

Abstract
Background:
The rationale for the evaluation of trametinib in advanced biliary cancer (BC) is based on the presence of mitogen-activated protein kinase alterations and on earlier promising results with MEK inhibitors in BC.
Methods:
Patients with histologically proven BC who progressed on gemcitabine/platinum were randomised to trametinib daily (arm 1) versus fluoropyrimidine therapy (infusional 5-fluorouracil or oral capecitabine, arm 2). The primary end-point was overall survival (OS). Secondary end-points included progression free survival (PFS) and response rate. A planned interim futility analysis of objective response was performed on the first 14 patients registered to the trametinib arm.
Results:
The study was stopped early based on the lack of measurable response in the trametinib arm. A total of 44 eligible patients were randomised (24 patients in arm 1 and 20 patients in arm 2). Median age was 62 years and the primary sites of tumour were cholangiocarcinoma (68%) and gallbladder (32%). The overall response rate was 8% (95% CI 0%–19%) in arm 1 versus 10% (95% CI 0%–23%) in arm 2 (p > .99) Median OS was 4.3 months for arm 1 and 6.6 months for arm 2. The median PFS was 1.4 months for arm 1 and 3.3 months for arm 2.
Conclusions:
This is the first prospective randomised study of a targeted agent versus chemotherapy for the second-line treatment of BC. In this unselected population, the interim analysis result of unlikely benefit with trametinib resulted in early closure.
Keywords: Advanced biliary cancer, MEK inhibitor, 5-Fluorouracil, Capecitabine
1. Introduction
Biliary cancer (BC) includes intra- and extrahepatic cholangiocarcinoma (EHCCA) and cancers of the gallbladder. In the United States of America (USA), an estimated 42,000 cases of liver and intrahepatic cholangiocarcinoma were diagnosed in 2017 [1], and annually, about 12,000 cases of extrahepatic bile duct cancer are diagnosed, of which two-thirds are gallbladder cancers [1]. Unfortunately, two-thirds of patients have unresectable/advanced disease at presentation, and those who undergo surgery often relapse [2]. ABC-02, a randomised phase III study of more than 400 patients, demonstrated an overall survival (OS) benefit for the combination of gemcitabine plus cisplatin compared to gemcitabine alone [3]. However, the survival for advanced BCs still rarely exceeds 1 year, and the 5-year survival is less than 10%.
The most commonly used second-line regimens in patients who have been previously treated with gemcitabine- or platinum-based chemotherapy are fluoropyrimidinesd—5-fluorouracil (5-FU) with leucovorin (LV) or capecitabine based on limited prospective data. The activity of these agents was only reported in several small trials that were primarily in first-line settings, either given as single agents or in combination chemotherapy [4,5]. Large retrospective series also reported very poor outcomes for second-line chemotherapy with median OS of around 6 months and progression free survival (PFS) ranging from 2 to 3 months [6,7]. However, in highly selected patients who were treated at academic medical centres, median OS was 11 months from the start of the second-line therapy [8]. It is also important to point out that there are prognostic model and scoring system that can identify patients who will unlikely benefit from second-line therapy such as peritoneal disease, poor baseline ECOG status or elevated CA 19/9 [9].
In the recent ABC-06 study, FOLFOX (fluorouracil + oxaliplatin with LV) was evaluated as second-line treatment after progression on gemcitabine plus cisplatin, and it demonstrated modest 1 month survival benefit over active symptom control [10].
Trametinib is a bioavailable, potent, and specific allosteric inhibitor of MEK 1/2. Aberrant activation of the Ras/Raf/mitogen-activated protein kinase (MAPK) pathway occurs in more than 60% of BCs, indicating the importance of these pathways in biliary carcinogenesis [11]. Comprehensive genomic profiling of biliary tract cancers revealed that the KRAS mutation can be as high as 42% in EHCCA compared to 22% in intrahepatic cholangiocarcinoma and 11% in gallbladder cancer. On the other hand, BRAF mutation can be found in 5% of the intrahepatic cholangiocarcinoma compared to 3% in EHCCA and 1% in gallbladder cancer [12]. Downstream of BRAF, the MAPK pathway appears active in 75% of BCs, as evidenced by phospho-MAPK immunostaining [13]. Furthermore, expression profiling across a panel of seven human BC cell lines demonstrated a number of RAS/MAPK pathway components and sensitivity to MEK inhibitors [14]. However, it is important to note that BC represents a heterogeneous group of tumours with variable frequency of alterations in specific molecular pathways based on site of origin (intrahepatic versus extrahepatic versus gallbladder cancer).
Given the important role of the MAPK pathway in biliary carcinogenesis and its relevance for the clinical activity of MEK inhibitors in small studies, the authors conducted a randomised phase II study of trametinib versus 5-FU/LV or capecitabine in refractory BC patients.
2. Patients and methods
2.1. Study design and patients
This was a multicentre, open-label randomised phase II trial comparing single-agent trametinib (arm 1) versus infusional 5-FU/LV or capecitabine (arm 2) in refractory BC patients completed within the National Cancer Institute’s National Clinical Trials Network groups SWOG, ECOG-ACRIN, and Alliance. If the patient was randomised to arm 2, then the patient and physician were able to choose between arms 2A (infusional 5-FU/LV) and 2B (capecitabine). Twenty-five US institutions participated, with SWOG being the coordin-ating group. The participating sites obtained institutional review board’s approval, and informed, written consent was obtained from all patients before enrolment.
