

Abstract
Objective
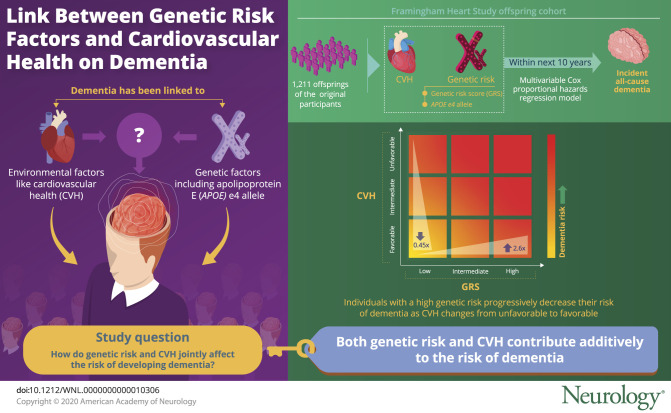
To determine the joint role of ideal cardiovascular health (CVH) and genetic risk on risk of dementia.
Methods
We categorized CVH on the basis of the American Heart Association Ideal CVH Index and genetic risk through a genetic risk score (GRS) of common genetic variants and the APOE ε4 genotype in 1,211 Framingham Heart Study (FHS) offspring cohort participants. We used multivariable Cox proportional hazards regression models to examine the association between CVH, genetic risk, and incident all-cause dementia with up to 10 years of follow-up (mean 8.4 years, 96 incident dementia cases), adjusting for age, sex, and education.
Results
We observed that a high GRS (>80th percentile) was associated with a 2.6-fold risk of dementia (95% confidence interval [CI] of hazard ratio [HR] 1.23–5.29; p = 0.012) compared with having a low GRS (<20th percentile); carrying at least 1 APOE ε4 allele was associated with a 2.3-fold risk of dementia compared with not carrying an APOE ε4 allele (95% CI of HR 1.49–3.53; p = 0.0002), and having a favorable CVH showed a 0.45-fold lower risk of dementia (95% CI of HR 0.20–1.01; p = 0.0527) compared to having an unfavorable CVH when all 3 components were included in the model. We did not observe an interaction between CVH and GRS (p = 0.99) or APOE ε4 (p = 0.16).
Conclusions
We observed that both genetic risk and CVH contribute additively to dementia risk.
Both genetic and environmental risks for Alzheimer disease (AD) and dementia have been identified. The APOE ε4 allele is the strongest established genetic risk factor for sporadic AD, associated with an ≈50% increase in risk of AD.1 Furthermore, genome-wide association studies have identified >20 single nucleotide polymorphisms (SNPs) associated with late-onset AD.2,3 A genetic risk score (GRS) of these SNPs is associated with an ≈16% increase in the late-onset, sporadic form of AD.4–6
There is increasing evidence that cardiovascular health (CVH) is associated not only with cardiovascular outcomes but also with late-life neurologic diseases, including dementia and AD.7–9 However, studies that have associated CVH with dementia and AD have not considered the full set of genetic risk factors. In addition, there is a need to identify target populations (e.g., those with a high genetic risk) in whom prevention efforts may have the greatest impact on reducing dementia burden. Our goals were to determine the joint relationship of genetic risk and CVH on risk of dementia and to determine whether having an ideal CVH mitigates the risk of dementia in those at high genetic risk.
Methods
Standard protocol approvals, registrations, and patient consents
All participants provided written informed consent. Study protocols and consent forms were approved by the institutional review board at the Boston University Medical Center.
Framingham Heart Study
The Framingham Heart Study (FHS) is a community-based, longitudinal cohort study that was initiated in 1948. The original cohort comprised 5,209 residents of Framingham, MA, and these participants have undergone up to 32 examinations, performed every 2 years, that have involved detailed history taken by a physician, a physical examination, and laboratory testing.10 In 1971, a total of 5,124 offspring of the participants in the original cohort and the spouses of these offspring were enrolled in an offspring cohort. The participants in the offspring cohort have completed up to 9 quadrennial examinations,11 with the latest examination cycle ending in 2014. We leveraged individuals in the FHS offspring cohort to investigate the joint association of CVH and genetic burden on 10-year risk of dementia.
