

Abstract
Commensal microorganisms are essential to the normal development and function of many aspects of animal biology. However, the dynamic shift patterns of the microbiota of different gut segments in sheep and the correlation between fat type large-tailed phenotype and microbiota remain poorly unknown. This study therefore sought to assess the composition and distribution of the intestinal microbiome, and compared the difference of gut microbiota from different gastrointestinal segments within breeds and same intestinal sections between breeds. For these analyses, 16S rRNA V4 regions from 4 gut sections prepared from each of six individuals (3 from each breed) were sequenced to detect the microbiome composition in these samples. These analyses revealed the presence of 51,173 operational taxonomic units distributed across 24 phyla and 420 genera in these samples, with Firmicutes and Bacteroidetes being the most prevalent phyla of microbes present in these samples. Moreover, the bacterial composition showed distinct microbial communities in different gastrointestinal segments within breed, but showed similar and relative fixed bacterial abundance in the same intestinal segments from individuals of different breeds. We also found that only a few bacterial species (Lachnospiraceae, Akkermansia) were needed to distinguish between Small-tailed Han sheep (STH) and Large-tailed Han sheep (LTH) and their metabolic process maybe influence the fat type large-tailed phenotype formation in sheep. The functional profile analysis revealed that the environment information processing, genetic information processing, and metabolic pathways were enriched in all samples. The main functional roles of the gut microbiota were amino acid metabolism, replication and repair, carbohydrate metabolism, and membrane transport. Finally, our findings suggested that distinguished gut species between STH and LTH have relative fixed and the potential correlation is existing between the intestinal microorganisms and the large-tailed phenotype trait formation of sheep, which may offer clues for further investigation to detect the roles of intestinal microbiota in the metabolism and fat deposition in the tail of sheep.
Electronic supplementary material
The online version of this article (10.1007/s12088-020-00885-4) contains supplementary material, which is available to authorized users.
Keywords: Sheep, Gut microbiota, Fat type large-tailed phenotype, Microbial diversity, Microbial structure
Introduction
Sheep (Ovis aries) are ruminants that belong to the suborder Ruminantia. They carry out the initial digestion of plant material through a special rumen digestive system. As one of the earliest domesticated livestock species, sheep are distributed in many countries, especially China [1]. Chinese indigenous sheep populations have distinct genetic differentiation, which might have been caused by geographical isolation, cross-breeding, and inbreeding [2]. This also may be the reason for diverse phenotype formation in Chinese indigenous breeds. All sheep worldwide can be classified as one of five tail phenotypes: short-fat tail (Mongolian, Small-tailed Han), short-thin tail (Tibetan), fat-rumped (Kazakh), long-fat tail (Large-tailed Han), and long-thin tail (Texel). Since wild sheep exhibit the thin tail phenotype, it suggesting that the tail phenotypes diversity was developed later on by natural and human selection.
The fat in the tail of the fat-tailed phenotype is regarded as stored energy, which is mobilized during the winter and migration and is considered as an adaptive response for the sheep to harsh environmental conditions. Moradi et al. [3] utilized FST methods to determine seven genomic regions in Iranian thin- and fat-tail breeds, and showed that the fat type large-tailed phenotype trait in sheep is heritable, and also provided principle evidence for the concept that genetic variants are associated with the tail phenotype trait. Moreover, Wang et al. [4] identified NELL1 and FMO3 genes in adipose tissues from the Kazak and Tibetan sheep breeds, which relevant to fat metabolism . Miao et al. [5, 6] identified 54 differentially expressed miRNAs and 602 differentially expressed genes responsible for fat deposition in the tail fat tissue from different sheep breeds. Xu et al. [7] identified four significantly associated autosomal SNPs and three approximately associated autosomal SNPs associated with fat deposition using phenotypes and genotypes of Small-tailed Han (STH) and Large-tailed Han (LTH) sheep breeds via a genome-wide association study. These findings provide novel insights into for the genetic mechanisms of fat type large-tailed phenotype trait formation in sheep. However, the correlation between the intestinal microbiota and the fat type large-tailed phenotype trait has not been reported in sheep.
Commensal microorganisms are essential to the normal development and function of many aspects of animal biology. Moreover, the gut microorganisms have genetic and metabolic functions, and some studies have shown that they are associated with obesity in humans [8–13]. The obesity-related changes of the intestinal microbiota were found in pigs [14], and the gut microbiome may be a major factor in regulating fat deposition [15, 16]. In Rex rabbits, the bacterial compositions of the gut were associated with fecal types and its body weight [17]. In chickens, the quantitative trait genetic selection of the host affected by the bacterial communities of the gut, e.g. the body weight growth trait [18]. In mice, the gut microbiota as an important environmental factor that affects diet digestion, nutrient absorption, and energy storage [19–23]. Furthermore, the ratio of the Firmicutes to Bacteroidetes are changing that influence microbiota obesity-related alterations in mice [24]. The gut microbiota produces metabolites in host circulation, including small organic acids, vitamins, bile acids, and lipids [25]. Dietary poly- and oligosaccharides as a source of carbon and energy for gut bacteria, which were digested in the mammalian host distal gut tract [26]. Wang et al. [27] showed that the body composition was regulated by microbiota through the circadian transcription factor NFIL3; moreover, the altered fat storage may be regulated by functional metabolites from bacteria but is less likely due to global nutritional differences between bacteria [28].
Until now, Wang et al. [29] reported the composition and diversity of the microbiota in the gastrointestinal tract sections of sheep. Wang et al. [30] conducted 16 s rRNA gene sequencing for studies on the age-related succession of rumen microbial communities from 27 Tibetan lambs at nine developmental stages. Using 16S rDNA genes Illumina sequencing, Chang et al. [31] dissect the specificity of gut microbiota among four breeds of sheep and find the difference between Tibetan sheep and other three types of sheep. However, for a long time, human and natural selection have fixed the gut microbial populations and shaped different tail phenotypes in sheep, moreover, the dynamic shift patterns of the microbiota of different gut segments in sheep and the relationship between the gut microbiota and the fat type large-tailed phenotype formation in sheep is remained unknown. Thus, we collected samples from different intestinal sections in LTH and STH sheep breeds with extreme divergent tail phenotypes. Then, we compared the microbial composition and community structure from samples of the four gut segments within and between the LTH and STH breeds by high-throughput sequencing techniques in order to investigate the dynamic shift patterns of the microbiota in different gut segments in sheep, as well as to evaluate the underlying association between the microbiota and the distinct large-tailed phenotype in sheep.
Materials and Methods
Animals and Sampling
In this study, a total of six unrelated adult individuals were selected from LTH and STH breeds (3 from each breed) located in Jia county in Henan province in China. All experimental sheep individuals were 2-year-old rams. Experimental animals of the same breeds were derived from similar genetic backgrounds and subjected to comparable husbandry practices. The test individuals have not shared a common ancestor for at least three generations within same breed. In addition, the test sheep were fed from weaning to slaughter daily with green hay and corn, which likely facilitated reductions in microbiota variability relative to test populations. Animals had free access to water, and all were healthy and not subjected to any antibiotic treatments. After sacrificing these six animals, the gastrointestinal tract was removed from each animal within 30 min of death and luminal contents were collected from each of the 4 indicated segments, including the rumen (R), small intestine (S), ileum (I), and cecum (C). These contents were specifically collected from the middle of each sample, with full disinfection of the experimental tools and work area being performed between samples to prevent any microbial cross-contamination. The rumen samples were fluid. The small intestine, ileum, and cecum samples were solids. The samples were collected in sterile cryopreservation tubes and quickly immersed in liquid nitrogen, and then the tubes were transferred to −80 °C until analysis. A total of 24 samples of fresh content were obtained and stored in liquid nitrogen prior to DNA and 16S rRNA gene profiling analysis.