Eligible patients were required to have histologically or cytologically documented carcinoma primary to the intra- or extrahepatic biliary system or gall bladder, with clinical and/or radiologic evidence of unresectable, locally advanced, or metastatic disease. Patients with ampullary carcinoma were not eligible. Patients had to be ≥ 18 years of age, have Eastern Cooperative Oncology Group performance score of 0 or 1, and have adequate bone marrow, renal and hepatic functions. All patients had to have measurable disease according to Response Evaluation Criteria in Solid Tumours version 1.1. All patients had to have experienced disease progression after no more than one prior regimen of systemic chemotherapy for advanced BC, have completed any prior chemotherapy at least 21 days before registration, and have recovered from any side-effects. Patients who had received adjuvant chemotherapy were allowed to participate if there was evidence of disease recurrence within 6 months of completion of the adjuvant treatment. Prior treatment with MEK inhibitors was not allowed. Prior 5-FU or capecitabine treatment was allowed only if given as a radiosensitiser concurrently with radiation therapy or if given as part of any adjuvant therapy regimen at least 12 months before study enrolment. The trial was registered with the National Cancer Institute (www.clinicaltrials.gov identifier NCT02042443.)
2.2. Treatment and assessment plan
Patients were randomised to either 2 mg oral trametinib once a day (arm 1), 2400 mg/m2 infusional 5-FU over 46 h + optional bolus 5-FU and LV every 2 weeks (arm 2A), or 1000 mg/m2 oral capecitabine on days 1–14 twice daily (arm 2B). One treatment cycle was 21 days for arms 1 and 2B and 28 days for arm 2A. Patients were seen and evaluated every cycle with appropriate laboratories, including complete blood count, complete metabolic panel and physical examination. Serum CA 19–9 was checked every 6 weeks. Toxicities were graded as per the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Patient response was assessed every 6 weeks with computed tomography of chest/abdomen/pelvis or magnetic resonance imaging using the Response Evaluation Criteria in Solid Tumours Classification 1.1. Treatment was continued until documented disease progression, development of unacceptable toxicity, withdrawal at the discretion of the treating investigator or withdrawal of consent. The full assessment schedule is provided in the trial protocol.
2.3. End-points and statistical analyses
The primary end-point was OS in patients with refractory advanced BC randomised to arm 1 compared to those randomised to arm 2. Secondary end-points were PFS and response rate (RR), which included complete response, partial response, unconfirmed complete response and unconfirmed partial response.
Approximately, 80 eligible patients (40 each for arms 1 and 2 [2A and 2B combined]) were required to detect an improvement in median OS from 5 months to 8.25 months (hazard ratio [HR] for experimental versus control 0.61), assuming a one-sided type-1 error of 10%, 80% power, 2 years of accrual and an additional year of follow-up. Accounting for the possibility of a 10% ineligibility rate, the study planned to accrue approximately 89 patients to achieve 80 eligible patients. Ran-domisation was stratified by planned chemotherapy (5-fluorouracil with LV versus capecitabine) and disease site (cholangiocarcinoma versus gallbladder).
An interim futility assessment was planned when the first 14 patients registered to the trametinib arm had been assessed for disease response. If none of these first 14 patients responded to treatment (corresponding to 95% confidence that the true RR was less than 20%), then the study would be closed to further accrual and the agent declared to be of no interest for further development in this setting. The trial allowed accrual to continue while the interim analysis was being conducted.
The primary analysis of OS was conducted in all eligible patients according to a modified intent-to-treat principle. Probabilities of OS and PFS were estimated using the Kaplan–Meier method. Statistical differences in event rates between treatment arms were assessed via stratified Cox regression model. RR was compared by treatment arm via chi-squared test. The significance of associations between high serum CA 19–9 decrease from baseline and RR, within each treatment arm, was also assessed by chi-squared test. Associations of high CA 19–9 decrease from baseline with OS, by treatment arm, were assessed via stratified Cox regression model. Statistical tests are reported as two-sided, and p < .05 is considered significant.
3. Results
3.1. Patients and treatment
A total of 53 patients (27 in arm 1 and 26 in arm 2) were randomised to the trial between February 2014 and March 2015 (Fig. 3 Consort diagram). Seven patients were ineligible for various reasons, including having received more than one prior line of chemotherapy and having no evidence of measurable disease. An additional two patients were excluded from analyses because they withdrew consent before starting protocol therapy. Patient characteristics are provided in Table 1. The median age was 62 years and 66% of patients were female. Fourteen patients (70%) who were randomised to chemotherapy received capecitabine and six (30%) received 5-FU/LV. All patients had previously failed one line of systemic therapy. Most patients (59%) had a diagnosis of intrahepatic cholangiocarcinoma, and 32% had a diagnosis of gallbladder cancer.
Fig. 3.
