

Abstract
Boron neutron capture therapy (BNCT) for cancer is on the rise worldwide due to recent developments of in-hospital neutron accelerators which are expected to revolutionize patient treatments. There is an urgent need for improved boron delivery agents, and herein we have focused on studying the biochemical foundations upon which a successful GLUT1-targeting strategy to BNCT could be based. By combining synthesis and molecular modeling with affinity and cytotoxicity studies, we unravel the mechanisms behind the considerable potential of appropriately designed glucoconjugates as boron delivery agents for BNCT. In addition to addressing the biochemical premises of the approach in detail, we report on a hit glucoconjugate which displays good cytocompatibility, aqueous solubility, high transporter affinity, and, crucially, an exceptional boron delivery capacity in the in vitro assessment thereby pointing toward the significant potential embedded in this approach.
Keywords: boron neutron capture therapy, cancer therapeutics, carbohydrates, glucose transporters, medicinal chemistry, drug delivery
1. Introduction
As one of the leading causes of morbidity and mortality on a global scale, cancer is a significant societal economic burden with annual global costs of 723–930 billion euros. Head and neck cancers account for up to 10% of all cancers with 630 000 new cases annually diagnosed worldwide.1,2 Despite traditional treatments featuring surgery, radiation, and chemotherapy, all of which are arduous for the patients, many of these cancers recur. In head and neck cancers, the inoperable recurrent ones are accompanied by a poor survival rate with a mean survival time of only a few months.3 A number of novel treatment strategies have recently gained ground. These include antibody–drug conjugates,4 proton therapy,5,6 and, especially, boron neutron capture therapy (BNCT).7 BNCT represents one of the most promising noninvasive binary treatment modalities for head and neck cancers since it can eradicate cancer cells while simultaneously sparing healthy cells (the basis of our approach is displayed in Figure 1).8,9 The selectivity in BNCT arises from a 2-fold effect. First, only cells with a sufficient concentration of 10B atoms are destroyed and, second, the external neutron beam can be applied to a narrow and highly specific area where malignant cells are present. Previously, the applications of, and interest in, clinical BNCT have been hampered by the need for nuclear reactors, as a neutron source, and the poor properties of clinically used boron delivery agents. In recent years, new in-hospital neutron accelerators10 have emerged thus revolutionizing the clinical aspects of patient treatments; a renewed interest in the BNCT field has been invoked—now, the final challenge to solve is that of developing improved boron delivery agents.
Figure 1.
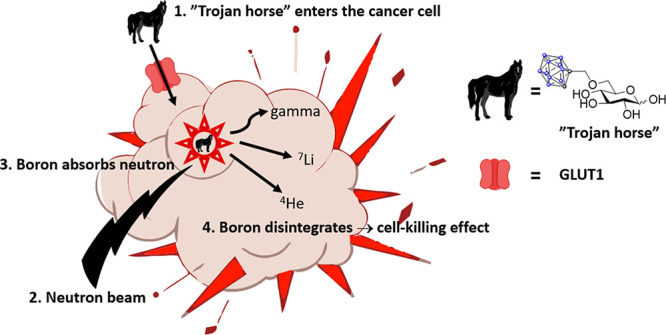
Principles of our approach to BNCT. Blue dots represent boron atoms while gray dots represent carbon atoms in the ortho-carboranylmethyl moiety.
An optimally functioning delivery agent for BNCT should display a minimal systemic toxicity, a cellular uptake of 20–35 μg/g of tumor (i.e., ppm range), and tumor/normal tissue (T/N) and tumor/blood (T/B) ratios above 3:1, with higher ratios naturally desirable. Combining these different aspects into one single delivery agent has proved challenging. Despite the large number of delivery agents (amino acids, carbohydrates, porphyrins, antibody–boron conjugates, polymers, peptides, liposomes, and nanoparticles) evaluated in the literature,11−13 only three are in clinical use. These are sodium borocaptate (BSH), boronophenylalanine (BPA and its fructose-complex) and decahydrodecaborate (GB-10). None of them exhibit optimal properties. BPA has poor water solubility, contains only one boron atom/delivered molecule, and gives poor T/B- and T/N-ratios.14,15 BSH16 and GB-1017 lack active targeting and uptake mechanisms and have an ionic nature which may cause undesired interactions with other biomolecules in a biological context.
The intrinsic properties of carbohydrates, i.e., high aqueous solubility, low systemic toxicity, and high biocompatibility make them seemingly ideal candidates for clinical BNCT. Polysaccharide and oligosaccharide carriers are, however, suboptimal from a BNCT-perspective: polysaccharides are constrained to the extracellular matrix while oligosaccharides display low lectin-binding affinities. Thus, we have chosen to focus on monosaccharides in combination with carbohydrate transporters, glucose transporters (GLUTs and SGLTs) in particular.
Glucose is an essential nutrient for mammalian cells. An increased expression of GLUTs and SGLTs, especially GLUT1, has been observed in head and neck cancers.18 The basis for this increase is the switch in glucose metabolism which in cancer cells proceeds by an inefficient aerobic glycolysis route in contrast to the oxidative metabolism in healthy noncancerous cells.19 This inefficient metabolic pathway leads to a substantial increase in glucose uptake which allows the cancer cells to grow rapidly and proliferate.20 Exploiting this “Warburg effect”, named after Nobel laureate Otto Heinrich Warburg, provides the foundation for the development of novel glucose-based “Trojan horses” for clinical BNCT. Before reaching the end stages of the development process (in vivo-studies with/without neutron sources), the biochemical foundations of the approach need to be addressed in detail. In this regard, it is important to note that concerns have been raised regarding the effects of glucoconjugates on glucose metabolism in healthy cells, the possible incorporation of metabolic products into other biomolecules, and the competition for the transporters with the high glucose levels found in blood.21 Therefore, for a GLUT1 targeting approach to be successful, it is crucial to address these issues already at the design stage.
To this end, we have designed and synthesized three glucoconjugates bearing an ortho-carboranylmethyl substituent. The carboranyl provides ten boron nuclei per delivery molecule in a charge-neutral, chemically stable form, and is thus highly suitable for the purposes of BNCT. In addition, a neutral and hydrophobic boron cluster should be advantageous when aiming for transport through a transmembrane protein since possible unfavorable interactions between charged boron clusters and amino acids can be avoided.22Figure 2 shows the three 6-O-carboranylmethyl glucoconjugates targeted: the hemiacetal and both methyl glycosides. The attachment of boron clusters at the sixth position in glucose is rare in the scientific literature, and conjugates with charge-neutral boron clusters have not been prepared earlier.23,24 A modification at this site will, however, remove the concerns regarding interference with glucose metabolism and incorporation into other biomolecules through the glycolysis route; the 6-O-carboranylmethyl glucoconjugates are no longer substrates for the glycolysis route in which the first transformation is a phosphorylation at the sixth position.25
Figure 2.
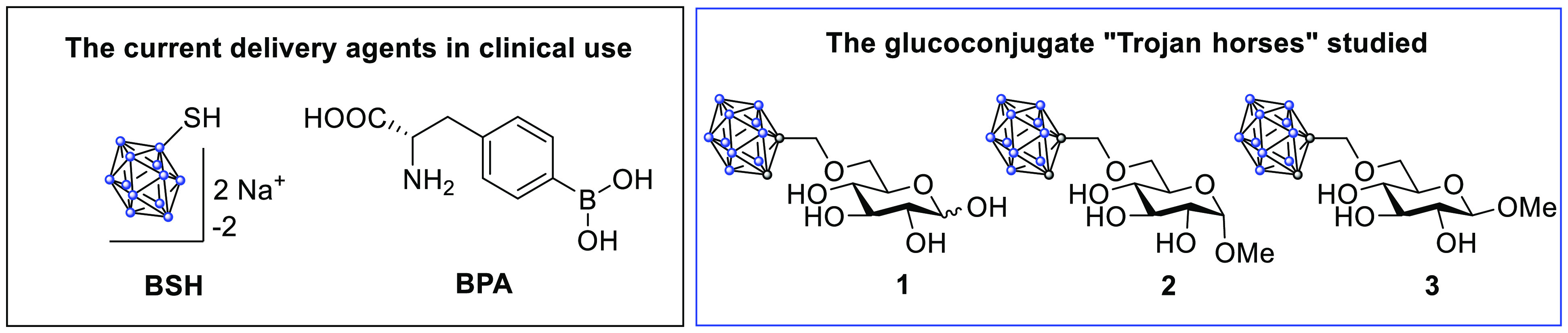
Two of the delivery agents in clinical use (left box) and the 6-O-ortho-carboranylmethyl glucoconjugates prepared in the current study (right box). Blue dots represent boron atoms while gray dots represent carbon atoms in the carboranyl moiety.
In addition to synthesizing the glucoconjugates and conducting the most detailed structural characterization of such conjugates to date, we have addressed the biochemical foundations of the GLUT1-targeting approach through a preliminary, yet, comprehensive in vitro evaluation study featuring cytotoxicity, computational/experimental receptor affinity, and cellular uptake experiments in the relevant human head and neck cancer cell line CAL 27 (oral adenosquamous carcinoma cell line). To our satisfaction, the new glucoconjugates display a significantly stronger binding affinity to GLUT1 than glucose. This shows that the previous fear regarding their competition with the high levels of free glucose in blood has been unfounded. Moreover, the glucoconjugates display a boron delivery capacity 40 times higher than the best agents currently in clinical use—showing that there is considerable potential embedded in this alternative approach.
2. Experimental Section
2.1. Synthesis and Structural Characterization
Reaction solvents were purified by the VAC vacuum solvent purification system prior to use when dry solvents were needed. All reactions containing moisture- or air-sensitive reagents were carried out under an argon atmosphere. All reagents were purchased from commercial sources. The NMR spectra were recorded with a Bruker Avance III NMR spectrometer operating at 500.13 MHz (1H: 500.13 MHz, 13C: 125.76 MHz, 11B: 160.46 MHz). The probe temperature during the experiments was kept at 23 °C. All products were characterized by utilization of the following 1D-techniques: 1H, 13C, 11B, and 1D-TOCSY and the following 2D-techniques: Ed-HSQC, HMBC, and COSY by using pulse sequences provided by the instrument manufacturer. Chemical shifts are expressed on the δ scale (in ppm) using TMS (tetramethylsilane), residual chloroform, methanol, or 15% BF3 in CDCl3 (11B NMR) as internal standards. Coupling constants have been obtained through spectral simulations with the Perch Peak Research software, are given in Hz, and are provided only once, when first encountered. Coupling patterns are given as s (singlet), d (doublet), t (triplet), etc. HRMS were recorded using Bruker Micro Q-TOF with ESI (electrospray ionization) operated in positive mode. The purity of the compounds was determined to be >95% in all cases. TLC was performed on aluminum sheets precoated with silica gel 60 F254 (Merck). Flash chromatography was carried out on silica gel 40. Spots were visualized by UV, followed by spraying the TLC plates with a solution of H2SO4:MeOH (1:4) and heating.
General Experimental Procedures
General Procedure for Selective Silylation of the 6-OH Group in Glucopyranose
tert-Butyldimethylsilyl chloride (1.35 equiv) was added portion-wise to a solution of d-glucose (1 equiv) in pyridine (10 mL/g of starting material) at 0 °C. The mixture was brought to rt and stirred for 21 h. The solvent was removed in vacuo, and the crude product was purified by column chromatography (DCM:MeOH 7:1), the solvents were removed, and the corresponding silylated glycopyranose was dried on the vacuum line.
General Procedure for Selective Silylation of the 6-OH Group in Glucopyranosides
The corresponding methyl d-glucopyranoside (1 equiv) was dissolved in dry DMF (10 mL/g of starting material) at 0 °C under an atmosphere of argon. Imidazole (1.5 equiv) and the tert-butyldimethylsilyl chloride (1.35 equiv) were added, and the reaction mixture was brought to rt. The mixture was stirred o/n. The reaction was quenched by the addition of MeOH (0.25 mL/g of starting material) and concentrated in vacuo. The crude product was purified by column chromatography (DCM:MeOH 7:1), the solvents were removed, and the corresponding silylated glycoside was dried on the vacuum line.
General Procedure for Alkylation of Free Hydroxyl Groups
The partially protected glucoside/glucopyranose (1 equiv) was dissolved in dry DMF (2 mL/100 mg of starting material) under an atmosphere of argon. The solution was cooled on an ice bath, and NaH (1.9 equiv) was added. The reaction mixture was stirred for 15 min and then brought to rt and stirred for a further 10 min. The corresponding bromide (1.5 equiv/free OH-group) was added, and the resulting mixture was stirred for 1–4 h, quenched with MeOH (0.4 mL/mmol of starting material), diluted with DCM (4 mL/100 mg), and washed with a satd. NaHCO3-solution. The organic phase was separated, and the aqueous phase was extracted with DCM (3 × 3 mL/100 mg). The organic phases were combined and washed with brine (3 mL/100 mg), dried over Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography (EtOAc:hexane 1:8), the solvents were removed, and the corresponding alkylated glycoside was dried on the vacuum line.
General Procedure for Deprotection of Silyl Protective Groups
To a solution containing the protected glycoside (1 equiv) in dry THF (3 mL/200 mg of starting material) at 0 °C, HF-pyridine (18 μL/0.03 mmol of starting material) was added. The resulting mixture was brought to rt and stirred for 20 h. The reaction mixture was diluted with DCM (30 mL/0.5 g of starting material) and quenched by the addition of a satd. NaHCO3-solution (20 mL/200 mg of starting material). The aqueous phase was extracted with DCM (3 × 20 mL), and the organic phases were combined and washed with brine (20 mL/500 mg of starting material). The combined organic phase was dried over Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography (hexane:EtOAc 2:1), the solvents were removed, and the corresponding deprotected glycoside was dried on the vacuum line.
