

Abstract
The spike protein receptor binding domain (S-RBD) is a necessary corona-viral protein for binding and entry of coronaviruses (COVs) into the host cells. Hence, it has emerged as an attractive antiviral drug target. Therefore, present study was aimed to target severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) S-RBD with novel bioactive compounds to retrieve potential candidates that could serve as anti-coronavirus disease 2019 (COVID-19) drugs. In this paper, computational approaches were employed, especially the structure-based virtual screening followed by molecular dynamics (MD) simulation as well as binding energy analysis for the computational identification of specific terpenes from the medicinal plants, which can block SARS-CoV-2 S-RBD binding to Human angiotensin-converting enzyme 2 (H-ACE2) and can act as potent anti-COVID-19 drugs after further advancements. The screening of focused terpenes inhibitors database composed of ~1000 compounds with reported therapeutic potential resulted in the identification of three candidate compounds, NPACT01552, NPACT01557 and NPACT00631. These three compounds established conserved interactions, which were further explored through all-atom MD simulations, free energy calculations, and a residual energy contribution estimated by MM-PB(GB)SA method. All these compounds showed stable conformation and interacted well with the hot-spot residues of SARS-CoV-2 S-RBD. Conclusively, the reported SARS-CoV-2 S-RBD specific terpenes could serve as seeds for developing potent anti-COVID-19 drugs. Importantly, the experimentally tested glycyrrhizin (NPACT00631) against SARS-CoV could be used further in the fast-track drug development process to help curb COVID-19.
Keywords: COVID-19, SARS-CoV-2, Terpenes, Computer aided drug design and discovery
Highlights
-
•
SARS-CoV-2 S-RBD attachment to ACE2 receptor is characterized to discover potent inhibitors.
-
•
Computational approaches identified potent inhibitors from medicinal plant's terpenes.
-
•
Selected inhibitors represent stable/strong binding with important binding-site residues.
-
•
Selected inhibitors can facilitate the development of natural therapeutic solution of COVID-19.
1. Introduction
Human civilization has faced many pathogenic outbreaks, in the past few decades. Among these, the occurrence and reoccurrence of CoVs related outbreaks have caused fetal respiratory disorders [1,2]. The positive-sense signal strand RNA CoVs have envelop/capsid around and poses high frequency of genomic mutation and recombination, hence, categorized as extremely evolving viruses [3,4]. Around six species of human coronavirus are known to exist (HCoV-OC34, HCoV-229E, MERS-CoV, HCoV-NL63, SARS-CoV, and HCoV-HKU1) that are responsible for many respiratory disorders [3,5,6]. At present, the world is fronting a severe respiratory coronavirus disease 2019 (COVID-19) pandemic mainly due to a newly evolved infectious CoV strain: SARS-CoV-2 [1,4,7,8]. Up till now (September 25, 2020), COVID-19 has affected ~32.1 M people worldwide and caused ~980 K deaths. This havoc has not stopped yet and still thousands of people are being affected on daily basis. Presently, none of the vaccine/drug is available in the markets for human use against COVID-19 [[9], [10], [11]], while many of the drugs are in the trial process and a few successful trials have been reported as well, but all are on the very initial stage [12]. Among these drugs, remdesivir has emerged as a promising drug [13] that is now being trailed/tested on patients suffering from severe disease [13,14].
The SARS-CoV-2 RNA enters into the human cell by a dense glycosylated spike (S) protein [15] that is basically a fusion protein belonging from the trimeric class I. The S protein normally occurs in a metastable prefusion structure that can be modified substantially for the fusion of viral membrane into the host cell [15,16]. The infection of COVID-19 is mainly directed through the interaction of the host cell receptor, CoV enveloped affixed S protein and angiotensin converting 2 (ACE2) enzyme [17,18]. The binding S-protein has further two subunits, S1: [the receptor-binding domain (RBD)] and S2 (carryout the union of viral and host cell membrane) [17,18]. The subunit S2 of SARS-CoV-2 is conserved and ~ 99% similar to the bat SARS-CoV and human SARS-CoV. In contrast, the S1 subunit is evolved differently (only ~70% similarity with human and bat SARS-CoVs) [18,19]. The affinity among S-RBD of SARS-CoV-2 and H-ACE2 is ten times higher than SARS-CoV S-RBD (year 2003), which infers that H-ACE2 is an explicit receptor accountable for the attachment of a virus to the host cell [19,20]. The inter-species transmission of CoVs can be attributed to the SARS-CoV S-RBD residues (Leu472, Tyr442, Asn479, Thr487, and Asp480) that undergo natural selection in SARS-CoV-2 [20,21]. The residues such as, Lys353 and Lys31, present on H-ACE2 are said to be liable for the virus-binding hotspot residues that ultimately bind S-protein [21,22]. The Lys31 of H-ACE2 receptor is composed of a salt bridge between Glu35 and Lys31, while hotspot 353 contains an alternative salt bridge between Asp38 and Lys353, bounded by hydrophobic surroundings [19,22]. H-ACE2 is recognized in SARS-CoV-2 by its residue (Leu455 and Gln493) that are liable for desirable molecular interfaces with hotspot 31, thus improving the binding of a virus to H-ACE2. Furthermore, other main residues of S-protein offer more backing for Lys31 (Leu455, Ser494, Phe486). In serine based residue (494) of SARS-CoV-2, strengthens the structural solidity of Lys353 [19,23]. Due to its crucial function the S protein, it is important for designing a drug. The computational approaches are helpful in drug development against first line of infection of H-ACE2 and SARS-CoV-2 S-RBD complex.
Phytochemicals act as secondary metabolites and active constituents in medicinal plants e.g., flavonoids, alkaloids, and terpenes peptides [[24], [25], [26]]. These biologically active molecules are parts of defense system in plants hence, in turn, reduce the infections [24,27,28]. As a part of the remedial function, these compounds forage, scavenge and obstruct the viral entry and DNA\RNA replication [27,28]. Among these, terpenes are most common and represent a large category of secondary metabolites that are composed of 5-C isoprene units that are interlinked in a complex manner [29]. Most of the terpenoids are biologically active and are widely used worldwide in order to treat many ailments such as, derivatives of Taxol used to treat cancer artemisinin has antimalarial effects [30].Moreover, for the FDA-approved antiviral drugs, numerous in-silico researches have been conducted to differentiate new phytochemical compounds against SARS-CoV-2/COVID-19 [4,[31], [32], [33], [34], [35], [36], [37]].