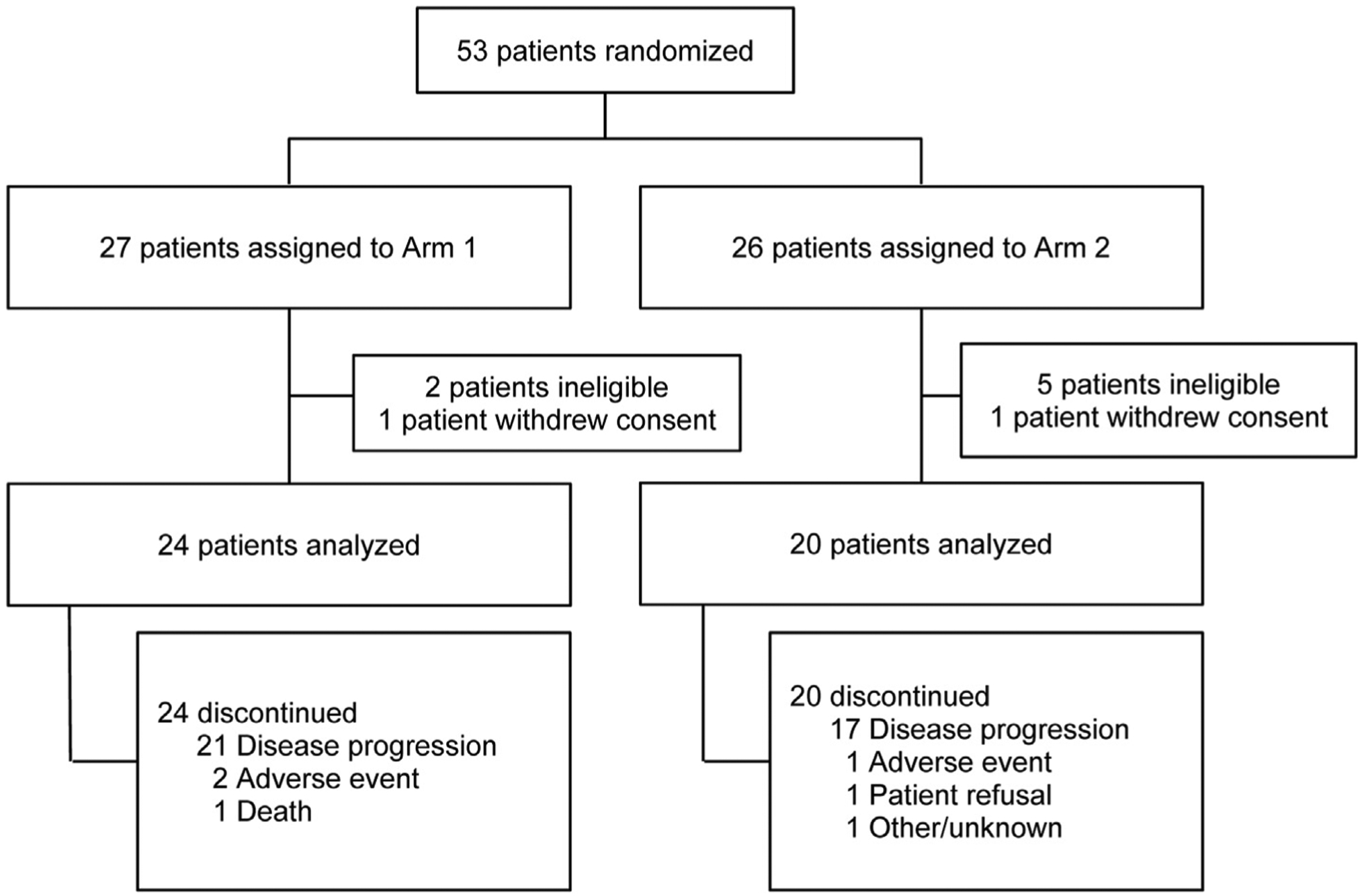
CONSORT flow diagram.
Table 1.
Patient characteristics.
| Baseline characteristic | Trametinib (n = 24) | Chemotherapy (n = 20) |
|---|---|---|
| Age, years | ||
| Median (range) | 63 (40–78) | 61 (41–81) |
| Sex, n (%) | ||
| Female | 18 (75) | 11 (55) |
| Male | 6 (25) | 9 (45) |
| Hispanic, n (%) | ||
| Yes | 3 (13) | 1 (5) |
| No | 20 (83) | 19 (95) |
| Unknown | 1 (4) | 0 (0) |
| Race, n (%) | ||
| White | 16 (67) | 15 (75) |
| Black | 5 (21) | 2 (10) |
| Asian | 2 (8) | 3 (15) |
| Unknown | 1 (4) | 0 (0) |
| Planned chemotherapy, n (%) | ||
| 5-FU + leucovorin | 7 (29) | 6 (30) |
| Capecitabine | 17 (71) | 14 (70) |
| Zubrod performance status | ||
| 0 | 8 (33) | 6 (30) |
| 1 | 16 (67) | 14 (70) |
| Primary site, n (%) | ||
| Gall bladder | 8 (33) | 6 (30) |
| Extrahepatic biliary system | 2 (8) | 2 (10) |
| Intrahepatic biliary system | 14 (58) | 12 (60) |
| Serum CA 19–9a, U/mL, n (%) | ||
| <37 | 6 (25) | 7 (35) |
| ≥37 | 17 (71) | 13 (65) |
| Median (range) | 1238 (44–36,911) | 260 (45–66,010) |
| Prior treatment for biliary cancer | ||
| Adjuvant therapy | 14 (58) | 8 (40) |
| Local therapy | 0 (0) | 0 (0) |
| Single-agent chemotherapy | 2 (8) | 2 (10) |
| Multiple-agent chemotherapy | 13 (54) | 12 (60) |
| Surgery | 9 (38) | 5 (25) |
FU, fluorouracil.
One patient missing baseline serum CA 19–9.
As of 1st April 2016, all patients had discontinued protocol treatment. The primary reason for treatment discontinuation was disease progression (88% in arm 1 and 85% in arm 2).
3.2. Efficacy
The planned interim analyses performed on 24th April 2015 showed that a lack of measurable response in the trametinib arm ruled out the prespecified treatment benefit with high confidence. Fifteen eligible patients in the trametinib arm were evaluated for disease response and none achieved partial response. The study perma-nently closed to accrual on 15th May 2015, with 53 patients enroled.