General Procedure for Coupling Reaction with Decaborane
B10H14 (1.8 equiv) in dry ACN (5 mL/150 mg) under argon was heated to 60 °C and stirred for 1 h. Meanwhile, the propargylated glycoside (1 equiv) was dissolved in dry toluene (5 mL/150 mg) and added after the first hour. The reaction mixture was stirred for 15–18 h at 80 °C. The mixture was quenched by the addition of dry methanol (1.8 mL/200 mg starting material) and allowed to stir for 30 min at 80 °C. The solvent was removed, and the crude product was purified by column chromatography (EtOAc:hexane 1:3), the solvents were removed, and the corresponding carboranyl glycoside was dried on the vacuum line.
General Procedure for Deprotection of Benzyl Groups
The corresponding protected glucoside was dissolved in EtOAc:MeOH 7:1 (1 mL/10 mg of starting material). Pd/C (10% Pd, 1 weight equiv) was added, and the reaction mixture was stirred in an autoclave under H2 (3–5 bar) for 4–6 h. The resulting mixture was filtered through Celite, washed with EtOAc:MeOH 7:1 (3 × 10 mL), and concentrated under vacuum. The crude product was purified by column chromatography (DCM:MeOH 5:1), the solvents were removed, and the product was dried on the vacuum line to give the corresponding deprotected glucoside/glucopyranose.
Substrate Specific Analytical Data
6-O-(tert-Butyldimethylsilyl)-d-glucopyranose
Synthesized from d-glucose (9.99 g/55.5 mmol), according to the general procedure for selective silylation of the 6-OH group in glucopyranoses. This reaction yielded an off-white powder (10.64 g, 69%; α:β 58:42). Rf = 0.61 (DCM:MeOH 5:1).
1H NMR of the α-anomer (500.13 MHz; CD3OD): δ 5.08 (d, 1H, J1,2 = 3.7 Hz, H-1), 3.85 (dd, 1H, J6a,5 = 2.1, J6a,6b = −11.2 Hz, H-6a), 3.84 (dd, 1H, J6b,5 = 4.6 Hz, H-6b), 3.75 (ddd, 1H, J5,4 = 9.8 Hz, H-5), 3.67 (dd, 1H, J3,4 = 9.1, J3,2 = 9.6 Hz, H-3), 3.35 (dd, 1H, H-4), 3.33 (dd, 1H, H-2), 0.90 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.08 and 0.07 (each s, each 3H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR of the α-anomer (125.76 MHz; CD3OD): δ 93.9 (C-1), 74.9 (C-3), 73.8 (C-2), 73.2 (C-5), 71.6 (C-4), 64.1 (C-6), 26.4 (6-OSi(CH3)2C(CH3)3), 19.3 (6-OSi(CH3)2C(CH3)3) and −5.0 and −5.1 (6-OSi(CH3)2C(CH3)3) ppm.
1H NMR of the β-anomer (500.13 MHz; CD3OD): δ 4.44 (d, 1H, J1,2 = 7.8 Hz, H-1), 3.94 (dd, 1H, J6a,5 = 2.0, J6a,6b = −11.2 Hz, H-6a), 3.78 (dd, 1H, J6b,5 = 5.4 Hz, H-6b), 3.33 (dd, 1H, J3,4 = 8.9, J3,2 = 9.8 Hz, H-3), 3.30 (dd, 1H, J4,5 = 9.4 Hz, H-4), 3.26 (ddd, 1H, H-5), 3.11 (dd, 1H, H-2), 0.90 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.08 and 0.07 (each s, each 3H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR of the β-anomer (125.76 MHz; CD3OD): δ 98.1 (C-1), 78.2 (C-3, C-5), 76.2 (C-2), 71.5 (C-4), 64.3 (C-6), 26.4 (6-OSi(CH3)2C(CH3)3), 19.3 (6-OSi(CH3)2C(CH3)3) and −5.1 (6-OSi(CH3)2C(CH3)3) ppm.
HRMS: m/z calcd. for C12H26O6SiNa [M + Na]+ 317.1397; found 317.1385.
1,2,3,4-Tetra-O-benzyl-6-O-(tert-butyldimethylsilyl)-d-glucopyranose (4)
Synthesized from 6-O-(tert-butyldimethylsilyl)-d-glucopyranose (0.95 g, 3.2 mmol), NaH (0.90 g, 37.4 mmol), and BnBr (4.90 g, 28.6 mmol) according to the general procedure for alkylation of free hydroxyl groups to give a white solid (1.85 g, 88%; α:β 31:69). TLC: Rf: 0.39 (EtOAc:Hex 1:8).
1H NMR of the α-anomer (500.13 MHz; CDCl3): δ 7.42–7.23 (m, 20H, arom. H), 5.00 and 4.84 (each d, each 1H, J = −10.6 Hz, 3-OCH2Ph), 4.89 and 4.66 (each d, each 1H, J = −11.0 Hz, 4-OCH2Ph), 4.83 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.69 and 4.56 (each d, each 1H, J = −11.9 Hz, 1-OCH2Ph), 4.66 and 4.58 (each d, each 1H, J = −11.7 Hz, 2-OCH2Ph), 4.06 (dd, 1H, J3,4 = 9.1, J3,2 = 9.8 Hz, H-3), 3.78 (dd, 1H, J6a,5 = 4.6, J6a,6b = −11.4 Hz, H-6a), 3.74 (dd, 1H, J6b,5 = 1.7 Hz, H-6b), 3.71 (ddd, 1H, J5,4 = 10.0 Hz, H-5), 3.56 (dd, 1H, H-4), 3.52 (dd, 1H, H-2), 0.90 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.06 and 0.05 (each s, each 3H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR of the α-anomer (125.76 MHz; CDCl3): δ 139.0–127.7 (arom. C), 95.2 (C-1), 82.3 (C-3), 80.5 (C-2), 78.0 (C-4), 76.0 (3-OCH2Ph), 75.2 (4-OCH2Ph), 73.1 (2-OCH2Ph), 71.9 (C-5), 68.8 (1-OCH2Ph), 62.3 (C-6), 26.1 (6-OSi(CH3)2C(CH3)3), 18.5 (6-OSi(CH3)2C(CH3)3) and −5.0 and −5.2 (6-OSi(CH3)2C(CH3)3) ppm.
1H NMR of the β-anomer (500.13 MHz; CDCl3): δ 7.42–7.23 (m, 20H, arom. H), 4.97 and 4.73 (each d, each 1H, J = −10.8 Hz, 2-OCH2Ph), 4.94 and 4.67 (each d, each 1H, J = −11.6 Hz, 1-OCH2Ph), 4.92 and 4.81 (each d, each 1H, J = −10.8 Hz, 3-OCH2Ph), 4.86 and 4.69 (each d, each 1H, J = −10.8 Hz, 4-OCH2Ph), 4.51 (d, 1H, J1,2 = 7.8 Hz, H-1), 3.90 (dd, 1H, J6a,5 = 1.7, J6a,6b = −11.4 Hz, H-6a), 3.86 (dd, 1H, J6b,5 = 4.4 Hz, H-6b), 3.65 (dd, 1H, J3,2 = 8.9, J3,4 = 9.0 Hz, H-3), 3.63 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.49 (dd, 1H, H-2), 3.30 (ddd, 1H, H-5), 0.93 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.12 and 0.10 (each s, each 3H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR of the β-anomer (125.76 MHz; CDCl3): δ 139.0–127.7 (arom. C), 102.4 (C-1), 84.9 (C-3), 82.7 (C-2), 77.8 (C-4), 76.0 (C-5, 3-OCH2Ph), 75.2 (4-OCH2Ph), 75.1 (2-OCH2Ph), 71.0 (1-OCH2Ph), 62.4 (C-6), 26.1 (6-OSi(CH3)2C(CH3)3), 18.5 (6-OSi(CH3)2C(CH3)3) and −4.8 and −5.2 (6-OSi(CH3)2C(CH3)3) ppm.
HRMS: m/z calcd. for C40H50O6SiNa [M + Na]+ 677.3275; found 677.3300.
1,2,3,4-Tetra-O-benzyl-d-glucopyranose
Synthesized from 1,2,3,4-tetra-O-benzyl-6-O-(tert-butyldimethylsilyl)-d-glucopyranose (1.73 g, 2.6 mmol) and HF-pyridine (1.6 mL, 17.8 mmol) according to the general procedure for deprotection of silyl protective groups to give a white solid (1.42 g, 99%; α:β 33:67). TLC: Rf: 0.8 (EtOAc:Hex 1:1).
1H NMR of the α-anomer (500.13 MHz; CDCl3): δ 7.41–7.25 (m, 20H, arom. H), 5.01 and 4.84 (each d, each 1H, J = −10.8 Hz, 3-OCH2Ph), 4.89 and 4.64 (each d, each 1H, J = −11.0 Hz, 4-OCH2Ph), 4.80 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.68 and 4.56 (each d, each 1H, J = −11.9 Hz, 2-OCH2Ph), 4.68 and 4.55 (each d, each 1H, J = −12.4 Hz, 1-OCH2Ph), 4.06 (dd, 1H, J3,4 = 9.2, J3,2 = 9.5 Hz, H-3), 3.71 (ddd, 1H, J5,6a = 2.7, J5,6b = 2.8, J5,4 = 9.5 Hz, H-5), 3.70 (ddd, 1H, J6a,6-OH = 5.7, J6a,6b = −11.1 Hz, H-6a), 3.68 (ddd, 1H, J6a,6-OH = 7.0 Hz, H-6b), 3.54 (dd, 1H, H-4), 3.50 (dd, 1H, H-2) and 1.57 (dd, 1H, 6-OH) ppm.
13C NMR of the α-anomer (125.76 MHz; CDCl3): δ 139.0–127.7 (arom. C), 95.7 (C-1), 82.1 (C-3), 80.2 (C-2), 77.6 (C-4), 75.9 (3-OCH2Ph), 75.2 (4-OCH2Ph), 73.2 (2-OCH2Ph), 71.1 (C-5), 69.4 (1-OCH2Ph) and 62.0 (C-6) ppm.
1H NMR of the β-anomer (500.13 MHz; CDCl3): δ 7.41–7.25 (m, 20H, arom. H), 4.95 and 4.73 (each d, each 1H, J = −10.9 Hz, 2-OCH2Ph), 4.93 and 4.81 (each d, each 1H, J = −10.9 Hz, 3-OCH2Ph), 4.92 and 4.69 (each d, each 1H, J = −11.9 Hz, 1-OCH2Ph), 4.86 and 4.64 (each d, each 1H, J = −11.0 Hz, 4-OCH2Ph), 4.57 (d, 1H, J1,2 = 7.9 Hz, H-1), 3.87 (ddd, 1H, J6a,5 = 2.9, J6a,6-OH = 5.8, J6a,6b = −11.5 Hz, H-6a), 3.70 (ddd, 1H, J6b,5 = 4.9, J6b,6-OH = 7.7 Hz, H-6b), 3.67 (dd, 1H, J3,2 = 9.2, J3,4 = 9.2 Hz, H-3), 3.57 (dd, 1H, J4,5 = 9.7 Hz, H-4), 3.49 (dd, 1H, H-2), 3.36 (ddd, 1H, H-5) and 1.84 (dd, 1 H, 6-OH) ppm.
13C NMR of the β-anomer (125.76 MHz; CDCl3): δ 139.0–127.7 (arom. C), 103.0 (C-1), 84.7 (C-3), 82.5 (C-2), 77.7 (C-4), 75.9 (3-OCH2Ph), 75.2 (C-5, 4-OCH2Ph), 75.1 (2-OCH2Ph), 71.8 (1-OCH2Ph) and 62.2 (C-6) ppm.
HRMS: m/z calcd. for C34H36O6Na [M + Na]+ 563.2410; found 563.2395.
1,2,3,4-Tetra-O-benzyl-6-O-propargyl-d-glucopyranose (5)
Synthesized from 1,2,3,4-tetra-O-benzyl-d-glucopyranose (1.42 g, 2.6 mmol, 1.0 equiv), NaH (0.15 g, 6.0 mmol), and propargyl-bromide (0.56 g, 4.7 mmol) according to the general procedure for alkylation of free hydroxyl groups to give a white solid (1.26 g, 83%; α:β 31:69).
1H NMR of the α-anomer (500.13 MHz; CDCl3): δ 7.42–7.23 (m, 20H, arom. H), 5.00 and 4.84 (each d, each 1H, J = −10.9 Hz, 3-OCH2Ph), 4.87 and 4.68 (each d, each 1H, J = −11.2 Hz, 4-OCH2Ph), 4.83 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.68 and 4.55 (each d, each 1H, J = −12.2 Hz, 1-OCH2Ph), 4.67 and 4.56 (each d, each 1H, J = −12.1 Hz, 2-OCH2Ph), 4.20 (dd, 1H, JCH2a,CH = −2.4, JCH2a,CH2b = −16.0 Hz, 6-OCH2aC≡CH), 4.12 (dd, 1H, JCH2b,CH = −2.4 Hz, 6-OCH2bC≡CH), 4.04 (dd, 1H, J3,4 = 9.0, J3,2 = 9.7 Hz, H-3), 3.83 (dd, 1H, J6a,5 = 3.4, J6a,6b = −10.0 Hz, H-6a), 3.81 (ddd, 1H, J5,6b = 1.6, J5,4 = 10.3 Hz, H-5), 3.63 (dd, 1H, H-4), 3.58 (dd, 1H, H-6b), 3.54 (dd, 1H, H-2) and 2.37 (dd, 1H, 6-OCH2C≡CH) ppm.