We assessed CVH as defined below from examination cycle 5 data collected between 1991 and 1995 (mean age 55 years). Follow-up for dementia began at examination cycle 7 (1998–2001). This allowed us to assess CVH before the onset of symptoms for dementia and to begin follow-up for dementia ≈7 years after the assessment of CVH. CVH before the start of follow-up (examination cycle 5) was previously shown to be associated with risk of incident dementia after examination cycle 7.8
Of the 5,124 FHS offsprings, 3,539 individuals attended examination cycle 7. We excluded participants <60 years of age (n = 1,288), those with prevalent dementia at examination cycle 7 (n = 8), and those with no follow-up for dementia (n = 87), which left 1,367 individuals. We in addition excluded individuals missing education level (n = 33) or genetic data (n = 123) leaving 1,211 participants available for analysis (table 1).
Table 1.
Characteristics of 1,211 individuals from the FHS at examination 5 contributing to the study

Definition of dementia
The surveillance methods and dementia tracking for the FHS have been published.12–14 In brief, cognitive status has been monitored at each examination cycle with the use of the Mini-Mental State Examination (MMSE).15 The MMSE was used to flag participants for dementia review if performance fell below education-based cutoff scores at any examination, a decline of >3 points was observed between consecutive examinations, or a decline of >5 points was observed from the participant’s highest obtained MMSE score. In addition, participants were screened for dementia in response to referrals or concern from participants, their family, their doctor, or other health professional. A dementia review panel, which includes a neurologist and a neuropsychologist, reviews possible cognitive decline and dementia. The panel determines whether a person had dementia, the dementia subtype, and the date of diagnosis using data from multiple sources.12 After a participant dies, a medical panel reviews medical and nursing records up to the date of death and includes an assessment of whether the participant might have had cognitive decline since his or her last examination.16 This medical panel refers any participants who might have had cognitive decline to the dementia review panel for postmortem review. A diagnosis of dementia is made in accordance with DSM-IV.
We considered a maximum of 10 years of follow-up from examination cycle 7. We focused our analysis on dementia given the limited sample size and observed number of cases of AD. A sensitivity analysis was performed with AD as the outcome.
Ideal CVH
The American Heart Association (AHA) Life's Simple 717,18 highlights the following 7 components to CVH: physical activity, cholesterol, healthy diet, blood pressure, healthy weight, blood glucose, and smoking status.
Ideal CVH score was calculated by summing together 7 optimal levels of CVH according to the AHA guidelines at examination 5. One point was added for each of the following optimal levels of the CVH score components: current self-reported nonsmoker, body mass index <25 and >18.5 kg/m2, adequate physical activity, a healthy diet, untreated total cholesterol <200 mg/dL, untreated resting blood pressure <120/<80 mm Hg, and fasting blood glucose <100 mg/dL. All factors were obtained from the FHS clinic except diet and physical activity, which were measured with the use of a validated food frequency and physical activity index questionnaire, respectively. Physical activity was calculated with the following formula as per previous FHS publications8,19: 1 × sleep hours/day + 1.1 × sedentary hours/day + 1.5 × slight activity hours/day + 2.4 × moderate activity hours/day + 5 × heavy activity hours/day. The top quartile of this score was used to indicate ideal physical activity, which corresponds qualitatively to the definition used by the AHA.17 Diet scores were adapted from the AHA guidelines,17 consistent with a previous FHS publication.8,19 Blood pressures were measured twice at the same examination by a physician, with the averaged systolic and diastolic values used in the ideal CVH score.
The ideal CVH score ranged from 0 to 7 and was used to define 3 categories9,19 of CVH: unfavorable CVH (0–2 optimal levels), intermediate CVH (3–4 optimal levels), and favorable CVH (≥5 optimal levels).
Genetics
FHS participants had DNA extracted and provided consent for genotyping in the 1990s. All available eligible participants were genotyped at Affymetrix (Santa Clara, CA) through an National Heart, Lung, and Blood Institute–funded SNP-Health Association Resource project using the Affymetrix GeneChip Human Mapping 500K Array Set and 50K Human Gene Focused Panel. Genotypes were imputed to the Haplotype Reference Consortium SNPs with the use of Mach/Minimac as done previously.2 In addition, APOE ε4 genotypes were obtained.
Twenty-three SNPs, not including APOE, have previously been aggregated into a GRS and shown to be associated with AD (table 2).5 We used these SNPs and their corresponding weights to define our GRS. We created a weight GRS for each individual i as . All SNPs had imputation quality >0.6 in our data.
Table 2.
SNPs and weights from van der Lee et al.5 contributing to the genetic risk score

We categorized individuals has having high genetic risk if they were above the 80th percentile of the GRS, as intermediate genetic risk if they were in the middle 60th percentile of the GRS distribution (20th–80th percentiles), and as low genetic risk if they were below the <20th percentile of the GRS distribution. We looked at APOE ε4 genotypes separately from the GRS.