The observed hazard rates for mortality and disease progression were higher for patients who received trametinib versus those who received chemotherapy (Figs.1 and 2). Median OS was 4.3 months in arm 1 and 6.6 months in arm 2 (HR 1.92, 95% confidence interval [CI] 0.98–3.77; p = .06). Median PFS was 1.4 months in the trametinib arm and 3.3 months in the chemotherapy arm (HR 2.77, 95% CI 1.30–5.92; p = .01).
Fig. 1.
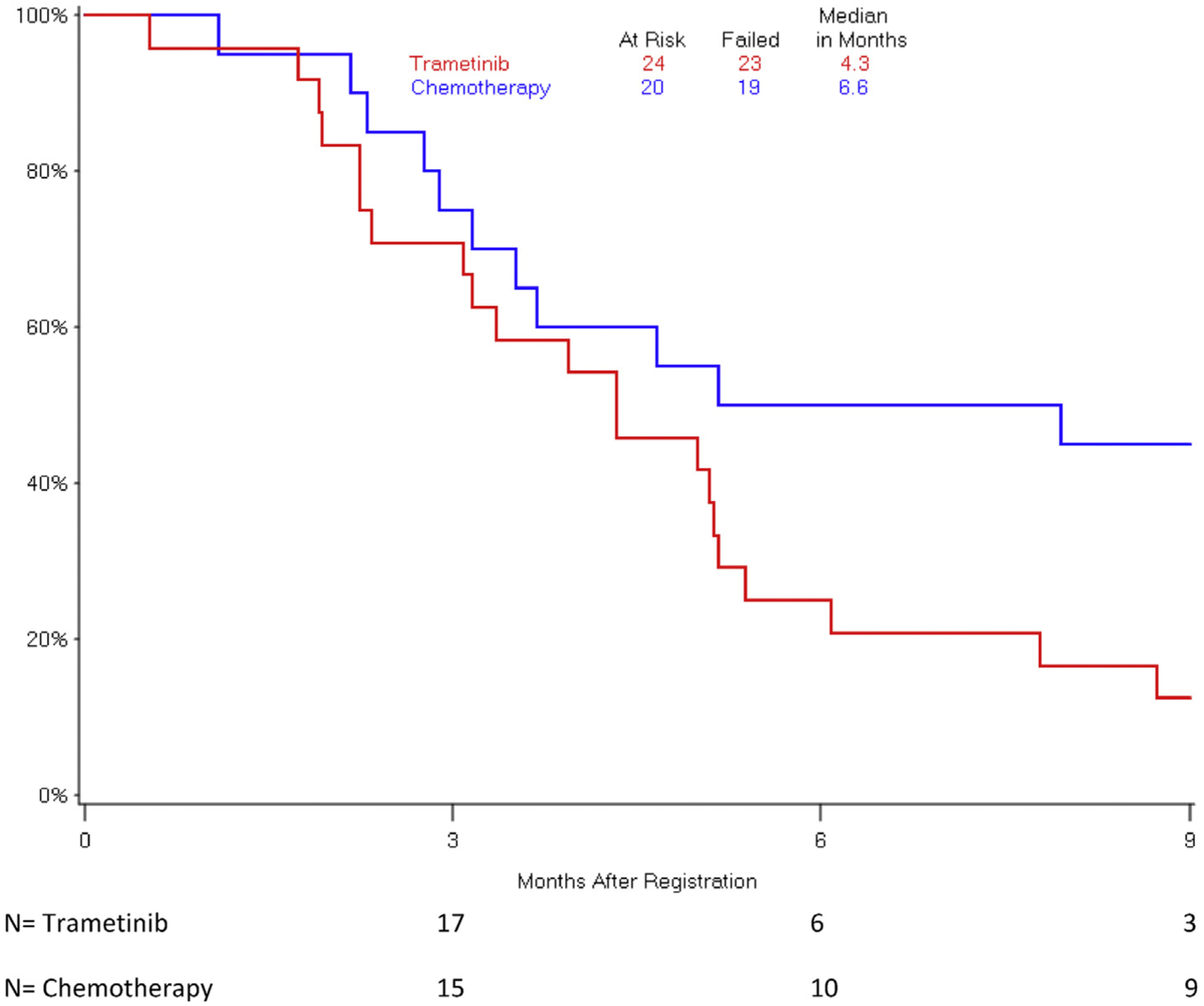
Overall survival by treatment arm.
Fig. 2.