13C NMR of the α-anomer (125.76 MHz; CDCl3): δ 139.1–127.7 (arom. C), 95.9 (C-1), 82.2 (C-3), 79.9 (C-2), 79.6 (6-OCH2C≡CH), 77.6 (C-4), 75.8 (3-OCH2Ph), 75.2 (6-OCH2C≡CH), 75.1 (4-OCH2Ph), 73.1 (2-OCH2Ph), 70.3 (C-5), 69.4 (1-OCH2Ph), 68.1 (C-6) and 58.7 (6-OCH2C≡CH) ppm.
1H NMR of the β-anomer (500.13 MHz; CDCl3): δ 7.41–7.25 (m, 20H, arom. H), 4.97 and 4.66 (each d, each 1H, J = −11.9 Hz, 1-OCH2Ph), 4.95 and 4.72 (each d, each 1H, J = −10.9 Hz, 2-OCH2Ph), 4.92 and 4.79 (each d, each 1H, J = −11.0 Hz, 3-OCH2Ph), 4.86 and 4.68 (each d, each 1H, J = −10.8 Hz, 4-OCH2Ph), 4.51 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.26 (dd, 1H, JCH2a,CH = −2.4, JCH2a,CH2b = −15.9 Hz, 6-OCH2aC≡CH), 4.20 (dd, 1H, JCH2b,CH = −2.4 Hz, 6-OCH2bC≡CH), 3.83 (dd, 1H, J6a,5 = 4.5, J6a,6b = −10.7 Hz, H-6a), 3.79 (dd, 1H, J6b,5 = 2.1 Hz, H-6b), 3.64 (dd, 1H, J3,4 = 9.0, J3,2 = 9.2 Hz, H-3), 3.62 (dd, 1H, J4,5 = 9.6 Hz, H-4), 3.51 (dd, 1H, H-2), 3.47 (ddd, 1H, H-5) and 2.39 (dd, 1H, 6-OCH2C≡CH) ppm.
13C NMR of the β-anomer (125.76 MHz; CDCl3): δ 139.0–127.7 (arom. C), 102.8 (C-1), 84.8 (C-3), 82.4 (C-2), 79.8 (6-OCH2C≡CH), 77.7 (C-4), 75.8 (3-OCH2Ph), 75.1–74.9 (C-2, 4-OCH2Ph, 2-OCH2Ph, 6-OCH2C≡CH), 74.8 (C-5), 71.3 (1-OCH2Ph), 68.5 (C-6) and 58.8 (6-OCH2C≡CH) ppm.
HRMS: m/z calcd. for C37H38O6Na [M + Na]+ 601.2566; found 601.2628.
1,2,3,4-Tetra-O-benzyl-6-O-(o-carboranylmethyl)-d-glucopyranose (6)
Synthesized from 1,2,3,4-tetra-O-benzyl-6-O-propargyl-d-glucopyranose (0.91 g, 1.6 mmol) and B10H14 (0.33 g, 2.7 mmol) according to the general procedure for coupling reaction of decaborane with propargylated glucosides to give a colorless oil (0.61 g, 55.1%; α:β 25:75). TLC: Rf: 0.55 (EtOAc:Hex 1:3).
1H NMR of the α-anomer (500.13 MHz; CDCl3): δ 7.39–7.23 (m, 20H, arom. H), 5.01 and 4.81 (each d, each 1H, J = −10.6 Hz, 3-OCH2Ph), 4.90 and 4.55 (each d, each 1H, J = −11.1 Hz, 4-OCH2Ph), 4.79 (d, 1H, J1,2 = 3.7 Hz, H-1), 4.68 and 4.56 (each d, each 1H, J = −11.8 Hz, 2-OCH2Ph), 4.66 and 4.53 (each d, each 1H, J = −12.2 Hz, 1-OCH2Ph), 4.04 (dd, 1H, J3,4 = 9.1, J3,2 = 9.4 Hz, H-3), 3.87 and 3.79 (each d, each 1H, J = −10.7 Hz, 6-OCH2-carborane), 3.81 (br s, 1H, carborane-CH), 3.74 (ddd, 1H, J5,6b = 1.7, J5,6a = 4.6, J5,4 = 10.1 Hz, H-5), 3.63 (dd, 1H, J6a,6b = −11.5 Hz, H-6a), 3.49 (dd, 1H, H-2), 3.46 (dd, 1H, H-6b), 3.40 (dd, 1H, H-4) and 2.99–1.50 (br m, 10H, carborane-BH) ppm.
13C NMR of the α-anomer (125.76 MHz; CDCl3): δ 138.7–127.9 (arom. C), 95.6 (C-1), 82.1 (C-3), 80.2 (C-2), 77.5 (C-4), 76.0 (3-OCH2Ph), 75.1 (4-OCH2Ph), 73.2 (2-OCH2Ph), 72.9 (6-OCH2-carborane), 71.0 (C-6, carborane-C), 70.6 (C-5), 69.4 (1-OCH2Ph) and 57.6 (carborane-CH) ppm.
1H NMR of the β-anomer (500.13 MHz; CDCl3): δ 7.39–7.23 (m, 20H, arom. H), 4.96 and 4.73 (each d, each 1H, J = −10.8 Hz, 2-OCH2Ph), 4.93 and 4.78 (each d, each 1H, J = −10.8 Hz, 3-OCH2Ph), 4.88 and 4.66 (each d, each 1H, J = −11.9 Hz, 1-OCH2Ph), 4.87 and 4.55 (each d, each 1H, J = −11.2 Hz, 4-OCH2Ph), 4.49 (d, 1H, J1,2 = 7.9 Hz, H-1), 3.93 and 3.87 (each d, each 1H, J = −10.7 Hz, 6-OCH2-carborane), 3.91 (br s, 1H, carborane-CH), 3.67 (dd, 1H, J6a,5 = 4.8, J6a,6b = −11.4 Hz, H-6a), 3.64 (dd, 1H, J6b,5 = 1.9 Hz, H-6b), 3.63 (dd, 1H, J3,4 = 9.0, J3,2 = 9.2 Hz, H-3), 3.47 (dd, 1H, H-2), 3.44 (dd, 1H, J4,5 = 9.9 Hz, H-4), 3.36 (ddd, 1H, H-5) and 2.99–1.50 (br m, 10H, carborane-BH) ppm.
13C NMR of the β-anomer (125.76 MHz; CDCl3): δ 138.7–127.9 (arom. C), 102.8 (C-1), 84.7 (C-3), 82.4 (C-2), 77.4 (C-4), 76.0 (3-OCH2Ph), 75.1 (2-OCH2Ph, 4-OCH2Ph), 74.8 (C-5), 72.9 (6-OCH2-carborane), 71.5 (1-OCH2Ph), 71.0 (C-6, carborane-C) and 57.7 (carborane-CH) ppm.
11B NMR (160.46 MHz; CDCl3): δ –2.56, –4.38, –8.64, –11.24 and −12.91 ppm.
HRMS: m/z calcd. for C37H48B10O6Na [M + Na]+ 721.4258; found 721.4327.
6-O-(o-Carboranylmethyl)-d-glucopyranose (1)
Synthesized from 1,2,3,4-tetra-O-benzyl-6-O-(o-carboranylmethyl)-d-glucopyranose (0.11 g, 0.02 mmol) and Pd/C (0.17 g, 0.02 mmol) according to the general procedure for hydrogenolysis to give a colorless oil (0.04 g, 79%; α:β 60:40). TLC: Rf: 0.56 (EtOAc:MeOH 5:1).
1H NMR of the α-anomer (500.13 MHz; CD3OD): δ 5.10 (d, 1H, J1,2 = 3.7 Hz, H-1), 4.59 (br s, 1H, carborane-CH), 4.04 and 4.02 (each d, each 1H, J = −10.8 Hz, 6-OCH2-carborane), 3.86 (ddd, 1H, J5,6b = 1.8, J5,6a = 4.9, J5,4 = 10.1 Hz, H-5), 3.75 (dd, 1H, J6a,6b = −11.3 Hz, H-6a), 3.73 (dd, 1H, H-6b), 3.66 (dd, 1H, J3,4 = 9.0, J3,2 = 9.6 Hz, H-3), 3.35(dd, 1H, H-2), 3.29 (dd, 1H, H-4) and 3.00–1.50 (br m, 10H, carborane-BH) ppm.
13C NMR of the α-anomer (125.76 MHz; CD3OD): δ 93.9 (C-1), 75.2 (carborane-C), 74.8 (C-3), 73.9 (6-OCH2-carborane), 73.7 (C-2), 72.5 (C-6), 72.2 (C-5), 71.6 (C-4) and 60.5 (carborane-CH) ppm.
1H NMR of the β-anomer (500.13 MHz; CD3OD): δ 4.63 (br s, 1H, carborane-CH), 4.46 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.05 and 4.03 (each d, each 1H, J = −10.8 Hz, 6-OCH2-carborane), 3.79 (dd, 1H, J6a,5 = 2.0, J6a,6b = −11.3 Hz, H-6a), 3.72 (dd, 1H, J6b,5 = 5.2 Hz, H-6b), 3.37 (ddd, 1H, J5,4 = 10.3 Hz, H-5), 3.34 (dd, 1H, J3,2 = 9.0, J3,4 = 9.3 Hz, H-3), 3.30 (dd, 1H, H-4), 3.12 (dd, 1H, H-2) and 3.00–1.50 (br m, 10H, carborane-BH) ppm.
13C NMR of the β-anomer (125.76 MHz; CD3OD): δ 98.2 (C-1), 78.0 (C-3), 76.9 (C-5), 76.2 (C-2), 75.2 (carborane-C), 73.9 (6-OCH2-carborane), 72.2 (C-6), 71.3 (C-4) and 60.6 (carborane-CH) ppm.
11B NMR (160.46 MHz; CD3OD): δ –2.09, –4.01, –8.31, –10.48 and −12.17 ppm.
HRMS: m/z calcd. for C9H24B10O6Na [M + Na]+ 361.2401; found 361.2382.
Methyl 6-O-tert-Butyldimethylsilyl-α-d-glucopyranoside
Synthesized from methyl-α-d-glucopyranoside (9.91 g, 51 mmol), according to the general procedure for selective silylation of the 6-OH group in glucopyranosides. This reaction yielded a white solid (12.61 g, 80%). Rf = 0.52 (DCM:MeOH 7:1).
1H NMR (500.13 MHz; CDCl3): δ 4.74 (d, 1H, J1,2 = 3.7 Hz, H-1), 3.88 (dd, 1H, J6a,5 = 5.0, J6a,6b = −10.8 Hz, H-6a), 3.83 (dd, 1H, J6b,5 = 5.0 Hz, H-6b), 3.74 (dd, 1H, J3,2 = J3–4 = 8.9, 9.6 Hz, H-3), 3.60 (ddd, 1H, J5,4 = 9.8 Hz, H-5), 3.52 (dd, 1H, H-2), 3.52 (dd, 1H, H-4), 3.42 (s, 3H, 1-OCH3), 0.91 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.10 (s, 6H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR (125.76 MHz; CDCl3): δ 99.3 (C-1), 74.8 (C-3), 72.4–72.3 (C-2, C-4), 70.6 (C-5), 64.3 (C-6), 55.4 (1-OCH3), 26.0 (6-OSi(CH3)2C(CH3)3), 18.4 (6-OSi(CH3)2C(CH3)3) and −5.3 (6-OSi(CH3)2C(CH3)3) ppm.
HRMS: m/z calcd. for C13H28O6SiNa [M + Na]+ 331.1553; found 331.1553.
Methyl 2,3,4-Tri-O-benzyl-6-O-tertbutyldimethylsilyl-α-d-glucopyranoside (7)
Synthesized from methyl 6-O-tertbutyldimethylsilyl-α-d-glucopyranoside (0.60 g, 1.93 mmol), according to the general procedure for the alkylation of free hydroxyl groups. This reaction yielded a white solid (0.70 g, 78%). Rf = 0.32 (EtOAc:hexane 1:8).
1H NMR (500.13 MHz; CDCl3): δ 7.37–7.26 (m, 15H, arom. H), 4.97 and 4.82 (each d, each 1H, J = −10.7 Hz, 3-OCH2Ph), 4.88 and 4.64 (each d, each 1H, J = −10.9 Hz, 4-OCH2Ph), 4.79 and 4.68 (each d, each 1H, J = −12.1 Hz, 2-OCH2Ph), 4.61 (d, 1H, J1,2 = 3.6 Hz, H-1), 3.99 (dd, 1H, J3,4 = 9.0, J3,2 = 9.6 Hz, H-3), 3.78 (m, 2H, H-6a and H-6b), 3.61 (ddd, 1H, J5,6a = 3.1, J5,6b = 3.1, J5,4 = 10.0 Hz, H-5), 3.53 (dd, 1H, H-4), 3.50 (dd, 1H, H-2) and 3.36 (s, 3H, 1-OCH3), 0.88 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.04 and 0.03 (each s, each 3H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR (125.76 MHz; CDCl3): δ 139.3–127.6 (arom. C), 98.0 (C-1), 82.3 (C-3), 80.4 (C-2), 77.9 (C-4), 76.0 (3-OCH2Ph), 75.1 (4-OCH2Ph), 73.5 (2-OCH2Ph), 71.6 (C-5), 62.4 (C-6), 55.0 (1-O-CH3), 26.1 (6-OSi(CH3)2C(CH3)3), 18.4 (6-OSi(CH3)2C(CH3)3) and −5.1 (6-OSi(CH3)2C(CH3)3) ppm.