There is a need to conduct a comprehensive analysis of the interactions of H-ACE2 receptor and S-RBD of SARS-CoV-2 in order to establish the remedies against COVID-19. Therefore, present study was conducted using high throughput screening (HTS) coupled with various computational techniques, including structure-based molecular docking approach, MD simulation approach and free binding energy MM/PBSA analysis, for the identification of novel medicinal plant based terpenes that can be helpful against SARS-CoV-2 infection. The comprehensive procedure and flowchart of the current study is given as Fig. 1 . The outcomes of this work are significant in the field of anti-COVID-19 terpenes drug efficacy and give a push to its progress as a drug against SARS-CoV-2.
Fig. 1.

A schematic flow chart of methodology used in this study.
2. Materials and methods
2.1. Ligand database preparation
For the development of Natural-based novel drug, a dataset of 1000 plant bioactive terpenes compounds with reported therapeutic potentials were downloaded from NPACT database [38] and MPD3 database [25]. All the compounds were optimized prior to docking using Autodock tools [39]. Polar hydrogens were added and gassteiger charges were computed of each compound in the library. Later, the compounds were saved in pdbqt format for docking purpose.
2.2. Receptor preparation and docking
Molecular docking of the terpenes dataset was conducted by Autodock Vina [40]. The of SARS-CoV-2 S-RBD crystal structure bound with the ACE2 receptor protein [41] was attained from the PDB database (PDB ID 6LZG) for docking purpose. All of the crystalline compounds were detached from the RBD structure prior to docking. The RBD structure was prepared using AutoDock tools [39,42]. Gassteiger charges were optimized and Polar hydrogens were added. The structure was saved in pdbqt format for docking. The Grid box was created with the size of 45 Å × 45 Å × 45 Å and centered on the important residues of RBD domain which binds to the ACE2 receptor. 20 poses for all compounds were created and results were analyzed on the basis of docking scores (kcal/mol).
2.3. Flexible docking
For flexible docking, the selection of top 100 compounds was made on the basis of docking scores. Flexible docking was also performed using Autodock Vina. The residues of binding pocket were retained flexible. Total 20 Confirmations were generated for every compounds. After docking top 50 compounds [based on docking scores (kcal/mol)] were chosen and visually inspected using PyMol [43].
2.4. In silico drug-likeness analysis and ADMET profiling
The available bioinformatics tool SwissADME (http://www.swissadme.ch/index.php) [44] was used for finding drug-likeness attributes. Lipinski's rule of five [45] was used to analyze the properties such as; hydrogen bond donor (HBD), hydrogen bond acceptor (HBA), molecular weight (MW), and lipophilicity (log P). PreADMET (https://preadmet.bmdrc.kr/) and pkCSM (http://biosig.unimelb.edu.au/pkcsm/) servers were employed to determine the ADMET (metabolism, distribution, excretion, absorption, and toxicity) attributes of target molecules.
2.5. Molecular dynamics simulations
Molecular dynamics simulations based on categorical solvent were conducted using GROMACS 5.1.4 (GROningen Machine for Chemical Simulations) to measure the binding and stability attributes of complex [46]. CHARMM36 force field was applied to achieve topology attributes of protein [47]. For force field ligand and topology parameters, CHARMM General Force Field (CGenFF) was employed [48]. Ligand-protein complexes resulted after docking were solvated in the cubical box through TIP3 water model. The neutralization of the system was performed by the addition of a suitable number of CL-ions. Energy was minimized in all systems through 10,000 steps of steepest descent approach. In order to balance/maintain the temperature of the system ranged from 0 to 300 K slowly for 250 ps, NVT ensemble was used. Moreover, simulation of the system was performed under NPT ensemble (at 1.0 bar and 300 K) for 250 ps. In order to quantify the long-range interfaces, Particle Mesh Ewald was used while for short-range Electrostatics and Van der Waals, a distance (10A) cut off was used [49]. For restraining all bonds, the use of LINCS (Linear Constraints Solver) algorithm was done [50]. Finally, 50 ns making run was performed and trajectories were saved after each 2 fs for all complex separately. Root Mean square fluctuations (RMSF), root mean square deviation (RMSD), SASA analysis and Radius of gyration (Rg) were done using GROMCAS modules.
2.6. Principle component analysis (PCA)
PCA/essential dynamics statistical tool was used to identify similarities and dissimilarities in a particular dataset and to detect patterns in a particular dataset [51]. The main idea lies in reducing the conformational space into essential subspace with varying degrees of freedom that comprises most of the positional atomic fluctuations. Thus, atomic fluctuations are expressed in the form of a covariance matrix and the transformation of the conformational space is done by diagonalization of the matrix. Only Cα atoms mean interatomic distance variation was applied to generate covariance matrix. Gmx covar module was used to construct gmx anaeig and covariance matrix module was employed to project trajectories on eigenvectors.
2.7. MM/PBSA binding free energy calculation
MM/PBSA was used to determine the binding free energies of protein-ligands [52] [53] [54]. G_mmpbsa module of GROMACS v.5.1.4 was employed to approximate the binding energies of simulated complex compounds [55]. The binding free energy snapshots for the last 30 ns of trajectory were estimated at 50 ps time interval. Overall, the extraction of 500 snapshots from the simulation trajectory (20 ns) was done for the MM/PBSA determinations. The binding free energies of the protein having ligand in liquid/solution were determined by following equations:
In the above equations, ΔGbind represents the net free binding energy, -TΔS corresponds to entropic energy, ΔEMM indicates molecular mechanics potential energy and can be split into van der Waals (ΔEvdw) energy and electrostatic (ΔEelec). The ΔEpolar and ΔESASA are used for polar contributions non-polar solvation energy, respectively. These methods have been well demonstrated in binding free energy calculations [56] for antiviral inhibitors [2,[57], [58], [59], [60], [61], [62], [63]].