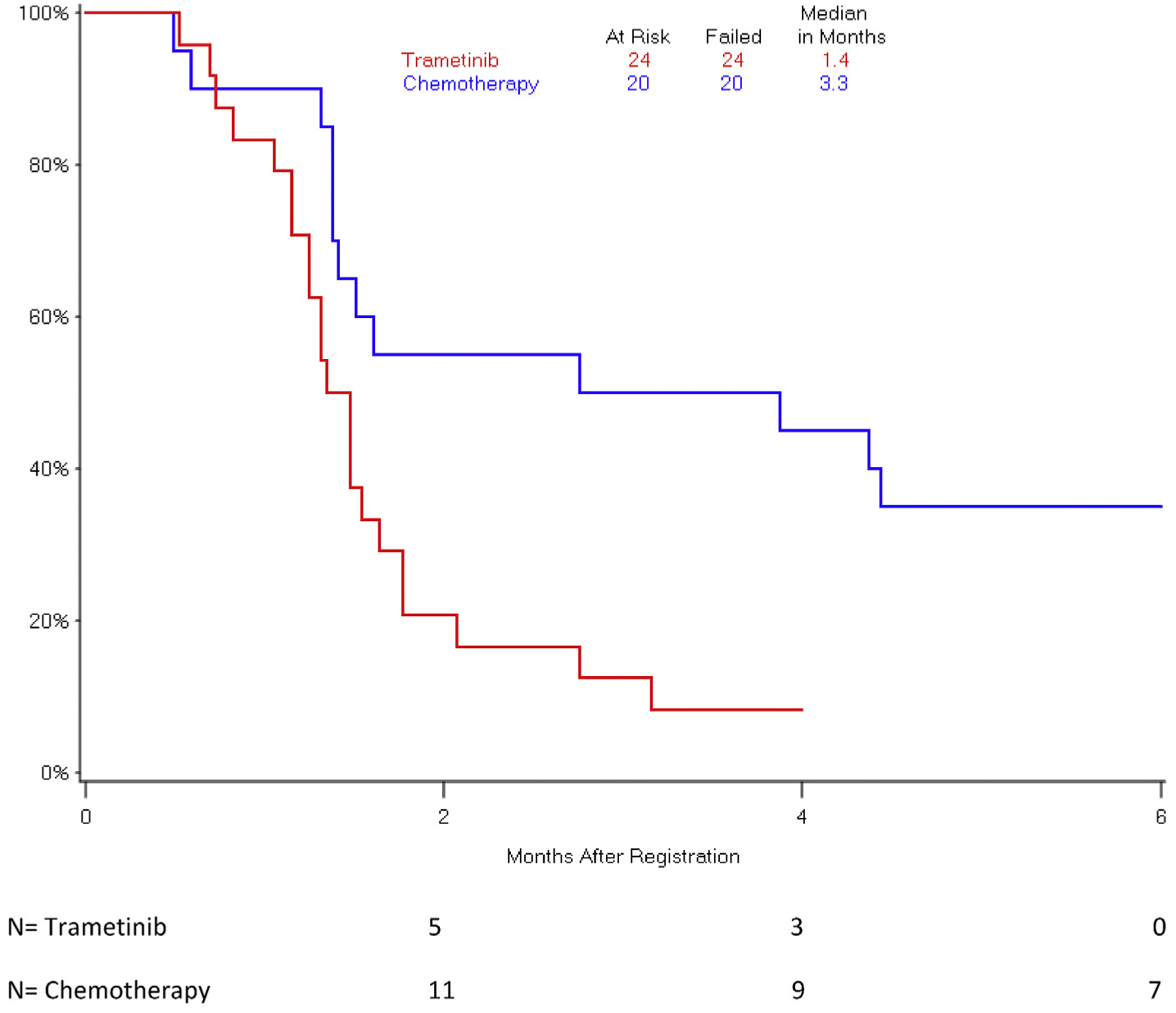
Progression-free survival by treatment arm.
The overall RR was 8% (95% CI–19%) in arm 1 versus 10% (95% CI 0–23%) in arm 2 (p > .99). The two unconfirmed partial responses in arm 1 were seen after the first 15 patients were evaluated. In arm 1, two patients (8%) had stable disease compared to nine patients (45%) in arm 2. Thus, the disease control rate was 17% in arm 1 versus 55% in arm 2. There were no complete responses in either group.
3.3. Safety
Twenty-three patients in arm 1 and 20 patients in arm 2 were assessed for adverse events (AEs; Table 2). More patients experienced serious treatment-related AEs in arm 1 than arm 2. There was one treatment-related death in arm 1 because of cardiac arrest; two patients experienced grade 4 events, including bilirubin increase and sepsis; and five additional patients experienced grade 3 events. In arm 2, seven patients (35%) experienced treatment-related grade 3 AEs, and there were no grade 4 or 5 AEs.
Table 2.
Selected treatment-related adverse events.
| Adverse events | Trametinib (n = 23) | Chemotherapy (n = 20) | ||
|---|---|---|---|---|
| All grades; n (%) | Grades ≥ 3; n (%) | All grades; n (%) | Grades ≥ 3; n (%) | |
| Rash acneiform | 14 (61) | 0 (0) | 1 (5) | 0 (0) |
| Fatigue | 7 (30) | 0 (0) | 8 (40) | 1 (5) |
| Vomiting | 6 (26) | 1 (4) | 3 (15) | 0 (0) |
| Anaemia | 5 (22) | 1 (4) | 6 (30) | 0 (0) |
| Nausea | 5 (22) | 0 (0) | 6 (30) | 0 (0) |
| Platelet count decreased | 5 (22) | 0 (0) | 5 (25) | 0 (0) |
| Anorexia | 4 (17) | 0 (0) | 3 (15) | 0 (0) |
| Diarrhoea | 3 (13) | 0 (0) | 7 (35) | 0 (0) |
| Papulopustular rash | 2 (9) | 0 (0) | 1 (5) | 0 (0) |
| Hand-foot syndrome | 2 (9) | 0 (0) | 8 (40) | 1 (5) |
| Pruritus | 3 (13) | 0 (0) | 0 (0) | 0 (0) |
| ALT increased | 2 (9) | 1 (4) | 0 (0) | 0 (0) |
| AST increased | 2 (9) | 1 (4) | 2 (10) | 0 (0) |
| Alopecia | 2 (9) | 0 (0) | 0 (0) | 0 (0) |
| Constipation | 2 (9) | 0 (0) | 2 (10) | 0 (0) |
| Dry mouth | 2 (9) | 0 (0) | 2 (10) | 0 (0) |
| Epistaxis | 2 (9) | 0 (0) | 0 (0) | 0 (0) |
| Blurred vision | 1 (4) | 0 (0) | 0 (0) | 0 (0) |
| Maximum grade any adverse event | 21 (91) | 8 (35) | 18 (90) | 7 (35) |
ALT: alanine aminotransferase; AST: aspartate aminotransferase.