HRMS: m/z calcd. for C34H46O6SiNa [M + Na]+ 601.2962; found 601.2983.
Methyl 2,3,4-Tri-O-benzyl-α-d-glucopyranoside
Synthesized from methyl 2,3,4-tri-O-benzyl-6-O-tert-butyldimethylsilyl-α-d-glucopyranoside (2.6 g, 4.49 mmol), according to the general procedure for deprotection of silyl protective groups. This reaction yielded a white solid (1.70 g, 82%). Rf = 0.60 (EtOAc:hexane 1:1).
1H NMR (500.13 MHz; CDCl3): δ 7.37–7.28 (m, 15H, arom. H), 4.99 and 4.84 (each d, each 1H, J = −10.9 Hz, 3-OCH2Ph), 4.88 and 4.64 (each d, each 1H, J = −11.0 Hz, 4-OCH2Ph), 4.80 and 4.67 (each d, each 1H, J = −12.1 Hz, 2-OCH2Ph), 4.56 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.00 (dd, 1H, J3,4 = 8.9, J3,2 = 9.6 Hz, H-3), 3.76 (ddd, 1H, J6a,5 = 2.7, J6a,6-OH = 5.3, J6a,6b = −11.9 Hz, H-6a), 3.69 (ddd, 1H, J6b,5 = 4.1, J6b,6-OH = 7.6 Hz, H-6b), 3.65 (ddd, 1H, J5,4 = 9.9 Hz, H-5), 3.52 (dd, 1H, H-4), 3.50 (dd, 1H, H-2), 3.37 (s, 3H, 1-OCH3) and 1.62 (dd, 1H, 6-OH) ppm.
13C NMR (125.76 MHz; CDCl3): δ 139.7–127.7 (arom. C), 98.3 (C-1), 82.1 (C-3), 80.1 (C-2), 77.5 (C-4), 75.9 (3-OCH2Ph), 75.2 (4-OCH2Ph), 73.6 (2-OCH2Ph), 70.8 (C-5), 62.0 (C-6) and 55.3 (1-OCH3) ppm.
HRMS: m/z calcd. for C28H32O6Na [M + Na]+ 487.2097; found 487.2084.
Methyl 2,3,4-Tri-O-benzyl-6-O-propargyl-α-d-glucopyranoside (8)
Synthesized from methyl 2,3,4-tri-O-benzyl-α-d-glucopyranoside (0.31 g, 0.67 mmol), according to the general procedure for the alkylation of free hydroxyl groups. This reaction yielded a yellow oil (0.30 g, 89%). Rf = 0.61 (EtOAc:hexane 1:2).
1H NMR (500.13 MHz; CDCl3): δ 7.37–7.26 (m, 15H, arom. H), 4.98 and 4.83 (each d, each 1H, J = −10.9 Hz, 3-OCH2Ph), 4.87 and 4.65 (each d, each 1H, J = −11.4 Hz, 4-OCH2Ph), 4.79 and 4.68 (each d, each 1H, J = −11.7 Hz, 2-OCH2Ph), 4.60 (d, 1H, J1,2 = 3.6 Hz, H-1), 4.19 (dd, 1H, JCH2a,CH = −2.4, JCH2a,CH2b = −16.0 Hz, 6-OCH2aC≡CH), 4.13 (dd, 1H, JCH2a,CH = −2.4 Hz, 6-OCH2bC≡CH), 3.98 (dd, 1H, J3,4 = 9.0, J3,2 = 9.7 Hz, H-3), 3.84 (dd, 1H, J6a,5 = 3.5, J6a,6b = −10.4 Hz, H-6a), 3.76 (ddd, 1H, J5,6b = 2.1, J5,4 = 10.1 Hz, H-5), 3.66 (dd, 1H, H-6b), 3.61 (dd, 1H, H-4), 3.54 (dd, 1H, H-2), 3.37 (s, 3H, 1-OCH3) and 2.37 (dd, 1H, 6-OCH2C≡CH) ppm.
13C NMR (125.76 MHz; CDCl3): δ 139.6–127.7 (arom. C), 98.5 (C-1), 82.2 (C-3), 79.9 (C-2), 79.6 (6-OCH2C≡CH), 77.6 (C-4), 75.9 (3-OCH2Ph), 75.2 (4-OCH2Ph), 75.1 (6-OCH2C≡CH), 73.6 (2-OCH2Ph), 70.0 (C-5), 68.2 (C-6), 58.7 (6-OCH2C≡CH) and 55.4 (1-OCH3) ppm.
HRMS: m/z calcd. for C31H34O6Na [M + Na]+ 525.2253; found 525.2258.
Methyl 2,3,4-Tri-O-benzyl-6-O-(o-carboranylmethyl)-α-d-glucopyranoside (9)
Synthesized from methyl 2,3,4-tri-O-benzyl-6-O-propargyl-α-d-glucopyranoside (0.37 g, 0.73 mmol), according to the general procedure for coupling with decaborane. This reaction yielded a white solid (0.27 g, 5 3%). Rf = 0.33 (EtOAc:hexane 1:3).
1H NMR (500.13 MHz; CDCl3): δ 7.38–7.25 (m, 15H, arom. H), 4.99 and 4.80 (each d, each 1H, J = −10.8 Hz, 3-OCH2Ph), 4.90 and 4.55 (each d, each 1H, J = −11.2 Hz, 4-OCH2Ph), 4.81 and 4.67 (each d, each 1H, J = −12.0 Hz, 2-OCH2Ph), 4.55 (d, 1H, J1,2 = 3.5 Hz, H-1), 3.97 (dd, 1H, J3,4 = 8.8, J3,2 = 9.6 Hz, H-3), 3.89 and 3.80 (each d, each 1H, J = −10.5 Hz, 6-OCH2-carborane), 3.81 (br s, carborane-CH), 3.67 (ddd, 1H, J5,6b = 1.8, J5,6a = 4.4, J5,4 = 9.7, Hz, H-5), 3.65 (dd, 1H, J6a,6b = −10.9 Hz, H-6a), 3.55 (dd, 1H, H-6b), 3.48 (dd, 1H, H-2), 3.39 (dd, 1H, H-4), 3.35 (s, 3H, 1-OCH3) and 2.62–1.61 (br m, 10H, carborane-BH) ppm.
13C NMR (125.76 MHz; CDCl3): δ 139.8–127.8 (arom. C), 98.2 (C-1), 82.1 (C-3), 80.1 (C-2), 77.2 (C-4), 76.1 (3-OCH2Ph), 75.1 (4-OCH2Ph), 73.6 (2-OCH2Ph), 72.9 (carborane-C), 71.0 (C-6), 70.3 (C-5), 60.5 (6-OCH2-carborane), 57.6 (carborane-CH) and 55.4 (1-OCH3) ppm.
11B NMR (160.46 MHz; CDCl3): δ −2.74, −4.74, −8.94, −11.52 and −13.06 ppm.
HRMS: m/z calcd. for C31H44B10O6Na [M + Na]+ 645.3966; found 645.3975.
Methyl 6-O-(o-Carboranylmethyl)-α-d-glucopyranoside (2)
Synthesized from methyl 2,3,4-tri-O-benzyl-6-O-(o-carboranylmethyl)-α-d-glucopyranoside (0.15 g, 0.25 mmol), according to the general procedure for the deprotection of benzyl groups. This reaction yielded a white solid (0.06 g, 72%). Rf = 0.47 (DCM:MeOH 7:1).
1H NMR (500.13 MHz; CD3OD): δ 4.66 (d, 1H, J1,2 = 3.8 Hz, H-1), 4.57 (br s, 1H, carborane-CH), 4.04 and 4.01 (each d, each 1H, J = −11.0 Hz, 6-OCH2-carborane), 3.76 (dd, 1H, J6a,5 = 1.8, J6a,6b = −11.3 Hz, H-6a), 3.71 (dd, 1H, J6b,5 = 5.3 Hz, H-6b), 3.60 (ddd, 1H, J5,4 = 9.7 Hz, H-5), 3.59 (dd, 1H, J3,4 = 9.2, J3,2 = 9.7 Hz, H-3), 3.40 (s, 3H, 1-OCH3), 3.37 (d, 1H, H-2), 3.26 (d, 1H, H-4) and 2.56–1.59 (br m, 10H, carborane-BH) ppm.
13C NMR (125.76 MHz; CD3OD): δ 101.3 (C-1), 75.2 (carborane-C), 75.0 (C-3), 74.0 (6-OCH2-carborane), 73.5 (C-2), 72.7 (C-5), 72.3 (C-6), 71.6 (C-4), 60.7 (carborane-CH) and 55.6 (1-OCH3) ppm.
11B NMR (160.46 MHz; CDCl3): δ –1.94, –3.92, –8.30, –10.44 and −12.05 ppm.
HRMS: m/z calcd. for C10H26B10O6Na [M + Na]+ 375.2558; found 375.2519.
Methyl 6-O-tert-Butyldimethylsilyl-β-d-glucopyranoside
Synthesized from methyl β-d-glucopyranoside (0.50 g, 2.59 mmol), according to the general procedure for selective silylation of the 6-OH group in glucopyranosides. This reaction yielded a white solid (0.66 g, 83%). Rf = 0.48 (DCM:MeOH 7:1).
1H NMR (500.13 MHz; CD3OD): δ 4.17 (d, 1H, J1,2 = 7.8 Hz, H-1), 3.99 (dd, 1H, J6a,5 = 2.1, J6a,6b = −11.4 Hz, H-6a), 3.83 (dd, 1H, J6b,5 = 5.3 Hz, H-6b), 3.53 (s, 3H, 1-OCH3), 3.36 (dd, 1H, J3,2 = 9.0, J3,4 = 9.2 Hz, H-3), 3.33 (dd, 1H, J4,5 = 9.7 Hz, H-4), 3.27 (ddd, 1H, H-5), 3.17 (dd, 1H, H-2), 0.90 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.09 (s, 6H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR (125.76 MHz; CD3OD): δ 105.3 (C-1), 78.1 (C-3, C-5), 75.0 (C-2), 71.4 (C-4), 64.1 (C-6), 57.2 (1-OCH3), 26.4 (6-OSi(CH2)2CH3), 19.3 (6-OSi(CH2)2CH3), –5.0 and −5.1 (each Si(CH3)2) (each 6-OSi(CH3)2C(CH3)3) ppm.
HRMS: m/z calcd. for C13H28O6SiNa [M + Na]+ 331.1553; found 331.1531.
Methyl 2,3,4-Tri-O-benzyl-6-O-tertbutyldimethylsilyl-β-d-glucopyranoside (10)
Synthesized from methyl 6-O-tert-butyldimethylsilyl-β-d-glucopyranoside (3.05 g, 9.89 mmol), according to the general procedure for the alkylation of free hydroxyl groups. This reaction yielded a white solid (4.62 g, 80%). Rf = 0.33 (EtOAc:hexane 1:8).
1H NMR (500.13 MHz; CDCl3): δ 7.38–7.27 (m, 15H, arom. H), 4.92 and 4.71 (each d, each 1H, J = −11.0 Hz, 2-OCH2Ph), 4.91 and 4.81 (each d, each 1H, J = −10.8 Hz, 3-OCH2Ph), 4.85 and 4.68 (each d, each 1H, J = −10.9 Hz, 4-OCH2Ph), 4.29 (d, 1H, J1,2 = 7.7 Hz, H-1), 3.88 (dd, 1H, J6a,5 = 1.7, J6a,6b = −11.5 Hz, H-6a), 3.83 (dd, 1H, J6b,5 = 4.3 Hz, H-6b), 3.64 (dd, 1H, J3,4 = 9.1, J3,2 = 9.2 Hz, H-3), 3.59 (dd, 1H, J4,5 = 9.7 Hz, H-4), 3.54 (s, 3H, 1-OCH3), 3.38 (dd, 1H, H-2), 3.28 (ddd, 1H, H-5), 0.90 (s, 9H, 6-OSi(CH3)2C(CH3)3) and 0.08 and 0.07 (each s, each 3H, 6-OSi(CH3)2C(CH3)3) ppm.
13C NMR (125.76 MHz; CDCl3): δ 139.6–127.7 (arom. C), 104.6 (C-1), 84.8 (C-3), 82.7 (C-2), 77.8 (C-4), 75.9 (3-OCH2Ph), 75.9 (C-5), 75.2 (4-OCH2Ph), 74.9 (2-OCH2Ph), 62.4 (C-6), 56.8 (1-OCH3), 26.1 (6-OSi(CH3)2C(CH3)3), 18.5 (6-OSi(CH3)2C(CH3)3), –4.9 and −5.1 (6-OSi(CH3)2C(CH3)3) ppm.
HRMS: m/z calcd. for C34H46O6SiNa [M + Na]+ 601.2962; found 601.2964.
Methyl 2,3,4-Tri-O-benzyl-β-d-glucopyranoside
Synthesized from methyl 2,3,4-tri-O-benzyl-6-O-tert-butyldimethylsilyl-β-d-glucopyranoside (2.94 g, 5.08 mmol), according to the general procedure for deprotection of silyl protective groups. This reaction yielded a white solid (2.09 g, 89%). Rf = 0.65 (EtOAc:hexane 1:1).
