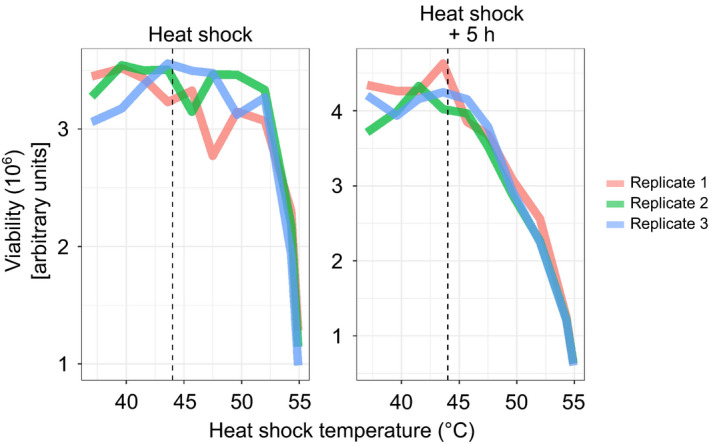
Cells were exposed to a heat shock for 10 min at different temperatures (37–55°C). Cell viability (based on ATP levels) was measured right after the heat shock and after 5 h of recovery at 37°C. Dashed vertical line indicates 44°C. Data shown for three technical replicates.Source data are available online for this figure.
After heat treatment, the cells were allowed to recover at 37°C (Fig 1A). Samples were collected directly after heat treatment and during recovery of 1, 2, 3, and 5 h. Samples were lysed with mild detergent (NP‐40) to preserve protein aggregates (Reinhard et al, 2015), and soluble protein fractions were collected. After tryptic digestion, peptides were labeled with TMT tags (McAlister et al, 2012; Werner et al, 2012; Fig 1B). Tagged samples were pooled, fractionated offline, and analyzed on an Orbitrap mass spectrometer (Fig 1B) to distinguish newly synthesized (heavy) and pre‐existing proteins (light) in the MS1 scan and different experimental conditions in the MS2 scan (Fig 1C).
Characterization of aggregation‐prone proteins
We collected mass spectrometry data for 7,226 proteins. To obtain a high‐quality dataset, we required that a protein had to be quantified in all conditions with at least two unique peptides in at least two biological replicates. The resulting high‐quality data included 4,786 light‐labeled (pre‐existing) and 1,269 heavy‐labeled (newly synthesized) proteins with high reproducibility (Appendix Figs S1 and S2).
The protein aggregation was analyzed from the pre‐existing (light) fraction. Directly after heat shock, the abundance of 300 proteins (< 7% of quantifiable proteome) decreased significantly (Benjamini–Hochberg‐adjusted P‐value < 0.05 in LIMMA analysis and fold change < 2/3) in the soluble fraction when compared with mock‐shocked sample (Fig 2A). We refer to those proteins as aggregators (Fig 2A). At the same time, the majority of proteins (4,486) remained soluble after heat shock (Fig 2A, soluble). While aggregators lost intensity in the soluble fraction, the total protein amount remained constant, as estimated from samples lysed with strong detergent (SDS) (Fig EV2A–C). This indicates that the observed loss of solubility is not an artifact of heat‐induced degradation.
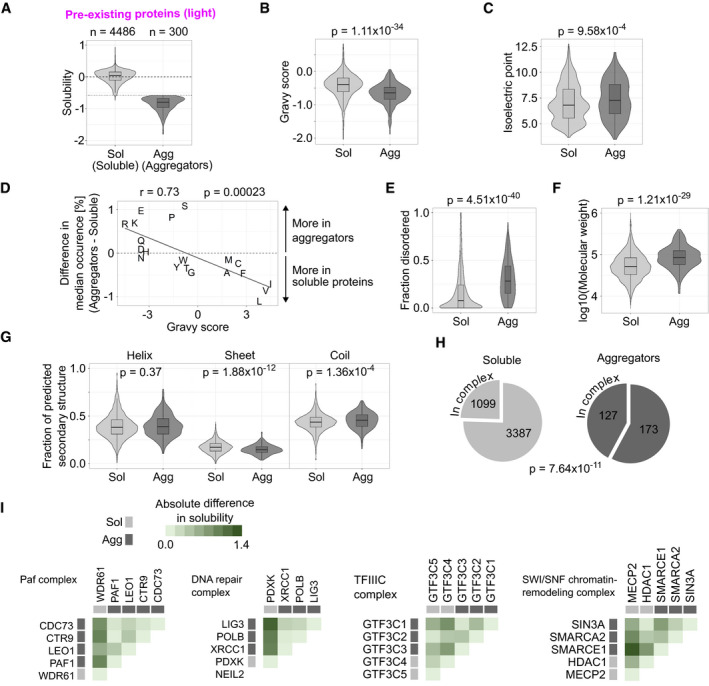
Figure 2. Characterization of proteins that aggregate in heat shock.
-
ADefinition of aggregating proteins. Solubility is measured as the log2‐transformed ratio of protein intensities in the soluble fraction between heat‐shocked and mock‐shocked samples. Proteins with significant reduction in solubility [Benjamini–Hochberg‐adjusted P‐value < 0.05 and solubility < log2(2/3)] are then considered as aggregators. Dotted horizontal line shows the cutoff at solubility of log2(2/3).
-
B–DComparisons of physicochemical properties between soluble proteins and aggregators. Hydrophobicity (gravy scores) (B) and isoelectric points (C) are shown as combined violin and boxplots (P‐values are for non‐parametric Wilcoxon test). Difference in median amino acid composition between soluble proteins and aggregators is compared with hydrophobicity (gravy score) for each amino acid (D). In (D), Pearson coefficient (r) with P‐value is shown for the correlation analysis.
-
E–GComparison of structural features between soluble proteins and aggregators. Fraction of protein sequence predicted to contain intrinsically disordered regions (E), molecular weight (F), and fraction of protein sequence predicted to contain secondary structure elements (alpha helix, beta sheet, or coil) (G) are compared. P‐values are for non‐parametric Wilcoxon test.
-
HProtein complex members in soluble proteins and aggregators. Pie charts show the fraction of proteins annotated to be a member of a protein complex. The number of proteins in each segment is indicated. P‐value is for Fisher's exact test.
-
IProtein complexes involving aggregators. Heatmaps show the absolute difference in solubility change after heat shock between each complex member. Protein complexes with at least 75% of its members quantified and containing at least 60% of its members as aggregators are shown. “DNA repair complex” = “DNA repair complex NEIL2‐PNK-Pol(beta)‐LigIII(alpha)‐XRCC1”.
Figure EV2. Protein abundance, total solubility, and aggregation upon heat shock.
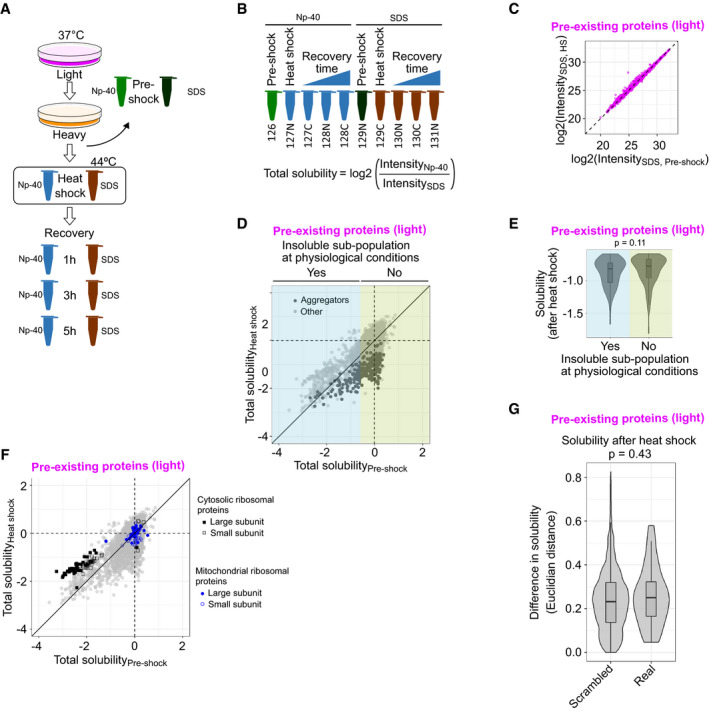
- Experimental design for measuring total protein abundance and total solubility.
- Sample labeling scheme. Total solubility is calculated as log2‐transformed ratio between protein intensity in the soluble fraction (NP‐40 lysis) and total protein abundance (SDS lysis).
- Scatterplot showing total protein abundance (intensity in SDS‐lysed samples) of pre‐existing proteins (light) after heat shock compared with pre‐shock control. Medians of normalized intensities (log2‐transformed).
- Total solubility before and after heat shock. Scatterplot comparing total solubility in pre‐shocked control and heat‐shocked sample. Aggregators (as defined in Fig 2A) are highlighted with darker color. In addition, proteins were assigned to contain an insoluble sub‐population at physiological conditions if the total solubility of the pre‐shocked sample was lower than −0.6 (highlighted with cyan at the left of the figure).
- Solubility after heat shock (see Fig 2A) of aggregators with or without an insoluble sub‐population at physiological conditions. P‐value is for non‐parametric Wilcoxon test. Boxplots indicate median, first and third quartiles with whiskers extended to 1.5 times the interquartile range out from each quartile. Violin plots show the data distribution. Data from at least two biological replicates.
- Total solubility before and after heat shock of ribosomal proteins. As in (D), except cytosolic and mitochondrial ribosomal proteins from large and small subunit are highlighted.
- Difference in solubility change after heat shock for aggregators in protein complexes (“Real”) compared with the same aggregators randomly distributed to complexes (“Scrambled”). The difference in aggregation within each complex is estimated by calculating mean of all Euclidian distances of solubility (see Fig 2A) between aggregators. P‐values are shown for non‐parametric Wilcoxon test. The analysis includes 32 protein complexes (“Real”) with at least 75% of members with good quality solubility data and include at least two aggregators. For the scrambled complex set, 10,000 complexes were created by randomly assigning aggregators from the 32 annotated complexes. The frequency distribution of aggregators in complexes was maintained in the scrambled set. Boxplots indicate median, first, and third quartiles with whiskers extended to 1.5 times the interquartile range out from each quartile. Violin plots show the data distribution. Solubility data used in the analysis are from at least two biological replicates.
Source data are available online for this figure.
Based on the readout of this experiment, we cannot state whether a decrease in solubility is caused by formation of amorphous aggregates (Wang et al, 2010), structured fibers (Wang et al, 2010; Knowles et al, 2014; Bauerlein et al, 2017), phase separation (Brangwynne et al, 2009; Riback et al, 2017), or any other homo‐ or heteromeric (Senohrabkova et al, 2019) protein assemblies [with or without other co‐assembling biomolecules, such as RNA (Saad et al, 2017)]. However, we assume that a decrease in solubility results in formation of an insoluble protein deposit that we from now on simply refer to as aggregation.
Comparison of the ratio of NP‐40‐extracted (soluble sub‐population) and SDS‐extracted (total) proteins reports on the solubility status of a protein. Under physiological conditions, proteins involved in phase separated membrane‐less nuclear organelles—such as the nucleolus—have been shown to contain an insoluble sub‐population (Becher et al, 2018; Sridharan et al, 2019). Analysis of NP‐40/SDS ratio of pre‐existing protein pool between pre‐shock and heat shock conditions showed that the aggregators included proteins that were completely soluble as well as proteins that have an insoluble sub‐population under physiological conditions (Fig EV2D). The extent of loss of solubility after heat shock was comparable between the two types of aggregators (Fig EV2E). We also observed that proteins from the cytosolic ribosome had an insoluble fraction in unstressed conditions (Fig EV2F). Similar observations have been made in yeast (Weids et al, 2016). We speculate that this insoluble fraction represents ribosomal proteins in the nucleolus, where the ribosomes are assembled. Contrary to cytosolic ribosomes, we found that proteins from mitochondrial ribosomes are fully soluble in unstressed conditions (Fig EV2F).
To gain a deeper view into the properties of aggregators, we analyzed their physicochemical characteristics (Fig 2B–D). Aggregators were more hydrophilic (Fig 2B; lower gravy score; P = 1.11 × 10−34) and positively charged (Fig 2C; higher isoelectric point; P = 9.58 × 10−4) when compared to proteins that stayed soluble after heat shock. We found a negative correlation (Pearson's r = −0.73, P = 0.00023) between amino acid hydrophobicity (gravy score) and amino acid composition in aggregators (Fig 2D). The increased isoelectric point was due to enrichment of positively charged arginine and lysine residues in aggregators; the negatively charged residues were either enriched (glutamate) or had similar occurrence in aggregators as in the soluble proteins (aspartate) (Fig 2D).