The most common AEs, affecting at least 20% of patients by arm, were anaemia, rash, thrombocytopenia, fatigue, nausea and vomiting in arm 1 and anaemia, diarrhoea, dry skin, fatigue, hand-foot skin reaction, nausea and thrombocytopenia in arm 2.
AE-related treatment discontinuations occurred in 8% versus 5% of patients in arms 1 and 2, respectively, and other AE-related dose modifications were required in 26% versus 50% of patients in arms 1 and 2, respectively.
3.4. Correlative studies: CA 19–9
Carbohydrate antigen 19–9 (CA 19–9) levels were checked at baseline and every 6 weeks. The maximum allowable percentage decrease in CA 19–9 from baseline was defined as high (≥50%) versus low (<50%). Of the 19 patients in arm 1 for whom follow-up data were available, one (5%) had a high decrease in CA 19–9. A high decrease was not associated with overall response (p = .11) or OS (p = .75). Of the 17 patients in arm 2 with follow-up data, four (24%) had high decreases in CA 19–9. High decreases were associated with overall response (p = .04) but not OS (p = .20).
4. Discussion
Currently, there is no standard treatment available for advanced BC after progression on standard first-line treatment even though FOLFOX may be an alternative option based on a recent phase III study (ABC-06) [10]. Furthermore, because there is no randomised trial, it is unclear if doublet of FOLFOX is better than single-agent fluoropyrimidine. However, based one large retrospective study, there was no PFS or OS benefit of fluoropyrimidin–eplatinum combination compared to fluoropyrimidine alone [15]. In this randomised phase II study, the authors explored the efficacy of MEK inhibitor trametinib as a treatment for refractory BC compared with fluoropyrimidine therapy (physician’s choice of the most commonly used second-line treatments, 5-FU or capecitabine).
This study was based on the hypothesis that targeting the RAS/RAF/MAPK (MEK)/extracellular signal-related kinase (ERK) pathway would result in superior survival and anti-tumour activity given the reported presence of MAPK alterations in BC. Furthermore, early phase trials with other MEK inhibitors had shown preliminary clinical activity in BC [16,17]. The first reported trial with selumetinib included 28 patients, 39% of whom had received one prior line of therapy [16]. Results were promising, as 12% of patients had a confirmed objective response that included one complete response. A second trial of another MEK inhibitor, binimetinib, demonstrated 10% objective response in BC [17].
This study was closed after the interim futility analysis, which showed that the trametinib arm failed to meet the predetermined criteria for efficacy based on RR. No responses were seen in the first 15 patients randomised to the trametinib arm. Two responses that were seen after the first 15 patients were both unconfirmed and lasted less than 2 months. The median PFS and OS of the trametinib arm were inferior to those of the control arm. Only 8% of patients achieved stable disease in the trametinib arm, compared to 45% in the chemotherapy arm.
The results of this study stand in stark contrast to those of a phase II study of MEK inhibitor selumetinib in the setting of metastatic BC, in which 68% of patients achieved stable disease with PFS of 3.7 months and OS of 9.8 months [16]. The differences may be due to lines of therapy (treatment in the SWOG 1310 study was second-line therapy) or possible inherent differences between the agents. In the selumetinib study, no clear molecular biomarkers were identified for response as none of the responders harboured RAS or RAF mutations. These biomarker-related findings were similar to those of a BC study of binimetinib, also a MEK inhibitor, as only one of the three patients who responded harboured an RAS mutation [17]. One hypothesis is that the patients who responded may have harboured MEK mutations, which have no correlations with RAF or RAS mutations [18,19]. Furthermore, it is notable that responses to MEK inhibitors as single agents are very rare, other than when they are used to treat melanoma [20,21].
MEK inhibitors, including binimetinib, have been studied with chemotherapy agents gemcitabine and cisplatin for the treatment of BC in the first-line setting, with encouraging results that included OS of 21 months and RR of 36% [22]. Furthermore, in a xenograft model, combination of an MEK inhibitor and gemcitabine was highly schedule dependent and more effective when the drugs were given sequentially then simultaneously [23]. However, a randomised II study of selumetinib with different schedule and cisplatin/gemcitabine in advanced BC did not demonstrate a significant benefit for the addition of selumetinib to the combination chemotherapy [24]. There were no treatment-group differences in RR, PFS or OS, but there was added toxicity with selumetinib. Another small study using binimetinib in combination with capecitabine has been reported, with median OS of 8 months and RR of 18% [25]. In this study, the patients with aberrations in the RAS/RAF/MEK/ERK pathway showed longer PFS and OS than those without such aberrations.