1H NMR (500.13 MHz; CDCl3): δ 7.38–7.27 (m, 15H, arom. H), 4.93 and 4.81 (each d, each 1H, J = −10.9 Hz, 3-OCH2Ph), 4.91 and 4.71 (each d, each 1H, J = −11.0 Hz, 2-OCH2Ph), 4.87 and 4.64 (each d, each 1H, J = −10.9 Hz, 4-OCH2Ph), 4.36 (d, 1H, J1,2 = 7.8 Hz, H-1), 3.88 (ddd, 1H, J6a,5 = 2.8, J6a,6-OH = 5.5, J6a,6b = −12.0 Hz, H-6a), 3.73 (ddd, 1H, J6b,5 = 4.5, J6b,6-OH = 7.7 Hz, H-6b), 3.67 (dd, 1H, J3,4 = 9.0, J3,2 = 9.2 Hz, H-3), 3.57 (dd, 1H, J4,5 = 9.8 Hz, H-4), 3.57 (s, 3H, 1-OCH3), 3.40 (dd, 1H, H-2), 3.37 (ddd, 1H, H-5) and 1.88 (dd, 1H, 6-OH) ppm.
13C NMR (125.76 MHz; CDCl3): δ 138.7–127.1 (arom. C), 105.0 (C-1), 84.6 (C-3), 82.5 (C-2), 77.6 (C-4), 75.8 (3-OCH2Ph), 75.3 (C-5), 75.1 (4-OCH2Ph), 75.0 (2-OCH2Ph), 62.2 (C-6) and 57.5 (1-OCH3) ppm.
HRMS: m/z calcd. for C28H32O6Na [M + Na]+ 487.2097; found 487.2089.
Methyl 2,3,4-Tri-O-benzyl-6-O-propargyl-β-d-glucopyranoside (11)
Synthesized from methyl 2,3,4-tri-O-benzyl-β-d-glucopyranoside (1.42 g, 3.02 mmol), according to the general procedure for the alkylation of free hydroxyl groups. This reaction yielded a yellow oil (1.44 g, 95%). Rf = 0.84 (EtOAc:hexane 1:1).
1H NMR (500.13 MHz; CDCl3): δ 7.35–7.26 (m, 15H, arom. H), 4.92 and 4.80 (each d, each 1H, J = −11.0 Hz, 3-OCH2Ph), 4.91 and 4.70 (each d, each 1H, J = −11.0 Hz, 2-OCH2Ph), 4.85 and 4.68 (each d, each 1H, J = −10.8 Hz, 4-OCH2Ph), 4.30 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.25 (dd, 1H, JCH2a,CH = −2.4, JCH2a,CH2b = −15.9 Hz, 6-OCH2aC≡CH), 3.64 (dd, 1H, JCHb,CH = −2.4 Hz, 6-OCH2bC≡CH), 3.83 (dd, 1H, J6a,5 = 4.4, J6a,6b = −10.7 Hz, H-6a), 3.78 (dd, 1H, J6b,5 = 2.0 Hz, H-6b), 3.64 (dd, 1H, J3,2 = 9.1, J3,4 = 9.1 Hz, H-3), 3.61 (dd, 1H, J4,5 = 10.0 Hz, H-4), 3.57 (s, 3H, 1-OCH3), 3.46 (ddd, 1H, H-5), 3.42 (dd, 1H, H-2) and 2.38 (dd, 1H, 6-OCH2C≡CH) ppm.
13C NMR (125.76 MHz; CDCl3): δ 128.6–127.7 (arom. C), 104.9 (C-1), 84.7 (C-3), 82.4 (C-2), 79.8 (6-OCH2C≡CH), 77.7 (C-4), 77.8 (3-OCH2Ph), 76.4 (6-OCH2C≡CH), 75.1 (4-OCH2Ph), 74.8 (2-OCH2Ph), 74.7 (C-5), 68.5 (C-6), 58.8 (6-OCH2C≡CH) and 57.3 (1-OCH3) ppm.
HRMS: m/z calcd. for C31H34O6Na [M + Na]+ 525.2253; found 525.2252.
Methyl 2,3,4-Tri-O-benzyl-6-O-(o-carboranylmethyl)-β-d-glucopyranoside (12)
Synthesized from methyl 2,3,4-tri-O-benzyl-6-O-propargyl-β-d-glucopyranoside (1.20 g, 2.38 mmol), according to the general procedure for coupling with decaborane. This reaction yielded a white solid (0.98 g, 66%). Rf = 0.26 (EtOAc:hexane 1:4).
1H NMR (500.13 MHz; CDCl3): δ 7.35–7.23 (m, 15H, arom. H), 4.93 and 4.78 (each d, each 1H, J = −10.8 Hz, 3-OCH2Ph), 4.91 and 4.71 (each d, each 1H, J = −11.1 Hz, 2-OCH2Ph), 4.87 and 4.54 (each d, each 1H, J = −11.1 Hz, 4-OCH2Ph), 4.28 (d, 1H, J1,2 = 7.1 Hz, H-1), 3.94 (br s, carborane-CH), 3.94 and 3.89 (each d, each 1H, J = −10.5 Hz, 6-OCH2-carborane), 3.66 (dd, 1H, J6a,5 = 2.3, J6a,6b = −10.5 Hz, H-6a), 3.65 (dd, 1H, J6b,5 = 4.4 Hz, H-6b), 3.63 (dd, 1H, J3,4 = 8.8, J3,2 = 9.3 Hz, H-3), 3.54 (s, 3H, 1-OCH3), 3.41 (dd, 1H, J4,5 = 10.0 Hz, H-4), 3.38 (dd, 1H, H-2), 3.37 (ddd, 1H, H-5) and 2.64–1.77 (br m, 10H, carborane-BH) ppm.
13C NMR (125.76 MHz; CDCl3): δ 138.7–127.9 (arom. C), 104.9 (C-1), 84.6 (C-3), 82.4 (C-2), 77.4 (C-4), 75.9 (3-OCH2Ph), 75.1 (4-OCH2Ph), 75.0 (2-OCH2Ph), 74.7 (C-5), 72.9 (6-OCH2-carborane and carborane-C), 71.1 (C-6), 57.7 (carborane-CH) and 57.3 (1-OCH3) ppm.
11B NMR (160.46 MHz; CDCl3): δ –2.83, –4.62, –8.89, –11.58 and −13.09 ppm.
HRMS: m/z calcd. for C31H44B10O6Na [M + Na]+ 645.3966; found 645.4025.
Methyl 6-O-(o-Carboranylmethyl)-β-d-glucopyranoside (3)
Synthesized from methyl 2,3,4-tri-O-benzyl-6-O-(o-carboranylmethyl)-β-d-glucopyranoside (0.15 g, 0.25 mmol), according to the general procedure for the deprotection of benzyl groups. This reaction yielded a white solid (0.068 g, 81%). Rf = 0.61 (DCM:MeOH 5:1).
1H NMR (500.13 MHz; CD3OD): δ 4.63 (br s, 1H, carborane-CH), 4.17 (d, 1H, J1,2 = 7.8 Hz, H-1), 4.06 (each d, each 1H, J = −11.1 Hz, 6-OCH2-carborane), 3.84 (dd, 1H, J6a,5 = 2.1, J6a,6b = −11.3 Hz, H-6a), 3.73 (dd, 1H, J6b,5 = 5.2 Hz, H-6b), 3.53 (s, 3H, 1-OCH3), 3.38 (ddd, 1H, J5,4 = 10.0 Hz, H-5), 3.35 (dd, 1H, J3,2 = 9.3, J3,4 = 9.3 Hz, H-3), 3.30 (dd, 1H, H-4), 3.16 (dd, 1H, H-2) and 2.54–1.54 (br m, 10H, carborane-BH) ppm.
13C NMR (125.76 MHz; CD3OD): δ 105.4 (C-1), 77.8 (C-3), 76.9 (C-5), 75.2 (carborane-C), 74.9 (C-2), 73.9 (6-OCH2-carborane), 72.2 (C-6), 71.2 (C-4), 60.5 (carborane-CH) and 57.4 (1-OCH3) ppm.
11B NMR (160.46 MHz; CDCl3): δ –2.06, –3.97, –8.30, –10.50 and −12.08 ppm.
HRMS: m/z calcd. for C10H26B10O6Na [M + Na]+ 375.2558; found 375.2557.
2.2. Molecular Modeling
The initial geometries of the ligands were optimized to a local minimum at the DFT level, using the dispersion-corrected hybrid Tao–Perdew–Scuseria–Staroverov functional TPSSh-D3(BJ),26−28 with the doubly polarized triple-ζ basis set def2-TZVPP.29 The structures of the different ligands were aligned so that geometries would be as similar as possible. Partial atomic charges were computed using the restrained electrostatic potential (RESP) protocol.30 For the RESP charge calculation, the molecule was divided into two parts, with one part consisting of the carborane and a linking carbon and the other part comprising the sugar. Partial charges of hydrogens bonded to the same carbon were constrained to be equal. The geometry optimizations were performed with Turbomole 7.3,31,32 and the RESP calculations with NWChem 6.8.33 Noncovalent interactions (NCI)34 between the ligands and protein were computed using the promolecular approach of NCIPLOT.35
Molecular docking studies were performed using AutoDock 4.2.6.36,37 All rotatable bonds in the carborane part were set to nonrotatable (inactive). For docking, the number of torsional degrees of freedom for the carboranes was set to 8 (torsdof 8). The docking studies were performed using the XylE inward-open 4QIQ and outward-open 6N3I PDB structures. The XylE protein structures were mutated using PyMOL, changing Gln-415 to Asn-415. The most probable rotamer, that is, the one with the least clashes with surrounding amino acids, as suggested by PyMOL was used. Each protein was prepared by removing the ligand and other superfluous small molecules (Zn for 4QIQ), adding hydrogens, merging them, and then computing Gasteiger partial charges. For all proteins, a grid of size 46 × 56 × 60 was used, with a grid spacing value of 0.375. The grid center was in the middle of the protein cavity, for the grid box to cover the binding site. During docking, the protein was kept rigid and only ligand torsional angles changed. For each ligand, 6000 (3 × 2000) independent search runs, each with max 2.5 million energy evaluations and population size of 150 with max 27000 generations, were performed using the default settings of the Lamarckian genetic algorithm (LGA), that is, a mutation rate of 0.02 and crossover rate of 0.8, with one top individual surviving to the next generation. Conformations were clustered (ranked) with a cluster RMS 2.0 Å.
Parameters for boron, missing from the standard distribution of Autodock, were added to the parameter file: R 2.285, Rii 4.57, epsilon 0.179, vol 49.9744; other parameters were set to their corresponding carbon values. R and epsilon were taken from Oda et al.,38 as reproduced by Couto et al.,39 and were used to calculate Rii and vol. The complete parameter definition was thus:
atom_par B 4.57 0.179 49.9744–0.00143 0.0 0.0 0 −1 −1 0 # Boron for Carborane
2.3. Cytotoxicity Studies
The CellTiter-Glo luminescent cell viability assay was purchased from Promega Corporation (Madison, WI, USA). The PierceTM BCA Protein Assay Kit was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Human CAL 27 squamous cell carcinoma was acquired from American Type Culture Collection (Manassas, VA, USA). The cell culturing flasks and 96-well plates were purchased from Corning (Corning, NY, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), Dulbecco’s phosphate buffer saline (10 × DPBS), fetal bovine serum (FBS), and Penicillin-Streptomycin (10,000 U/mL) were obtained from Gibco (Life Technologies, Carlsbad, CA, USA).
The in vitro cell cytotoxicity was carried out using a commercial CellTiter-Glo luminescent cell viability assay. The human epithelial CAL 27 squamous carcinoma cell line was used as a head-and-neck cancer cell model in this experiment. The cells were plated on a 96-well plate at 15,000 cells per well in 100 μL DMEM supplemented with 10% FBS and 1% Penicillin-Streptomycin, and cells were allowed to attach overnight. The medium was removed and replaced with 100 μL of the glucoconjugates 1, 2, 3, or sodium borocaptate (BSH) solution in complete cell culture medium at concentrations of 5 μM, 25 μM, 50 μM, 125 μM, and 250 μM. Fresh medium and 1% (v/v) Triton X-100 were used as positive and negative controls of cell viability, respectively. The incubation time points of the compounds were set at 6 and 24 h in a temperature and humidity controlled incubator (37 °C, 95% relative humidity and 5% CO2). At predetermined time points, the plates were equilibrated to room temperature for 30 min. The incubated solutions were discarded, and the cells were washed twice with 1 × PBS. Then, 50 μL of both 1 × PBS and CellTiter-Glo reagent were added to the wells. The plates were protected from light with aluminum foil and placed on an orbital shaker for 2 min before luminescence measurement with a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). All measurements were done in quadruplicate.
Furthermore, the total protein content in each sample was quantified using the colorimetric bicinchoninic acid (BCA) assay (Pierce, Thermo Fisher Scientific, Waltham, MA, USA). The procedures were carried out according to the manufacturer protocol. Cell lysates (25 μL each) from the cytotoxicity assay was transferred to a new 96-well plate. Working reagent (200 μL) was subsequently added to each sample at a 1:8 ratio. The plates were kept in the dark with aluminum foil and gently mixed on an orbital shaker for 30 s before proceeding to incubate at 37 °C for 30 min. The absorbance was read at 562 nm on a plate reader, and the protein content was determined against a bovine serum albumin (BSA) standard curve (0–2000 μg/mL). The total protein content results were used to normalize the cell viability from the CellTiter-Glo assay by dividing the luminescence value in each sample by the total protein content (μg) in the same sample before the percent cell viability determination. The experiment was carried out in quadruplicate and the statistical significance of the mean viability was determined using an unpaired Student’s t-test against the negative control for cell viability.