DNA Extraction
A TIANamp Stool DNA Kit (DP328) (Tiangen BioTech Beijing, China) was used to isolate luminal bacterial DNA based on provided directions, after which a NanoDrop One Microvolume Spectrophotometer (Thermo Fisher Scientific, DE, USA) was used to assess DNA concentrations, while 0.8% agarose gel electrophoresis was used to assess DNA quality and purity.
16S rRNA Gene PCR Amplification, Purification, and Sequencing
All amplifications were performed on a T100™ thermal cycler for PCR (Bio-Rad, Hercules, CA, USA). Briefly, the bacterial 16S rRNA V4 region was amplified using a primer pair (16S V4: 515F: 5′-GTGCCAGCMGCCGCGGTAA-3′; 806R: 5′-GGACTACHVGGGTWTCTAAT-3′). To facilitate multiplexed sequencing, samples were barcoded with 7-bp tags that were specific for each sample. All PCR reactions were performed with Phusion® High-Fidelity PCR Master Mix (New England BioLabs Inc., Ipswich, MA, USA). The reaction was carried out in a total volume of 30 µL containing 10 ng DNA template (1 ng/µL), 15 µL Phusion Master Mix, 3 µL primer (2 µM), and 2 µL ddH2O. After denaturation at 95 °C for 1 min, 30 amplification cycles were performed comprising a denaturation step at 95 °C for 10 s, an annealing step at 50 °C for 30 s, an extension at 72 °C for 30 s, followed by a last extension at 72 °C for 5 min. Next, 2% agarose gel electrophoresis was used to confirm that amplicon sizes were consistent with expectations (~ 500 bp) and that samples were of good quality and purity. The ~ 500 bp sample band was then subjected purification with a GeneJET Gel Extraction Kit (Thermo Scientific, USA) based on provided directions. A Next Ultra DNA Library Prep Kit for Illumina (New England BioLabs) was then used for library preparation based on provided directions, with a Qubit@ 2.0 Fluorometer (Thermo Fischer Scientific) and an Agilent Bioanalyzer 2100 machine used to evaluate library quality. Sequencing of the resultant library was performed on an Illumina MiSeq platform, generating 250-bp paired-end reads.
Taxonomy Classification and Statistical Analysis
Sequencing data was processed using the Quantitative Insights Into Microbial Ecology (QIIME, v1.8.0) pipeline [32]. Briefly, any sequencing reads that exactly matched barcodes were assigned as valid sequences to the corresponding samples. Any low quality reads were then filtered to remove reads meeting the following criteria: sequences that were < 150 bp long, had average Phred scores < 20, contained ambiguous bases, or mononucleotide repeats > 8 bp long. FLASH was used for paired-end read assembly [33]. High-quality sequences that remained after chimera detection were grouped using UCLUST into operational taxonomic units (OTUs) at the 97% sequence identity level [34]. For each OTU, a representative sequence was then selected based on default parameters to facilitate BLAST-mediated taxonomic classification with the Greengenes Database [35]. The abundance of the OTUs in a given sample were compiled in an OTU table. Subsequent analyses were performed based on the compiled data. Rarefaction and rank abundance curves were generated based on these three metrics via R (Version 2.15.3) software drawing.
QIIME [32] and R packages (v3.2.0) were used to analyze sequencing data. We used the Observed Species, Chao1, Abundance-based coverage estimator (ACE), Shannon, Simpson, and Good’s Coverage alpha diversity indices, which were calculated using QIIME [32], in order to assess species diversity and complexity among samples. The abundance of microbes at the different category levels were compared between samples with Metastats analysis [36]. In order to identify those OTUs differing significantly among the four sample regions, we utilized the linear discriminant analysis coupled with the effect size (LEfSe) algorithm based upon relative OTU abundance [37]. Briefly, this algorithm first used a non-parametric factorial Kruskal–Wallis (KW) sum-rank test to identify OTUs that were present at significantly different levels, after which pairwise Wilcoxon tests were used to assess biological consistency between groups. Linear discriminant analysis (LDA) scores were then used to yield an estimated effect size for each differentially abundant feature. UniFrac distance metrics were used to assess variations in beta diversity among samples corresponding to structural differences in microbial community composition, and were calculated by QIIME software [38]. These metrics were visualized using principal component analysis (PCA), principal coordinate analysis (PCoA), and unweighted pair-group method with arithmetic means (UPGMA) hierarchical clustering.
Microbial Function Prediction
PICRUSt was used to predict microbial function according to high-quality sequences [39]. The OTUs were mapped to databases by QIIME software [32]. The predicted genes and their function were aligned to the KEGG database and the differences between samples were identified by STAMP (Statistical Analysis of Metagenomic Profiles) software package [40]. The significant differences among bacterial classes or KEGG functional groups were performed using the Wilcoxon Signed-Ranks test with the FDR correction [36].
Results
Sequencing Data Overview
We were able to successfully amplify the 16S rRNA sequences from luminal samples collected from 4 different gut regions from each of LTH and STH breeds (3 from each breed). All 24 of the resultant samples were sequenced, yielding 1,556,033 raw paired-end reads (S1 file). The sequenced raw data sets have been submitted in the NCBI databases (SRP114799). Quality control was performed. After reads containing incorrect primer or barcode sequences were removed, the resultant raw tags, clean tags, and effectives tags are shown in an additional file (S1 file). The sequencing average length was 253 bp for each sequence read. The average sequence reads were 61,480 for each sample (S1 file). Finally, a total of 1,475,539 validated sequence reads (712,774 for LTH and 760,765 for STH) were used for further investigation. QIIME processing grouped these samples into 51,173 operational taxonomic units (OTUs) (S2 file). In order to confirm whether all characterized 51,173 OTUs appeared in the datasets, and we then performed the rarefaction and rank-abundance curve analyses to confirm the presence of these OTUs within each of all samples. The rarefaction and rank-abundance curve patterns were similar across samples, suggesting that most detectable bacterial species were present in most or all samples (Fig. 1).
Fig. 1.

Rarefaction (left) and rank-abundance curve (right) analysis of the different gastrointestinal tract samples at 97% sequences identity. STH Small-tailed Han, LTH Large-tailed Han, R Rumen samples, S Small intestine samples, I ileum samples, C cecum samples. If the curves reach or nearly reach a plateau, it indicates that most of the species present in all samples have been observed
Diversity Analysis
To explore the composition of microbial communities in different regions of the intestinal tract of the two sheep breeds, we initially assessed the alpha diversity of the microbiome in the 4 tested regions (S3 file).We observed significant differences in the Good’s coverage diversity index among these sections, with the higher Good’s coverage diversity index being present in Large-tailed Han ileum (LI) relative to the Large-tailed Han rumen (LR) in the LTH sheep breed (P < 0.05), in STH sheep breed, with the higher Good’s coverage diversity index in the Small-tailed Han small intestine (SS) relative to in Small-tailed Han ileum (SI) (P < 0.05), and the Good’s coverage diversity index in LI was also significantly higher than in SS (P < 0.05) between different sheep breeds (Fig. 2). For the other five alpha-diversity values, no significant differences (P > 0.05) in these index were detected among regions of the intestinal tract within same sheep breed (SR (Small-tailed Han rumen) and SS, SR and SI, SR and SC (Small-tailed Han cecum), SS and SI, SS and SC, SI and SC, or LR and LS (Large-tailed Han small intestine), LR and LI, LR and LC (Large-tailed Han cecum), LS and LI, LS and LC, LI and LC). Similarly, for all of the alpha-diversity values in the same gut segments of the different sheep breeds, no significant differences (P > 0.05) were also detected between SR and LR, SS and LS, SI and LI, or SC and LC.