3. Results
3.1. Structure of the RBD domain of the SARS-CoV-2
In order to obtain understanding about the interaction between H-ACE2 and SARS-CoV-2 S-RBD, first we analyzed the interface of S-RBD and H-ACE2 complex (Fig. 2A). We found as mentioned in literature, the overall interface is arbitrated essentially by polar interactions. A lengthy loop section of the RBD spans the arch-shaped α1 helix of the ACE2 similar to a bridge. The α2 helix and a ring that links the β3 and β4 anti-parallel strands, stated as loop 3–4, of the ACE2 also make partial assistances to the synchronization of the RBD. Overall, 14 potent residues of S-RBD (Phe486, Asn487, Ala475, Glu484, Tyr489, Leu455, Gln493, Tyr453, Ser494, Tyr449, Gln498, Gly496, Gly446 and Asn501) were identified that were making significant binding with ACE2 and contributing the stability of complex (Fig. 2B).
Fig. 2.

Interface of SARS-CoV-2 S-RBD and H-ACE2 receptor. (A) S-RBD & ACE2 complex 3D representation and (B) S-RBD residues responsible for interacting with ACE2 receptor.
3.2. Molecular docking
In the present study, a library of terpenes was screened against the SARS-CoV-2 S-RBD domain. The approach is on the basis of computationally fitting molecules for a target protein using 3D structure of the active site, subsequently the ranking of the docked compounds and interaction analysis. The residues located at the interface section of SARS-CoV-2 RBD-ACE2 were targeted in order to screen the terpene compounds using AutoDock Vina. The two step docking experiments were performed. In the first step of docking, the RBD was retained rigid and 1000 terpene compounds were docked and filtered on the basis of the docking score. In the second step of docking, top 100 compounds were selected and docked flexibly to the targeted sites. Only important interface residues of the RBD were treated flexibly in the flexible docking experiment. After flexible docking, 50 top hits were visually inspected on the basis of the interactions. 3-beta-O-[alpha-L-rhamnopyranosyl-(1- > 2)-alpha-L-arabinopyranosyl]olean-12-ene-28-O-[alpha-L-rhamnopyranosyl-(1- > 4)-beta-d-glucopyranosyl-(1- > 6)-beta-d-glucopyranosyl] ester (NPACT01552), 3-O-[beta-d-glucopyranosyl (1- > 3)-beta-D-galactopyranosyl(1- > 2)]-beta-D-glucuronopyranosyl-oleanolic acid-28-O-beta-D-glucopyranoside (NPACT01557) and Glycyrrhizin (NPACT00631) hits having energies −11 kcal/mol, −10.3 kcal/mol and − 9.5 kcal/mol, respectively, were selected for more analysis. The structures and characteristics of three finally screened compounds are displayed in Table 1 .
Table 1.
Details of the terpenes selected as top hits.
| IDs | Names | Source | Chemical Structure | Chemical Formula | Mwt (g/mol) | LogP | HBD | HBA |
|---|---|---|---|---|---|---|---|---|
| NPACT01552 | 3-beta-O-[alpha-L-rhamnopyranosyl-(1→2)-alpha-L-arabinopyranosyl]olean-12-ene-28-O-[alpha-L-rhamnopyranosyl-(1- > 4)-beta-d-glucopyranosyl-(1- > 6)-beta-d-glucopyranosyl] ester | Trevesia Palmata | 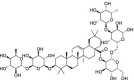 |
C59H96O26 | 1220.62 | −0.4 | 15 | 25 |
| NPACT01557 | 3-O-[beta-d-glucopyranosyl (1- > 3)-beta-D-galactopyranosyl(1- > 2)]-beta-D-glucuronopyranosyl-oleanolic acid-28-O-beta-D-glucopyranoside | Aralia Dasyphylla | 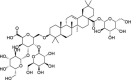 |
C55H88O24 | 1132.57 | −0.93 | 14 | 23 |
| NPACT00631 | Glycyrrhizin | Glycyrrhiza glabra | 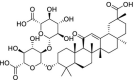 |
C42H62O16 | 822.9 | 3.13 | 8 | 16 |
3.3. Interaction analysis between RBD and compounds
Discovery studio software was used to prepare two-dimensional plot of the molecular interaction network of ligands with S-RBD while the docked poses for each of these molecules are portrayed in Fig. 3 . Out of 1000 terpenes, NPACT01552, NPACT01557 and NPACT00631 showed highest binding energies (−11 kcal/mol, −10.3 kcal/mol and − 9.5 kcal/mol, respectively) and interact with RBD remains through five, five and six hydrogen bonds, respectively. As mentioned above, RBD domain binds with the ACE2 receptor protein through these important interface residues Gly502, Gly496, Thr500, Gln498, Tyr449, Ala475, Asn487, lys417, Tyr489, Phe486, Glu484, Gln493, Ser494, Leu455 and Phe486. In the docked conformations, NPACT01552 displayed hydrogen bond interactions with Gly496, Tyr453, GLn493, Ser494 and Glu484 out of which Gly496, GLn493, Ser494, are important interface interacting residues. Moreover, NPACT01552 exhibited highest number of hydrophobic interactions with residues, Lys417, Leu452, Tyr489, Phe456 and Leu455. Out of these residues, Lys417, Tyr489 and Leu455 are important in the binding with the ACE2 receptor. NPACT01557 interacts with Thr500, Asn501, Gln498, TYr449 and Glu484 through H-bond and with Leu455 through hydrophobic interactions. NPACT00631 interacts with Asn501, Gln498, Gly496, Tyr449, Tyr453 and Glu484 through hydrogen bond interactions and interacts with Tyr489, Phe456 and Leu455 through hydrophobic interactions. All the compounds exhibited hydrogen bonds and hydrophobic interactions with the important interface residues. Table 2 summarizes the energy scores and interacting residues which interact with compounds of selected terpenes.
Fig. 3.

Illustration of the selected terpenes occupying the potent active site residues of SARS-CoV-2 S-RBD. (A) Surface representation of the binding mode of NPACT01552 (purple), NPACT01557 (pink) and NPACT00631 (yellow) to SARS-CoV-2 S-RBD (green). (B) The mechanism of molecular interactions of (a-b) NPACT01552 (purple), (c-d) NPACT01557 (pink) and (e-f) NPACT00631 (yellow) to SARS-CoV-2 S-RBD in 3D and 2D representations. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 2.