Next, we looked at structural features of aggregators (Fig 2E–G). Aggregators were enriched in high proportion of intrinsically disordered regions (Fig 2E; P = 4.51 × 10−40) and high molecular weight (Fig 2F; P = 1.21 × 10−29). To expand the structural view, we calculated the fraction of predicted secondary structure elements in proteins (Fig 2G). Aggregators and soluble proteins contained similar amounts of alpha helices (P = 0.37) while aggregators contained less beta sheets (P = 1.88 × 10−12). In accordance with the results for disordered regions, aggregators were enriched in (random) coil‐like structures (Fig 2G; P = 1.36 × 10−4).
Almost half (> 42%) of aggregators were annotated to be part of a protein complex while the same holds true for only a quarter (< 25%) of soluble protein (Fig 2H, P = 7.64 × 10−11). Protein complexes that had at least 60% of the members aggregating are shown in Fig 2I. All of them (Paf complex, TFIIIC complex, DNA repair complex, and SWI/SNF chromatin‐remodeling complex) are nuclear complexes that operate on chromatin. To analyze whether the complexes had truly distinct aggregating parts, we compared the heat shock‐induced solubility changes between all members in each complex (heatmaps in Fig 2I). The complexes had similar solubility changes between aggregators that were distinct from the soluble proteins. This suggests that protein complexes composed mainly of aggregators contain distinct and unstable sub‐structures rather than proteins with a continuum of different stabilities. However, the coherent aggregation was not evident when all complexes with at least two aggregators were analyzed (Fig EV2G).
We performed GO term enrichment analysis for the aggregators using all quantified proteins as background. The aggregators were enriched in nuclear proteins involved in DNA binding, chromatin organization, and transcription regulator activity (Fig EV3A). The enrichment of nuclear proteins in aggregators was complementary observed by analyzing protein localization annotations (Fig EV3B); the analysis also indicated that soluble proteins were enriched in cytoplasmic proteins. Since the aggregators are defined from a comparison between heat‐ and mock‐shocked samples, all detergent‐based or other technical biases related to the lysis conditions would be cancelled out.
Figure EV3. Functions and localizations of aggregators and heat shock‐induced solubility changes of stress granule proteins.
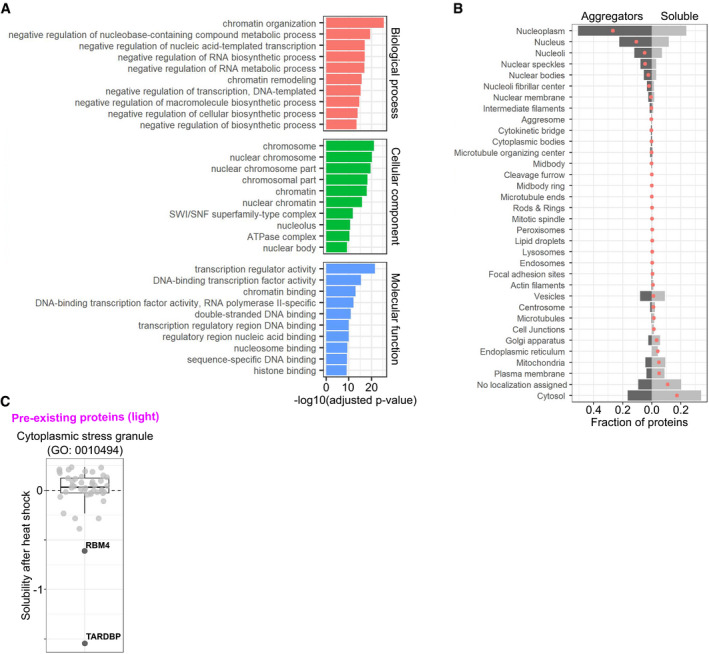
- Gene ontology (GO) enrichment of aggregators. Bar plot showing 10 most enriched (lowest Benjamini–Hochberg‐adjusted P‐value in hypergeometric test) terms from each GO domain.
- Localization annotations for aggregators and soluble proteins. Bar plot showing the fraction of aggregating or soluble proteins having particular localization annotation. Red dots indicate the difference between aggregators and soluble proteins.
- Solubility changes of stress granule proteins after heat shock. Heat shock‐induced solubility changes of proteins with a GO term “cytoplasmic stress granule” (GO:0010494). Aggregators are labeled and highlighted with darker color.
Source data are available online for this figure.
One feature observed in heat and other stresses is the formation of cytoplasmic stress granules (Collier & Schlesinger, 1986; Collier et al, 1988; Ivanov et al, 2018). Stress granule‐forming factors related to translation have been observed to aggregate in yeast upon heat stress (Grousl et al, 2013; Wallace et al, 2015). Interestingly, from 44 proteins assigned to a GO term “cytoplasmic stress granule” that we could quantify in our dataset, only two were found to aggregate (TARDBP and RBM4), while the solubility of the other 42 was not affected by the heat shock (Fig EV3C). The different results could stem from technical experimental differences or biological dissimilarities in the core structures between human and yeast stress granules (Jain et al, 2016).
Many features enriched in aggregators (such as the high amount of disordered regions) could be a result of them being also enriched in nuclear or DNA‐binding protein. When analyzing the features presented in Fig 2 for similar enrichment or depletion as observed for aggregators, nuclear and DNA‐binding proteins are enriched or depleted in the same features (Appendix Fig S3). However, the molecular weight was similar between nuclear and cytosolic proteins. In addition, predicted alpha helical content was significantly lower for DNA‐binding and nuclear proteins. These results suggest that within nuclear proteins, large proteins with lower alpha helical content tend to aggregate.
Chromosome duplications can lead to gene overexpression. It has been shown that the protein overproduction is counteracted by aggregation (Brennan et al, 2019). As K562 cells contain aneuploidic chromosomes, we analyzed the frequency of aggregators in each chromosome to test whether aggregators would be over‐represented in duplicated chromosomes. We found no difference in the occurrence of aggregators or soluble proteins in any of the 23 chromosome (Appendix Table S1) suggesting that protein overproduction does not contribute to aggregation propensity upon heat shock.
To analyze whether aggregators would have stronger preference for chaperones, we searched for Hsp70 binding motifs in them. A Hsp70‐binding motif has been reported to contain four or five hydrophobic residues flanked by positively charged residues (Rüdiger et al, 1997). We found a slightly higher occurrence of the binding motifs in aggregators but the difference was not significant (P = 0.09582, Appendix Fig S4A). In addition, we could not find differences in the amino acid composition within the binding motif of aggregators as compared to the soluble proteins (Appendix Fig S4B).
Supersaturation (high concentration relative to solubility) has been shown to correlate with aggregation propensity (Ciryam et al, 2013, 2015). We found that supersaturation scores (Ciryam et al, 2013) were not higher for aggregators than for soluble proteins (Appendix Fig S5A). The supersaturation score has two components: protein concentration and structurally corrected aggregation propensity score (based on Zyggregator method (Tartaglia et al, 2008); Ciryam et al, 2013). While the supersaturation score was not higher for aggregators, we found a higher Zyggregator‐based aggregation propensity score for them (Appendix Fig S5B). This suggests that the aggregation propensity in heat shock is more related to structural features of proteins rather than to supersaturation.
In summary, we observed heat shock‐induced aggregation of nuclear, hydrophilic proteins with high molecular weight and intrinsically disordered regions. In addition, proteins that aggregated were more likely to be part of protein complexes.
Disaggregation of heat‐induced protein aggregates during recovery from heat shock
To monitor protein solubility during recovery, we measured protein intensities of the pre‐existing proteins (light) in the soluble (NP‐40 extractable) fraction. We sampled multiple time points and had a time‐matched mock‐shocked reference for each one of them (Fig 1A). Therefore, this approach allowed for fine‐controlled measure of the solubility during recovery with high temporal resolution. The ratios between heat‐shocked and mock‐shocked samples were calculated at each time point and the log2‐transformed ratios for pre‐existing proteins (light) are shown in Fig 3A.
Figure 3. Disaggregation of aggregated proteins during recovery from heat shock.
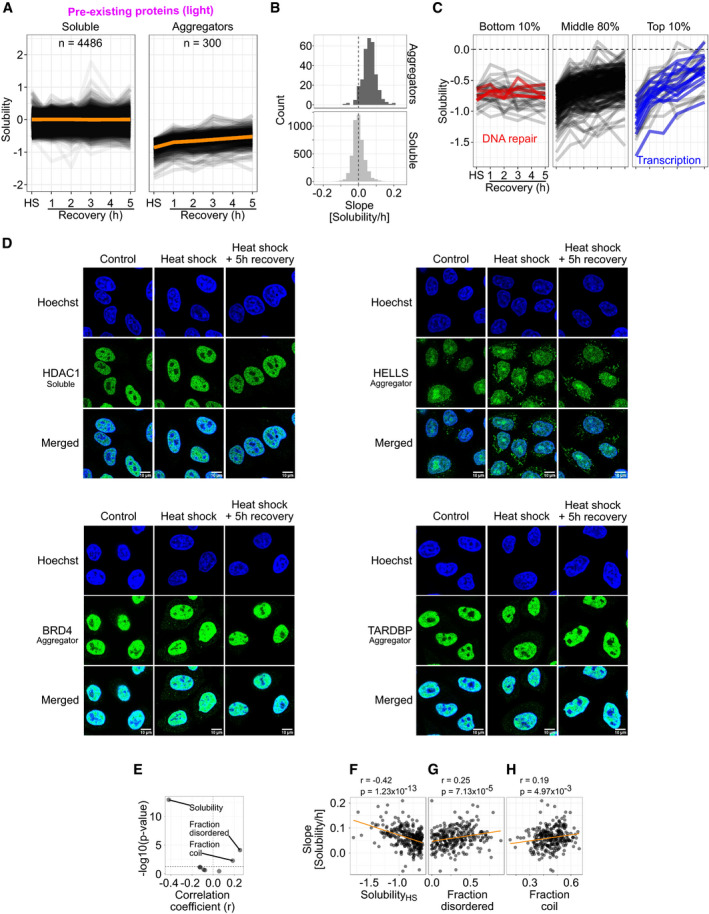
-
ALine graphs showing solubility after heat shock (HS) and during different time points of recovery. Each line corresponds to one protein. Orange lines show the mean solubility.
-
BQuantification of disaggregation rates. Disaggregation rate for each protein is estimated as a slope from linear fits of data used in (A). Histograms of slopes (binwidth = 0.02 Solubility/h) are shown for aggregators and soluble proteins.
-
CDifferent disaggregation profiles for aggregating proteins. Solubility line graphs as in (A) shown for aggregators with the bottom 10%, middle 80%, and top 10% of disaggregation rates. Proteins with bottom 10% of disaggregation rates and related to DNA repair are highlighted with red. Proteins with top 10% disaggregation rates and directly related to transcription are highlighted with blue.
-
DProtein localization upon heat shock and during recovery as analyzed by immunofluorescence microscopy. HeLa cells were fixed and immunostained with antibodies against indicated target proteins (green) at different conditions [control, after heat shock (10 min at 44°C) and after 5 h of recovery from the heat shock]. DNA staining (Hoechst) is shown in blue.
-
E–HComparison of disaggregation rates with different protein characteristics enriched in aggregators (presented in Fig 2B, C, and E–G). Volcano plot presenting correlation coefficients and Benjamini–Hochberg‐adjusted P‐values (E; horizontal dashed line shows a P‐value of 0.05). Scatterplot comparing disaggregation slopes and solubility change after heat shock (F), fraction of intrinsically disordered regions (G), and fraction of (random) coil‐like secondary structure (H). In (F–H), scatterplots are shown for correlations with a P‐value lower than 0.05. Correlation coefficients (r) with P‐values are shown for Spearman's rank‐order correlation.
Proteins that stayed soluble after heat shock remained largely soluble during the recovery period (Fig 3A). However, most aggregators regained solubility during recovery from heat shock (Fig 3A). To quantitatively analyze dynamics of disaggregation, a linear model was fitted for each protein and the slope was used as an estimate for the disaggregation rate. The distributions of slopes (Fig 3B) indicate the steady solubility maintained with proteins that stay soluble after heat shock. In addition, the disaggregation of aggregators is evident from a positive shift of the slope values (Fig 3B). Similar observations were made with yeast (Wallace et al, 2015). Together, these results indicate that the disaggregation is the main strategy to deal with aggregates.
The total protein intensity remained constant during the recovery (Fig EV4A). This indicated that the increased intensity in the soluble fraction during recovery for aggregators stemmed from increasing solubility. In addition, this also showed that heat shock did not induce protein degradation.
Figure EV4. Disaggregation and protein synthesis during recovery from heat shock.