Several challenges may have influenced the results of this study. Cancers of the biliary tract and gallbladder represent a heterogeneous group of tumours with variable frequency of alterations in specific molecular pathways based on site of origin (intrahepatic versus extrahepatic versus gallbladder cancer) [26]. In addition, the population included in this study was not selected or enriched based on specific molecular alterations because there were no established predictive biomarkers of trametinib. It has become evident in several solid tumours that the usage of single agents to target RAF or MEK does not yield significant anti-tumour activity because of the induction of alternative signalling pathways. In advanced colon cancer, single-agent RAF or MEK inhibitors have failed to demonstrate activity in KRAS or BRAF mutant tumours [27,28]. The data suggest that RAF or MEK inhibitors alone cannot sustain MAPK pathway inhibition likely due to feedback signals which can reactivate MAPK pathway. The major limitation to our trial was lack of correlatives. The lack of significant clinical activity and the small accrual numbers prevented the authors from being able to obtain funding for the correlatives. Although molecular profiling of these tumours may have helped the authors with the interpretation of the results, it is also important to note that it would have been unlikely to reach definitive conclusions related to biomarkers and activity given the heterogeneity of the disease, the small number of responders and the small accrual overall.
The control arm in this study manifested a level of activity that is comparable to published reports. For example, the largest series from Korea which included 255 patients evaluating fluoropyrimidine monotherapy after failure of gemcitabine and cisplatin, demonstrated PFS of 1.8 months and OS of 6.5 months [6].
In conclusion, S1301 was the first randomised study in refractory BC after failure of first-line systemic chemotherapy. In an unselected population of patients with BCs, trametinib was generally well tolerated but did not demonstrate clinical activity. However, in BC, we now have series of successful trials demonstrating success of molecular-guided personalised therapy. For example, in the MOSCATO-01 trial, molecular analyses were performed on biopsies to drive the administration of molecular-targeted agents [29]. Based on molecular profiling, 53% of patients in the study were found to have actionable mutations such as fibroblast growth factor receptor (FGFR) fusions, IDH 1 mutations and ERBB amplifications and derived clinical benefit. Recently, encouraging results have been observed in tyrosine kinase inhibitors targeting FGFR receptors in patients who harbour FGFR fusions including RR up to 25% [30,31].
Therefore, a further exploration of biomarkers is needed to identify a group of BC patients who may benefit from MEK inhibitors, and to develop potential rational combinations.
Funding
This work was funded by National Institutes of Health, National Cancer Institute grant awards CA180888, CA180819, CA180826, CA189858, CA180801, CA180846, CA189954, CA189821, CA180850, CA189960, CA46368, CA189830, CA46282, CA11083, CA189971, CA189972, CA180818, CA180818, CA189872, CA180798, CA180835, CA46113, CA58723, CA12644, CA180830; and in part by GlaxoSmithKline (Novartis Pharmaceuticals Corporation). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
RK has received honoraria from Bayer, Bristol Myer Squib (BMS) and Lilly for consulting; research funding from Bayer, BMS and Eisai. AE has received honoraria from BMS, Bayer, Merck, Eisai, Exelixis, Genentech/Roche, Astra Zeneca, Astex, Agenus, Novartis, EMD serono and Cytomx for consulting/advisory role; research funding from Astra Zeneca and Astex. TB has received honoraria from AbbVie, Bayer, Genentech, Glenmark, Imugene and Immuneering for consulting. SMS has received honoraria from Merck, BMS, Genentech and Eisai for advisory boards. VS has received honoraria from Celegene, Ipsen and Halozyme for consulting; research funding from Celgenem BMS, Halozyme and Medimmune. EK has received honoraria from Vicus, Armo, Bluepath Solutions and Guidepoint for advisory board and consulting; speaker fee from Guardant Health; research funding from BMS, Sanumed, Boston Biomedical, Halozyme, Merck, Celegene, Oncomed, EpicentRx and Astellas Pharma. ADB has received fees from BMS, Eli Lilly and Amgen for speaker’s bureau. AS currently is a full-time employee of Merck since August 2015.
Footnotes
Conflict of interest statement
SM, GPK, AB, SG, MJ and HH declare no conflicts of interest.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Ca - Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- [2].Jarnagin WR, Fong Y, DeMatteo RP, Gonen M, Burke EC, Bodniewicz BJ, et al. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann Surg 2001;234: 507–17. discussion 17–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–81. [DOI] [PubMed] [Google Scholar]
- [4].Choi CW, Choi IK, Seo JH, Kim BS, Kim JS, Kim CD, et al. Effects of 5-fluorouracil and leucovorin in the treatment of pancreatic-biliary tract adenocarcinomas. Am J Clin Oncol 2000; 23:425–8. [DOI] [PubMed] [Google Scholar]
- [5].Verderame F, Russo A, Di Leo R, Badalamenti G, Santangelo D, Cicero G, et al. Gemcitabine and oxaliplatin combination chemotherapy in advanced biliary tract cancers. Ann Oncol 2006; 17(Suppl 7):vii68–72. [DOI] [PubMed] [Google Scholar]