Binding energy score and details of interacting residues.
| Compound ID | Energy score (kcal/mol) | Hydrogen bond interacting residues | Hydrophobic interactions |
|---|---|---|---|
| NPACT01552 | −11 | Gly496, Tyr453, GLn493, Ser494, Glu484 | Lys417, Leu452, Tyr489, Phe456, Leu455 |
| NPACT01557 | −10.3 | Thr500, Asn501, Gln498, TYr449, Glu484 | Leu455 |
| NPACT00631 | −9.5 | Asn501, Gln498, Gly496, Tyr449, Tyr453, Glu484 | Tyr489, Phe456, Leu455 |
3.4. ADMET/Drug scan results
For further validation of the drug likeliness capability of selected compounds, all three terpene molecules were subjected to the Lipinski's rule of five: absorption, distribution, metabolism, excretion, and toxicity testing. These parameters were evaluated using different thresholds. All three hit compounds passed the threshold of drug ability (Table 1, Table 3 ).
Table 3.
ADMET Profiling for absorption, metabolism and toxicity parameters of identified compounds.
| Model | NPACT01552 | NPACT01557 | NPACT00631 |
|---|---|---|---|
| A. Absorption and distribution | |||
| Blood-brain barrier | No | No | No |
| Human intestinal absorption | High | High | High |
| Caco-2 permeability | Yes | Yes | No |
| P-glycoprotein inhibitor | Inhibitor | Inhibitor | Non inhibitor |
| Renal Organic cation transporter | Non inhibitor | Non inhibitor | Non inhibitor |
| B. Metabolism | |||
| CYP450 2C9 | Non inhibitor | Non inhibitor | Non inhibitor |
| CYP450 2D6 | Non inhibitor | Non inhibitor | Non inhibitor |
| CYP450 3A4 | Non inhibitor | Non inhibitor | Non inhibitor |
| CYP450 1A2 | Non inhibitor | Non inhibitor | Non inhibitor |
| CYP450 2C19 | Non inhibitor | Non inhibitor | Non inhibitor |
| C. Toxicity | |||
| AMES (mutagenicity) | Non ames toxic | Non ames toxic | Non ames toxic |
| Carcinogens | Non carcinogens | Non carcinogens | Non carcinogens |
3.5. Molecular dynamics simulations
Molecular dynamics (MD) simulation is the most widely used technique to assess the micro interaction between ligand structure and protein. Simulation of all complexes was done by 50 ns MD in order to access further its stability and dynamics. After MD simulations, RMSD, RMSF, Rg, and SASA were assessed.
3.5.1. Root mean square deviations (RMSD)
Root mean square deviations of backbone atoms of all the systems were analyzed to observe the stability of systems. The RMSD calculation of a structure along its trajectory relative to the reference structure tells the conformational changes in the structure. The RMSD curve indicates that the RBD-Apo, NPACT01552, and NPACT01557 complexes remained stable throughout the simulation with the average RMSD 0.12, 0.11 and 0.13 nm, respectively. There were no significant differences observed in NPACT01552, and NPACT01557 complexes. As a whole, the results of RMSD signify that the binding of these two compounds with RBD of S-protein is steady. However, compound NPACT00631 was stable in the start, but after 20 ns this compound fluctuated up to 0.24 nm RMSD until the end of simulation, which may indicate that this compound is more flexible as can be seen from Fig. 4A.
Fig. 4.

Comparison and detail representation of (A) RMSD: Root mean square deviations, (B) RMSF: Root mean square fluctuations, (C) Rg: Radius of gyration and (D) SASA: solvent-accessible surface area, over the time of 50 ns of reference RBD-Apo and all the selected terpenes complexes.
3.5.2. Root mean square fluctuations (RMSF)
Root means square fluctuations (RMSF) were measured in order to further compute the residual flexibility over 50 ns time. The variability in the conformation of trajectories can be monitored to calculate the RMSF for individual atoms. The average RMSF of overall positions of RBD-Apo, NPACT01552, and NPACT01557 is 0.10 nm while the average RMSF of NPACT00631 0.13, which is larger than the other two compounds, and Apo. Significant fluctuations were observed at the N-terminal and C-terminal in all trajectories. The overall residual fluctuations of the RBD-APO and complexes are shown in Fig. 4B. The total fluctuation of RBD-Apo, NPACT01552, and NPACT01557 was roughly the same except few residual fluctuations, such as, residue 386–389 and 477–479 exhibit higher fluctuations in all the systems. Besides, there were few residues, which showed more flexibility in the RBD-APO as compared to both complexes as evident from figure. Complex NPACT00631 has a larger conformational flexibility as compared to the RBD-Apo, complex NPACT01552, and NPACT01557 as evident from Fig. 4B.
3.5.3. Radius of gyration (Rg)
The radius of gyration (Rg) depicts the system density, and eventually impacts the folding rate as well as proteins stability. Rg was used to assess the complexes compactness and it was revealed that Rg of all systems was steady with the RMSD of system. This depicts that protein was stable and compressed throughout the 50 ns time Fig. 4C.
3.5.4. Solvent accessible surface area (SASA)
Solvent accessible surface area (SASA) is alternative method in order to retain the protein folding and stability. The calculated SASA values for wild type and mutants are displayed in Fig. 4D. The average SASA values for RBD-Apo, NPACT01552, NPACT01557 and NPACT00631 were found to be 109, 108, 108 and 109 nm2, respectively signifying that there were no notable differences in the available area of all the systems throughout the simulation process.
3.6. Principal component analysis (PCA)
All the simulation systems covered the same conformational space except complex NPACT00631. The trace of the covariance matrix and eigenvalues for RBD-Apo, NPACT01552, NPACT01557 and NPACT00631 was 1.47, 1.33, 1.47 and 2.84 nm2 respectively. The eigenvalues for RBD-Apo, NPACT01552 and NPACT01557 were nearly same, which indicate that these systems did not undergo random fluctuations, which can also be observed from the 2D projection of trajectories in Fig. 5 . In the Figure, each corresponds to one frame in the trajectory. Complex NPACT00631 has the highest eigenvalues, which indicate that this complex may have some random fluctuations. The distribution of the conformation of Complex NPACT00631 was broader than the RBD-Apo, NPACT01552 and NPACT01557 on both eigenvectors.