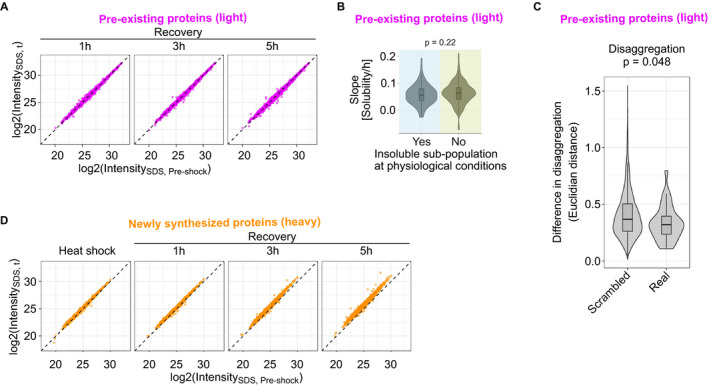
- Total protein abundances of pre‐existing proteins (light) during recovery from heat shock. Scatterplot showing medians of normalized intensities (log2‐transformed) of heat‐shocked samples compared with pre‐shocked control.
- Disaggregation slopes of aggregators with or without an insoluble sub‐population at physiological conditions. P‐value is for non‐parametric Wilcoxon test. Boxplots indicate median, first, and third quartiles with whiskers extended to 1.5 times the interquartile range out from each quartile. Violin plots show the data distribution. Data from at least two biological replicates.
- Difference in disaggregation for aggregators in protein complexes (“Real”) compared with the same aggregators randomly distributed to complexes (“Scrambled”). The difference in disaggregation within each complex is estimated by calculating mean of all Euclidian distances of solubility (see Fig 2A) between aggregators in each time point. To examine only the disaggregation, solubility in each recovery time point is normalized to the initial loss of solubility after heat shock prior to the distance calculation. P‐values are shown for non‐parametric Wilcoxon test. The analysis includes 32 protein complexes (“Real”) with at least 75% of members with good quality solubility data and include at least two aggregators. For the scrambled complex set, 10,000 complexes were created by randomly assigning aggregators from the 32 annotated complexes. The frequency distribution of aggregators in complexes was maintained in the scrambled set. Boxplots indicate median, first, and third quartiles with whiskers extended to 1.5 times the interquartile range out from each quartile. Violin plots show the data distribution. Solubility data used in the analysis are from at least two biological replicates.
- Total protein abundances of newly synthesized proteins (heavy) during recovery from heat shock. Scatterplot showing medians of normalized intensities (log2‐transformed) of heat‐shocked samples compared with pre‐shocked control.
Source data are available online for this figure.
We took a more detailed look at the disaggregation patterns by concentrating on aggregators with the top or bottom deciles of slope values. Examination of aggregators with the lowest 10% of slope values revealed a small subset that were not disaggregated within 5 h of recovery (Fig 3C). These included proteins related to DNA damage: TDP1, FANCI, POLE, RIF1, and TIMELESS. Almost half of the aggregators with the highest 10% of slope values were transcription factors (FOXK2, MGA, ARID3A) or proteins closely related to transcription (TAF4, TCEB3, ELL, TRIM24, PRAME, BRD4, SMARCD2, SMARCE1, DAXX, and SCML2).
We validated the aggregation and disaggregation propensities of a few proteins from our dataset using immunofluorescence. HDAC1, a non‐aggregating protein, showed nuclear localization in control conditions and remained unchanged upon heat shock and during recovery (Fig 3D). On the other hand, aggregators HELLS, BRD4, and TARDBP (all localized in the nucleus) showed increased intensity in the cytoplasm upon heat shock (Fig 3D). The increased cytoplasmic signal was strongest for HELLS (the most aggregating protein in the mass spectrometry experiment) while the cytoplasmic signal was weaker for BRD4 and TARDBP. HELLS seemed to form foci in the cytoplasm. Interestingly, prolonged heat shock caused the cytoplasmic and nuclear HELLS to localize and form foci at nuclear membranes (Appendix Fig S6). The cytoplasmic BRD4 formed bigger foci during prolonged heat shock while TARDBP formed nuclear foci or the intensity from non‐foci proteins decreased (Appendix Fig S6).
After 5 h of recovery, a reduction in the cytoplasmic signal of BRD4 and TARDBP was observed, while the cytoplasmic HELLS remained in foci (Fig 3D). These findings corroborated the observations from the mass spectrometry experiment, where BRD4 and TARDBP disaggregated while HELLS remained aggregated in the insoluble fraction during the recovery. The solubility changes of nuclear proteins observed with mass spectrometry upon heat shock and during recovery coincide with protein transport to cytoplasm and foci formation.
Since aggregators were enriched in certain molecular features (Fig 2B–G), we wondered if these features would also be related to the rate of disaggregation. By analyzing the correlations between disaggregation slope and each of the features (Fig 3E), we found a significant (Benjamini–Hochberg‐adjusted P‐value < 0.05) negative correlation between disaggregation slope and the loss of solubility after heat shock (Fig 3F; P = 1.23 × 10−13; r = −0.42). The proportion of disordered regions (Fig 3G; P = 7.13 × 10−5; r = 0.25) and fraction of (random) coil‐like secondary structure (Fig 3H; P = 4.97 × 10−3; r = 0.19) had a weak but significant correlation with the disaggregation slope. The disaggregation rates were independent on whether or not the aggregators had an insoluble sub‐population at physiological conditions (Fig EV4B).
The correlation between solubility and disaggregation slopes could be a result of ratio compression during TMT quantification, which may stem from peptide co‐fragmentation (Savitski et al, 2013). We therefore analyzed signal to interference values (which are lower for proteins prone for ratio compression) in the context of aggregation and disaggregation. We could not find any signs of lowered signal to interference values for aggregators (Appendix Fig S7A). In addition, no correlation was observed between signal to interference and solubility (Appendix Fig S7B) nor disaggregation slope (Appendix Fig S7C). Therefore, it is unlikely that ratio compression played a role in the observed correlation between solubility and disaggregation.
As mentioned earlier, aggregators were enriched in protein complex members (Fig 2H). Next, we explored their disaggregation as protein complex members in the recovery period. Within a protein complex (n = 32), aggregators had a weak trend for more similar disaggregation profiles when compared to scrambled complexes (n = 10,000) containing the same aggregators randomly re‐distributed (Fig EV4C; P = 0.048). However, this coupling within complexes is not evident for the initial loss of solubility after heat shock (Fig EV2G; P = 0.43) suggesting that aggregators in complexes aggregate to different extent but can disaggregate similarly. However, as discussed earlier, complexes with a majority of aggregating members did aggregate coherently (Fig 2I).
To conclude, aggregated proteins were disaggregated during recovery from heat shock. The disaggregation rates dependent mainly on how much a protein aggregated in heat shock. In addition, larger extent of intrinsically disordered regions in a protein associated with faster disaggregation.
Reversible stall in protein synthesis after heat shock
The dynamic SILAC approach allowed to analyze also newly synthesized proteins (Fig 1A). To monitor protein synthesis, we quantified newly synthesized proteins (heavy) from soluble fractions and compared each time point after heat treatment to a control collected before heat shock (Fig 1A). This approach allowed us to follow the accumulation of heavy‐labeled proteins during recovery.
After mock shock, a steady rate of protein synthesis was observed (Fig 4A). The apparently fast synthesis rate—protein amount approximately doubled in 5 h—most probably stemmed from low starting amounts of heavy‐labeled proteins: a small increase in the absolute protein amount will result in a large increase in relative amount.
Figure 4. Newly synthesized proteins after heat shock.
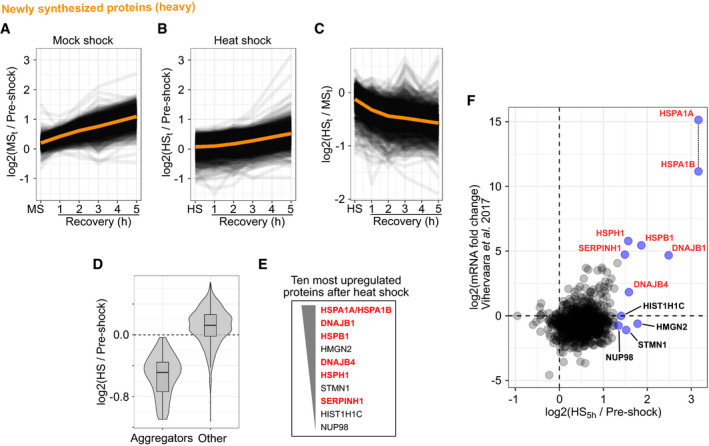
-
A, BAccumulation of newly synthesized proteins after mock shock (A) and heat shock (B). Line graphs showing the accumulation of signal intensity (log2‐transformed ratios to pre‐shock control) in the soluble fraction. Orange lines show the mean ratio.
-
CAmount of newly synthesized proteins after heat shock compared with mock shock. Line graphs show the heat shock to mock shock ratio during recovery. Orange line shows the mean ratio.
-
DCombined violin and boxplots show the log2‐transformed ratio to pre‐shock of newly synthesized proteins in the soluble fraction after heat shock. Proteins that aggregated in the pre‐existing (light) fraction are compared with all other proteins. Boxplots indicate median, first, and third quartiles with whiskers extended to 1.5 times the interquartile range out from each quartile. Violin plots show the data distribution. Data from at least two biological replicates.
-
ETen proteins with the highest intensity at 5 h after heat shock (i.e., strongest upregulation) are listed in order of decreasing intensity. Heat shock proteins are highlighted with red.
-
FComparison of heat shock‐induced protein synthesis and mRNA synthesis after heat shock (Vihervaara et al, 2017). Ten most upregulated proteins in the proteomics analysis are labeled and highlighted in blue; heat shock proteins are labeled red. HSPA1A and HSPA1B could not be distinguished from one another in the proteomics analysis and are plotted as they would have the same intensity (indicated with a vertical dashed line).
After heat shock, the accumulation of newly synthesized proteins slowed down globally (Figs 4B and C, and EV4D), in line with previous studies (Holcik & Sonenberg, 2005; Kirstein‐Miles et al, 2013; Shalgi et al, 2013). However, during recovery the synthesis rates slowly increased and approached approximately the mock shock levels at the late time points of the recovery with exception of few proteins (Fig 4A and B).
The early medium switch in dynamic SILAC (90 min before heat shock) allowed incorporation of some heavy‐labeled amino acids to newly synthesized proteins before heat treatment. Therefore, we could observe aggregation of newly synthesized proteins (Fig 4B and D). Aggregators identified from the pre‐existing fraction (light) were predominantly the same proteins that aggregated in the newly synthesized fraction (heavy) (Fig 4D, Appendix Fig S8A). In addition, the solubility change upon heat shock was very similar for aggregators in both SILAC fractions (Appendix Fig S8A). The small differences between both protein populations did not correlate with disordered regions (Appendix Fig S8B).
At the end of the recovery period, few proteins showed a sharp increase in the newly synthesized fraction (Fig 4B and E). To analyze the upregulation of their synthesis, we looked at the protein intensities at the last time point, where the effect is most evident. The 10 most upregulated proteins included many heat shock proteins (Hsp): HSPA1B‐HSPA1A (Hsp70), DNAJB1 (Hsp40), DNAJB4 (Hsp40), HSPB1 (Hsp27), HSPH1 (Hsp105), and SERPINH1 (Hsp47). Since HSPA1A and HSPA1B share high sequence similarity, we could not distinguish between the two paralogs in the mass spectrometry analysis and the results reflect a combination of the two. Interestingly, both highly upregulated Hsp40s (DNAJB1 and DNAJB4) belonged to class B of Hsp40s which are involved in protein disaggregation (Nillegoda et al, 2015). Although other Hsp40s were also upregulated (Appendix Fig S9), this suggested that the response to heat shock involved an increase in disaggregation capacity by upregulated protein synthesis.
Since some proteins that aggregated in heat hock were not disaggregated (Fig 3C), they possibly were replaced by upregulated protein synthesis. We could not find correlation between disaggregation and protein synthesis (Appendix Fig S10). However, it should be noted that the sample size in the analysis is relatively low due to the lower coverage of the newly synthesized protein fraction.
Next, we analyzed how the regulation of protein synthesis would match to transcriptional regulation after heat shock. We compared our results with previously reported changes of mRNA levels after heat shock (30 min at 42°C) in K562 cells (Vihervaara et al, 2017). Upregulation on mRNA level matched with upregulation on protein level only with the most upregulated transcripts (Fig 4F). From our 10 most upregulated proteins, majority of them were also upregulated on mRNA level (Fig 4F). We noticed that the upregulated heat shock proteins were the only ones with upregulation also on mRNA level; non‐heat shock proteins were upregulated only on the protein level, suggesting that their upregulation was not driven by mRNA levels. Similar findings have been made with yeast (Muhlhofer et al, 2019).