- [6].Kim BJ, Yoo C, Kim KP, Hyung J, Park SJ, Ryoo BY, et al. Efficacy of fluoropyrimidine-based chemotherapy in patients with advanced biliary tract cancer after failure of gemcitabine plus cisplatin: retrospective analysis of 321 patients. Br J Canc 2017; 116:561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fornaro L, Vivaldi C, Cereda S, Leone F, Aprile G, Lonardi S, et al. Second-line chemotherapy in advanced biliary cancer progressed to first-line platinum-gemcitabine combination: a multicenter survey and pooled analysis with published data. J Exp Clin Canc Res 2015;34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goff LW, Lowery MA, Jordan E, Wang R, Bocobo AG, Chou JF, et al. Second-line chemotherapy (CTx) outcomes in advanced biliary cancers (ABC): a retrospective multicenter analysis. J Clin Oncol 2016;34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Neuzillet C, Casadei Gardini A, Brieau B, Vivaldi C, Smolenschi C, Brandi G, et al. Prediction of survival with second-line therapy in biliary tract cancer: actualisation of the AGEO CT2BIL cohort and European multicentre validations. Eur J Canc 2019;111:94–106. [DOI] [PubMed] [Google Scholar]
- [10].Lamarca A, Palmer DH, Wasan HS, Ross PJ, May YT, Arora A. ABC-06 | A randomised phase III, multi-centre, open-label study of Active Symptom Control (ASC) alone or ASC with oxaliplatin/5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/gemcitabine (CisGem) chemotherapy. In: 2019. ASCO Annual Meeting; Chicago, IL; 2019. [Google Scholar]
- [11].Jimeno A, Rubio-Viqueira B, Amador ML, Grunwald V, Maitra A, Iacobuzio-Donahue C, et al. Dual mitogen-activated protein kinase and epidermal growth factor receptor inhibition in biliary and pancreatic cancer. Mol Canc Therapeut 2007;6: 1079–88. [DOI] [PubMed] [Google Scholar]
- [12].Ross JS, Wang K, Javle MM, Catenacci DVT, Shroff RT, Ali SM, et al. Comprehensive genomic profiling of biliary tract cancers to reveal tumor-specific differences and frequency of clinically relevant genomic alterations. J Clin Oncol 2015;33. [Google Scholar]
- [13].Hori H, Ajiki T, Mita Y, Horiuchi H, Hirata K, Matsumoto T, et al. Frequent activation of mitogen-activated protein kinase relative to Akt in extrahepatic biliary tract cancer. J Gastroenterol 2007;42:567–72. [DOI] [PubMed] [Google Scholar]
- [14].Jimeno A, Rubio-Viqueira B, Amador ML, Grunwald V, Maitra A, Iacobuzio-Donahue C, et al. Dual mitogen-activated protein kinase and epidermal growth factor receptor inhibition in biliary and pancreatic cancer. Mol Canc Therapeut 2007;6: 1079–88. [DOI] [PubMed] [Google Scholar]
- [15].Kim BJ, Yoo C, Kim KP, Hyung J, Park SJ, Ryoo BY, et al. Efficacy of fluoropyrimidine-based chemotherapy in patients with advanced biliary tract cancer after failure of gemcitabine plus cisplatin: retrospective analysis of 321 patients. Br J Canc 2017; 116:561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol 2011;29:2357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bendell JC, Javle M, Bekaii-Saab TS, Finn RS, Wainberg ZA, Laheru DA, et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Canc 2017;116:575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and BRAF inhibition. Proc Natl Acad Sci U S A 2009;106:20411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Canc Res 2010;70:2264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rosen L, LoRusso P, Ma WW, Goldman J, Weise A, Colevas AD, et al. A first-in-human phase 1 study to evaluate the MEK1/2 inhibitor GDC-0973 administered daily in patients with advanced solid tumors. Canc Res 2011:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Delord J, Houde N, Awada A, Taamma A, Faivre SJ, Besse-Hammer T, et al. First-in-human phase I safety, pharmacokinetic (PK), and pharmacodynamic (PD) analysis of the oral MEK-inhibitor AS703026 (two regimens [R]) in patients (pts) with advanced solid tumors. J Clin Oncol 2010;28. [Google Scholar]
- [22].Lowery MA, O’Reilly EM, Harding JJ, Salehi E, Hollywood E, Bradley M, et al. A phase I trial of binimetinib in combination with gemcitabine (G) and cisplatin (C) patients (pts) with untreated advanced biliary cancer (ABC). J Clin Oncol 2015;33. [Google Scholar]
- [23].Xu J, Knox JJ, Ibrahimov E, Chen E, Serra S, Tsao M, et al. Sequence dependence of MEK inhibitor AZD6244 combined with gemcitabine for the treatment of biliary cancer. Clin Canc Res 2013;19:118–27. [DOI] [PubMed] [Google Scholar]
- [24].Doherty M, Tam V, McNamara M, Hedley D, Dhani N, Chen E, et al. Selumetinib (Sel) and cisplatin/gemcitabine (CisGem) for advanced biliary tract cancer (BTC): a randomized trial. J Clin Oncol 2018;36. [Google Scholar]
- [25].Kim J, Lee K, Kim J, Suh K, Bang J, Bang Y, et al. Phase Ib study of binimetinib (MEK162) in combination with capecitabine in gemcitabine-pretreated advanced biliary tract cancer. 2018. [Google Scholar]
- [26].Sohal DPS, Shrotriya S, Abazeed M, Cruise M, Khorana A. Molecular characteristics of biliary tract cancer. Crit Rev Oncol-Hematol 2016;107:111–8. [DOI] [PubMed] [Google Scholar]
- [27].Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol 2015;33:4032–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012;379:1893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Verlingue L, Malka D, Allorant A, Massard C, Ferte C, Lacroix L, et al. Precision medicine for patients with advanced biliary tract cancers: an effective strategy within the prospective MOSCATO-01 trial. Eur J Canc 2017;87:122–30. [DOI] [PubMed] [Google Scholar]
- [30].Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2018; 36:276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Javle M, Kelley RK, Roychowdhury S, Weiss KH, Abou-Alfa GK, Macarulla T, et al. AB051. P-19. A phase II study of infigratinib (BGJ398) in previously-treated advanced cholangiocarcinoma containing FGFR2 fusions. Hepatobiliary Surg Nutr 2019;8:AB051. [Google Scholar]