Fig. 5.

Two-dimensional principle component analysis (PCA) projections of trajectories obtained from 50 ns MD simulations. (A) RBD-Apo-black, (B) NPACT01552-red, (C) NPACT01557-blue and (D) NPACT00631-green. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.7. MM/PBSA binding free energy calculation
To clarify the energetics of the binding of compounds NPACT01552, NPACT01557 and NPACT00631 with the S-RBD quantitatively, we calculated the MM/PBSA calculations. Table 4 shows the binding free energies and energy components. The calculated bind free energy of complex NPACT01552 is −23.234 ± 41.377 kcal/mol, NPACT01557 is −62.565 ± 39.091 kcal/mol and of complex NPACT00631 is −38.100 ± 35.383 kcal/mol. The van der Waals energy term (ΔEvdw) and electrostatic interaction energy (ΔEele) were the major contributors to the binding free energy in complexes NPACT01552 and NPACT01557.
Table 4.
Binding Free energy (kcal/mol) analysis. ΔEvdw: van der Waals interaction energy; ΔEelec: electrostatic interaction energy; -TΔS: entropic energy; ΔESASA: SAS energy; ΔEpolar: polar solvation energy; ΔGbind: binding energy.
| Energy Terms (kcal/mol) | NPACT01552 | NPACT01557 | NPACT00631 |
|---|---|---|---|
| ΔEvdw | −78.347 ± 16.388 | −99.349 ± 14.024 | −74.056 ± 21.368 |
| ΔEelec | −29.915 ± 26.018 | −23.100 ± 14.708 | −9.993 ± 10.287 |
| -TΔS | 36.02 | 39.43 | 33.90 |
| ΔESASA | −11.464 ± 2.354 | −12.930 ± 2.115 | −9.965 ± 2.647 |
| ΔEpolar | 96.492 ± 57.747 | 72.814 ± 47.032 | 55.915 ± 38.991 |
| ΔGbind | −23.234 ± 41.377 | −62.565 ± 39.091 | −38.100 ± 35.383 |
3.8. Per-residue energy decomposition analysis
The energy contributions of the active residues to the total binding energies were also measured for each terpene-SRBD complex to comprehend the residues that contributed significantly to the interactions of inhibitor-protein. The values for binding energy of the residues at the interface that significantly contributed to the total binding energy are displayed in Fig. 6 . Residues Leu455, Phe456, Ala475, Phe486 and Tyr 489 contributed significantly in the total binding energy.
Fig. 6.

Per-residue energy decomposition analysis of potential interface-binding site residues of SARS-CoV-2 S-RBD.
4. Discussion
The comprehensive knowledge of the viral receptor identification mechanism is a key to understand the pathogenic, infectious, and the host specific nature of COVID-19 that is further important for developing remedial therapies, drugs and antiviral cures [19]. Till now, none of the drug or therapy can be specifically prescribed against SARS-CoV-2, while the application/development of new drugs is expensive and requires a lots of trial steps, hence, time consuming [4,64,65]. More worse is the prediction of WHO that COVID-19 may become endemic, which is an alarm in the scientific world that it has become more crucial to develop drugs against it immediately [66]. In this regard, the computational methods are scientifically sound to design extremely specific inhibitor for viral proteins, as an antiviral drug development/discovery. Recently, structural modeling has predicted a few drugs against SARS-CoV-2 proteins along with many other antiviral drugs [2,64,65,67].
The life cycle of the virus is consisted of four main phases, in the first phase, virus is attached to the host cell by identifying the specific receptors. In the second phase, it is fused with the host cell membrane and viral genetic material (RNA) is transferred into the host cell, (in next stage) later on, the viral genetic material gets a hold of host genome and start making structural and non-structural proteins and finally the viral parts are assembled and phages are ejaculated from host cell that are ready to attack other host cells [19,68]. In order to cure the infections caused by RNA viruses such as dengue virus, chikungunya virus, SARS, Ebola virus, Sindbis virus MERS, etc., it is necessary to target these viruses at any of the stage of their life cycle [19,69]. The drugs that block the entry of the virus into the host cell or inhibit the viral replication have already been introduced in the market for chikungunya, dengue, and other RNA viruses [19,70]. Inhibition of the capsid synthesis can also be a key for preventing viral infection at a budding stage [19,71]. Similarly, such drugs can be developed against SARS-CoV-2 for targeting/inhibition the viral growth at any stage of its life cycle. The envelop of SARS-CoV-2 contains S-protein that is liable for the interaction with ACE2 receptor on the host cells through RBD unit [18]. This is the basic and first step for the interaction of the SARS-CoV-2 with host cell and creation of infection. The antiviral drug that can target S-RBD–ACE2 interaction that is able to render the entry of virus into the host cell and hence, offer a fast resolution to combat SARS-CoV-2 inflammation [19,72].
Till now, there is no precise antiviral specific strategy for the treatment of COVID-19 (vaccine/drug), supportive care such as, supplementation with the combination of broad-spectrum antivirals, antibiotics, corticosteroids as well as convalescent plasma is widely employed [73]. More than 3/4th quarter of the world's population is using herbal medicines [25,27]. Out of the total 2,50,000 species of flora present on the earth ~80,000 poses medicinal attributes [24,27]. Many bioactive compounds are the part of plant biomass such as alkaloids, peptides, terpenes and flavonoids [4,28]. In all of these compounds, terpenes are the most abundant and largest group of 5-C secondary compounds/metabolites that consisted of isoprene subunits that can be interlinked with each other in 1000 s of ways [74]. The in silico research has proposed the screening of many novel phytochemical that can be used as inhibiting agent for many of key protease of SARS-CoV-2 [4,[31], [32], [33], [34], [35], [36], [37]].