To summarize, heat shock stalled translation. However, as the heat stress was removed the translation rates recovered accompanied by protein level upregulation including many heat shock proteins.
Heat shock‐induced changes in thermal stability
Next, we moved our focus to proteins that remain soluble after heat shock. We analyzed immediate heat shock‐induced responses by applying two‐dimensional thermal proteome profiling (2D‐TPP) (Savitski et al, 2014; Becher et al, 2016; Mateus et al, 2017; Fig 5A). With this technique, we measured the propensity of heat‐induced denaturation of soluble proteins after heat shock. In brief, after heat treatment, aliquots of cells were exposed to a short temperature gradient (3 min) that went well beyond the heat shock temperature (up to 66.3°C) eventually denaturing and aggregating all proteins in the sample (Fig 5A). Proteins from the soluble fraction were quantified and differences between heat shock and mock shock above 44°C (the heat shock temperature) indicated a heat shock‐induced change in thermal stability (Fig 5A). We collected high‐quality data (quantified with at least two unique peptides from minimum of six different temperatures) for 5,319 proteins with high reproducibility (Appendix Fig S11).
Figure 5. Heat shock‐induced changes in protein thermal stability.
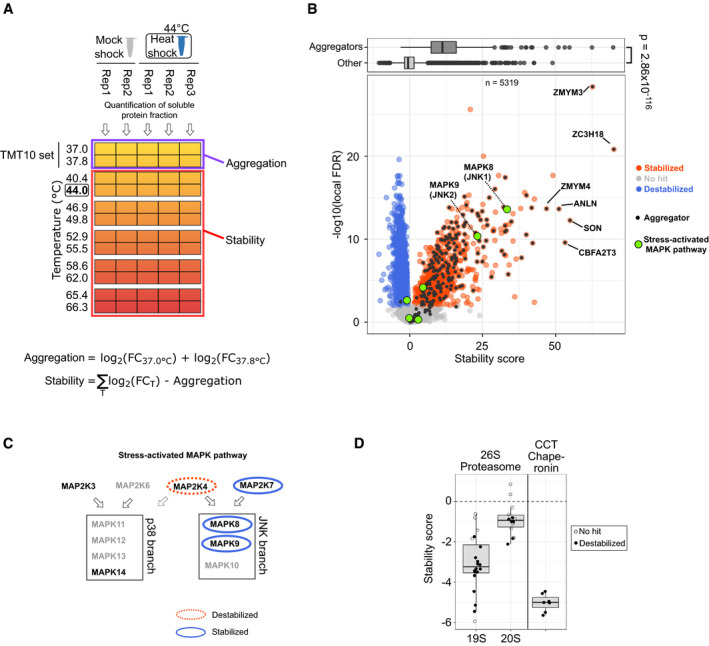
- Experimental design to measure changes in thermal stability. Samples treated with either heat shock or mock shock are aliquot for treatment with temperatures that denature and aggregate all proteins. Samples are lysed with mild detergent (NP‐40) and after tryptic digestion peptides from soluble protein fraction are labeled with TMT labels. The labeling is conducted so that samples from two adjacent temperature treatments are pooled and each TMT set is analyzed by mass spectrometer. At each temperature, the amount of protein still soluble is quantified and the fold change (FC) between heat shock and mock shock is calculated (see “Materials and Methods” for details). Fold changes in each temperature are summed after adjustment for heat shock‐induced aggregation. Fold change‐based thermal stability measures are finally transformed to scores for thermal stability (see “Materials and Methods” for details).
- Volcano plot for the score for thermal stability. Significant (see “Materials and Methods”) heat shock‐induced stabilization and destabilization are indicated with blue and red, respectively. Proteins that aggregate in heat shock (as defined in Fig 2A) are shown in black—aggregators with the strongest stabilization are annotated. Members of stress‐activated MAPK pathway highlighted in green—alternative names for MAP kinases are shown in parenthesis. Boxplot on top compares stability scores of aggregators (as defined in Fig 2A and other proteins). The P‐value is for non‐parametric Wilcoxon test.
- Schematic for the structure of stress‐activated MAPK pathway (adapted from Hotamisligil & Davis, 2016 and Davis, 2000) with the two branches of MAP kinases (p38 and JNK) and their upstream kinases (MKK3, MKK4, MKK6, and MKK7). Arrows show the target MAPK branch for each MAPKK. The role of MAP2K4 in p38 phosphorylation is unclear (Hotamisligil & Davis, 2016) and is presented with dashed line. Changes in thermal stability after heat shock are marked with ovals.
- Thermal stability changes in protein complex members. Scores for thermal stability shown for proteins from 26S proteasome (separately for 19S regulatory and 20S core particles) and CCT chaperonin complex. Boxplots indicate median, first, and third quartiles with whiskers extended to 1.5 times the interquartile range out from each quartile. Data used in stability score calculation is from two (mock shock) and three (heat shock) biological replicates.
Strikingly, a large fraction of thermally stabilized proteins corresponded to aggregators (Fig 5B) including almost 90% of aggregators that were quantified in the assay (210 out of 234). Since the measurement was performed only on proteins that were soluble after heat treatment (mock shock or heat shock), the thermally stabilized proteins reflected the soluble remnants of aggregators. In other words, a sub‐population of aggregators remained soluble after heat shock and required higher temperatures to aggregate.
Among the soluble remnants of aggregators, the strongest thermal stabilization was observed for ZC3H18 and ZMYM3, both proteins containing a zinc‐finger domain (Fig 5B). Another zinc‐finger containing protein (ZMYM4) was among the most thermally stabilized proteins.
When exploring the changes in thermal stability of proteins that were not aggregators, we observed some of the strongest effects for two mitogen‐activated protein kinases (MAPKs) MAPK8 and MAPK9 (Fig 5B). They both belong to the c‐Jun N‐terminal kinases (JNK) group of MAP kinases, a branch in stress‐activated MAPK pathway (Fig 5C) (Davis, 2000; Hotamisligil & Davis, 2016). We also observed changes in thermal stability of upstream kinases that are specific for JNKs (Fig 5C). We did not observe changes in thermal stability for p38 branch of stress‐activated MAP kinases nor their specific upstream kinases (Fig 5C), although only one protein from each kinase level could be quantified. These results suggested that the activation of these pathways led to pronounced stability changes of proteins involved in them.
We detected thermal destabilization of RNA polymerase II subunits (Fig EV5) which was recently linked to detachment from DNA (Becher et al, 2018). This observation would be in line with the global down‐regulation of transcription upon heat shock (Vihervaara et al, 2017).
Figure EV5. Stability changes of histone H1 variants and DNA polymerase II proteins.
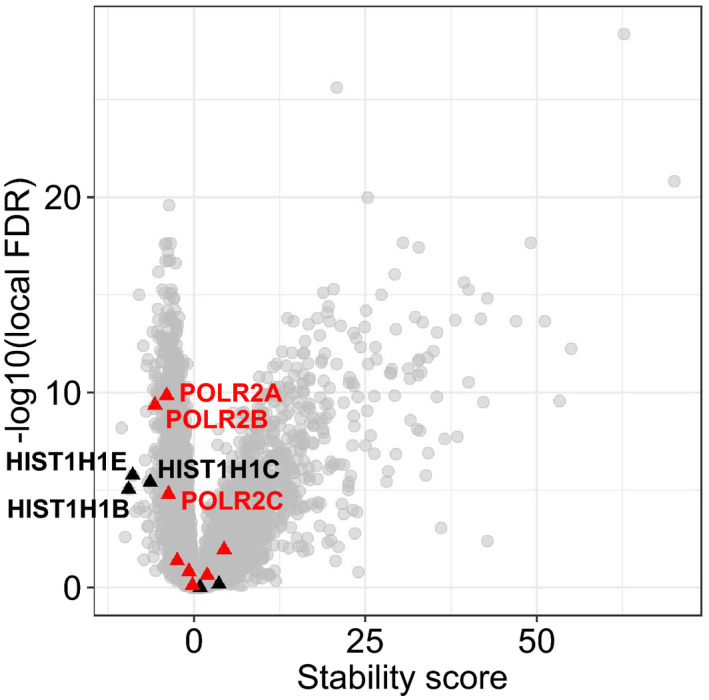
Volcano plot for stability score. Histone H1 (black triangles) and DNA polymerase II (red triangles) proteins are highlighted. Destabilized proteins are labeled.Source data are available online for this figure.
Some of the strongest thermal destabilizations were measured for H1 histones (Fig EV5), proteins that link nucleosomes together in compacted chromatin (Hergeth & Schneider, 2015). C‐terminus of H1 histones is largely unstructured but it folds when bound to DNA (Roque et al, 2005). We reasoned that the thermal destabilization of H1 histones would correspond to partial unfolding and detachment from DNA upon heat shock—possibly resulting in opening of compact chromatin. In yeast, the human H1 histone homolog Hho1p was indeed reported to detach from repressed DNA upon heat shock (Zanton & Pugh, 2006).
Protein complex members showed thermal stability patterns that were possibly related to complex (de)activation or (dis)assembly as exemplified with 26S proteasome and CCT chaperonin complex (Fig 5D), both essential components of protein quality control. The thermal stability of both complexes decreased in heat shock. With the 26S proteasome the thermal destabilization was stronger for 19S regulatory particle than for the 20S core particle (Fig 5D). Interestingly, proteins from the 19S regulatory particle were thermally stabilized when ATP was added to cell lysates (Sridharan et al, 2019). However, the ATP levels were not altered during the heat shock (Fig EV1: viability measurements based on ATP quantification). The thermal destabilization of 19S regulatory particle could be linked to an impairment of ATP‐driven proteasome activation observed in acute heat shock (Kuckelkorn et al, 2000). However, the inhibiting effect might be only a temporary response, since prolonged exposure to repeated heat shocks increases proteasomal activity (Beedholm et al, 2004).
Overall, we found a sub‐pool of aggregation‐prone proteins that resisted aggregation in heat shock. In addition, changes in thermal stability were observed in different cellular processes, such as stress signaling, DNA binding and with complexes related to protein quality control.
Discussion
We developed a platform to study protein aggregation and disaggregation in human cells in situ after non‐lethal heat shock. We found that heat shock induced the aggregation of proteins enriched in nuclear localization, intrinsically disordered regions, high molecular weight, and hydrophilic character.
The nuclear localization of aggregators could be linked to large extents of disordered regions found in the underlying protein, particularly since DNA‐binding proteins are known to contain disordered regions (Fuxreiter et al, 2011; Vuzman & Levy, 2012). Previous studies have shown that components of Hsp70 chaperone system localize to nuclear organelles, such as nucleolus (Welch & Feramisco, 1984) and nuclear speckles (Deane & Brown, 2017) upon heat shock, suggesting for higher need of quality control measures at nuclear sites.
Previously, when mapping melting points of the human proteome, DNA‐binding proteins were found to be the most unstable proteins (Savitski et al, 2014). Similarly, in bacteria, proteins with the lowest melting points include topoisomerases and proteins involved in DNA replication (Mateus et al, 2018). Therefore, unstable proteins might specifically relate to DNA.
Aggregation in the nucleus could mean several different things. For example, during stress, proteins have been reported to enter the nucleoli (Frottin et al, 2019) and increase abundance at chromatin (Aprile‐Garcia et al, 2019). These could be linked to our results of increased thermal stability for soluble sub‐pools of aggregators that did not aggregate in heat shock (Fig 5B); the most strongly thermally stabilized proteins were indeed zinc‐finger containing DNA‐binding proteins (Fig 5B).
We found that disordered regions are not only enriched in aggregators (Fig 2E), but the amount of disordered regions in a protein sequence correlated with the disaggregation rates (Fig 3G). Proteins containing disordered regions have been previously reported to be prone for aggregation (Uemura et al, 2018) and enriched in aggregates formed in in vivo models (Walther et al, 2015; Hosp et al, 2017). Walther et al (2015) propose that it could be a protective mechanism to actively sequester proteins with disordered regions, since these are often associated with aggregation diseases. In addition, the involvement of disordered regions in dosage sensitivity has been demonstrated (Vavouri et al, 2009; Bolognesi et al, 2016). Indeed, overexpressed disordered proteins from aneuploidic chromosomes are sequestered to aggregates (Brennan et al, 2019), degraded faster when present in complexes with super‐stoichiometric amounts (McShane et al, 2016) and form toxic cytoplasmic granules when present with high concentration (Bolognesi et al, 2016). Therefore, aggregation of disordered proteins upon heat shock could protect cells from their potentially toxic effects.