Statistical analysis
We used multivariable Cox proportional hazards regression models to examine the association of ideal CVH and genetic risk with incident all-cause dementia using up to 10 years of follow-up. We present hazard ratios (HRs) accompanied by 95% confidence intervals (CIs). In addition, we calculated cumulative hazards of dementia over 10 years. We examined the assumption of proportionality of hazards using statistical tests and graphic diagnostics based on scaled Schoenfeld residuals. We adjusted all models for age at examination 5, sex, and education (coded as at least high school degree [1] or not [0]). Genetic principal components of ancestry are routinely used to adjust for population structure in genetic analyses.20,21 These are well defined and have been used previously in genetic analyses within the FHS. We observed that none of the top 5 principal components were associated with dementia status and did not include them in our models. We determined the absolute risk increase/reduction and the number needed to harm/treat using methods appropriate for Cox models.22,23
We considered results statistically significant if p < 0.05. We conducted all analyses in SAS 9.3 (SAS Institute Inc, Cary, NC) or R-3.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
Data availability
Data are available through database of Genotypes and Phenotypes (accession No. phs000007.v30.p11).
Results
Of the 1,211 persons with dementia at follow-up, 96 (7.9%) developed dementia within 10 years (mean follow-up 8.4 years). Of the 96 cases of dementia, 67 were clinically consistent with AD. We observed that cases with dementia were older at examination cycle 5 (67 vs 61 years old), were more likely to be women (63% vs 52%), and were less likely to have a high school education (88% vs 94%) compared with those who did not develop dementia. Cases with incident dementia also had a higher rate of type 2 diabetes mellitus (52% vs 38%) compared with those who did not develop dementia after adjustment for age and sex (table 1). We found that 10% (24) of the sample had both a favorable CVH and a high GRS, with 23 of these 24 not developing dementia. On the other hand, we found that 28% (68) of the sample had an unfavorable CVH and a low GRS, which was 36% of those with dementia and 28% of those without dementia.
We first associated CVH and genetic risk (both APOE ε4 carriers and a GRS) individually with dementia. We observed that after adjustment for age, sex, and education, having a high GRS was associated with a 2.6-fold hazard of dementia compared with having a low GRS (95% CI 1.27–5.45; p = 0.009) and carrying at least 1 APOE ε4 allele was associated with a 2.3-fold higher hazard of dementia compared with not carrying an APOE ε4 allele (95% CI 1.49–3.54; p = 0.0002) (table 3). We observed that the absolute risk increase of having a high GRS is 1.5% with a number needed to harm of 65 compared to having a low GRS at 5 years, while the absolute risk increase of having an APOE ε4 allele is 5.7% with a number needed to harm of 18 at 5 years. On the other hand, a favorable CVH showed a 0.41-fold lower hazard of dementia (95% CI 0.18–0.91, p = 0.03) compared to an unfavorable CVH. We observed that the absolute risk reduction of having a favorable CVH is 1.4% with a number needed to treat of 70 at 5 years. We observed that when we included genetic risk factors (both APOE ε4 carriers and a GRS) and CVH in the same model, the effect estimates remained consistent with the effect estimates seen when the factors were analyzed separately (table 3).
Table 3.
CVH and genetics on risk of dementia

In a sensitivity analysis with incident AD as the outcome (67 cases and 1,144 individuals who did not develop AD), we observed that having a high GRS was associated with a 4.3-fold higher hazard of AD compared with having a low GRS (95% CI 1.39–13.29; p = 0.01), carrying at least 1 APOE ε4 allele was associated with a 3.0-fold higher hazard of AD compared with not carrying an APOE ε4 allele (95% CI 1.81–4.96; p < 0.0001), and having a favorable CVH was associated with a nonsignificant decreased hazard of AD compared to an unfavorable CVH (HR 0.47, 95% CI 0.18–1.24; p = 0.13). Furthermore, we found similar effects of the GRS, APOE ε4, and CVH on risk of dementia when maternal family history was included in a model with a sample of 479 individuals (26 with dementia) with information on maternal dementia status by 80 years of age (results not shown).