In accordance with the results of 1000 focused terpenes with reported therapeutic potentials library screening, molecular docking was executed to obtain binding mode insights and crucial molecular interfaces of the designated terpenes and S-RBD protein obtained from SARS-CoV-2. Likewise, compounds were evaluated through MD approach, MM-PBSA and per-residue energy disintegration revealed that the relative significance of amino acid responsible for the binding reinforced under the influence of various molecules having binding free energy. NPACT01552, NPACT01557 and NPACT00631 were projected to be possible inhibitors against S-RBD of SARS-CoV-2 as these compounds raise the binding energies and as a result can interact and block the residues of S-RBD that recognize the attachment sites on the host cell. Furthermore, these complexes also exhibit successful ADMET attributes and these compounds are also side-effect free/have very low risk. The other drugs having poor ADMET can produce toxic metabolites [49] or may not be able to cross cell membranes barriers [50], and the potential solubilities were also approximated within the suitable range [51]. Many of the compounds were proposed as non-inhibitors for the isozymes, such as, cytochrome P450, but these drugs play a very crucial role in metabolism and enzymes inhibition that may leads to toxicity and deprived defecation in liver [52].
Among all 3 compounds, NPACT01552 (3-beta-O-[alpha-L-rhamnopyranosyl-(1- > 2)-alpha-L-arabinopyranosyl]olean-12-ene-28-O-[alpha-L-rhamnopyranosyl-(1- > 4)-beta-d-glucopyranosyl-(1- > 6)-beta-d-glucopyranosyl] ester) is most effective in hydrogen bond formation with Gly496, Tyr453, GLn493, Ser494 and Glu484 residues of S-RBD, with the energy score of −11 kcal/mol. NPACT01552 usually obtained from Trevesia Palmata and have significant anti-cancer activities reported [75]. NPACT01557 (3-O-[beta-d-glucopyranosyl (1- > 3)-beta-D-galactopyranosyl(1- > 2)]-beta-D-glucuronopyranosyl-oleanolic acid-28-O-beta-D-glucopyranoside) is the 2nd most effective having the energy score of −10.3 kcal/mol, whereas NPACT00631 (Glycyrrhizin) has the energy score of −9.5 kcal/mol and is at number three. NPACT01557 is extracted from Aralia Dasyphylla and well documented to have anticancer effects as well [76]. NPACT00631 is also known as Glycyrrhizin that is present in Glycyrrhiza glabra and has been successfully used to treat the HIV-1 and chronic hepatitis C in past [[77], [78], [79], [80], [81]]. Before this, Cinatl et al., [78] has determined the antiviral attributes of mycophenolic acid, ribavirin, 6-azauridine, glycyrrhizin, and pyrazofurin clinical isolates of coronavirus (FFM-2 and FFM-1). Glycyrrhizin (having the selectivity index of 67) is the most promising inhibitor the replication of SARS-CV in Vero cells. It also inhibits the penetration and adsorption of virus in the early steps of viral lifecycle. But at the same time, Glycyrrhizin was more effective once applied after the adsorption phase rather than during adsorption. It was also effective once applied at both phases before and after adsorption. In another study, Luo et al., has also discussed the medicinal properties of glycyrrhizin against COVID-19 with the following mechanisms; a) Binding S-RBD/ACE2 to inhibit SARS-CoV-2, b) control proin-flammatory cytokines, c) preventing the intracellular ROS buildup of, d) controlling thrombi,. e) preventing the fast production of airway exudates, and f) Persuading endogenous interferon against SARS-CoV-2 [82]. The current study has recognized the importance of glycyrrhizin in binding S-RBD and inhibiting the SARS-CoV-2 S-protein attachment with H-ACE2 receptor. Furthermore, selected hits in present study showed stronger binding energies, higher docking scores, and more closer interactions with the S-RBD residues than the compounds reported in earlier studies against SARS-CoV-2 S-RBD [64,[83], [84], [85], [86]]. More precisely, our work provides a detailed and integrate approach for the reorganization of novel terpenes based on COVID-19 inhibitors and can act as a starting point for the vitro testing foe the efficacy of these compounds. The in-depth explanation of interrelating remains together with the vibrant conformations approved over 50 ns MD simulations of recognized compounds presented the technique to develop safe and more actual inhibitors for the treatment of SARS-CoV-2.
5. Conclusions
The terpenes from plant source act as an inhibitor of the interaction of SARS-CoV-2 S-RBD and H-ACE2 receptors challenges the ability of SARS-CoV-2 to enter host cells as the main goal of our study. Our study has produced three new terpene hits: 3 NPACT01552, NPACT01557 and NPACT00631 as potential anti-COVID-19 drug candidates. All compounds exhibited affinity with a strong bond with S-RBD. These compounds have a hot residue of S-RBD that inhibits it. Therefore, it is necessary to explore more about these complexes through in vivo and other analyses. Current research offers a strong potential for the development of new, cost-effective and safe plant/nature-based medicines against SARS-CoV-2.
Abbreviations
- SARS-CoV-2
Severe Acute Respiratory Syndrome Corona virus 2
- COVID-19
Coronavirus disease 2019
- S
Spike
- RBS
Receptor Binding Domain
- ACE2
Angiotensin-Converting Enzyme 2
- MMPBSA
Molecular Mechanics Poisson-Boltzmann Surface Area.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (31770333, 31370329, 11631012), the Program for New Century Excellent Talents in University (NCET-12-0896), and the Fundamental Research Funds for the Central Universities (GK201403004).
CRediT authorship contribution statement
Ziyad Tariq Muhseen: Data curation, Visualization, Investigation, Writing- Original draft preparation. Alaa R. Hameed: Data curation, Validation, Writing- Reviewing and Editing. Halah M.H. Al-Hasani: Data curation, Validation, Writing- Reviewing and Editing. Muhammad Tahir ul Qamar: Software, Validation, Writing- Reviewing and Editing. Guanglin Li: Conceptualization, Methodology, Supervision.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Authors would like to acknowledge Shaanxi Normal University, Xi'an, China for providing facilities for this study.
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R.J. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alamri M.A., Tahir ul Qamar M., Mirza M.U., Bhadane R., Alqahtani S.M., Muneer I., Froeyen M., Salo-Ahen O.M. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1782768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y., Liu Q., Guo D. J. Med. Virol. 2020;92:418. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tahir ul Qamar M., Alqahtani S.M., Alamri M.A., Chen L.-L. Journal of Pharmaceutical Analysis. 2020;10:313–319. doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Wit E., Van Doremalen N., Falzarano D., Munster V.J. Nat. Rev. Microbiol. 2016;14:523. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ul Qamar M.T., Saleem S., Ashfaq U.A., Bari A., Anwar F., Alqahtani S. J. Transl. Med. 2019;17:362. doi: 10.1186/s12967-019-2116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., Yuan M.-L., Zhang Y.-L., Dai F.-H., Liu Y., Wang Q.-M., Zheng J.-J., Xu L., Holmes E.C., Zhang Y.-Z. Nature. 2020;579:265. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. Nature. 2020;579:270. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.G. Li, E. De Clercq, Nature Publishing Group, 2020.