Interestingly, the disordered regions per se are water soluble and might not contribute to the aggregation propensity of proteins (Uemura et al, 2018). Therefore, we speculate whether disordered regions could serve other functions not related to sequestering proteins to aggregates. The correlation between disordered regions and disaggregation rates could be explained by weaker intra‐molecular interactions between proteins in aggregates. Disordered regions could also facilitate disaggregation by providing flexible loop regions for disaggregase(s) to act up upon. It has been proposed that, with amyloid fibers, Hsp70 disaggregase acts through flexible regions to exert pulling forces to the aggregated proteins (Wentink et al, 2019).
Enrichment of high molecular weight proteins in aggregates has been reported in different stress conditions, for example in yeast (Weids et al, 2016) and mice (Hosp et al, 2017). In addition, proteins were observed to lose more solubility when exposed to common precipitants if they had high molecular weight (Kramer et al, 2012). Together with our findings (Fig 2F), these results suggest that high molecular weight proteins are aggregation‐prone and this is probably more due to their biophysical properties rather than to any biological reasons.
The formation of cytoplasmic foci that coincide with the aggregation detected in mass spectrometry experiment suggests that nuclear proteins do not aggregate in the nucleus, but rather translocate and aggregate in cytoplasm. It should be noted that the immunofluorescence detection cannot distinguish between aggregate formation and protein accumulation. In addition, the link between foci formation and loss of solubility is not trivial. Small aggregates that are beyond the detection limit of microscopy could contribute to a large fraction of the solubility decrease observed for aggregators (Mogk et al, 2018). In addition, majority of proteins related to cytoplasmic stress granules remained soluble (Fig EV3C). Therefore, we would hesitate to conclude that loss of solubility could be explained by cytoplasmic foci formation. Although some evidence connects foci formation and solubility change in mass spectrometry‐based assays (Wallace et al, 2015), more focused studies would be required to investigate the issue.
We observed that the majority of aggregated proteins disaggregated during recovery (Fig 3A and B). These findings indicate that, as observed with yeast (Wallace et al, 2015), the main strategy for human cells to handle aggregates is disaggregation. However, it should be noted that proteasomal degradation, as well requires aggregate re‐solubilization prior to degradation (Nillegoda et al, 2018). Therefore, it remains an open question whether the disaggregated proteins are destined for re‐folding or degradation. Based on our data, at least in the 5 h following heat shock no degradation was observed (Fig EV4A).
Based on earlier work done with human disaggregase in vitro (Nillegoda et al, 2015) and in yeast (Wallace et al, 2015), one might expect a full disaggregation to take place in a time scale from minutes to approximately an hour. However, even after 5 h of recovery most of the aggregators are still on their way to being fully disaggregated (Fig 3A). Therefore, disaggregation in human cells seems much slower than in yeast. The reasons for this might stem from different heat shock conditions and how severe they are for human and yeast. Biologically, however, the simplest explanation could be the Hsp100 disaggregation system present in yeast that can be more efficient than the human Hsp70‐based system (Rampelt et al, 2012). Similar to humans, in Caenorhabditis elegans (another metazoan without the Hsp100‐like disaggregase system), minute‐scale disaggregation rates were observed for aggregated luciferase in vitro while traces of luciferase aggregates were found even days after heat shock in vivo (Kirstein et al, 2017). These results suggest that, although capable to disaggregate, metazoan disaggregase system is less efficient also in real cellular context than yeast Hsp100‐based system.
We noted that the most upregulated heat shock proteins could, in theory, form a Hsp70‐based disaggregase if assuming that the components would be co‐expressed. These proteins include a Hsp70 (HSPA1A|HSPA1B) with a nucleotide exchange factor (HSPH1) and two Hsp40 proteins (DNAJB1 and DNAJB4). To speculate further, the two class B Hsp40s could reflect an adjustment toward clearance of larger aggregates, as it has been suggested for class B in contrast to class A (which are involved in clearing smaller size aggregates) (Nillegoda et al, 2015). However, it should be noted that other Hsp40s are upregulated too, for example DNAJC8, that has no known link to disaggregation. Further studies would help to understand the co‐expression of disaggregase components and the possible adjustment of its composition to reflect the aggregation load.
The thermal stabilization of soluble remnants of aggregating proteins could reflect an instant post‐translational mechanism of induced thermotolerance, although it should be noted that we cannot conclude how much of the increased stability signal is stemming from more stable protein sub‐population that was already present in the sample before the heat shock and how much of the signal reflects heat shock‐induced stability changes. For the heat shock‐induced stabilization, the 2D‐TPP assay, as we applied it here, can be viewed as a way to measure instantly gained thermotolerance without transcriptional or translational regulation. This is achieved by concentrating purely on protein solubility (direct measure of heat “sensitivity”) and having no recovery time between the two heating steps (i.e., the heat/mock shock and the temperature gradient applied to aliquots). We speculate that the heat shock‐induced stability could be done by a network of kinases, or other modifying enzymes, that modify proteins making them more stable. It would also be tempting to speculate that our results reflect the actions of chaperone networks [i.e., the epichaperome (Rodina et al, 2016)]. The limitation of the method is that it does not contain information about the reasons behind the stability changes. Therefore, it remains unsolved whether the stability changes are because of direct changes in proteins (e.g., post‐translational modifications) or interactions with other molecules (e.g., chaperones or DNA). While the 2D‐TPP assay discussed here reflects the response of mainly pre‐existing proteins, it would be interesting to develop the method further to analyze heat‐induced stability changes of newly synthesized proteins. For example, one could speculate that the stability of newly synthesized proteins could be affected by incomplete folding or chaperone binding. Extending the analysis by using chaperone inhibitors could help to understand these processes.
To conclude, we mapped protein solubility after non‐lethal heat shock and during recovery in situ by hyperplexed proteomics. This allowed not only to characterize stress‐induced aggregation but to study disaggregation patterns as well. Complementary, non‐aggregating proteins were studied with 2D‐TPP which allowed to explore solubility‐independent heat shock‐induced processes. While we introduced these techniques here to study heat‐induced solubility changes, in the future it would be interesting to adapt the same approach to study and compare different stress condition that lead to protein aggregation such as chemical or other environmental stress conditions.
Materials and Methods
Reagents and Tools table
| Reagent/resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| K‐562 human chronic myelogenous leukemia cell line | ATCC | CCL‐243 |
| HeLa cells | S. Narumiya | CVCL_1922 |
| Antibodies | ||
| Rabbit polyclonal anti‐HELLS | Sigma | HPA063242‐100UL |
| Mouse monoclonal anti‐TARDBP | Novus Biological | NBP1‐92695SS |
| Rabbit polyclonal anti‐BRD4 | Sigma | HPA015055‐100UL |
| Rabbit polyclonal anti‐HDAC1 | Sigma | HPA029693 |
| Goat anti‐rabbit AlexaFluor‐488 | Thermo Fisher | A‐11008 |
| Goat anti‐mouse AlexaFluor‐488 | Thermo Fisher | A‐11001 |
| Reagents | ||
| l‐lysine (heavy) 13C615N2 | Thermo Fisher Scientific | 88209 |
| l‐arginine (heavy) 13C615N4 | Thermo Fisher Scientific | 89990 |
| cOmplete EDTA‐free Protease inhibitor cocktail | Roche | 11873580001 |
| PhosSTOP (phosphatase inhibitor cocktail) | Roche | 4906845001 |
| NP‐40 (IGEPAL CA‐630) | Sigma‐Aldrich | 18896 |
| Benzonase Nuclease | Millipore | 71206‐3 |
| Hydroxylamine | Sigma‐Aldrich | 438227 |
| TMT10 | Thermo Fisher Scientific | 90111 |
| TMT11 | Thermo Fisher Scientific | A37724 |
| Chloroacetamide | Sigma‐Aldrich | C0267 |
| Tris(2‐carboxyethyl)phosphine hydrochloride | Sigma‐Aldrich | C4706 |
| Trypsin | Promega | V5111 |
| Lys‐C | FUJIFILM Wako | 125‐05061 |
| DMSO | Sigma‐Aldrich | 276855 |
| 16% Paraformaldehyde | Thermo Fisher Scientific | 28908 |
| Software | ||
| isobarQuant | Franken et al (2015) | |
| https://github.com/protcode/isob | ||
| Mascot | Matrix Science | |
| http://www.matrixscience.com/ | ||
| R | R Core Team | |
| https://www.R-project.org | ||
| Fiji | ImageJ | |
| https://imagej.net/Fiji/ | ||
| Databases | ||
| UniProt | UniProt Consortium (2019) | |
| https://www.uniprot.org/ | ||
| Human Protein Atlas | Thul et al (2017) | |
| http://www.proteinatlas.org | ||
| Protein complexes | Ori et al (2016) | |
| http://www.bork.embl.de/Docu/variable_complexes/ | ||
| Database of Disordered Protein Predictions | Oates et al (2013) | |
| http://d2p2.pro | ||
| Gene Ontology Annotation Database | Huntley et al (2015) | |
| Commercial kits and consumables | ||
| CellTiter‐Glo Luminescent Viability Assay | Promega | G7571 |
| Optiplate‐96 Luminescence plate | PerkinElmer | |
| Filter plates (0.45 μm) | Merck Millipore | MSHVN4550 |
| Filter plates (0.22 μm) | Merck Millipore | MSHVN2250 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | 23225 |
| Carboxylate modified magnetic particles, hydrophilic | Sigma‐Aldrich | 45152105050250 |
| Carboxylate modified magnetic particles, hydrophobic | Sigma‐Aldrich | 65152105050250 |
| Trapping cartridge. Acclaim PepMap 100 C18 LC column; 5 μm particles with 100 Å pores; 5 mm column with 300 μm inner diameter | Thermo Fisher Scientific | |
| Analytical column. nanoEase HSS C18 T3, 75 μm × 25 cm, 1.8 μm, 100 Å | Waters | |
| Other | ||
| UltiMate 3000 RSLC nano LC system | Thermo Fisher Scientific | |
| Q Exactive Plus Orbitrap mass spectrometer | Thermo Fisher Scientific | |
| Orbitrap Fusion Lumos mass spectrometer | Thermo Fisher Scientific | |
| SureCycler 8800 Thermal Cycler | Agilent | |
| Infinite M1000 PRO plate reader | TECAN | |
| 1290 Infinity (for HPLC fractionation) | Agilent | |
Methods and Protocols
Cell culture
K‐562 cells (ATCC CCL‐243) were cultured in SILAC RPMI 1640 medium (Thermo Fisher) supplemented with 2 mM l‐glutamine, 0.96 mM l‐lysine (light) (Thermo Fisher), 0.48 mM l‐arginine (light) (Thermo Fisher), and 10% dialyzed FBS at +37°C (5% CO2). For heavy SILAC medium, 13C6 15N2 l‐lysine (Thermo Fisher) and 13C6 15N4 l‐arginine (Thermo Fisher) were used keeping their molar concentration same as in light. The K562 cells were chosen based on their tolerance to a 45°C heat shock (Mivechi, 1989; Fig EV1) and easy handling (suspension cells).
Heat shock and recovery with dynamic SILAC
-
Experiment 1:
-
○
Analysis of soluble protein fraction after 0, 1, 2, 3, and 5 h of recovery from heat shock (10 min, 44°C)
-
○
Heat‐shocked samples compared with time‐matched mock controls
-
○
Samples lysed with weak non‐ionic detergent (NP‐40)
-
○
-
Experiment 2:
-
○
Analysis of soluble and total protein fraction before and after 0, 1, 3, and 5 h from heat shock (10 min, 44°C)
-
□
Soluble fraction: samples lysed with weak non‐ionic detergent (NP‐40)
-
□
Total protein fraction: samples lysed with strong ionic detergent (SDS)
-
□
-
○
The protocol for both Experiments is as follows (differences between Experiment 1 and 2 are point out):
Medium switch:
Transfer cells with fully adapted light amino acids to a conical centrifugation tube
Approximately 2.5 × 106 cells needed for each replicate and Experiment
Pellet cells [190 × g, 3 min, room temperature (RT)]
Gently aspirate supernatant
Re‐suspend cells to 1 ml of pre‐warmed (37°C) heavy SILAC medium
Pellet cells (190 × g, 3 min, RT)
Gently aspirate supernatant
Re‐suspend cells to pre‐warmed (37°C) heavy SILAC medium to a final cell density of 5 × 105 cells/ml.