Fig. 2.
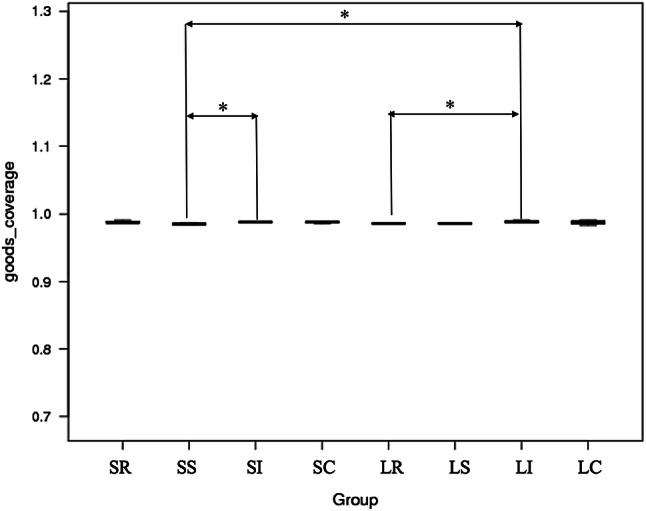
Alpha diversity measures (using Good’s coverage diversity) were compared between the different gastrointestinal tract microbiome samples of Large-tailed Han (LTH) and Small-tailed Han (STH) breeds (Wilcoxon test *0.05, **0.001, ***0.0001)
The Analysis of Community Composition
At the phylum level, we assessed the taxonomic distributions of the most abundant bacterial OTUs in each sample region, 24 phyla were observed; ten main phyla of the 24 phyla were detected (Fig. 3a). We compared the compositional structure of the microbiota in different gut segments of the same breed as well as the same gut segments of different breeds (Fig. 3a). We found that the Firmicutes was the most prevalent phylotype in the LR (51.2%), LS (60.5%), LI (52.6%), and LC (57.3%) of the LTH, Bacteroidetes was the second most populous bacteria and was observed in the LR (40.4%), LS (19.1%), LI (37.6%), and LC (32.5%) microbiota content of the LTH. The proportion of predominant phyla in the LR was varied among the rumen, small intestine, ileum, and cecum samples. In the LI, the composition of microbes was also more similar to that of the LC at the phylum level than in the LR and LS. However, Bacteroidetes was the richest phylum in the SR of the STH, accounting for approximately 47.1%, followed by Firmicutes, comprising approximately 41.5%. Conversely, in the SS, SI, and SC segments of the STH, a higher percentage (65.2%, 49.3%, and 56.6%) of the sequences belonged to Firmicutes, whereas Bacteroidetes was the second largest phylum in these intestinal segments, comprising approximately 13.5%, 35.6%, and 31.8% in the SS, SI, and SC segments, respectively. For all the intestinal samples, Firmicutes and Bacteroidetes phyla were pronounced and accounted for more than 86%. Moreover, the combination of Firmicutes and Bacteroidetes phyla was 91.5% in the LR samples. Furthermore, we found that the proportion of Firmicutes and Bacteroidetes showed dynamic patterns by comparison to different gastrointestinal samples. In some other phyla, there was a marked difference in the proportion of Spirochaetes and Actinobacteria between the different gut segments for LTH and STH; the proportion of Spirochaetes bacteria increased, whereas the proportion of Actinobacteria bacteria decreased in the four gut segments of the STH as compared to LTH. These results showed that the microbial community structure was varied in different gut regions along the sheep gastrointestinal tract.
Fig. 3.
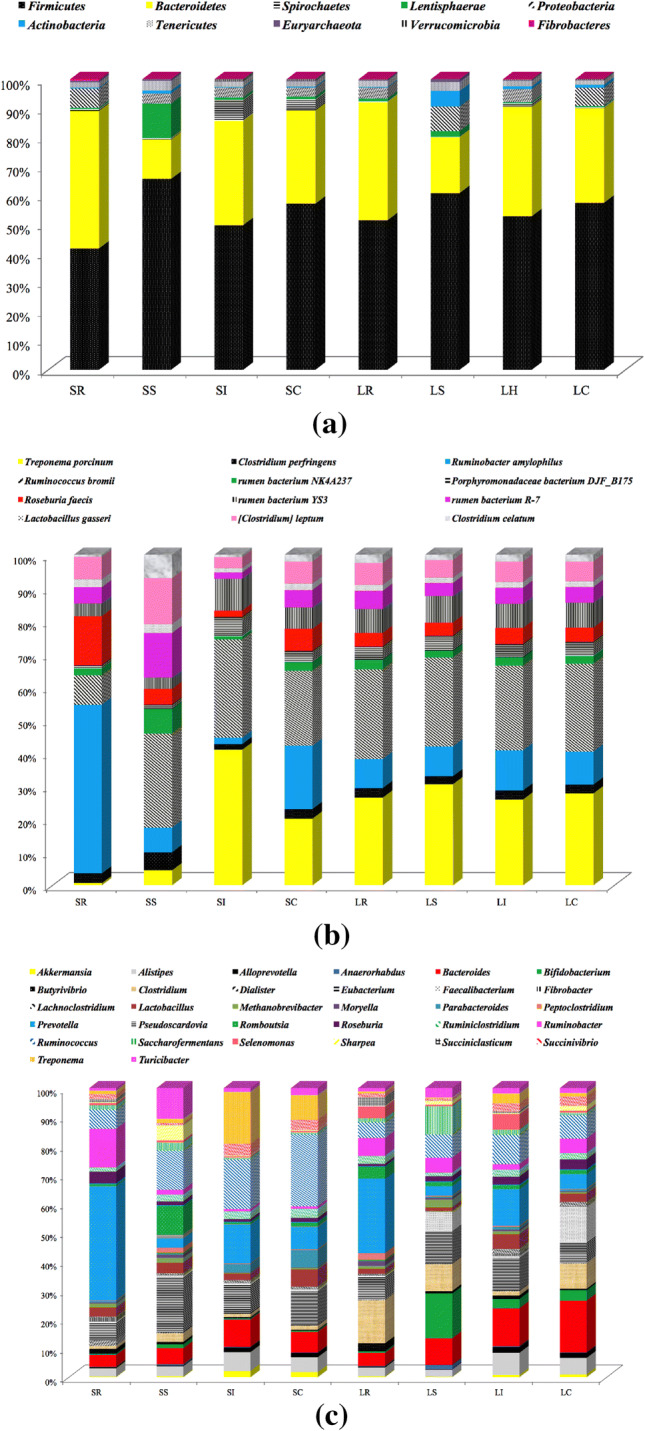
Relative abundance of sequences belonging to different bacterial phyla (a), families (b) and genera (c). Sequences that could not be classified into any known group were assigned as ‘Unknown’
By comparing microbes in samples from the same intestinal segments of the different sheep breeds, we observed that the microbe profiles of the gastrointestinal segments of the LTH were quite different from those of the STH. We found that the abundance of Firmicutes and Bacteroidetes was not existing significant difference (P > 0.05) in either the rumen and small intestine or the ileum and cecum between LTH and STH breeds. However, the SS microbiota of the STH (65.2%) contained higher abundance of Firmicutes than the LS of the LTH (60.5%). LTH and STH had a higher richness of Firmicutes and lower richness of Bacteroidetes in the small intestine. Overall, the LTH and STH displayed different microbial abundances in the same sections of their intestines.