- 10.Tahir ul Qamar M., Rehman A., Ashfaq U.A., Awan M.Q., Fatima I., Shahid F., Chen L.-L. 2020. bioRxiv. (2020.02.28.970343) [Google Scholar]
- 11.ul Qamar M.T., Shahid F., Aslam S., Ashfaq U.A., Aslam S., Fatima I., Fareed M.M., Zohaib A., Chen L.-L. Infectious Diseases of Poverty. 2020;9(1) doi: 10.1186/s40249-020-00752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu R.-f., Gao R.-l., Robert S.-H., Gao J.-p., Yang S.-g., Zhu C. 2020. medRxiv. [Google Scholar]
- 13.Cao Y.-c., Deng Q.-x., Dai S.-x. Travel Med. Infect. Dis. 2020:101647. doi: 10.1016/j.tmaid.2020.101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hillaker E., Belfer J.J., Bondici A., Murad H., Dumkow L.E. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy. 2020;40:592–598. doi: 10.1002/phar.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Cell. 2020;181:281–292. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Science. 2020;367:1444. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Nature. 2020;581:221. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choudhary S., Malik Y.S., Tomar S., Tomar S. 2020. Chemrxiv. [Google Scholar]
- 20.Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. Nat. Med. 2020;26:450. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi C., Sun X., Ye J., Ding L., Liu M., Yang Z., Lu X., Zhang Y., Ma L., Gu W. Cell. Mol. Immunol. 2020:1. doi: 10.1038/s41423-020-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu K., Li W., Peng G., Li F. Proc. Natl. Acad. Sci. 2009;106:19970. doi: 10.1073/pnas.0908837106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan Y., Shang J., Graham R., Baric R.S., Li F. J. Virol. 2020;94 doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.U.A. Ashfaq, A. Mumtaz, T. ul Qamar, T. Fatima, Bioinformation 9 (2013) 993. [DOI] [PMC free article] [PubMed]
- 25.Mumtaz A., Ashfaq U.A., Ul Qamar M.T., Anwar F., Gulzar F., Ali M.A., Saari N., Pervez M.T. Nat. Prod. Res. 2017;31:1228. doi: 10.1080/14786419.2016.1233409. [DOI] [PubMed] [Google Scholar]
- 26.Malik A., Manan A., Mirza M.U. Trop. J. Pharm. Res. 2017;16:67. [Google Scholar]
- 27.Tahir ul Qamar M., Ashfaq U.A., Tusleem K., Mumtaz A., Tariq Q., Goheer A., Ahmed B. Pak. J. Pharm. Sci. 2017;30:2119. [PubMed] [Google Scholar]
- 28.ul Qamar M.T., Maryam A., Muneer I., Xing F., Ashfaq U.A., Khan F.A., Anwar F., Geesi M.H., Khalid R.R., Rauf S.A. Sci. Rep. 2019;9:1. doi: 10.1038/s41598-018-38450-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perveen S., Al-Taweel A. IntechOpen; 2018. Terpenes and Terpenoids. [Google Scholar]
- 30.Paduch R., Kandefer-Szerszeń M., Trytek M., Fiedurek J. Arch. Immunol. Ther. Exp. 2007;55:315. doi: 10.1007/s00005-007-0039-1. [DOI] [PubMed] [Google Scholar]
- 31.S. Adem, V. Eyupoglu, I. Sarfraz, A. Rasul, M. Ali, (2020).
- 32.V. Chandel, S. Raj, B. Rathi, D. Kumar, (2020).
- 33.Kumar A., Choudhir G., Shukla S.K., Sharma M., Tyagi P., Bhushan A., Rathore M. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1772112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gentile D., Patamia V., Scala A., Sciortino M.T., Piperno A., Rescifina A. Marine Drugs. 2020;18:225. doi: 10.3390/md18040225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.L.A. Gonzalez-Paz, C.A. Lossada, L.S. Moncayo, F. Romero, J. Paz, J. Vera-Villalobos, A.E. Pérez, E. San-Blas, Y.J. Alvarado, (2020).
- 36.S. Khaerunnisa, H. Kurniawan, R. Awaluddin, S. Suhartati, S. Soetjipto, Prepr. doi10 20944 (2020) 1.
- 37.N. Sung, W.-L. Wong, J. Guo, (2020).
- 38.Mangal M., Sagar P., Singh H., Raghava G.P., Agarwal S.M. Nucleic Acids Res. 2013;41:D1124. doi: 10.1093/nar/gks1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. J. Comput. Chem. 2009;30:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trott O., Olson A.J. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.-Y. Cell. 2020;181:894–904. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noor Z.I., Ahmed D., Rehman H.M., Qamar M.T., Froeyen M., Ahmad S., Mirza M.U. Biology. 2019;8:92. doi: 10.3390/biology8040092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeLano W.L. CCP4 Newsletter on Protein Crystallography. 2002;40:82. [Google Scholar]
- 44.Daina A., Michielin O., Zoete V. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipinski C.A. Drug Discov. Today Technol. 2004;1:337. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. J. Comput. Chem. 2005;26:1701. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 47.Brooks B.R., Brooks C.L., III, Mackerell A.D., Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. J. Comput. Chem. 2009;30:1545. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanommeslaeghe K., Ghosh J., Polani N.K., Sheetz M., Pamidighantam S.V., Connolly J.W., mackerell A.D. Biophys. J. 2011;100:611a. [Google Scholar]
- 49.T. Darden, Chem. Phys. 98 10089.