Transfer cells to a cell culture flask and incubate 90 min at +37°C (5% CO2)
Heat treatment:
Prior to heat treatment, collect pre‐shocked sample (see “Sample collection”)
-
Distribute cells to 96‐well PCR plates (200 μl/well, 105 cells/well)
-
○
Experiment 1: two plates (mock and heat shock) with five samples on each plate
-
○
Experiment 2: one plate (heat shock) with 10 samples on the plate
-
○
Seal plates with aluminum foil
-
Place plates on pre‐warmed heat blocks for 10 min
-
○
Experiment 1 and 2: Heat shock at 44°C
-
○
Experiment 1: Mock shock at 37°C
-
○
Recovery from heat shock:
After heat treatment, remove the aluminum foil
Seal plates with vent filter membrane
Incubate cells at 37°C (5% CO2)
Sample collection:
-
Collect samples right after heat shock and 1, 2, 3 and 5 h after recovery
-
○
Experiment 2: skip the sample collection at 2 h of recovery
-
○
Remove vent filter membrane and transfer cells to 0.2 ml strip tubes
-
Wash cells:
-
○
Pellet cells down (1,000 × g, 1 min, RT)
-
○
Remove 90% of the supernatant (i.e., 180 μl)
-
○
Re‐suspend cells to 180 μl of ice cold PBS
-
○
Repeat the washing
-
○
Pellet cells down (1,000 × g, 1 min, RT)
-
○
Remove 90% of the supernatant (i.e., 180 μl)
-
○
Snap‐freeze cells in liquid nitrogen
-
○
The samples can be stored in −80°C for later processing
-
○
Cell lysis:
Thaw cells on ice
-
add 30 μl of 5/3× concentrated lysis buffer to a final concentration of
-
○
50 mM HEPES
-
○
0.8% NP‐40
-
□
Experiment 2: to collect total protein fraction, use 1% SDS
-
□
-
○
1.5 mM MgCl2
-
○
1× protease inhibitor cocktail
-
○
1× phosphatase inhibitor cocktail
-
○
0.25 U/μl benzonase
-
○
pH ≈ 7.4
-
○
-
Incubate at 4°C on a shaker for 1 h
-
○
Experiment 2: incubate total protein fractions 30 min at RT to avoid SDS precipitation
-
○
Two‐dimensional thermal proteome profiling
Preparing cells for the experiment:
-
Transfer cells to a conical centrifugation tube
-
○
Approximately 1.5 × 107 cells needed for each replicate
-
○
Pellet cells (190 × g, 3 min, RT)
Gently aspirate supernatant
Re‐suspend cell to medium to a final cell density of 5 × 105 cells/ml.
Heat treatment:
Distribute cells to two 96‐well PCR plates (200 μl/well, 3 × 105 cells/well)
Seal plates with aluminum foil
-
Place plates on pre‐warmed heat blocks for 10 min
-
○
Heat shock at 44°C
-
○
Mock shock at 37°C
-
○
Thermal shift assay:
After heat treatment, pool mock‐ and heat‐shocked samples to separate conical centrifugation tubes
Pellet cells (180 × g, 3 min, RT)
-
Wash cells:
-
○
Gently aspirate supernatant
-
○
Re‐suspend cells to 30 ml of PBS (RT)
-
○
Pellet cells (180 × g, 3 min, RT)
-
○
Gently aspirate supernatant
-
○
Re‐suspend cells to PBS (RT) to a final cell density of 5.5 × 106 cells/ml
-
Aliquot cells to a 96‐well PCR plate (100 μl/well, 5.5 × 105 cells/well)
-
○
Mock‐ and heat‐shocked samples each in separate rows (e.g., mock‐shocked samples aliquot to row A and heat‐shocked to row B)
-
○
Pellet cells on the plate (390 × g, 2 min, RT)
Gently aspirate 80% of the supernatant (80 μl)
Carefully re‐suspend pelleted cells to the remaining PBS
-
Put the plate on a thermal cycler for 3 min
-
○
Temperature gradient: 37.0; 37.8; 40.4; 44.0; 46.9; 49.8; 52.9; 55.5; 58.6; 62.0; 65.4; and 66.3 °C
-
○
Equilibrate samples for 3 min at RT before placing the plate on ice
Cell lysis:
-
add 30 μl of 5/3× concentrated lysis buffer to a final concentration of
-
○
50 mM HEPES
-
○
0.8% NP‐40)
-
○
0.25 U/μl
-
○
1.5 mM MgCl2
-
○
1× protease inhibitor cocktail
-
○
1× phosphatase inhibitor cocktail
-
○
pH ≈ 7.4
-
○
Incubate at 4°C on a shaker for 1 h
Lysates can be stored in −80°C for later processing
Extraction of soluble protein fraction
-
Pre‐wet 96‐well filter plate (0.45 μm):
-
○
Add 50 μl of lysis buffer to each well
-
○
Place filter plate of top of a 96‐well collection plate
-
○
Centrifuge lysis buffer through the filter (500 g, 5 min, 4°C)
-
○
Discard the collection plate
-
○
Transfer lysates on to a pre‐wet 96‐well filter plate.
Place filter plate of top of a 96‐well collection plate
Centrifuge lysates through the filter (500 g, 5 min, 4°C)
-
The flow‐through on the collection plate contains the soluble fraction
-
○
Optional: measure protein concentration (with, e.g., BCA assay) or dry samples on a vacuum concentrator
-
○
Samples can be stored in −80°C for later processing
Viability assay
Cell viability after heat shock and during recovery was estimated with CellTiter‐Glo Luminescent Cell Viability Assay (Promega). Cells were pelleted, density was adjusted to 5 × 104 cells/ml with fresh medium and aliquot to 96‐well PCR plates (104 cells/well). Cells were exposed to a heat treatment for 10 min in thermal cycler (Agilent SureCycler 8800) with different temperature for each aliquot (37.0; 39.6; 41.5; 43.6; 45.7; 47.5; 49.6; 52.0; 54.3, and 54.9°C).
After heat treatment, cells were allowed to recover at 37°C (5% CO2). Samples were collected after heat shock and after 5 h of recovery. Half of each sample (5 × 103 cells) was transferred to a luminescence plate (PerkinElmer Optiplate‐96). Cells from the remaining half were pelleted, and equal volume (to match the sample) of supernatant was transferred to the luminescence plate (as blank measures). Equal volume of CellTiter‐Glo reagent was added to each well, plate was mixed on a shaker for 2 min, and luminescence was recorded after 10 min. Luminescence was measured with TECAN Infinite M1000 PRO.
Blank measurement was subtracted from each sample. All samples were done as technical triplicates.
Protein extraction and digestion
For protein extraction, we used a modified version of SP3 sample preparation protocol (Hughes et al, 2014; Moggridge et al, 2018; Smits et al, 2019). Samples (5–15 μg of protein depending on sample availability) were mixed with 47.6% ethanol/2.4% formic acid binding buffer containing carboxylate modified magnetic particles (Sera‐Mag SpeedBead Carboxylate Modified Magnetic Particles, Hydrophilic Ref#45152105050250, Hydrophobic Ref#65152105050250). Proteins were let to bind to particles for 15 min at RT on shaker. Particle‐bound proteins were transferred to a filter plate (Millipore MultiScreen, 0.22 μm, MSGVN2250) and centrifuged (1,000 g, 1 min, RT) to remove binding buffer. Proteins were washed four times with 70% ethanol and digested over‐night in a digestion solution [90 mM HEPES, 5 mM CAA, 1.25 mM TCEP, 200 ng/sample trypsin (Promega, V5111), 200 ng/sample Lys‐C (FUJIFILM Wako, 125‐05061)] at RT on a shaker.
After digestion, peptides were collected by centrifugation (1,000 g, 1 min, RT). Residual particle‐bound peptides were eluted with 2% DMSO, collected by centrifugation (1,000 g, 1 min, RT), and added to the original peptide sample. Samples were dried.
TMT labeling
Peptides were dissolved in water and TMT labels (Thermo Fisher TMT10plex, TMT11‐131C) (dissolved in acetonitrile) were added (with final acetonitrile concentration of 28.6%). Labeling reaction was conducted at RT for 1 h on a shaker. The reaction was quenched with 1.1% hydroxylamine for 15 min. Labeled samples were pooled and diluted with 0.05% formic acid to decrease acetonitrile concentration below 5%.
The labeling scheme for recovery assay with 11 TMT labels was as follows: mock‐shocked samples (mock shock, 1, 2, 3, and 5 h of recovery), pre‐shock control and heat‐shocked samples (heat shock, 1, 2, 3, and 5 h of recovery) given in the order of increasing TMT reporter ion mass (i.e., from 126 to 131C) (see Fig 1B).
The labeling scheme for 2D‐TPP with 10 TMT labels was as follows: mock shock replicate one (temperature one: TMT126, temperature two: TMT129N), mock shock replicate two (127N, 129C), heat shock replicate one (127C, 130N), heat shock replicate two (128N, 130C), and heat shock replicate three (128C, 131). In other words, samples from two adjacent temperatures of the temperature gradient were combined in each TMT set (see Fig 5A).
Peptide desalting
Samples were transferred to an OASIS microplate (Waters HLB μElution plate, 186001828BA) for desalting. After binding peptides to the columns, they were washed two times with 0.05% formic acid and finally eluted with 0.05% formic acid/80% acetonitrile. Peptides were dried.
Offline fractionation
Samples were dissolved in 20 mM ammonia and injected for reverse phase fractionation under high pH conditions. Samples were fractionated to 32 fraction and partially pooled to reduce the amount of fractions to 12. Fractions were dried.
Quantitative mass spectrometry
Peptides were dissolved in 0.1% formic acid and subjected to liquid‐chromatography using an UltiMate 3000 RSLC nano LC system (Thermo Fisher Scientific). The LC system was equipped with a trapping cartridge (Acclaim PepMap 100 C18 LC column: 5 μm particles with 100 Å pores, 5 mm column with 300 μm inner diameter) for online desalting and an analytical column (Waters nanoEase HSS C18 T3, 75 μm × 25 cm, 1.8 μm, 100 Å) for separation. Peptides were loaded on the trapping cartridge for 3 min with 0.05% TFA in LC‐MS grade water at a flow rate of 30 μl/min. Peptides were eluted using buffers A (0.1% formic acid in LC‐MS grade water) and B (0.1% formic acid in LC‐MS grade acetonitrile) using increasing concentrations of buffer B at a flow rate of 0.3 μl/min. During a total analysis time of 120 min, the concentration of buffer B increased from initial 2–4% in the first 4 min, to 8% in the next 2 min, to 28% in the next 96 min, and finally to 40% in the next 10 min, followed by a 4 min washing step at 85% B before returning to initial conditions.
Peptides were injected to either a Q Exactive Plus Orbitrap (QE Plus, Thermo Fisher Scientific) or Orbitrap Fusion Lumos (FL) both using a Nanospray Flex ion source. In the following, the parameters are given for QE Plus and in parenthesis for FL. Mass spectrometers were operated in positive ion mode with spray voltage of 2.3 kV (2.4 kV) and capillary temperature of 275°C (300°C). Full scan MS spectra were acquired for a mass range of 375–1,200 m/z (375–1,500 m/z) were in profile mode with a resolution of 70,000 (120,000) with a maximum fill time of 250 ms (64 ms) or automatic gain control with a maximum of 3 × 106 ions (4 × 105 ions).
On the MS scan, data‐dependent acquisition was applied by selectively fragmenting top 10 peptide peaks (3 s cycle time) with a charge state of 2–4 (2–7) using dynamic exclusion window of 30 s (60 s) and mass window of 0.7 m/z (0.7 m/z) for isolation. Selected peptides were fragmented with normalized collision energy of 32 (38). MS/MS spectra were acquired in profile mode with a resolution of 35,000 (30,000) and an automatic gain control target of 2 × 105 ions (1 × 105 ions). The first mass was set to 100 m/z.
Data analysis
Raw mass spectrometry data were processed with isobarQuant (Franken et al, 2015). For protein identification (against human database in UniProt), Mascot search engine was used with the following search parameters: digestion with trypsin, maximum of three missed cleavages, 10 ppm peptide tolerance, and 0.02 Da MS/MS tolerance; carbamidomethylation of cysteines and TMT labels on lysine as fixed modifications; acetylation of N‐terminus, methionine oxidation, and TMT label on N‐terminus as variable modifications. For SILAC‐TMT data, two separate Mascot searches were conducted: first with the settings described above for light and then a slightly modified search for heavy. The heavy search included a 10 Da heavier arginine and 8 Da heavier TMT label attached to lysine as fixed modifications. The rationale for using heavier TMT label on lysine was to mimic 8 Da heavier lysine since Mascot does not allow for two separate modifications for one amino acid—in this case a heavy lysine and a TMT tag.
After peptide and protein identification with Mascot, peptide level quantification (based on TMT reporter intensities) was conducted and peptide intensities were summed to protein level with isobarQuant (Franken et al, 2015). The isobarQuant output (protein level data) was imported to R (https://www.R-project.org). Proteins identified as contaminants or reverse database hits were filtered out. In addition, only proteins quantified with at least two unique peptides were kept for the following analysis. Protein intensities were then log2‐transformed.
Batch effects were removed from protein intensities of each TMT channel with R package limma (Ritchie et al, 2015) using removeBatchEffect function. Resulting intensities were normalized using variance stabilization (vsn) method with R package vsn (Huber et al, 2002). Missing values were imputated with R package MSnbase (Gatto & Lilley, 2012) using impute function.