We next calculated the cumulative hazards of dementia over 10 years and at the 5-year point by genetic risk and CVH. Individuals with a high GRS and unfavorable CVH had the highest cumulative hazards of dementia, followed by those with intermediate GRS and unfavorable CVH (figure 1A). On the other hand, those with low GRS and favorable CVH had the lowest cumulative hazards of dementia, followed by those with low GRS and intermediate CVH. We observed that individuals with a high genetic risk have a greater reduction in 5-year cumulative hazards when moving from an unfavorable to favorable CVH (reduction of 0.016) compared to individuals with a low genetic risk (0.0063) (figure 1B). A similar pattern can be seen with cumulative hazards by APOE ε4 status and CVH (figure 2). APOE ε4–positive individuals with either unfavorable or intermediate CVH had the highest cumulative hazards of dementia, while APOE ε4–positive individuals with a favorable CVH had a lower cumulative hazards than individuals who were ε4 negative with unfavorable CVH (figure 2).
Figure 1. Hazards of dementia by GRS and CVH.

(A) Cumulative hazards of dementia over 10 years of follow-up by genetic risk score (GRS) category (1 = low [dotted line], 2 = intermediate [dashed line], and 3 = high [solid line]) and cardiovascular health (CVH) category (favorable [pink], intermediate [green], and unfavorable [blue]). (B) Cumulative hazard estimates at 5 years of follow-up (middle of maximum follow-up) by GRS and CVH. Error bars represent 95% confidence interval of estimated cumulative hazards at 5 years.
Figure 2. Hazards of dementia by APOE ε4 and CVH.

(A) Cumulative hazards of dementia over 10 years of follow-up by APOE ε4 status (APOE4+ [solid line] and APOE4− [dotted line]) and cardiovascular health (CVH) category (favorable [pink], intermediate [green], and unfavorable [blue]). (B) Cumulative hazard estimates at 5 years of follow-up (middle of maximum follow-up) by APOE ε4 status and CVH category. Error bars represent 95% confidence interval of estimated cumulative hazards at 5 years.
We did not find an interaction between CVH categories and GRS groups (p = 0.99) or APOE ε4 status (p = 0.16), indicating that CVH does not appear to modify the association of genetic risk on dementia. We calculated the least detectable effect of an interaction using Quanto 1.2.4 given our data and the observed effects for CVH and APOE ε4 on dementia. We assumed a population dementia frequency of 5% and set α = 0.05, power of 80%, case n = 96, and a control:case ratio of 11.61. Under these parameters, the least detectable odds ratio for the interaction was 3.5. Therefore, we were powered to detect only a strong interaction.
Discussion
In a large community-based sample, we show that both genetics and modifiable CVH are independently associated with risk of dementia. We did not find a statistically significant interaction between CVH categories and GRS groups or APOE ε4 status, although we were underpowered to detect such effects. Our results suggest that CVH and genetic factors additively contribute to AD risk.
Both genetic and cardiovascular risk factors for dementia have been described. Estimates suggest that ≈35% of cases of dementia may be attributable to modifiable risk factors, including CVH.13 Indeed, the AHA Ideal CVH guidelines have been touted as a mechanism to promote optimal brain health and to reduce the risk of cognitive impairment.8,9 In our study, having a favorable, relative to unfavorable, CVH score was associated with a 59% decrease in the risk of incident dementia. In a prior publication,8 it was found that ideal CVH ≈7 years before incident events was inversely associated with both incident all-cause dementia (HR 0.80, p = 0.02) and clinical AD (HR 0.79, p = 0.006) in 1,364 individuals in the FHS offspring cohort. Furthermore, in ≈6,600 older individuals (mean age 74 years), ideal CVH was associated with lower risk of dementia and lower rates of cognitive decline.10 Our results are consistent with these finding for dementia. We find that favorable CVH is associated with lower risk of dementia (95% CI of HR 0.18–0.91, p = 0.03) without genetic factors in the model (table 3). When genetic factors are included in the model, the effect estimates remain consistent, but the CIs become slightly wider. We in addition report how these hazards of dementia change according to genetic risk factors. Due to the reduced sample size when the genetic data were incorporated, we did not find a significant association between favorable CVH and incident AD, as was previously reported,8 but we observed similar effect sizes.
Beyond the effects of age, carriage of the APOE ε4 allele is the strongest risk factor for late-onset dementia. Although several other common genetic variants have small individual effects, their combination also modifies the risk of incident dementia over and above the effects of APOE.5 Indeed, we confirm that both APOE ε4 carriage and a polygenic risk score of common genetic variants are associated with a higher risk of incident dementia, particularly clinical AD.