- 50.Hess B., Bekker H., Berendsen H., Fraaije J. 1997. CO. [Google Scholar]
- 51.Van Aalten D.M., De Groot B.L., Findlay J.B., Berendsen H.J., Amadei A. J. Comput. Chem. 1997;18:169. [Google Scholar]
- 52.Yang T., Wu J.C., Yan C., Wang Y., Luo R., Gonzales M.B., Dalby K.N., Ren P. Proteins: Structure, Function, and Bioinformatics. 2011;79:1940. doi: 10.1002/prot.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W. Acc. Chem. Res. 2000;33:889. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 54.Hou T., Wang J., Li Y., Wang W. J. Chem. Inf. Model. 2011;51:69. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumari R., Kumar R., O.S.D.D. Consortium, Lynn A. J. Chem. Inf. Model. 2014;54:1951. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 56.Hou T., Wang J., Li Y., Wang W. J. Chem. Inf. Model. 2011;51:69. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srivastava H.K., Sastry G.N. J. Chem. Inf. Model. 2012;52:3088. doi: 10.1021/ci300385h. [DOI] [PubMed] [Google Scholar]
- 58.Tan J.J., Chen W. Zu, Wang C.X. J. Mol. Struct. THEOCHEM. 2006;766:77. [Google Scholar]
- 59.Mirza M.U., Ahmad S., Abdullah I., Froeyen M. Comput. Biol. Chem. 2020:107376. doi: 10.1016/j.compbiolchem.2020.107376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mirza M.U., Saadabadi A., Vanmeert M., Salo-Ahen O.M., Abdullah I., Claes S., De Jonghe S., Schols D., Ahmad S., Froeyen M. Eur. J. Pharm. Sci. 2020:105537. doi: 10.1016/j.ejps.2020.105537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alamri M.A., ul Qamar M.T., Mirza M.U., Alqahtani S.M., Froeyen M., Chen L.-L. Journal of Pharmaceutical Analysis. 2020 doi: 10.1016/j.jpha.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirza M.U., Vanmeert M., Ali A., Iman K., Froeyen M., Idrees M. J. Med. Virol. 2019;91:2029. doi: 10.1002/jmv.25357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mirza M.U., Vanmeert M., Froeyen M., Ali A., Rafique S., Idrees M. Sci. Rep. 2019;9:1. doi: 10.1038/s41598-019-43129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X. Acta Pharm. Sin. B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Cell Discovery. 2020;6:1. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li R., Pei S., Chen B., Song Y., Zhang T., Yang W., Shaman J. Science. 2020;368:489. doi: 10.1126/science.abb3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aftab S.O., Ghouri M.Z., Masood M.U., Haider Z., Khan Z., Ahmad A., Munawar N. J. Transl. Med. 2020;18(1) doi: 10.1186/s12967-020-02439-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chazal N., Gerlier D. Microbiol. Mol. Biol. Rev. 2003;67:226. doi: 10.1128/MMBR.67.2.226-237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaur R., Mudgal R., Jose J., Kumar P., Tomar S. Virology. 2019;526:91. doi: 10.1016/j.virol.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 70.Singh H., Mudgal R., Narwal M., Kaur R., Singh V.A., Malik A., Chaudhary M., Tomar S. Biochimie. 2018;149:51. doi: 10.1016/j.biochi.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 71.Sharma R., Fatma B., Saha A., Bajpai S., Sistla S., Dash P.K., Parida M., Kumar P., Tomar S. Virology. 2016;498:265. doi: 10.1016/j.virol.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 72.Adedeji A.O., Severson W., Jonsson C., Singh K., Weiss S.R., Sarafianos S.G. J. Virol. 2013;87:8017. doi: 10.1128/JVI.00998-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang Y., Islam M.S., Wang J., Li Y., Chen X. Int. J. Biol. Sci. 2020;16:1708. doi: 10.7150/ijbs.45538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Muhseen Z.T., Li G. Molecules. 2020;25:155. doi: 10.3390/molecules25010155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Tommasi N., Autore G., Bellino A., Pinto A., Pizza C., Sorrentino R., Venturella P. J. Nat. Prod. 2000;63:308. doi: 10.1021/np990231n. [DOI] [PubMed] [Google Scholar]
- 76.Xiao K., Yi Y.-H., Wang Z.-Z., Tang H.-F., Li Y.-Q., Lin H.-W. J. Nat. Prod. 1999;62:1030. doi: 10.1021/np9805185. [DOI] [PubMed] [Google Scholar]
- 77.Xiao X.-y., Hao M., Yang X.-y., Ba Q., Li M., Ni S.-j., Wang L.-S., Du X. Cancer Lett. 2011;302:69. doi: 10.1016/j.canlet.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 78.Cinatl J., Morgenstern B., Bauer G., Chandra P., Rabenau H., Doerr H. Lancet. 2003;361:2045. doi: 10.1016/S0140-6736(03)13615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sato H., Goto W., Yamamura J.-i., Kurokawa M., Kageyama S., Takahara T., Watanabe A., Shiraki K. Antivir. Res. 1996;30:171. doi: 10.1016/0166-3542(96)00942-4. [DOI] [PubMed] [Google Scholar]
- 80.Rossum T.V., Man R.D. Aliment. Pharmacol. Ther. 1998;12:199. doi: 10.1046/j.1365-2036.1998.00309.x. [DOI] [PubMed] [Google Scholar]
- 81.Ito M., Sato A., Hirabayashi K., Tanabe F., Shigeta S., Baba M., De Clercq E., Nakashima H., Yamamoto N. Antivir. Res. 1988;10:289. doi: 10.1016/0166-3542(88)90047-2. [DOI] [PubMed] [Google Scholar]
- 82.LuoLiu P., Li J. Int. J. Antimicrob. Agents. 2020:105995. doi: 10.1016/j.ijantimicag.2020.105995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hall D.C., Jr., Ji H.-F. Travel Med. Infect. Dis. 2020:101646. doi: 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choudhary S., Malik Y.S., Tomar S. Front. Immunol. 2020;11:1664. doi: 10.3389/fimmu.2020.01664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pandey P., Rane J.S., Chatterjee A., Kumar A., Khan R., Prakash A., Ray S. J. Biomol. Struct. Dyn. 2020:1. doi: 10.1080/07391102.2020.1794969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baig M.S., Alagumuthu M., Rajpoot S., Saqib U. Drugs in R&D. 2020;20:161. doi: 10.1007/s40268-020-00312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]