For SILAC data in recovery assay, we used a normalization approach where protein intensities from light‐labeled proteins (pre‐existing proteins) were first normalized and these normalization coefficients were applied to heavy‐labeled (newly synthesized) proteins. We justify this approach since light‐labeled proteins generally should have consistent intensities throughout the time course while the intensities of heavy‐labeled proteins should increase over time. Therefore, these patterns are preserved throughout the analysis. In addition, it is expected that the intensity and coverage of heavy labeled proteins is relatively low and thus any normalization based on them would be strongly biased toward high abundant protein species. It is worth mentioning that any background degradation in pre‐existing fraction (light) present in both, mock‐ and heat‐shocked samples, is masked away by the normalization approach, although reported protein half‐lives are generally much longer than 5 h (Schwanhausser et al, 2011; Mathieson et al, 2018).
Ratios between heat shock and mock shock were calculated for each time point. For heavy‐labeled proteins, a ratio against pre‐shocked control was calculated separately for heat‐shocked and mock‐shocked samples.
To statistically examine the solubility changes in heat shock, a LIMMA analysis was used to test for difference in heat shock/mock shock ratios (referred to as solubility in the main text) in light‐labeled proteins. A difference was assigned significant if Benjamini–Hochberg‐adjusted P‐value was below 0.05 and fold change below 2/3. These proteins are referred to as aggregators in the main text.
2D‐TPP data were analyzed as described before (Becher et al, 2018). Briefly, within all conditions, each temperature was normalized with vsn separately and ratio against 37°C sample in the temperature gradient was calculated for each temperature. The principle behind calculating scores for thermal stability is based on summing up differences between heat‐ and mock‐shocked samples in every temperature point; to correct for the aggregation already taken place in heat‐shocked sample, the average difference in the first two temperature points (37.0 and 37.8°C) were subtracted from all temperature points. When correcting for the aggregation, we assume that no aggregation has taken place in the mock‐shocked sample in these temperatures. In practice, an iterative bootstrapping approach was used for each protein: data from one replicate were randomly selected for each temperature and scores for thermal stability were calculated within 500 rounds. These iterated scores were transformed to z‐scores, and their mean was tested for deviation from zero (i.e., no change in thermal stability). From that comparison, Benjamini–Hochberg‐adjusted P‐value was calculated for each protein to estimate local false discovery rate (FDR). Finally, the means from every protein were transformed to z‐scores and depict the final score for thermal stability. R package fdrtool (Strimmer, 2008) was used to calculate global FDRs. Proteins quantified from less than six temperatures were filtered out. Protein was assigned as hit if both, local and global FDR, were below 0.01; destabilized hits had a negative score for thermal stability and stabilized hits had a positive score for thermal stability.
Protein localization annotations were from Human Protein Atlas (www.proteinatlas.org) (Thul et al, 2017). Annotation with reliability levels of “Approved”, “Supported” or “Validated” were included.
Gravy score for each protein was calculated as a sum of the values assigned to each amino acid in a protein sequence: arginine (−4.5), lysine (−3.9), asparagine (−3.5), aspartate (−3.5), glutamine (−3.5), glutamate (−3.5), histidine (−3.2), proline (−1.6), tyrosine (−1.3), tryptophan (−0.9), serine (−0.8), threonine (−0.7), glycine (−0.4), alanine (1.8), methionine (1.9), cysteine (2.5), phenylalanine (2.8), leucine (3.8), valine (4.2), and isoleucine (4.5).
Isoelectric points and molecular weights were calculated using R package Peptides (Osorio et al, 2015).
The predicted fraction of intrinsically disordered regions in proteins was from D2P2 database (Oates et al, 2013).
Protein secondary structure prediction was done with R package DECIPHER (Wright, 2016) using default settings. For each secondary structure element (sheet, helix, or coil), the predicted proportion in the protein sequence was calculated.
The amino acid sequence of the canonical isoform for each protein was used as input for calculating gravy score, isoelectric point, molecular weight, or predicting secondary structure elements.
For protein complex annotations, a manually curated database integrated from multiple sources (Ori et al, 2016) was used (including complexes with minimum of five distinct proteins).
Gene ontology (GO) term enrichments were conducted with R package clusterProfiler (Yu et al, 2012). Enrichment analysis was based on hypergeometric test with a cutoff of 0.05 for Benjamini–Hochberg‐adjusted P‐value.
List of human proteins linked to GO term “cytoplasmic stress granule” (GO:0010494) was collected from Gene Ontology Annotation Database (Huntley et al, 2015).
In comparisons between means of distributions (Figs 2B, C, E–G and 5C, and EV4) and correlation analysis (Figs 2D and 3E–H), the normality of distributions was estimated with a Shapiro–Wilk test. A distribution was assigned to be normally distributed if P‐value in the test was at or above 0.05. In comparisons between two normally distributed data, a parametric test was used (t‐test, Pearson correlation); otherwise a non‐parametric alternative was used (Wilcoxon test, Spearman correlation). The used tests are indicated in the figure legends.
Immunofluorescence microscopy
HeLa cells were grown in Dulbecco's minimal essential medium containing 2 mM l‐Glutamine and 10% fetal calf serum at +37°C (5% CO2). 20,000 cells in 200 μl of medium were seeded on each well of 8‐well LabTek chambered imaging plates and incubated at +37°C with 5% CO2 for 24 h. The media was changed 30 min before the start of the heat shock experiment. For heat shock, cells were transferred to an incubator set to +44°C (5% CO2) for 10 min. For recovery, cells were transferred back to +37°C (5% CO2). The control cells remained in +37°C (5% CO2) throughout the experiment.
Cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Subsequently, the cells were permeabilized with 0.05% Triton X (10 min at room temperature) and washed twice with PBS. Non‐specific binding was blocked with 3%BSA in PBST (0.1% Tween 20 in PBS) for 1 h at room temperature. Following primary antibody, dilutions were prepared in 3%BSA containing PBST; 1:1,000 anti‐BRD4, 1:1,000 anti‐HDAC1, 1:500 anti‐HELLS, and 1:1,000 anti‐TARDBP. After blocking, the cells were incubated with the diluted primary antibodies for 22–24 h at 4°C. Goat anti‐mouse and Goat anti‐rabbit cross‐adsorbed secondary antibodies tagged with AlexaFluor‐488 were diluted at 1:2,000 ratio in 3%BSA containing PBST. Hoechst 33342 was added to the diluted secondary antibody solution at a final concentration of 5 μg/ml. Cells were washed three times with PBST before incubating them with Hoechst‐containing secondary antibodies. Following an incubation at room temperature for 90 min, the cells were washed three times with PBST and stored PBS for imaging.
Cells were imaged on Zeiss 780 NLO confocal microscope using a 63×/1.4 oil immersion objective and argon laser. The images were acquired with the following settings for the different fluorophores: Hoechst—Ex: 405 nm, Em: 410–479 nm and AlexaFluor‐488—Ex: 488 nm, Em: 489–585 nm. The microscope was controlled using Zen 2012 software. The images were processed using Fiji, ImageJ.
Author contributions
MMS supervised the study. MMS, TAM, MR, and FS designed the study. TAM, MR, and DH performed the experiments. SS designed, performed, and analyzed immunofluorescence microscopy experiments. MMS, TAM, NK, and FS analyzed the data. MMS and TAM wrote the manuscript. All authors critically reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Expanded View
Review Process File
Acknowledgements
We thank members of the Savitski laboratory for discussions. We thank André Mateus for insightful and critical review of the manuscript. We thank the Proteomics Core Facility at the European Molecular Biology Laboratory (EMBL) for expert support. This work was supported by the EMBL. Open access funding enabled and organized by Projekt DEAL.
Mol Syst Biol. (2020) 16: e9500
Data availability
The datasets produced in this study are available in the following database: Proteomics data: PRIDE PXD017291 (https://www.ebi.ac.uk/pride/archive/projects/PXD017291).
References
- Aggarwal S, Talukdar NC, Yadav AK (2019) Advances in higher order multiplexing techniques in proteomics. J Proteome Res 18: 2360–2369 [DOI] [PubMed] [Google Scholar]
- Aprile‐Garcia F, Tomar P, Hummel B, Khavaran A, Sawarkar R (2019) Nascent‐protein ubiquitination is required for heat shock‐induced gene downregulation in human cells. Nat Struct Mol Biol 26: 137–146 [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer‐Hartl M, Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353: aac4354 [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Lemeer S, Savitski MM, Kuster B (2012) Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem 404: 939–965 [DOI] [PubMed] [Google Scholar]
- Bauerlein FJB, Saha I, Mishra A, Kalemanov M, Martinez‐Sanchez A, Klein R, Dudanova I, Hipp MS, Hartl FU, Baumeister W et al (2017) In situ architecture and cellular interactions of PolyQ inclusions. Cell 171: 179–187.e10 [DOI] [PubMed] [Google Scholar]
- Becher I, Werner T, Doce C, Zaal EA, Togel I, Khan CA, Rueger A, Muelbaier M, Salzer E, Berkers CR et al (2016) Thermal profiling reveals phenylalanine hydroxylase as an off‐target of panobinostat. Nat Chem Biol 12: 908–910 [DOI] [PubMed] [Google Scholar]
- Becher I, Andres‐Pons A, Romanov N, Stein F, Schramm M, Baudin F, Helm D, Kurzawa N, Mateus A, Mackmull MT et al (2018) Pervasive protein thermal stability variation during the cell cycle. Cell 173: 1495–1507 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beedholm R, Clark BF, Rattan SI (2004) Mild heat stress stimulates 20S proteasome and its 11S activator in human fibroblasts undergoing aging in vitro . Cell Stress Chaperones 9: 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B, Lorenzo Gotor N, Dhar R, Cirillo D, Baldrighi M, Tartaglia GG, Lehner B (2016) A concentration‐dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Rep 16: 222–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, Hyman AA (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324: 1729–1732 [DOI] [PubMed] [Google Scholar]
- Brennan CM, Vaites LP, Wells JN, Santaguida S, Paulo JA, Storchova Z, Harper JW, Marsh JA, Amon A (2019) Protein aggregation mediates stoichiometry of protein complexes in aneuploid cells. Genes Dev 33: 1031–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciryam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M (2013) Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep 5: 781–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciryam P, Kundra R, Morimoto RI, Dobson CM, Vendruscolo M (2015) Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol Sci 36: 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier NC, Schlesinger MJ (1986) The dynamic state of heat shock proteins in chicken embryo fibroblasts. J Cell Biol 103: 1495–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier NC, Heuser J, Levy MA, Schlesinger MJ (1988) Ultrastructural and biochemical analysis of the stress granule in chicken embryo fibroblasts. J Cell Biol 106: 1131–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2007) Is proteomics the new genomics? Cell 130: 395–398 [DOI] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C (2010) Widespread protein aggregation as an inherent part of aging in C. elegans . PLoS Biol 8: e1000450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Deane CA, Brown IR (2017) Components of a mammalian protein disaggregation/refolding machine are targeted to nuclear speckles following thermal stress in differentiated human neuronal cells. Cell Stress Chaperones 22: 191–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Gygi SP (2012) Hyperplexing: a method for higher‐order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci Signal 5: rs2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, Beynon RJ (2009) Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J Proteome Res 8: 104–112 [DOI] [PubMed] [Google Scholar]
- Doyle SM, Genest O, Wickner S (2013) Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol 14: 617–629 [DOI] [PubMed] [Google Scholar]
- Franken H, Mathieson T, Childs D, Sweetman GM, Werner T, Togel I, Doce C, Gade S, Bantscheff M, Drewes G et al (2015) Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat Protoc 10: 1567–1593 [DOI] [PubMed] [Google Scholar]
- Frottin F, Schueder F, Tiwary S, Gupta R, Korner R, Schlichthaerle T, Cox J, Jungmann R, Hartl FU, Hipp MS (2019) The nucleolus functions as a phase‐separated protein quality control compartment. Science 365: 342–347 [DOI] [PubMed] [Google Scholar]
- Fuxreiter M, Simon I, Bondos S (2011) Dynamic protein‐DNA recognition: beyond what can be seen. Trends Biochem Sci 36: 415–423 [DOI] [PubMed] [Google Scholar]
- Gatto L, Lilley KS (2012) MSnbase‐an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28: 288–289 [DOI] [PubMed] [Google Scholar]
- Grousl T, Ivanov P, Malcova I, Pompach P, Frydlova I, Slaba R, Senohrabkova L, Novakova L, Hasek J (2013) Heat shock‐induced accumulation of translation elongation and termination factors precedes assembly of stress granules in S. cerevisiae . PLoS ONE 8: e57083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergeth SP, Schneider R (2015) The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep 16: 1439–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcik M, Sonenberg N (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6: 318–327 [DOI] [PubMed] [Google Scholar]