Despite the available information on CVH and genetic variants as dementia risk factors, the potential for CVH to modify high genetic risk of dementia has not been well described. Genetic risk factors have been shown to lower the age at dementia onset24 and to associate with more rapid accumulation of AD pathology.25 Therefore, enrichment of individuals with high genetic risk is one strategy used to increase power in clinical trials of emerging therapies.26 However, this enrichment strategy would be viable for primary prevention trials only if individuals with high genetic risk for AD had the potential for dementia risk modification. Our data suggest that this is indeed the case because the potential for dementia prevention via cardiovascular interventions is independent of genetic risk for late-onset AD. Therefore, persons at highest genetic risk would have the greatest absolute reduction in risk.
In the absence of curative treatment for dementia, there is considerable interest in the risks and benefits of disclosing genetic information (e.g., APOE ε4 status) to research participants.27–29 Our data suggest that persons with high genetic risk, i.e., individuals with either an APOE ε4 allele or a high GRS, may gain by adopting healthy CVH behaviors. This is consistent with observations suggesting that both APOE ε4 and cardiovascular risk factors, in conjunction, aid in predicting cognitive decline.30,31
Our results are consistent with those previously reported,32 which looked at >196,383 participants from the UK Biobank study. This study did not include data on body mass index, total cholesterol, blood pressure, and glucose in their measurement of the healthy lifestyle score, as we did here. In addition, they found that a majority of UK Biobank participants followed a favorable lifestyle, while a majority of the participants in our study had an unfavorable or intermediate CVH profile. Despite these differences, our results are consistent with those from the UK Biobank study. The advantages of our study include careful and comprehensive adjudicated outcomes based on up to 10 years of longitudinal follow-up. We used a validated measure of CVH17–19 and CVH measured before the onset of symptoms of dementia, which diminishes the impact that dementia has on changes in CVH, limiting reversed causality.
Limitations of our study include, first, that the FHS is composed of individuals of European descent; therefore, these results may not be generalizable to non-European populations. Samples of European ancestry were used to derive the weights for the GRS2; thus, the GRS is most appropriately applied to a European ancestry sample. Second, we were limited by our small sample size, which leads to large error bars in the estimates of cumulative hazards. Third, some of the genetic markers may influence both CVH and dementia, although we observed that including only 1 factor or both factors in the model did not change the results.
In a community-based sample, we observed that both genetic risk and CVH are additively associated with incident dementia. Further studies are needed to replicate these results in larger and ethnically diverse samples.
Acknowledgment
The authors thank the FHS participants, the study team, and the investigators and staff of the neurology team.
Glossary
- AD
Alzheimer disease
- AHA
American Heart Association
- CI
confidence interval
- CVH
cardiovascular health
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, 4th edition
- FHS
Framingham Heart Study
- GRS
genetic risk score
- HR
hazard ratio
- MMSE
Mini-Mental State Examination
- SNP
single nucleotide polymorphism
Appendix. Authors

Footnotes
CME Course: NPub.org/cmelist
Study funding
The FHS is supported by the National Heart, Lung, and Blood Institute (contract 75N92019D00031, N01-HC-25195, HHSN268201500001) and by grants from the National Institute on Aging (R01 AG054076, R01 AG049607, R01 AG033193, U01 AG049505, U01 AG052409, RF1 AG059421) and the National Institute of Neurologic Disorders and Stroke (NS0 17950 and UH2 NS100605). Funding for SNP-Health Association Resource Affymetrix genotyping was provided by National Heart, Lung, and Blood Institute contract N02-HL-64278. M.P. Pase is funded by a National Heart Foundation of Australia Future Leader Fellowship (102052).