At the family level, a total of 168 families were characterized in all the samples. The 19 most dominant families (> 1%) of microbial contained 90.31% of all microbial, including Lachnospiraceae, Ruminococcaceae, Prevotellaceae, Bacteroidales_S24-7_group, Rikenellaceae, Christensenellaceae, Bacteroidaceae, Erysipelotrichaceae, Succinivibrionaceae, Spirochaetaceae, Enterobacteriaceae, Peptostreptococcaceae, Bacteroidales_BS11_gut_group, Bifidobacteriaceae, Lactobacillaceae, Clostridiaceae_1, and an unknown family belonging to the Bacteroidales and Mollicutes_RF9 order, and the Lentisphaerae_RFP12_gut_group class (Fig. 3b). A higher percentage of Peptococcaceae (P < 0.05) and Rhodospirillaceae (P < 0.05) was found in cecum samples from STH than LTH (S4 file). Another abundance of Peptococcaceae (P < 0.01) and Lachnospiraceae (P < 0.05) was also found in ileum samples from STH which was greater than that of LTH (S4 file). The relative proportion of Rikenellaceae and Peptococcaceae was clearly different in LS than in the LI samples from LTH (S4 file). In contrast, the relative abundance of the Bacteroidales_BS11_gut_group (belonging to Firmicutes) was significantly different in SS than that in SI and SC samples from STH (P < 0.01) (S4 file).
Additionally, we identified 430 genera at the genus level, and the distribution abundance and variation of the genera was greater; the 99 genera accounted for 94.9% of the total genera. The most dominant genera were unknown, and these unknown genera were assigned to the orders Bacteroidales and Clostridiales and families Bacteroidales_S24-7_group (8.33%) and Lachnospiraceae (8.09%). The 32 most predominant genera (Fig. 3c), comprising more than 84% of the entire sequences, were Akkermansia, Alistipes, Alloprevotella, Anaerorhabdus, Bacteroides, Bifidobacterium, Butyrivibrio, Clostridium, Dialister, Eubacterium, Faecalibacterium, Fibrobacter, Lachnoclostridium, Lactobacillus, Methanobrevibacter, Moryella, Parabacteroides, Peptoclostridium, Prevotella, Pseudoscardovia, Romboutsia, Roseburia, Ruminiclostridium, Ruminobacter, Ruminococcus, Saccharofermentans, Selenomonas, Sharpea, Succiniclasticum, Succinivibrio, Treponema, and Turicibacter. The bacterial abundance of the 32 genera was different in the different gastrointestinal segments of the sheep. The number of genera in the rumen, small intestine, ileum, and cecum sites was respectively 254, 288, 250, and 258. Among the four different gut segments, 183 genera were shared.
In some genera attributed to the phylum Firmicutes, they were clearly higher in the STH small intestine microorganisms, such as genera attributed to the families Lachnospiraceae and Ruminococcaceae. The STH had a higher proportion of Ruminococcaceae_UCG-013 in the cecum compared with the LTH (P < 0.05); the small intestine microorganisms of STH had some higher proportion of Bacteroides compared with the LTH, such as the Lachnospiraceae_NK4A136_group (P < 0.05), Ruminococcaceae_UCG-002 (P < 0.01), and Lachnoclostridium_10 (P < 0.01). One genus, Akkermansia, is a member of the phylum Verrucomicrobia. The percentage of Akkermansia was relative higher in the microbiota of the small intestine, ileum, and cecum sites of STH as compared to the LTH (2.04, 3.97, and 2.07, respectively), with the exception of the rumen. Although there was no significantly statistical difference between the same intestinal segments of Akkermansia.
At the phylum, family, and genus levels, the samples from the same intestinal segments were found to have similar a microbial community structure; a pattern that was consistent until the species level. In addition, microbial diversity and proportions were also analyzed by class and order. A total of 32, 37, 33, and 32 classes and 52, 63, 50, and 52 orders were identified in the rumen, small intestine, ileum, and cecum sites, respectively. The most dominant class and order shared in the four regions were Clostridia (48.91%) and Bacteroidia (31.13%), respectively. Moreover, inter-sheep variations were found in the phylum of the OTU levels; the observed results indicated that the variation levels in the small intestinal and ileum samples were higher compared with the rumen and cecum samples of the inter-sheep. Although STH and LTH breeds originated from the same progenitor population, they underwent long-term natural and human selection for the large- or small-tailed phenotype. The quantitative genes may control the tail fat deposition trait, further suggesting that the host quantitative inheritance background also influences the community structure and composition of intestinal microbiota in different classification levels.
To identify those bacterial species most characteristic of the 4 tested gut regions with the same sheep breeds, we conducted an LEfSe analysis of the taxa with LDA scores > 4. In the STH breed, four bacterial taxa were distinctly more abundant in the rumen samples of STH (p_Bacteroidetes, o_Bacteroidetes, c_Bacteroidia, and f_ Bacteroidetes_BS11_gut_group. P < 0.05), while only one taxa was represented in ileum samples of the STH (g_Ruminococcaceae_UCG_005, P < 0.05) (Fig. 4a). We also carried out LEfSe to identify bacterial taxa between the same intestinal segments of different sheep breeds. Two and one taxa were represented in ileum samples (s_Ruminococcus_bromii, g_Ruminococcus_2, and f_Lachnospiraceae, P < 0.05) of STH and LTH, respectively (Fig. 4b). Eight taxa were distinguished as having higher abundance in the cecum samples (g_Ruminococcus_2, s_Ruminococcus_bromii, p_Spirochaetes, _unidentified_Spirochaetes, f_Spirochaetaceae, o_Spirochaetes, g_Treponema_2, _coprostanoligenes_group, P < 0.05) of the STH group, which was not found in the cecum samples of LTH (Fig. 4c).
Fig. 4.
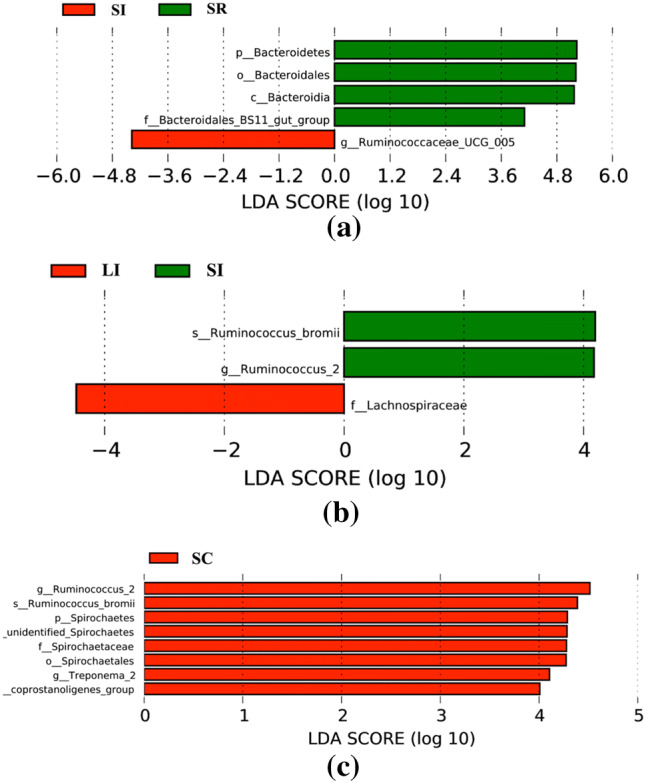
Bacterial taxa were significantly differentiated between the same gastrointestinal sites with different sheep breeds and different gastrointestinal tract sites with same sheep breeds, identified by linear discriminant analysis coupled with effect size (LEfSe) using the default parameters. a Shows different taxa within STH in the ileum (I) and rumen (R) samples; b, c show different taxa between STH and LTH in the ileum (I) and cecum (C) samples, respectively
The Analysis of PCA, PCoA, and UPGMA
Cluster analysis was preceded by principal component analysis (PCA). The results indicated that the microbiota of the rumen and small intestine were distinctly different from the ileum and cecum samples at the four different segments of the gastrointestinal tract within STH or LTH breeds (Fig. 5). Cluster heatmap results further validated the PCA results (S5 file). PCoA results illustrated the relationships among the community structures of the sheep gut microbiota quite well (S6 file). The UPGMA trees further showed that the Firmicutes, Bacteroidetes, Tenericutes, and Proteobacteria were obviously dominant and significant in clustering of the small intestine, rumen, ileum, and cecum microbiota (Fig. 6). These results suggested that the abundance and proportion of microbiota in different gastrointestinal segments have variation, whereas those between the ileum and cecum samples were not significantly different within breeds, and the microbial community structures in the same intestinal segments (rumen and small intestine) were similar from individuals of different breeds.