2.4. GLUT1 Affinity and Cellular Uptake Studies
CAL 27 squamous cell carcinoma cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) or supplied by the University of Helsinki. The CAL 27 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, ThermoFisher Scientific, Waltham, MA, USA) supplemented with l-glutamine (2.0 mM; ThermoFisher Scientific, Waltham, MA, USA), heat-inactivated fetal bovine serum (10%; Gibco, ThermoFisher Scientific, Waltham, MA, USA), penicillin (50 U/mL), streptomycin (50 μg/mL) solution (ThermoFisher Scientific, Waltham, MA, USA). The CAL 27 cells (passages 7–20) were seeded at the density of 5 × 105 cells/wells onto 24-well plates. The cells were used in the affinity and uptake studies 2 days after seeding. The culture medium was removed, and the cells were washed with prewarmed HBSS (Hank’s balance salt solution) without glucose (pH 7.4). The cells were then incubated with HBSS at 37 °C for 10 min before the experiments. Additional information on the experimental protocols is supplied in the Supporting Information.
In order to determine the ability of the compounds to bind to GLUT1 in the CAL 27 cell line, the cells were cultured, seeded, and preincubated as described above. The HBSS was removed, and the ability of the compounds to inhibit the uptake of the known GLUT1 substrate, [14C]-d-glucose (PerkinElmer, Waltham, MA, USA), was studied by incubating the cells at rt for 5 min in a buffer with a pH 7.4 (250 μL) and further containing 1.8 μM (0.1 mCi/ml) of [14C]-d-glucose. The compounds were studied at concentrations ranging from 0.1–1800 μM and the HBSS was used as a blank. After incubation, the experiment was ended by the addition of ice-cold HBSS and the cells were washed twice with ice-cold HBSS. The cells were then lysed with 250 μL of 0.1 M sodium hydroxide, the lysate was mixed with 1.0 mL of Emulsifier safe cocktail (PerkinElmer, Waltham, MA, USA), and the radioactivity was measured by liquid scintillation counter (MicroBeta2 counter, PerkinElmer, Waltham, MA, USA). The inhibition of [14C]-d-glucose in the presence of the boron containing compounds compared to the control (HBSS) was calculated as percentages (%). See Supporting Information Figure 44.
The concentration-dependent uptake studies of the glucoconjugates were performed by adding 10–400 μM of the compounds in 250 μL of prewarmed HBSS buffer on the cell layer. The incubation times for each compound were 5 and 30 min. After incubation the cells were washed and lysed as described above. The lysate from 4 wells was combined in a Eppendorf tube, centrifuged at 4 °C, and 800 μL of the supernatant was collected and digested in 1.0 mL of conc. HNO3 (TraceMetal grade, Fisher Chemical) for 24 h. After sample digestion, Milli-Q water (USF Elga Purelab Ultra) was added in order to reach a total volume of 10 mL, and the boron concentrations were analyzed by inductively coupled plasma mass spectrometry (ICP-MS).
The boron concentrations were analyzed by ICP-MS using a NeXION 350D ICP-MS instrument (PerkinElmer Inc., Waltham, MA, USA) and ESI PrepFAST autosampler (Elemental Scientific, Omaha, NE, USA). For sample injection a peristaltic pump and nebulizer were used. The instrument was operated with an RF power of 1.6 kW and with nebulizer gas, auxiliary gas, and plasma gas flows of 0.90, 18, and 1.2 l/min, respectively. The sample uptake rate was 3.5 mL/min, and dwell times were set at 100 ms per AMU. To remove polyatomic interferences, a triple-quadrupole reaction system operating in collision mode with kinetic energy discrimination (KED) was used (with He as the cell gas (3.7 mL/min)). An internal standard, 89Y, was mixed online with the samples to compensate for matrix effects and instrument drift. Boron was determined against a certified multielement calibration standard (TraceCERT Periodic Table Mix 1, Sigma-Aldrich) under acid conditions (6.7% HNO3, TraceMetal grade, Fisher Chemical). The calibration range used for 11B was 4–400 μg/lL, and the detection limit (LOD) was 1.0 μg/L. Three replicates were obtained for each sample. The data was processed using the PerkinElmer Syngistix Data Analysis Software.
3. Results
3.1. Synthesis and Structural Characterization of 6-O-Carboranylmethyl Glucoconjugates
The construction of the targeted glucoconjugates requires insights in boron cluster chemistry and carbohydrate chemistry. A significant amount of progress has been achieved in both areas over a considerable timespan, and robust reaction methodologies can be found in the existing literature.23,24 Yet, each new synthetic target requires the development of a suitable strategy, and unlike the 3-O-carboranylmethyl,40 the carboranylmethyl-glucosides,41 and other types of glucoconjugates previously evaluated,23,42,43 the 6-O-carboranylmethyl glucoconjugates (Figure 2) have been explicitly designed for clinical BNCT of head and neck cancers. The methyl glucopyranosides were included in order to evaluate if there is a difference between the affinity and cellular uptake of the two anomers since the hemiacetal exists as a mixture of both. In addition, the methyl group is minimally intrusive which is beneficial since information on the substrate tolerance of GLUT1 is limited.44
It was important to account for the susceptibility of decaborane to free hydroxyl groups and the possibility for carboranes to undergo degradation under strongly basic conditions when planning the synthesis of the 6-O-carboranylmethyl glucoconjugates. With these issues in mind, we developed the multistep synthetic routes to the three 6-O-carboranylmethyl glucoconjugates 1, 2, and 3. The synthesis and structural characterization discussion will herein be limited to glucoconjugate 1 (Scheme 1). The reaction routes to 2 and 3 are displayed in Supporting Information Scheme 1, and the synthesis and characterization of 2 and 3, and all intermediates on these routes, were conducted in a similar fashion as described below for 1.
Scheme 1. Reaction Route Leading to 1.
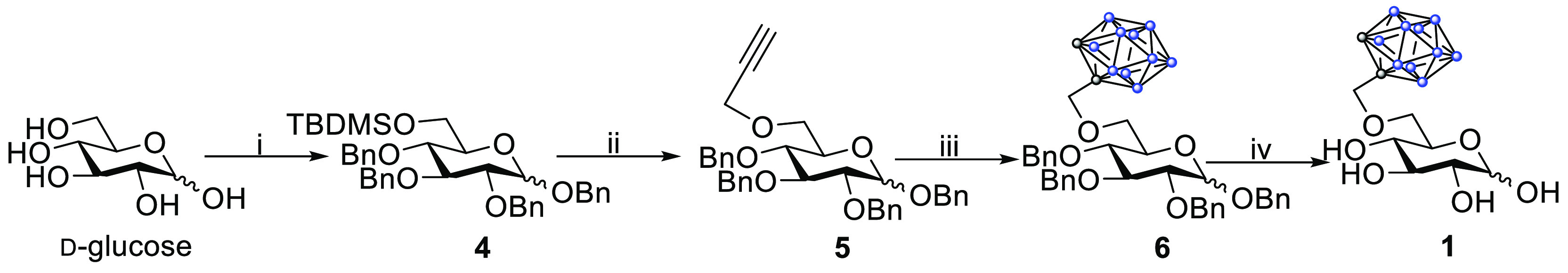
Reagents and conditions: (i) (1) TBDMSCl, pyridine, rt, 24 h, 69%; (2) BnBr, NaH, DMF, rt, 4 h, 88%; (ii) (1) HF-pyridine, THF, rt, 18 h, 99%; (2) propargyl bromide, NaH, DMF, rt, 15 h, 83%; (iii) (1) B10H14, acetonitrile, 60 °C, 1 h; (2) 5, toluene, 80 °C, 16 h, 55%; (iv) H2, 10% Pd/C, EtOAc:MeOH 7:1, 3–5 bar, rt, 4–6 h, 79%.
In short, the synthesis commenced from d-glucose. In the first step, the sterically least hindered primary hydroxyl group was temporarily protected as a bulky silyl ether with TBDMSCl in pyridine in an acceptable yield. The remaining hydroxyl groups were benzylated, through standard alkylation protocols,45 using BnBr and NaH in an 88% isolated yield. The temporary silyl protective group was removed with Olah’s reagent in excellent yield,46 followed by the alkylation of the unmasked hydroxyl group with propargyl bromide and NaH in an 87% yield. The coupling between decaborane (B10H14) and the terminal alkyne was achieved by first forming a decaborane–ACN complex47 followed by a substitution reaction with 5. In the pioneering work of Tietze et al., which encompassed the synthesis of carboranyl C-glycosides, the removal of benzyl groups was reported to proceed in high yields (61%–quant.).48 In our first attempts, we encountered challenges regarding the debenzylation reaction, and d-glucose was formed in considerable amounts as a side product (>40%). These observations were consistent regardless of the employed transition metal catalyst. While we did not optimize the reaction conditions fully, we did note that performing the reaction at a lower substrate concentration (40 mg/mL vs 14 mg/mL) led to a marked increase in the isolated yields (59% → 79%). The isolated yields on the synthetic routes were high throughout and the overall yield for the synthesis of 1 was 22%.
In order to understand how the glucoconjugates interact with GLUT1, insights on their structural properties were required. As a result, we performed a detailed conformational characterization of the synthesized molecules by NMR-spectroscopy (1H, 13C, 11B, 1D-TOCSY, DQF-COSY, ed-HSQC, and HMBC) further coupled with spectral simulations by quantum mechanical optimization utilizing the PERCH peak research software. The 1H NMR spectrum of 1 was challenging to solve because the hemiacetal exists as an anomeric mixture (59% α, 41% β) and the signals overlap in several parts of the spectrum. In order to overcome these challenges, 1D-TOCSY was utilized.49,50 The well separated H-1α (5.10 ppm) and H-1β-protons (4.46 ppm) were irradiated, and a mixing time of 300 ms was applied in order to ensure the transfer of magnetization throughout the spin-systems. This experiment resulted in information on the chemical shifts of all proton signals on both residues. By the use of standard 2D-NMR techniques, all of the 1H and 13C NMR signals of both anomers could be assigned. The coupling constants which provide information on the angles between adjacent protons and constitute the basis of a conformational characterization could not be reliably extracted from the 1H NMR spectra alone. By use of the PERCH-software, the 1H NMR spectrum was simulated and the coupling constants were obtained. The coupling constants confirmed that the glucoconjugates exist primarily in the 4C1-conformation and that the gg and gt rotamers (C5–C6-bond) are dominating in solution (JH-5,H-6a = 1.8–2.0 Hz, JH-5,H-6b = 4.9–5.2 Hz and JH-6a,H-6b = −11.3 Hz).51
The last step on the road to a complete NMR-spectroscopic characterization was to confirm that the boron cluster had remained intact. To this end, we measured decoupled 11B NMR spectra and the signals appearing in the 0 to −30 ppm region confirmed52 that the carboranyl cluster was indeed intact. In addition, we assigned all the signals (carboranylmethyl moiety, protective groups, and carbohydrate) in the 1H and 13C NMR spectra of all compounds and verified their structural identity and purity also by high resolution mass spectrometry.
3.2. Experimental and Computational GLUT1 Affinity Studies
We were interested in understanding how the glucoconjugates interact with GLUT1 since this provides the biochemical foundation for their potential use in the intended application. We sought inspiration from the previous work of Lippard et al. focusing on cytotoxic glucose-platinum conjugates.53 While the requirements for a successful GLUT1 targeting approach are similar, there is a significant conceptual difference between the therapeutic approaches of using either nontoxic boron delivery agents (with a targeted external neutron beam) or delivery agents containing cytotoxic compounds since the glucoconjugates are likely to be transported into all cells expressing GLUT1—albeit in different amounts. In order to study the GLUT1 affinities of the glucoconjugates, an experimental cis-inhibition assay was devised using the human CAL 27 cell line. The CAL 27 cell line represents a head and neck cancer type amenable to treatment with BNCT. The overexpression of GLUT1 in CAL 27 is responsible for the aberrant growth of these tumors,54,55 and therefore the GLUT1 targeting approach is warranted. Before conducting the assays, we validated the GLUT1 function of CAL 27 (see Supporting Information). The cis-inhibition assay was devised as a competition experiment between the glucoconjugates and [14C]-d-glucose, with d-glucose serving as a control. This experiment accurately mimics the situation that the delivery agents would face in a biological context. It has been previously speculated and shown that 6-O-substituted glucoconjugates display a higher affinity to GLUT1 than free d-glucose.53,56 In our current study, the affinities for the glucoconjugates 1–3 were in the low μM range in contrast to the low mM-affinity displayed by free d-glucose. The exact GLUT1 IC50-values were determined to be 43.96 μM for 1, 262.4 μM for 2, 15.2 μM for 3, and >1 mM for free d-glucose. The 4–67 times stronger affinity displayed by the glucoconjugates confirm that at least glucoconjugates 1 and 3 are capable of targeting GLUT1 in the intended application despite the high glucose levels found in blood (6 mM).
In order to elucidate the interactions between the glucoconjugates and the transporters on a molecular level, we next turned to molecular modeling. We focused on the differences in binding to GLUT1 and studied the protein–ligand interactions of the glucoconjugates 1–3 in the outward- and inward-open conformations of the transporter, i.e., on the outside and inside of the cell. To set up a computational model, an experimental structure of the transporter was required. However, only the crystal structure of the inward-open conformation of GLUT1 has been reported.57 In order to perform the docking studies on equal footing for both the inward and outward-open conformations, we created a model based on XylE, a d-xylose-proton symporter found in E. coli for which crystal structures of both the inward-open (PDB ID 4QIQ),58 and outward open (PDB ID 6N3I)59 conformations exist. XylE is structurally very similar to the GLUT1–4 proteins (29% sequence identity and 49% similarity).60 Importantly, the binding site residues are identical to GLUT1 except for the Gln415 in XylE which is Asn411 in GLUT1.61,62 After virtual mutation of this residue, we estimated binding energies by molecular docking studies. In our models, the glucoconjugates 1–3 bind significantly stronger to both the outward- and inward-open binding sites of the transporter than d-glucose, in line with the experimental observations above. For the outward-open conformation, the binding free energy difference is estimated to be up to 5 kcal/mol in favor of the glucoconjugates, corresponding to a binding affinity increase on the order of 103 (see Supporting Information for details).