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Tsai MS, Tangalos EG, Petersen RC, et al. Apolipoprotein E: risk factor for Alzheimer disease. Am J Hum Genet 1994;54:643–649. [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet 2019;51:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chouraki V, Reitz C, Maury F, et al. Evaluation of a genetic risk score to improve risk prediction for Alzheimer's disease. J Alzheimers Dis 2016;53:921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Lee SJ, Wolters FJ, Ikram MK, et al. The effect of APOE and other common genetic variants on the onset of Alzheimer's disease and dementia: a community-based cohort study. Lancet Neurol 2018;17:434–444. [DOI] [PubMed] [Google Scholar]
- 6.Escott-Price V, Sims R, Bannister C, et al. Common polygenic variation enhances risk prediction for Alzheimer's disease. Brain 2015;138:3673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet 2017;390:2673–2734. [DOI] [PubMed] [Google Scholar]
- 8.Pase MP, Beiser A, Enserro D, et al. Association of ideal cardiovascular health with vascular brain injury and incident dementia. Stroke 2016;47:1201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samieri C, Perier MC, Gaye B, et al. Association of cardiovascular health level in older age with cognitive decline and incident dementia. JAMA 2018;320:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawber TR, Kannel WB, Lyell LP. An approach to longitudinal studies in a community: the Framingham Study. Ann NY Acad Sci 1963;107:539–556. [DOI] [PubMed] [Google Scholar]
- 11.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families: the Framingham Offspring Study. Am J Epidemiol 1979;110:281–290. [DOI] [PubMed] [Google Scholar]
- 12.Seshadri S, Beiser A, Au R, et al. Operationalizing diagnostic criteria for Alzheimer's disease and other age-related cognitive impairment, part 2. Alzheimers Dement 2011;7:35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seshadri S, Wolf PA, Beiser A, et al. Lifetime risk of dementia and Alzheimer's disease: the impact of mortality on risk estimates in the Framingham study. Neurology 1997;49:1498–1504. [DOI] [PubMed] [Google Scholar]
- 14.Satizabal CL, Beiser AS, Chouraki V, Chene G, Dufouil C, Seshadri S. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med 2016;374:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State:” a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 16.Au R, Seshadri S, Knox K, et al. The Framingham Brain Donation Program: neuropathology along the cognitive continuum. Curr Alzheimer Res 2012;9:673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd-Jones DM, Hong Y, Labarthe D, et al. Defining and setting national goals for cardiovascular health promotion and disease reduction: the American Heart Association's strategic Impact Goal through 2020 and beyond. Circulation 2010;121:586–613. [DOI] [PubMed] [Google Scholar]
- 18.Sacco RL. The new American Heart Association 2020 goal: achieving ideal cardiovascular health. J Cardiovasc Med 2011;12:255–257. [DOI] [PubMed] [Google Scholar]
- 19.Xanthakis V, Enserro DM, Murabito JM, et al. Ideal cardiovascular health: associations with biomarkers and subclinical disease and impact on incidence of cardiovascular disease in the Framingham Offspring Study. Circulation 2014;130:1676–1683. [DOI] [PubMed] [Google Scholar]
- 20.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 22.Altman DG, Andersen PK. Calculating the number needed to treat for trials where the outcome is time to an event. BMJ 1999;319:1492–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Austin PC. Absolute risk reductions and numbers needed to treat can be obtained from adjusted survival models for time-to-event outcomes. J Clin Epidemiol 2010;63:46–55. [DOI] [PubMed] [Google Scholar]
- 24.Sando SB, Melquist S, Cannon A, et al. APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer's disease; a case control study from central Norway. BMC Neurol 2008;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim YY, Mormino EC; Alzheimer's Disease Neuroimaging Initiative. APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology 2017;89:1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer's Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis 2011;26(suppl 3):321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hietaranta-Luoma HL, Luomala HT, Puolijoki H, Hopia A. Using ApoE genotyping to promote healthy lifestyles in Finland: psychological impacts: randomized controlled trial. J Genet Couns 2015;24:908–921. [DOI] [PubMed] [Google Scholar]
- 28.Kane RA, Kane RL. Effect of genetic testing for risk of Alzheimer's disease. N Engl J Med 2009;361:298–299. [DOI] [PubMed] [Google Scholar]
- 29.Cohn-Hokke PE, van Swieten JC, Pijnenburg YAL, Tibben A, Meijers-Heijboer H, Kievit A. The effect of predictive testing in adult-onset neurodegenerative diseases on social and personal life. J Genet Couns 2018;27:947–954. [DOI] [PubMed] [Google Scholar]
- 30.Haan MN, Shemanski L, Jagust WJ, Manolio TA, Kuller L. The role of APOE epsilon4 in modulating effects of other risk factors for cognitive decline in elderly persons. JAMA 1999;282:40–46. [DOI] [PubMed] [Google Scholar]
- 31.Falsetti L, Viticchi G, Buratti L, Grigioni F, Capucci A, Silvestrini M. Interactions between atrial fibrillation, cardiovascular risk factors, and ApoE genotype in promoting cognitive decline in patients with Alzheimer's disease: a prospective cohort study. J Alzheimers Dis 2018;62:713–725. [DOI] [PubMed] [Google Scholar]
- 32.Lourida I, Hannon E, Littlejohns TJ, et al. Association of lifestyle and genetic risk with incidence of dementia. JAMA 2019;322:430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available through database of Genotypes and Phenotypes (accession No. phs000007.v30.p11).