Fig. 5.
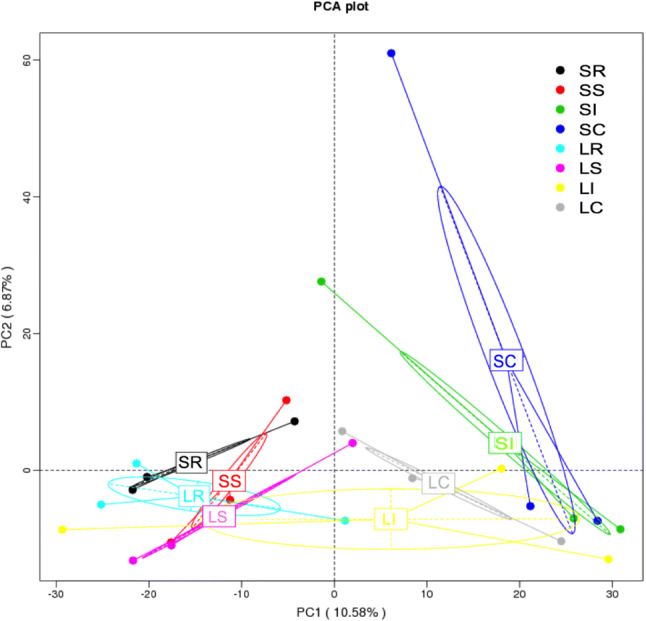
The principal component analysis plot of samples from different gastrointestinal tracts in two sheep breeds
Fig. 6.

Differences in microbial communities among different gastrointestinal samples as shown by weighted UniFrac distances
OTU Overlap Across Different Gut Segments Within and Between Breeds
Venn diagrams showed the numbers of OTUs shared among different gastrointestinal segments within breed were 1824 and 1854 (Fig. 7a, b), and the percentage of shared OTUs in STH (57.56%) was higher than LTH (55.36%), which suggested that the STH-correlated microbiota have a relatively higher abundance compared with LTH. Furthermore, 2222, 2173, 2150, and 2090 OTUs were shared by the rumen, small intestine, ileum, and cecum samples between breeds, respectively (Fig. 7c, d, e, f). The percentage of shared OTUs between rumen samples (74.16%) was higher than between small intestine (73.36%), ileum (73.78%), and cecum (73.10%) samples, which also suggested that the rumen-correlated microbiota was also relatively higher abundance compared with that of the small intestine, ileum, and cecum.
Fig. 7.

Venn diagrams demonstrating 97% OTU cluster overlap among different gastrointestinal sites within breed (a, b) and in the same gastrointestinal tract sites between breeds (c, d, e, f)
Analysis of Microbial Function
We next used PICRUSt in an effort to develop an understanding of the metagenomic activity of the identified bacteria across our samples, as such functional assessments of the microbiome may offer more meaningful insights into the spatial distinctions in metabolic activity across the length of the intestinal tract. These metagenomic inferences were made based upon available annotations for the detected OTUs in this study. The genes identified through this metagenomic analysis were then aligned to the KEGG database to gain functional insights. Through this approach, we identified 5839 KEGG genes that were assigned to 278 pathways (S7 file). In comparing between groups with different segments of the gastrointestinal tract, seven pathways were distinguished in the four gut segments. Genetic information processing, environmental information processing, and metabolism were the most enriched function modules at the KEGG A class in all samples (S8 file). The prevalence of pathways at the KEGG B class level was similar in the different samples (S8 file). Interestingly, five metabolism pathways (nucleotide, cofactors and vitamins, energy, carbohydrate, amino acid), one environmental information processing pathway (member transport), and two genetic information processing pathways (translation, replication and repair) were enriched and more abundant in all samples (Fig. 8, S7 file). We identified 278 gene families in all samples, and these families were predicted to be mainly involved in transporters, general function prediction, ABC transporters, DNA repair and recombination proteins, ribosome, purine metabolism, peptidases, pyrimidine metabolism, and biosynthesis of amino acids (Fig. 8, S7 file). The clustered heatmap indicated that the rumen samples were well distinguished from the small intestinal, ileum, and cecum samples (S9 file). Together these findings suggest that the functional metabolic activity of the microbiome varies over the length of the intestinal tract, with certain pathways being preferentially engaged in a spatially-defined manner.
Fig. 8.

Heatmap clustered by KEGG pathway based on functional prediction of the different gastrointestinal segments microbiota in sheep
Discussion
Several studies have been conducted on the gut microbial composition and its relation to obesity and fat deposition based on high-throughput sequencing in humans [8–13], pigs [14–16], and mice [19–24]. To our knowledge, the features of distribution proportion and community structure along the gastrointestinal tract of sheep poorly reported. Moreover, the correlation between the gut microbiota and the fat type large-tailed phenotype trait in sheep has also not been completely analyzed. In this study, we compared the microbial composition and community structure in four gut locations, characterized the functional profiles of intestinal microbiota, and investigated the potential correlation of gut microbiota with the large-tailed phenotype in sheep.
First, a total of 1,475,539 validated read numbers were acquired from 24 samples, and the average sequence reads was 61,480 for each sample, and the read numbers were greater than previously reported for gut microbes in sheep [41, 42]. Since different parts of the mammalian gastrointestinal tract have different environmental conditions. The stomach and small intestine have too harsh for microbes to grow, the bacterial diversity was increased along the animal intestinal tract. In our results, based on the Good’s coverage index values (Fig. 2), the microbiota diversity in the LI and SI were obviously higher than in the LR and SS (P < 0.05) in same sheep breed, and the microbiota in the LI was also clearly higher than in the SS (P < 0.05) between different sheep breeds. In other studies, changes in microbial diversity were found in human and horse stomachs [43–45]. A more important phenomenon is that the gut microbiota diversity varied in different gastrointestinal segments, perhaps due to the transient microorganisms. Due to the harsh environment conditions of the host stomach, the transient microorganisms are easy to inactivate by low pH stimulation. Only a few of the transient microbiota quickly escapes from the stomach and reaches other parts of the gastrointestinal tract (duodenum, small intestine, ileum, and cecum). The ileum and cecum are far from the stomach, and offer a better environment for growing transient microbiota. These might be the reasons that the ileum and cecum samples had higher and more stable microbiota populations than those from the rumen and small intestine; these results are consistent with the results of Eckburg et al. [46] and Gu et al. [47].