There are a few observations of importance from the BNCT delivery agent perspective. The estimated binding affinity of each glucoconjugate is an order of magnitude lower in the inward-open binding pocket than in the corresponding outward-open one; that is, the ligand binds more tightly on the outside. This is beneficial from a functional point-of-view, as it implies that after the conformational change of the protein from the outward-open to the inward-open conformation, the ligand is more readily released to the inside of the cell. For d-glucose, this difference in binding energy between the inward- and outward-open conformations is absent.
For the outward-open structure, two major binding poses for the glucoconjugates were identified. In one, the sugar end of the glucoconjugate overlaps with the most favorable binding pose of free glucose, while the carboranyl end extends toward the hydrophobic end of the binding pocket; see Figure 3. This agrees with the hydrophobic nature of the carboranes,22 as manifested by the almost neutral partial atomic charges of both boron and hydrogen in the B–H bonds (see Supporting Information). In the second pose, the glucoconjugate is slightly rotated. This pose is estimated to bind with practically equal affinity (see Table S1).
Figure 3.
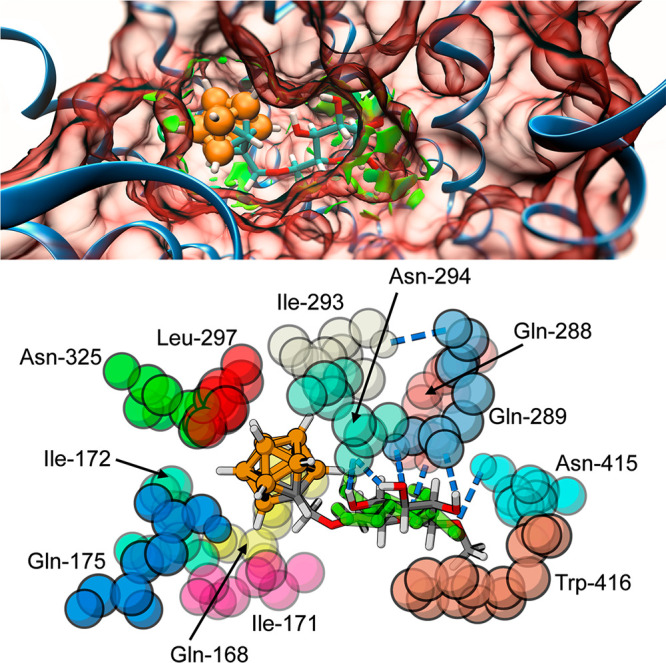
Glucoconjugate 3 bound to the outward-open conformation of the transporter. Top: Green areas indicate intramolecular noncovalent interactions between 3 and the protein. Bottom: The closest amino acids surrounding the ligands, using PDB 6N3I numbering; hydrogen bonds shown as blue dashes, the binding pose of β-Glc superimposed in green color over the glucoconjugate.
In general, the binding pocket seems rather accommodating from a structural point-of-view, with enough space for the bulky carboranylmethyl substituent at the sixth position in addition to the sugar. In order to corroborate this, we performed a quantum mechanical noncovalent interaction (NCI) analysis34 on the ligand/transporter complex. The analysis revealed favorable interactions between ligand and protein at both ends of the glucoconjugate (Figure 3, top). Importantly, no repulsive intramolecular steric interactions are identified; even the largest of the boron cluster conjugates fits snugly to the transporter pocket. The inward-open conformation, on the other hand, displays greater flexibility in the binding site, which leads to a number of binding poses for both glucose and the glucoconjugates. This might also explain the lower binding affinity for the glucoconjugates to this site.
3.3. Cytotoxicity and Cellular Uptake Studies
With the biochemical foundations of the GLUT1 targeting approach investigated, we continued by addressing the cellular uptake and cytotoxicity of the glucoconjugates since these are essential properties of boron delivery agents and important factors for eventual translation into the clinics. The CAL 27 cell line was used in these studies because of its clinical relevance. In the cytotoxicity assays, the glucoconjugates 1, 2, and 3 were incubated with the cells at concentrations of 5 μM, 25 μM, 50 μM, 125 μM, and 250 μM for 6 and 24 h. These concentrations were chosen based on the affinity results and the time points were selected with the clinical perspective related to intravenous administration of the boron carriers in mind. In these studies, the clinically deployed BSH was used as a reference. BPA was omitted because its IC50-value has been previously reported to be in the low mM range.14 The cell viability was quantified by the detection of ATP metabolic activity-generated luminescence from the viable cells after incubation using a commercially available Cell-Titer Glo assay. The glucoconjugates 1–3 displayed IC50-values in the μM range and were consistently less toxic than BSH. The IC50-values were obtained from nonlinear regression fitting of the cell viability data and were found to be 214.8 μM for 1, 196.1 μM for 2, 276.6 μM for 3, and 98.7 μM for BSH (see Figure 4). From a toxicity standpoint, there is therefore no objection to their use as delivery agents in BNCT.
Figure 4.

Cell cytotoxicity studies in CAL 27 cells after incubation with negative (cell culture medium) and positive (1% Triton X-100) controls, and glucoconjugates 1, 2, 3, and BSH at 5 μM, 25 μM, 50 μM, 125 μM, and 250 μM for 6 and 24 h. Error bars represent the mean ± s.d. (n = 4) in comparison with the negative control. The statistical hypothesis was evaluated by unpaired Student’s t-test where the significant probabilities were set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Our final focus in this study was to determine if the glucoconjugates are transported into the cells through GLUT1 or if they, at the very least, remain attached to the cells. This is within the critical range required in clinical BNCT for the generated alpha particles to exert a cell-killing effect. This information was obtained by determining the boron content in the CAL 27 cell lysates after incubation with the compounds and careful washing. The development of a functioning protocol featuring suitable incubation times, compound concentrations, workup protocols, and methods for the robust analysis of boron content required an extensive number of trials. In the end, incubation times of 5 and 30 min were selected based on the optimal performance of [14C]-d-glucose under these conditions, and the concentration range 10–400 μM was selected based on the GLUT1 affinity results. The ICP-MS instrument used in determination of the boron content was found to be somewhat insensitive, and cells from four wells were combined and digested in order to obtain results of high reliability. The results are summarized in Figure 5.
Figure 5.

Cell uptake studies in the CAL 27 cell line after incubation with glucoconjugates 1 (▲), 2 (■), 3 (●), BPA (⧫), and BSH (▼) in the 10–400 μM range for 5 min (A; n = 3) and 30 min (B; n = 3). The Michaelis–Menten kinetic parameters for glucoconjugates when available: At 5 min incubation time (A), glucoconjugate 2: Vmax = 2.791; Km = 80.49. At 30 min incubation time (B), glucoconjugate 1: Vmax = 16.29; Km = 630.5, glucoconjugate 2: Vmax = 6.417; Km = 488.3, glucoconjugate 3: Vmax = 16.89; Km = 894.2, and BPA: Vmax = 3.625; Km = 3737.
In addition to studying the uptake of glucoconjugates 1–3, both BSH and BPA were included as representatives of delivery agents in clinical use. All glucoconjugates delivered a significantly higher boron content to the CAL 27 cells than BPA and BSH across the entire concentration range. To a certain degree, the observations may be explained by the different uptake mechanisms of the glucoconjugates, BPA, and BSH. Nevertheless, according to our preliminary assessment, targeting GLUT1 translates into a competitive strategy for BNCT—outperforming the passive transport of BSH and the LAT1-targeting approach of BPA in the in vitro cellular uptake model used. Glucoconjugates 1 and 3 were found to have the best boron delivery capacity, with 3 being slightly better at the 5 min mark and 1 being considerably better at the 30 min mark. We note that the correlation to the GLUT1 affinity results is perhaps weaker than one would expect. The difference is natural when considering that the two methods provide complementary insights on two separate properties: the ability to compete with other GLUT1 substrates for the transporter and the ability to remain attached to the cell or internalized.
Lastly, in order to distinguish which of our two prime candidates, glucoconjugates 1 and 3, would be better suited for future in vivo and potential preclinical BNCT studies, we determined their aqueous solubility. This is an important factor from the formulation and treatment perspective, as exemplified by BPA which is administered as a fructose complex due to the low aqueous solubility of BPA itself. Surprisingly, glucoconjugate 1 displayed a significantly higher aqueous solubility than 3 (1 mg/mL vs <1 mg/500 mL). When this property is further coupled with its high GLUT1 affinity, low cytotoxicity, and outstanding in vitro delivery capacity, we are pleased to report that 6-O-(ortho-carboranylmethyl)-d-glucopyranose, our “Trojan horse”, has considerable potential as a delivery agent for BNCT.
4. Discussion
Head and neck cancers account for up to 10% of all cancers, and the recurrent ones are accompanied by a poor survival rate in patients.3 BNCT has been successfully applied to the treatment of head and neck cancers63,64 and is currently attracting large investments on a global scale due to the recent development of in-hospital neutron accelerators which is a game-changer from a patient treatment perspective.
In this work, we have designed and synthesized molecular-scale “Trojan horses”, i.e. conjugates of glucose and boron clusters containing a high boron content, and studied the biochemical foundations of a GLUT1-targeting strategy to BNCT. In more detail, we have used a chemistry-based approach featuring both experimental and computational methodologies. From the onset, important factors such as the possible interference with glucose metabolism through the glycolysis route was accounted for. In addition to addressing the biochemical foundations of this approach, we have identified a hit molecule which displays good cytocompatibility, sufficient aqueous solubility, and high cellular uptake in the relevant human CAL 27 head and neck cancer cell line. In our in vitro assessment, glucoconjugate 1 was able to outperform the current delivery agents in clinical use (BPA and BSH) in terms of boron delivery capacity while simultaneously having a sufficiently high affinity to GLUT1 to permit competition with the high levels of glucose found in blood. Therefore, in addition to providing a missing link on the biochemical foundations of a GLUT1-targeting strategy to BNCT—we have identified a potentially promising new glucoconjugate for clinical BNCT. With the solid basis reported herein, the development of a suitable formulation featuring the 10B-enriched version of glucoconjugate 1 with accompanied in vivo-testing is soon to follow. The results from these studies will be reported in due course.
Acknowledgments
Jussi Kärkkäinen (University of Eastern Finland), Ronja Rättö (University of Helsinki), and Joonas Waaramaa (University of Helsinki) are acknowledged for laboratory assistance, and Hélder A. Santos and Alexandra Correia (University of Helsinki) are acknowledged for their contributions to the cytotoxicity studies. CSC-The Finnish IT Center for Science and the Finnish Grid and Cloud Infrastructure (urn:nbn:fi:research-infras-2016072533) are acknowledged for providing the computational resources required for this work. Further, the authors are grateful for financial support from the Jane and Aatos Erkko foundation, the Cancer foundation, the Swedish Cultural foundation, the Ruth and Nils-Erik Stenbäck foundation, the Finnish Academy of Science and Letters, the University of Helsinki research funds, the Academy of Finland (projects: 308329, 318422, 289179, and 319453), and the Waldemar von Frenckell foundation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.0c00630.