- Hosp F, Gutierrez‐Angel S, Schaefer MH, Cox J, Meissner F, Hipp MS, Hartl FU, Klein R, Dudanova I, Mann M (2017) Spatiotemporal proteomic profiling of Huntington's disease inclusions reveals widespread loss of protein function. Cell Rep 21: 2291–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Davis RJ (2016) Cell signaling and stress responses. Cold Spring Harb Perspect Biol 8: a006072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18(Suppl 1): S96–S104 [DOI] [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J (2014) Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10: 757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley RP, Sawford T, Mutowo‐Meullenet P, Shypitsyna A, Bonilla C, Martin MJ, O'Donovan C (2015) The GOA database: gene Ontology annotation updates for 2015. Nucleic Acids Res 43: D1057–D1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibstedt S, Sideri TC, Grant CM, Tamas MJ (2014) Global analysis of protein aggregation in yeast during physiological conditions and arsenite stress. Biol Open 3: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Kedersha N, Anderson P (2018) Stress granules and processing bodies in translational control. Cold Spring Harb Perspect Biol 11: a032813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase‐modulated stress granules contain a diverse proteome and substructure. Cell 164: 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Arnsburg K, Scior A, Szlachcic A, Guilbride DL, Morimoto RI, Bukau B, Nillegoda NB (2017) In vivo properties of the disaggregase function of J‐proteins and Hsc70 in Caenorhabditis elegans stress and aging. Aging Cell 16: 1414–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein‐Miles J, Scior A, Deuerling E, Morimoto RI (2013) The nascent polypeptide‐associated complex is a key regulator of proteostasis. EMBO J 32: 1451–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15: 384–396 [DOI] [PubMed] [Google Scholar]
- Kramer RM, Shende VR, Motl N, Pace CN, Scholtz JM (2012) Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys J 102: 1907–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuckelkorn U, Knuehl C, Boes‐Fabian B, Drung I, Kloetzel PM (2000) The effect of heat shock on 20S/26S proteasomes. Biol Chem 381: 1017–1023 [DOI] [PubMed] [Google Scholar]
- Mateus A, Maatta TA, Savitski MM (2017) Thermal proteome profiling: unbiased assessment of protein state through heat‐induced stability changes. Proteome Sci 15: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus A, Bobonis J, Kurzawa N, Stein F, Helm D, Hevler J, Typas A, Savitski MM (2018) Thermal proteome profiling in bacteria: probing protein state in vivo . Mol Syst Biol 14: e8242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, Becher I et al (2018) Systematic analysis of protein turnover in primary cells. Nat Commun 9: 689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD et al (2012) Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem 84: 7469–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane E, Sin C, Zauber H, Wells JN, Donnelly N, Wang X, Hou J, Chen W, Storchova Z, Marsh JA et al (2016) Kinetic analysis of protein stability reveals age‐dependent degradation. Cell 167: 803–815.e21 [DOI] [PubMed] [Google Scholar]
- Miller SB, Mogk A, Bukau B (2015) Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J Mol Biol 427: 1564–1574 [DOI] [PubMed] [Google Scholar]
- Mivechi NF (1989) Heat sensitivity, thermotolerance, and profile of heat shock protein synthesis of human myelogenous leukemias. Can Res 49: 1954–1958 [PubMed] [Google Scholar]
- Moggridge S, Sorensen PH, Morin GB, Hughes CS (2018) Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J Proteome Res 17: 1730–1740 [DOI] [PubMed] [Google Scholar]
- Mogk A, Bukau B, Kampinga HH (2018) Cellular handling of protein aggregates by disaggregation machines. Mol Cell 69: 214–226 [DOI] [PubMed] [Google Scholar]
- Mosser DD, Ho S, Glover JR (2004) Saccharomyces cerevisiae Hsp104 enhances the chaperone capacity of human cells and inhibits heat stress‐induced proapoptotic signaling. Biochemistry 43: 8107–8115 [DOI] [PubMed] [Google Scholar]
- Muhlhofer M, Berchtold E, Stratil CG, Csaba G, Kunold E, Bach NC, Sieber SA, Haslbeck M, Zimmer R, Buchner J (2019) The heat shock response in yeast maintains protein homeostasis by chaperoning and replenishing proteins. Cell Rep 29: 4593–4607.e8 [DOI] [PubMed] [Google Scholar]
- Nillegoda NB, Bukau B (2015) Metazoan Hsp70‐based protein disaggregases: emergence and mechanisms. Front Mol Biosci 2: 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Kirstein J, Szlachcic A, Berynskyy M, Stank A, Stengel F, Arnsburg K, Gao X, Scior A, Aebersold R et al (2015) Crucial HSP70 co‐chaperone complex unlocks metazoan protein disaggregation. Nature 524: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nillegoda NB, Wentink AS, Bukau B (2018) Protein disaggregation in multicellular organisms. Trends Biochem Sci 43: 285–300 [DOI] [PubMed] [Google Scholar]
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztanyi Z, Uversky VN, Obradovic Z, Kurgan L et al (2013) D(2)P(2): database of disordered protein predictions. Nucleic Acids Res 41: D508–D516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell JD, Tsechansky M, Royall A, Boutz DR, Ellington AD, Marcotte EM (2014) A proteomic survey of widespread protein aggregation in yeast. Mol BioSyst 10: 851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386 [DOI] [PubMed] [Google Scholar]
- Ori A, Iskar M, Buczak K, Kastritis P, Parca L, Andres‐Pons A, Singer S, Bork P, Beck M (2016) Spatiotemporal variation of mammalian protein complex stoichiometries. Genome Biol 17: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio D, Rondon‐Villarreal P, Torres R (2015) Peptides: a package for data mining of antimicrobial peptides. R J 7: 4–14 [Google Scholar]
- Parsell DA, Kowal AS, Singer MA, Lindquist S (1994) Protein disaggregation mediated by heat‐shock protein Hsp104. Nature 372: 475–478 [DOI] [PubMed] [Google Scholar]
- Rampelt H, Kirstein‐Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B (2012) Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J 31: 4221–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard FB, Eberhard D, Werner T, Franken H, Childs D, Doce C, Savitski MF, Huber W, Bantscheff M, Savitski MM et al (2015) Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat Methods 12: 1129–1131 [DOI] [PubMed] [Google Scholar]
- Reis‐Rodrigues P, Czerwieniec G, Peters TW, Evani US, Alavez S, Gaman EA, Vantipalli M, Mooney SD, Gibson BW, Lithgow GJ et al (2012) Proteomic analysis of age‐dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell 11: 120–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback JA, Katanski CD, Kear‐Scott JL, Pilipenko EV, Rojek AE, Sosnick TR, Drummond DA (2017) Stress‐triggered phase separation is an adaptive. Evolutionarily tuned response. Cell 168: 1028–1040 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Wang T, Yan P, Gomes ED, Dunphy MP, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H et al (2016) The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538: 397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque A, Iloro I, Ponte I, Arrondo JL, Suau P (2005) DNA‐induced secondary structure of the carboxyl‐terminal domain of histone H1. J Biol Chem 280: 32141–32147 [DOI] [PubMed] [Google Scholar]
- Rüdiger S, Germeroth L, Schneider‐Mergener J, Bukau B (1997) Substrate specificity of the DnaK chaperone determined by screening cellulose‐bound peptide libraries. EMBO J 16: 1501–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad S, Cereghetti G, Feng Y, Picotti P, Peter M, Dechant R (2017) Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat Cell Biol 19: 1202–1213 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Lindquist SL (1990) HSP104 required for induced thermotolerance. Science 248: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Savitski MM, Mathieson T, Zinn N, Sweetman G, Doce C, Becher I, Pachl F, Kuster B, Bantscheff M (2013) Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J Proteome Res 12: 3586–3598 [DOI] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FB, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S et al (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346: 1255784 [DOI] [PubMed] [Google Scholar]
- Savitski MM, Zinn N, Faelth‐Savitski M, Poeckel D, Gade S, Becher I, Muelbaier M, Wagner AJ, Strohmer K, Werner T et al (2018) Multiplexed proteome dynamics profiling reveals mechanisms controlling protein homeostasis. Cell 173: 260–274.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Senohrabkova L, Malcova I, Hasek J (2019) An aggregation‐prone mutant of eIF3a forms reversible assemblies escaping spatial control in exponentially growing yeast cells. Current Genet 65: 919–940 [DOI] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Krykbaeva I, Taipale M, Lindquist S, Burge CB (2013) Widespread regulation of translation by elongation pausing in heat shock. Mol Cell 49: 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J (2011) The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell‐free system. PLoS ONE 6: e26319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits AH, Ziebell F, Joberty G, Zinn N, Mueller WF, Clauder‐Munster S, Eberhard D, Falth Savitski M, Grandi P, Jakob P et al (2019) Biological plasticity rescues target activity in CRISPR knock outs. Nat Methods 16: 1087–1093 [DOI] [PubMed] [Google Scholar]
- Sridharan S, Kurzawa N, Werner T, Gunthner I, Helm D, Huber W, Bantscheff M, Savitski MM (2019) Proteome‐wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat Commun 10: 1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K (2008) fdrtool: a versatile R package for estimating local and tail area‐based false discovery rates. Bioinformatics 24: 1461–1462 [DOI] [PubMed] [Google Scholar]
- Sui X, Pires DEV, Ormsby AR, Cox D, Nie S, Vecchi G, Vendruscolo M, Ascher DB, Reid GE, Hatters DM (2020) Widespread remodeling of proteome solubility in response to different protein homeostasis stresses. Proc Natl Acad Sci USA 117: 2422–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia GG, Pawar AP, Campioni S, Dobson CM, Chiti F, Vendruscolo M (2008) Prediction of aggregation‐prone regions in structured proteins. J Mol Biol 380: 425–436 [DOI] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM et al (2017) A subcellular map of the human proteome. Science 356: eaal3321 [DOI] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B (2010) Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 11: 777–788 [DOI] [PubMed] [Google Scholar]
- Uemura E, Niwa T, Minami S, Takemoto K, Fukuchi S, Machida K, Imataka H, Ueda T, Ota M, Taguchi H (2018) Large‐scale aggregation analysis of eukaryotic proteins reveals an involvement of intrinsically disordered regions in protein folding. Sci Rep 8: 678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan JS, Lindquist S (2014) Mechanisms of protein‐folding diseases at a glance. Dis Model Mech 7: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, Semple JI, Garcia‐Verdugo R, Lehner B (2009) Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138: 198–208 [DOI] [PubMed] [Google Scholar]
- Vihervaara A, Mahat DB, Guertin MJ, Chu T, Danko CG, Lis JT, Sistonen L (2017) Transcriptional response to stress is pre‐wired by promoter and enhancer architecture. Nat Commun 8: 255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuzman D, Levy Y (2012) Intrinsically disordered regions as affinity tuners in protein‐DNA interactions. Mol BioSyst 8: 47–57 [DOI] [PubMed] [Google Scholar]
- Wallace EW, Kear‐Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM et al (2015) Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162: 1286–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M et al (2015) Widespread proteome remodeling and aggregation in aging C. elegans . Cell 161: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Schubert D, Sawaya MR, Eisenberg D, Riek R (2010) Multidimensional structure‐activity relationship of a protein in its aggregated states. Angew Chem Int Ed Engl 49: 3904–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weids AJ, Ibstedt S, Tamas MJ, Grant CM (2016) Distinct stress conditions result in aggregation of proteins with similar properties. Sci Rep 6: 24554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch WJ, Feramisco JR (1984) Nuclear and nucleolar localization of the 72,000‐dalton heat shock protein in heat‐shocked mammalian cells. J Biol Chem 259: 4501–4513 [PubMed] [Google Scholar]
- Wentink A, Nussbaum‐Krammer C, Bukau B (2019) Modulation of amyloid states by molecular chaperones. Cold Spring Harb Perspect Biol 11: a033969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T, Becher I, Sweetman G, Doce C, Savitski MM, Bantscheff M (2012) High‐resolution enabled TMT 8‐plexing. Anal Chem 84: 7188–7194 [DOI] [PubMed] [Google Scholar]
- Wright ES (2016) Using DECIPHER v2.0 to analyze big biological sequence data in R. R J 8: 352–359 [Google Scholar]
- Yu G, Wang L, Han Y, He Q (2012) ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanton SJ, Pugh BF (2006) Full and partial genome‐wide assembly and disassembly of the yeast transcription machinery in response to heat shock. Genes Dev 20: 2250–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Expanded View
Review Process File
Data Availability Statement
The datasets produced in this study are available in the following database: Proteomics data: PRIDE PXD017291 (https://www.ebi.ac.uk/pride/archive/projects/PXD017291).