The community structure and composition of the intestinal microbiota had already been investigated in mice [47], pigs [48], horse [49], donkey [50], zebrafish [51], chickens [52], and camels [53]. In our study, the community structure and composition distribution of gut microorganisms was also varied along the sheep gastrointestinal tract. However, the entire taxonomic populations represented within the sheep gastrointestinal tract were similar in all samples. Ten main phyla were distributed in the different gut segments of LTH and STH, respectively (Fig. 3a). The abundances of the each bacterial differed in the four intestinal segments at the phyla level. The highest proportion of Firmicutes and Bacteroidetes were existing in all the samples. In the gut microbiota of other mammals, the proportion of Firmicutes and Bacteroidetes were also more than 98% [47, 54–57]. However, Bacteroidetes was the most predominant phylum, and Firmicutes was the second largest phylum in the SR. Conversely, in the SS, SI, and SC segments, a higher percentage was belonged to Firmicutes. Bacteroidetes was the second largest phylum in the SS, SI, and SC segments, respectively. Furthermore, the composition structure of microbiota displayed a significant similarity between the ileum and cecum of LTH and STH. The rumen and small intestine microbiome communities were quite different from those of the ileum and cecum samples (Figs. 5, 6, Fig S3). In pigs and cows, the similar microbial composition and community structure were found between swine jejunum and ileum [16], and between bovine jejunum and ileum [58]. The different distribution of small intestinal and ileum microbes in different species was influenced by the many factors, such as species, environment, and diets. The different gut segments had physicochemical conditions, which is a prerequisite for the presence of animal gut microorganisms, including pH, intestinal motility, nutrient, and host secretions [59–61]. We observed major compositional shifts among gut segments within the same breeds when environment and diet remained constant; it is likely that the different parts of the sheep gastrointestinal tract have different environmental conditions that is a major factor affecting microbial flora changes.
Moreover, OTU overlap analyses revealed that the core microbiota exists in different gut segments of the sheep (Fig. 7). This finding also showed that the physicochemical conditions and selective pressures of the gut microbes play an important role in intestinal microorganism formation. OTU overlap analyses also discovered that unique microorganisms exist in different gastrointestinal segments (Fig. 7a, b), and we thought that these unique microbial could be markers for each different gut segments. However, it is well known that the majority of the gut microbes influence animal physiology and nutrition, and some research has been reported the microbial communities in rumen of the sheep and cows [41, 42, 62–66]. In our study, we found some overlap of OTUs among the different gut samples (Fig. 7c–f). Furthermore, the rumen sample shared more common OTUs compared with that of the ileum, small intestine, and cecum samples. We thought that these shared microorganisms were the permanent residents; moreover, were formed during the evolution of the host.
Until now, the correlation between fat type large-tailed phenotype and microbiota in sheep remain poorly unknown. In this study, we also tested the potential relationship of sheep gut microbiota with large-tailed phenotype trait, and that distinct microbial community variations were found in same gut segments between STH and LTH. This difference may be caused by sheep breed. Just as in humans [8–13], pigs [14–16], and mice [19–24], and the LTH showed a higher abundance of Firmicutes and a lower abundance of Bacteroidetes than STH with small-tailed phenotype in the rumen. We also identified that Lachnospiraceae was overrepresented in the ileum samples of LTH (Fig. 4b), which is abundant in the gastrointestinal tracts of many mammals. Members of this family have been linked to obesity and protection from colon cancer in humans [67]. Furthermore, Lachnospiraceae can induce and significantly increases blood glucose levels, liver and mesenteric adipose tissue weights in germ free mice [68]. At the genus level, a higher proportion of Akkermansia was found in three gut segments of STH as compared to the LTH, except for the rumen, although there is no significant difference between the same intestinal segments. Researchers have discovered Akkermansia to be a mucin-degrading bacterium that is beneficial to the formation a healthy gut microbiota environment [69]. Moreover, Akkermansia muciniphila could reduce type 2 diabetes and obesity by cross-talk with intestinal epithelium [70] and was found to prevent type 1 diabetes incidence in mice [71]. The Methanovibribacter species co-occurs with other species and has been connected to a lean phenotype [72, 73]. A. muciniphila was associated with metabolic health; however, the interaction between gut microbiota ecology and A. muciniphila evidences further investigation [74]. Therefore, the higher abundance of Akkermansia exists in the intestinal microbiota of the sheep, which may influence the metabolism of sheep as well as the fat tissue development in the sheep tail, but further study would be needed to verify this theory. Overall, these findings suggested that the potential correlation is existing between the intestinal microorganisms (e.g. Lachnospiraceae, Akkermansia) and the large-tailed phenotype trait formation of sheep.
The changes we observed in the community composition between breeds needs careful consideration as to the tail phenotypic genetic context in studies of host-microbe interactions. We used consistent breed and different intestinal segments of the sampled hosts to make comparisons across studies, and found individuals of two breeds for which only a few bacterial species were distinguished between STH and LTH samples. These differences of bacterial species in the gut anatomy and their metabolic process maybe involved in the tail phenotype trait formation of sheep. However, we observed the sheep tail phenotype difference in two sheep breeds is, in part, attributed to changes in the microbiota. At present, in humans and other animal hosts, the finding that abundance and composition of the gut microbiota influences host phenotype formation processes has been reported [75–80]. Our identification of gut microbiota dynamics across different intestinal segments in sheep and the genomes of representative members provide an important information for future studies.
We also conducted a metagenomic analysis of the functional capacity of the intestinal microbiome in sheep, leading us to identify several significantly differentially enriched pathways across spatial regions within all the samples (Fig. 8). The main metagenomic activities of the rumen microbiome in sheep was related to metabolism, including nucleotide metabolism, metabolism of cofactors and vitamins, energy metabolism, carbohydrate metabolism, and amino acid metabolism. These pathways in the rumen confirm that the gut microorganisms in the rumen of the sheep have important food digestion and nutrition absorption functions. In contrast, microbiota function involved in metabolic pathways in the small intestine of the sheep have lower richness compared to those in the rumen. However, the relevance of the application of PICRUSt to predict bacterial activities needs further confirmation [16]. Furthermore, a majority of the OTUs were not matched to the database, thus their functions were not well predicted [16]. Nevertheless, PICRUSt provided an important basis for identifying the gut bacterial community functions of the sheep. These analysis results thus suggest that the gut microbiome exhibits distinct functional and spatial organization that helps to facilitate the rapid degradation and utilization of diverse nutrient sources by local bacterial species that are able to proliferate and maintain gut homeostasis. Further work, however, will be needed to confirm our results which are largely predictive in nature.
Conclusions
Taken together, our study demonstrated a distinct microbial composition and structure among the rumen, small intestine, ileum, and cecum in LTH and STH with extreme divergent tail phenotypes, and identified bacterial species that were different in four segments of the gastrointestinal tract within breed, and were similar and relatively fixed in the same gastrointestinal segments from individuals of different breeds. We also found that only a few bacterial species (Lachnospiraceae, Akkermansia) were needed to distinguish between STH and LTH, and their metabolic process may be involved in the tail phenotype trait formation of sheep, these results suggested that the potential correlation is existing between the intestinal microorganisms and the large-tailed phenotype trait formation of sheep. In future work, we will integrate multi-omics (metagenomic, metatranscriptomic and metaproteomic) approaches to resolve the association between the microbiota and the distinct large-tailed phenotype in sheep. These results may provide novel insight into the complexity of the sheep intestinal microbial community and act as an important target for further investigation to detect their roles in the metabolism and fat tissue development in the tail of sheep.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The sequencing service was provided by the Beijing Novogene Bioinformation Technology Co., Ltd. This study was supported by the He’nan Research Program of Foundation and Advanced Technology of China (102300410143, 132300410398), the Foundation of He’nan Educational Committee of China (12B230011, 14B230017), and the Cooperation Project of Henan Science and Technology Department of China (182107000041).
Authors Contributions
GLY and ML conceived and designed the project. YL, QKW, XYL, and YFY collected the samples. YL, JH, ZQL extracted the DNA. GLY and SHZ analyzed the data, and wrote the paper. All authors reviewed and approved the manuscript.