Information on additional synthetic routes, NMR-spectra of the synthesized compounds, details on the molecular docking studies and ligand PDBQT files, as well as additional information on the characterization of the GLUT1 function in the CAL 27 cell line and affinity/uptake studies (PDF)
Author Contributions
ϕ J.M. and J.J. contributed equally. F.S.E. and O.A. conceived the idea and F.S.E. designed the project and led the initiate with input from O.A., A.A., M.P.J., M.S., and J.R. J.M., H.C.B., R.M.F., and F.S.E. contributed to the synthesis and structural characterization of the glucoconjugates. M.S. and S.I. were responsible for the cytotoxicity testing. M.P.J. and I.K.S. were responsible for the molecular modeling aspects and analysis. J.J., K.H., J.T., S.P., and J.R. were responsible for the experimental affinity and cellular uptake studies. The manuscript was written with contributions from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Parkin D. M.; Bray F.; Ferlay J.; Pisani P. Global Cancer Statistics, 2002. Ca-Cancer J. Clin. 2005, 55 (2), 74–108. 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Vigneswaran N.; Williams M. D. Epidemiologic Trends in Head and Neck Cancer and Aids in Diagnosis. Oral Maxillofac. Surg. Clin. N. Am. 2014, 26 (2), 123–141. 10.1016/j.coms.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong L. Y.; Wei W. I.; Lam L. K.; Yuen A. P. W. Salvage of Recurrent Head and Neck Squamous Cell Carcinoma after Primary Curative Surgery. Head Neck 2003, 25 (11), 953–959. 10.1002/hed.10310. [DOI] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaïa N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Yuan T.-Z.; Zhan Z.-J.; Qian C.-N. New Frontiers in Proton Therapy: Applications in Cancers. Cancer Commun. 2019, 39 (1), 61. 10.1186/s40880-019-0407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yock T. I.; Tarbell N. J. Technology Insight: Proton Beam Radiotherapy for Treatment in Pediatric Brain Tumors. Nat. Clin. Pract. Oncol. 2004, 1 (2), 97–103. 10.1038/ncponc0090. [DOI] [PubMed] [Google Scholar]
- Hosmane N. S.Boron Science: New Technologies and Applications; CRC Press, 2011. [Google Scholar]
- Locher G. L. Biological Effects and Therapeutic Possibilities of Neutrons. Am. J. Roentgenol Radium Ther. 1936, 36, 1–13. [Google Scholar]
- Barth R. F.; Zhang Z.; Liu T. A Realistic Appraisal of Boron Neutron Capture Therapy as a Cancer Treatment Modality. Cancer Commun. 2018, 38 (1), 36. 10.1186/s40880-018-0280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiner A. J.; Bergueiro J.; Cartelli D.; et al. Present Status of Accelerator-Based BNCT. Rep. Pract. Oncol. Radiother. 2016, 21 (2), 95–101. 10.1016/j.rpor.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth R. F.; Mi P.; Yang W. Boron Delivery Agents for Neutron Capture Therapy of Cancer. Cancer Commun. 2018, 38 (1), 35. 10.1186/s40880-018-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloway A. H.; Tjarks W.; Barnum B. A.; et al. The Chemistry of Neutron Capture Therapy. Chem. Rev. 1998, 98 (4), 1515–1562. 10.1021/cr941195u. [DOI] [PubMed] [Google Scholar]
- Hey-Hawkins E.; Teixidor C. V.. Boron-Based Compounds: Potential and Emerging Applications in Medicine; John Wiley & Sons, 2018. [Google Scholar]
- Nemoto H.; Cai J.; Iwamoto S.; Yamamoto Y. Synthesis and Biological Properties of Water-Soluble p-Boronophenylalanine Derivatives. Relationship between Water Solubility, Cytotoxicity, and Cellular Uptake. J. Med. Chem. 1995, 38 (10), 1673–1678. 10.1021/jm00010a012. [DOI] [PubMed] [Google Scholar]
- Kankaanranta L.; Seppälä T.; Koivunoro H.; et al. L-Boronophenylalanine-Mediated Boron Neutron Capture Therapy for Malignant Glioma Progressing After External Beam Radiation Therapy: A Phase I Study. Int. J. Radiat. Oncol., Biol., Phys. 2011, 80 (2), 369–376. 10.1016/j.ijrobp.2010.02.031. [DOI] [PubMed] [Google Scholar]
- Wittig A.; Collette L.; Appelman K.; et al. EORTC Trial 11001: Distribution of Two 10 B-Compounds in Patients with Squamous Cell Carcinoma of Head and Neck, a Translational Research/Phase 1 Trial. J. Cell. Mol. Med. 2009, 13 (8b), 1653–1665. 10.1111/j.1582-4934.2009.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivillin V. A.; Garabalino M. A.; Colombo L. L.; et al. Biodistribution of the Boron Carriers Boronophenylalanine (BPA) and/or Decahydrodecaborate (GB-10) for Boron Neutron Capture Therapy (BNCT) in an Experimental Model of Lung Metastases. Appl. Radiat. Isot. 2014, 88, 94–98. 10.1016/j.apradiso.2013.11.115. [DOI] [PubMed] [Google Scholar]
- Szablewski L. Expression of Glucose Transporters in Cancers. Biochim. Biophys. Acta, Rev. Cancer 2013, 1835 (2), 164–169. 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324 (5930), 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the Origin of Cancer Cells. Science 1956, 123 (3191), 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- König M.; Bulik S.; Holzhütter H.-G. Quantifying the Contribution of the Liver to Glucose Homeostasis: A Detailed Kinetic Model of Human Hepatic Glucose Metabolism. PLoS Comput. Biol. 2012, 8 (6), e1002577 10.1371/journal.pcbi.1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann P.; Gozzi M.; Kuhnert R.; et al. New Keys for Old Locks: Carborane-Containing Drugs as Platforms for Mechanism-Based Therapies. Chem. Soc. Rev. 2019, 48 (13), 3497–3512. 10.1039/C9CS00197B. [DOI] [PubMed] [Google Scholar]
- Satapathy R.; Dash B. P.; Mahanta C. S.; et al. Glycoconjugates of Polyhedral Boron Clusters. J. Organomet. Chem. 2015, 798, 13–23. 10.1016/j.jorganchem.2015.06.027. [DOI] [Google Scholar]
- Luderer M. J.; de la Puente P.; Azab A. K. Advancements in Tumor Targeting Strategies for Boron Neutron Capture Therapy. Pharm. Res. 2015, 32 (9), 2824–2836. 10.1007/s11095-015-1718-y. [DOI] [PubMed] [Google Scholar]
- Bar-Even A.; Flamholz A.; Noor E.; Milo R. Rethinking Glycolysis: On the Biochemical Logic of Metabolic Pathways. Nat. Chem. Biol. 2012, 8 (6), 509–517. 10.1038/nchembio.971. [DOI] [PubMed] [Google Scholar]
- Staroverov V. N.; Scuseria G. E.; Tao J.; Perdew J. P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 2003, 119 (23), 12129–12137. 10.1063/1.1626543. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132 (15), 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Becke A. D.; Johnson E. R. A Density-Functional Model of the Dispersion Interaction. J. Chem. Phys. 2005, 123 (15), 154101. 10.1063/1.2065267. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7 (18), 3297. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97 (40), 10269–10280. 10.1021/j100142a004. [DOI] [Google Scholar]
- Ahlrichs R.; Bär M.; Häser M.; et al. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162 (3), 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Eichkorn K.; Weigend F.; Treutler O.; Ahlrichs R. Auxiliary Basis Sets for Main Row Atoms and Transition Metals and Their Use to Approximate Coulomb Potentials. Theor. Chem. Acc. 1997, 97 (1–4), 119–124. 10.1007/s002140050244. [DOI] [Google Scholar]
- Valiev M.; Bylaska E. J.; Govind N.; et al. NWChem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun. 2010, 181 (9), 1477–1489. 10.1016/j.cpc.2010.04.018. [DOI] [Google Scholar]
- Johnson E. R.; Keinan S.; Mori-Sánchez P.; et al. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132 (18), 6498–6506. 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-García J.; Johnson E. R.; Keinan S.; et al. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7 (3), 625–632. 10.1021/ct100641a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huey R.; Morris G. M.; Olson A. J.; Goodsell D. S. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J. Comput. Chem. 2007, 28 (6), 1145–1152. 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; et al. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda A.; Fukuyoshi S.; Nakagaki R.; Takahashi O. Determination of AMBER Force Field Parameters for Thioester by Quantum Chemical Calculations. Chem. Lett. 2013, 42 (10), 1206–1208. 10.1246/cl.130517. [DOI] [Google Scholar]
- Couto M.; García M. F.; Alamón C.; et al. Discovery of Potent EGFR Inhibitors through the Incorporation of a 3D-Aromatic-Boron-Rich-Cluster into the 4-Anilinoquinazoline Scaffold: Potential Drugs for Glioma Treatment. Chem. - Eur. J. 2018, 24 (13), 3122–3126. 10.1002/chem.201705181. [DOI] [PubMed] [Google Scholar]
- Tjarks W.; Anisuzzaman A. K. M.; Liu L.; et al. Synthesis and in Vitro Evaluation of Boronated Uridine and Glucose Derivatives for Boron Neutron Capture Therapy. J. Med. Chem. 1992, 35 (9), 1628–1633. 10.1021/jm00087a019. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; Bothe U. Ortho-Carboranyl Glycosides of Glucose, Mannose, Maltose and Lactose for Cancer Treatment by Boron Neutron-Capture Therapy. Chem. - Eur. J. 1998, 4 (7), 1179–1183. . [DOI] [Google Scholar]
- Satapathy R.; Dash B. P.; Bode B. P.; et al. New Classes of Carborane-Appended 5-Thio-D-Glucopyranose Derivatives. Dalton Trans. 2012, 41 (29), 8982–8988. 10.1039/c2dt30874f. [DOI] [PubMed] [Google Scholar]
- Hosmane N. S.; Eagling R.. Handbook of Boron Science; World Scientific; (Europe: ), 2018. [Google Scholar]
- Zhao F.-Q.; Keating A. Functional Properties and Genomics of Glucose Transporters. Curr. Genomics 2007, 8 (2), 113–128. 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekholm F. S.; Ardá A.; Eklund P.; et al. Studies Related to Norway Spruce Galactoglucomannans: Chemical Synthesis, Conformation Analysis, NMR Spectroscopic Characterization, and Molecular Recognition of Model Compounds. Chem. - Eur. J. 2012, 18 (45), 14392–14405. 10.1002/chem.201200510. [DOI] [PubMed] [Google Scholar]
- Olah G. A.; Welch J. T.; Vankar Y. D.; et al. Synthetic Methods and Reactions. 63. Pyridinium Poly(Hydrogen Fluoride) (30% Pyridine-70% Hydrogen Fluoride): A Convenient Reagent for Organic Fluorination Reactions. J. Org. Chem. 1979, 44 (22), 3872–3881. 10.1021/jo01336a027. [DOI] [Google Scholar]
- Valliant J. F.; Guenther K. J.; King A. S.; et al. The Medicinal Chemistry of Carboranes. Coord. Chem. Rev. 2002, 232 (1–2), 173–230. 10.1016/S0010-8545(02)00087-5. [DOI] [Google Scholar]
- Tietze L. F.; Griesbach U.; Schuberth I.; et al. Novel Carboranyl C-Glycosides for the Treatment of Cancer by Boron Neutron Capture Therapy. Chem. - Eur. J. 2003, 9 (6), 1296–1302. 10.1002/chem.200390148. [DOI] [PubMed] [Google Scholar]
- Ekholm F. S.; Sinkkonen J.; Leino R. Fully Deprotected β-(1→2)-Mannotetraose Forms a Contorted α-Helix in Solution: Convergent Synthesis and Conformational Characterization by NMR and DFT. New J. Chem. 2010, 34 (4), 667. 10.1039/b9nj00702d. [DOI] [Google Scholar]
- Berger S.; Braun S.. 200 and More NMR Experiments: A Practical Course, 3rd rev. and expanded ed.; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- Stenutz R.; Carmichael I.; Widmalm G.; Serianni A. S. Hydroxymethyl Group Conformation in Saccharides: Structural Dependencies of 2JHH, 3JHH, and 1JCH Spin–Spin Coupling Constants. J. Org. Chem. 2002, 67 (3), 949–958. 10.1021/jo010985i. [DOI] [PubMed] [Google Scholar]
- Hermanek S. Boron-11 NMR Spectra of Boranes, Main-Group Heteroboranes, and Substituted Derivatives. Factors Influencing Chemical Shifts of Skeletal Atoms. Chem. Rev. 1992, 92 (2), 325–362. 10.1021/cr00010a007. [DOI] [Google Scholar]
- Patra M.; Johnstone T. C.; Suntharalingam K.; Lippard S. J. A Potent Glucose-Platinum Conjugate Exploits Glucose Transporters and Preferentially Accumulates in Cancer Cells. Angew. Chem., Int. Ed. 2016, 55 (7), 2550–2554. 10.1002/anie.201510551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-D.; Li S.-J.; Liao J.-X. Inhibition of Glucose Transporter 1 (GLUT1) Chemosensitized Head and Neck Cancer Cells to Cisplatin. Technol. Cancer Res. Treat. 2013, 12 (6), 525–535. 10.7785/tcrt.2012.500343. [DOI] [PubMed] [Google Scholar]
- Li S.; Yang X.; Wang P.; Ran X. The Effects of GLUT1 on the Survival of Head and Neck Squamous Cell Carcinoma. Cell. Physiol. Biochem. 2013, 32 (3), 624–634. 10.1159/000354466. [DOI] [PubMed] [Google Scholar]
- Gynther M.; Ropponen J.; Laine K.; et al. Glucose Promoiety Enables Glucose Transporter Mediated Brain Uptake of Ketoprofen and Indomethacin Prodrugs in Rats. J. Med. Chem. 2009, 52 (10), 3348–3353. 10.1021/jm8015409. [DOI] [PubMed] [Google Scholar]
- Deng D.; Xu C.; Sun P.; et al. Crystal Structure of the Human Glucose Transporter GLUT1. Nature 2014, 510 (7503), 121–125. 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- Wisedchaisri G.; Park M.-S.; Iadanza M. G.; et al. Proton-Coupled Sugar Transport in the Prototypical Major Facilitator Superfamily Protein XylE. Nat. Commun. 2014, 5 (1), 4521. 10.1038/ncomms5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Wu J.; Ke M.; et al. Engineered XylE as a Tool for Mechanistic Investigation and Ligand Discovery of the Glucose Transporters GLUTs. Cell Discovery 2019, 5 (1), 14. 10.1038/s41421-019-0082-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L.; Zeng X.; Yan C.; et al. Crystal Structure of a Bacterial Homologue of Glucose Transporters GLUT1–4. Nature 2012, 490 (7420), 361–366. 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- Deng D.; Sun P.; Yan C.; et al. Molecular Basis of Ligand Recognition and Transport by Glucose Transporters. Nature 2015, 526 (7573), 391–396. 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- Park M.-S. Molecular Dynamics Simulations of the Human Glucose Transporter GLUT1. PLoS One 2015, 10 (4), e0125361 10.1371/journal.pone.0125361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankaanranta L.; Seppälä T.; Koivunoro H.; et al. Boron Neutron Capture Therapy in the Treatment of Locally Recurred Head-and-Neck Cancer: Final Analysis of a Phase I/II Trial. Int. J. Radiat. Oncol., Biol., Phys. 2012, 82 (1), e67–e75. 10.1016/j.ijrobp.2010.09.057. [DOI] [PubMed] [Google Scholar]
- Wang L.-W.; Chen Y.-W.; Ho C.-Y.; et al. Fractionated Boron Neutron Capture Therapy in Locally Recurrent Head and Neck Cancer: A Prospective Phase I/II Trial. Int. J. Radiat. Oncol., Biol., Phys. 2016, 95 (1), 396–403. 10.1016/j.ijrobp.2016.02.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.