Compliance with Ethical Standards
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The animal care and use guidelines put forth by the Ministry of Science and Technology of China (Guidelines on Ethical Treatment of Experimental Animals (2006) No. 398) were followed for this study, with the Ethics Committee of Shangqiu Normal University having approved all experiments herein (Shang (2017) No. 168).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Guangli Yang and Shuhong Zhang are as co-first authors.
Contributor Information
Guangli Yang, Email: yangguangli_502@hotmail.com.
Ming Li, Email: liming@henau.edu.cn.
References
- 1.Du LX, Li JQ, Ma N, Ma YH, Wang JM, Ying CA, et al. China National Commission of animal genetic resources, animal genetic resources in China, sheep and goats. Beijng: China Agriculture Publishers; 2011. [Google Scholar]
- 2.Yang GL, Fu DL, Lang X, Yan YF, Luo YZ. Genetic variation of 5 SNPs of MC1R gene in Chinese indigenous sheep breeds. Russ J Genet. 2014;50:1048–1059. doi: 10.1134/S1022795414100159. [DOI] [PubMed] [Google Scholar]
- 3.Moradi MH, Nejati-Javaremi A, Moradi-Shahrbabak M, Dodds KG, McEwan JC. Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genet. 2012;13:10. doi: 10.1186/1471-2156-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang XL, Zhou GX, Xu XC, Geng RQ, Zhou JP, Yang YX, Chen YL. Transcriptome profile analysis of adipose tissues from fat and short-tailed sheep. Gene. 2014;549:252–257. doi: 10.1016/j.gene.2014.07.072. [DOI] [PubMed] [Google Scholar]
- 5.Miao XY, Luo QM, Qin XY, Guo YT. Genome-wide analysis of microRNAs identifies the lipid metabolism pathway to be a defining factor in adipose tissue from different sheep. Sci Rep. 2015;5:18470. doi: 10.1038/srep18470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miao XY, Luo QM, Qin XY, Guo YT, Zhao HJ. Genome-wide mRNA-seq profiling reveals predominant down-regulation of lipid metabolic processes in adipose tissues of Small Tail Han than Dorset sheep. Biochem Biophys Res Commun. 2015;467:413–420. doi: 10.1016/j.bbrc.2015.09.129. [DOI] [PubMed] [Google Scholar]
- 7.Xu SS, Ren X, Yang GL, Xie XL, Zhao YX, Zhang M, Shen ZQ, Ren YL, Gao L, Shen M, Kantanen J, Li MH. Genome-wide association analysis identifies the genetic basis of fat deposition in the tails of sheep (Ovis aries) Anim Genet. 2017;48:560–569. doi: 10.1111/age.12572. [DOI] [PubMed] [Google Scholar]
- 8.Zhao L. The gut microbiota and obesity: from correlation to causality. Nat Rev Microbiol. 2013;11:639–647. doi: 10.1038/nrmicro3089. [DOI] [PubMed] [Google Scholar]
- 9.Million M, Lagier JC, Yahav D, Paul M. Gut bacterial microbiota and obesity. Clin Microbiol Infect. 2013;19:305–313. doi: 10.1111/1469-0691.12172. [DOI] [PubMed] [Google Scholar]
- 10.Angelakis E, Armougom F, Million M, Raoult D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012;7:91–109. doi: 10.2217/fmb.11.142. [DOI] [PubMed] [Google Scholar]
- 11.Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, Shi J, Zhao S, Liu W, Wang X, Xia H, Liu Z, Cui B, Liang P, Xi L, Jin J, Ying X, Wang X, Zhao X, Li W, Jia H, Lan Z, Li F, Wang R, Sun Y, Yang M, Shen Y, Jie Z, Li J, Chen X, Zhong H, Xie H, Zhang Y, Gu W, Deng X, Shen B, Xu X, Yang H, Xu G, Bi Y, Lai S, Wang J, Qi L, Madsen L, Wang J, Ning G, Kristiansen K, Wang W. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med. 2017;23:859–868. doi: 10.1038/nm.4358. [DOI] [PubMed] [Google Scholar]
- 12.Beaumont M, Goodrich JK, Jackson MA, Yet I, Davenport ER, Vieira-Silva S, Debelius J, Pallister T, Mangino M, Raes J, Knight R, Clark AG, Ley RE, Spector TD, Bell JT. Heritable components of the human fecal microbiome are associated with visceral fat. Genome Biol. 2016;17:189. doi: 10.1186/s13059-016-1052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Roy CI, Beaumont M, Jackson MA, Steves CJ, Spector TD, Bell JT. Heritable components of the human fecal microbiome are associated with visceral fat. Gut Microbes. 2018;9:61–67. doi: 10.1080/19490976.2017.1356556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedersen R, Ingerslev HC, Sturek M, Alloosh M, Cirera S, Christoffersen BØ, Moesgaard SG, Larsen N, Boye M. Characterisation of gut microbiota in Ossabaw and Göttingen minipigs as models of obesity and metabolic syndrome. PLoS ONE. 2013;8:e56612. doi: 10.1371/journal.pone.0056612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He M, Fang S, Huang X, Zhao Y, Ke S, Yang H, Li Z, Gao J, Chen C, Huang L. Evaluating the contribution of gut microbiota to the variation of porcine fatness with the cecum and fecal samples. Fron Microbiol. 2016;7:2108. doi: 10.3389/fmicb.2016.02108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang H, Huang XC, Fang SM, Xin WS, Huang LS, Chen CY. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci Rep. 2016;6:27427. doi: 10.1038/srep27427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng B, Han S, Wang P, Wen B, Jian W, Guo W, Yu Z, Du D, Fu X, Kong F, Yang M, Si X, Zhao J, Li Y. The bacterial communities associated with fecal types and body weight of rex rabbits. Sci Rep. 2015;5:9342. doi: 10.1038/srep09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng H, Zhang Y, Zhao L, Zhao W, He C, Honaker CF, Zhai Z, Sun Z, Siegel PB. Body weight selection affects quantitative genetic correlated responses in gut microbiota. PLoS ONE. 2014;9:e89862. doi: 10.1371/journal.pone.0089862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M, Gu D, Xu N, Lei F, Du L, Zhang Y, Xie W. Gut carbohydrate metabolism instead of fat metabolism regulated by gut microbes mediates high-fat diet-induced obesity. Benefic Microbes. 2014;5:335–344. doi: 10.3920/BM2013.0071. [DOI] [PubMed] [Google Scholar]
- 23.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 26.den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54:2325–2340. doi: 10.1194/jlr.r036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science. 2017;357:912–916. doi: 10.1126/science.aan0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin CJ, Wang MC. Microbial metabolites regulate host lipid metabolism through NR5A–Hedgehog signaling. Nat Cell Biol. 2017;19:550–557. doi: 10.1038/ncb3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Fan H, Han Y, Zhao JZ, Zhou ZJ. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Aust J Anim Sci. 2017;30:100–110. doi: 10.5713/ajas.16.0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Zhang K, Zhang CG, Feng YZ, Zhang XW, Wang XL, Wu GF. Dynamics and stabilization of the rumen microbiome in yearling Tibetan sheep. Sci Rep. 2019;9:19620. doi: 10.1038/s41598-019-56206-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang JJ, Yao XT, Zuo CX, Qi YX, Chen DK, Ma WT. The Gut microbiota in sheep of different breeds in Qinghai Province. Anim Sci. 2020 doi: 10.21203/rs.2.15821/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5:e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, Beiko RG, Huttenhower C. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26:715–721. doi: 10.1093/bioinformatics/btq041. [DOI] [PubMed] [Google Scholar]
- 41.De Barbieri I, Hegarty RS, Li L, Oddy VH. Association of wool growth with gut metabolism and anatomy in sheep. Livest Sci. 2015;173:38–47. doi: 10.1016/j.livsci.2014.12.018. [DOI] [Google Scholar]
- 42.De Barbieri I, Gulino L, Hegarty RS, Oddy VH, Maguire A, Li L, Klieve AV, Ouwerkerk D. Production attributes of Merino sheep genetically divergent for wool growth are reflected in differing rumen microbiotas. Livest Sci. 2015;178:119–129. doi: 10.1016/j.livsci.2015.05.023. [DOI] [Google Scholar]
- 43.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, Perez-Perez G, Blaser MJ, Relman DA. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perkins GA, den Bakker HC, Burton AJ, Erb HN, McDonough SP, McDonough PL, Parker J, Rosenthal RL, Wiedmann M, Dowd SE, Simpson KW. Equine stomachs harbor an abundant and diverse mucosal microbiota. Appl Environ Microbiol. 2012;78:2522–2532. doi: 10.1128/AEM.06252-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu S, Chen D, Zhang JN, Lv X, Wang K, Duan LP, Nie Y, Wu XL. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE. 2013;8:e74957. doi: 10.1371/journal.pone.0074957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Looft T, Allen HK, Cantarel BL, Levine UY, Bayles DO, Alt DP, Henrissat B, Stanton TB. Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014;8:1566–1576. doi: 10.1038/ismej.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costa MC, Arroyo LG, Allen-Vercoe E, Stampfli HR, Kim PK, Sturgeon A, Weese JS. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3–V5 region of the 16S rRNA gene. PLoS ONE. 2012;7:e41484. doi: 10.1371/journal.pone.0041484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Fan H, Ding X, Hong Z, Nei Y, Liu Z, Li G, Guo H. Analysis of the gut microbiota by high- throughput sequencing of the V5–V6 regions of the 16S rRNA gene in donkey. Currt Microbiol. 2014;68:657–662. doi: 10.1007/s00284-014-0528-5. [DOI] [PubMed] [Google Scholar]
- 51.Stephens WZ, Burns AR, Stagaman K, Wong S, Rawls JF, Guillemin K, Bohannan BJ. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016;10:644–654. doi: 10.1038/ismej.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi JH, Kim GB, Cha CJ. Spatial heterogeneity and stability of bacterial community in the gastrointestinal tracts of broiler chickens. Poult Sci. 2014;93:1942–1950. doi: 10.1038/ismej.2015.140. [DOI] [PubMed] [Google Scholar]
- 53.He J, Yi L, Hai L, Ming L, Gao W, Ji R. Characterizing the bacterial microbiota in different gastrointestinal tract segments of the Bactrian camel. Sci Rep. 2018;8:654. doi: 10.1038/s41598-017-18298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 56.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mao S, Zhang M, Liu J, Zhu W. Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep. 2015;5:16116. doi: 10.1038/srep16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hopkins MJ, Sharp R, Macfarlane GT. Variation in human intestinal microbiota with age. Dig Liver Dis. 2002;34:S12–S18. doi: 10.1016/S1590-8658(02)80157-8. [DOI] [PubMed] [Google Scholar]
- 61.Zoetenda EG, Akkermans ADL, Akkermans-van Vliet WM, de Visser JAGM, de Vos WM. The host genotype affects the bacterial community in the human gastronintestinal tract. Microb Ecol Health Dis. 2001;13:129–134. doi: 10.1080/089106001750462669. [DOI] [Google Scholar]
- 62.Omoniyi LA, Jewell KA, Isah OA, Neumann AP, Onwuka CFI, Onagbesan OM, Suen G. An analysis of the ruminal bacterial microbiota in West African Dwarf sheep fed grass- and tree-based diets. J Appl Microbiol. 2014;116:1094–1105. doi: 10.1111/jam.12450. [DOI] [PubMed] [Google Scholar]
- 63.Hernandez-Sanabria E, Goonewardene LA, Wang Z, Zhou M, Moore SS, Guan LL. Influence of sire breed on the interplay among rumen microbial populations inhabiting the rumen liquid of the progeny in beef cattle. PLoS ONE. 2013;8:e58461. doi: 10.1371/journal.pone.0058461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lopes LD, de Souza Lima AO, Taketani RG, Darias P, da Silva LR, Romagnoli EM, Louvandini H, Abdalla AL, Mendes R. Exploring the sheep rumen microbiome for carbohydrate-active enzymes. Antonie Van Leeuwenhoek. 2015;108:15–30. doi: 10.1007/s10482-015-0459-6. [DOI] [PubMed] [Google Scholar]
- 65.Mateos I, Ranilla MJ, Saro C, Carro MD. Comparison of fermentation characteristics and bacterial diversity in the rumen of sheep and in batch cultures of rumen microorganisms. J Agr Sci. 2015;153:1097–1106. doi: 10.1017/S0021859615000167. [DOI] [Google Scholar]
- 66.Li F, Henderson G, Sun X, Cox F, Janssen PH, Guan LL. Taxonomic assessment of rumen microbiota using total RNA and targeted amplicon sequencing approaches. Front Microbiol. 2016;7:987. doi: 10.3389/fmicb.2016.00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meehan CJ, Beiko RG. A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol Evol. 2014;6:703–713. doi: 10.1093/gbe/evu050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kameyama K, Itoh K. Intestinal colonization by a Lachnospiraceae Bacterium contributes to the development of diabetes in obese mice. Microbes Environ. 2014;29:427–430. doi: 10.1264/jsme2.ME14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belzer C, de Vos WM. Microbes inside-from diversity to function: the case of Akkermansia. ISME J. 2012;6:1449–1458. doi: 10.1038/ismej.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansen C, Krych L, Nielsen D, Vogensen F, Hansen L, Sørensen SJ, Buschard K, Hansen AK. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. 2012;55:2285–2294. doi: 10.1007/s00125-012-2564-7. [DOI] [PubMed] [Google Scholar]
- 72.Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS ONE. 2009;4:e7125. doi: 10.1371/journal.pone.0007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, Goodfellow J, Zaneveld JR, McDonald DT, Goodrich JA, Heath AC, Knight R, Gordon JI. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci USA. 2011;108:4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dao MC, Everard A, Aron-Wisnewsky J, Sokolovska N, Prifti E, Verger EO, Kayser BD, Levenez F, Chilloux J, Hoyles L, Dumas ME, Rizkalla SW, Doré J, Cani PD, Clément K. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut. 2016;65:426–436. doi: 10.1136/gutjnl-2014-308778. [DOI] [PubMed] [Google Scholar]
- 75.Ding T, Schloss PD. Dynamics and associations of microbial community types across the human body. Nature. 2014;509:357–360. doi: 10.1038/nature13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, MetaHIT consortium. Bork P, Wang J, Ehrlich SD, Pedersen O. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 77.Lukovac S, Belzer C, Pellis L, Keijser BJ, de Vos WM, Montijn RC, Roeselers G. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio. 2014;5:e01438–14. doi: 10.1128/mBio.01438-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, Zhernakova DV, Jankipersadsing SA, Jaeger M, Oosting M, Cenit MC, Masclee AA, Swertz MA, Li Y, Kumar V, Joosten L, Harmsen H, Weersma RK, Franke L, Hofker MH, Xavier RJ, Jonkers D, Netea MG, Wijmenga C, Fu J, Zhernakova A. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48:1407–1412. doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 79.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, Guttman DS, Griffiths A, Panaccione R, Otley A, Xu L, Shestopaloff K, Moreno-Hagelsieb G, Paterson AD, Croitoru K. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet. 2016;48:1413–1417. doi: 10.1038/ng.3693. [DOI] [PubMed] [Google Scholar]
- 80.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. Cross-species comparisons of host genetic associations with the microbiome. Science. 2016;352:532–535. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.