

Abstract
Purpose
This phase 1 study evaluated the effect of hepatic impairment on pharmacokinetics and safety of crizotinib in patients with advanced cancer.
Methods
Patients were dosed according to hepatic function classified by modified National Cancer Institute Organ Dysfunction Working Group criteria and group assignment [normal (A1 and A2), mild (B), moderate (C1 and C2), or severe (D)]. Primary pharmacokinetic endpoints included area under the concentration–time curve as daily exposure (AUCdaily) and maximum plasma concentration (Cmax) at steady state. Safety endpoints included types, incidence, seriousness, and relationship to crizotinib of adverse events.
Results
The AUCdaily and Cmax in patients with normal liver function were 7107 ng h/mL and 375.1 ng/mL (A1) and 5422 ng h/mL and 283.9 ng/mL (A2), respectively. The AUCdaily and Cmax ratios of adjusted geometric means for Groups B, C2, and D versus Group A1 were 91.12 and 91.20, 114.08 and 108.87, and 64.47 and 72.63, respectively. Any grade treatment-related adverse events (TRAEs) occurred in 75% of patients; grade 3/4 TRAEs occurred in 25%, including fatigue (6%), hyponatremia (5%), and hyperbilirubinemia (3%).
Conclusions
No adjustment to the approved 250 mg twice daily (BID) dose of crizotinib is recommended for patients with mild hepatic impairment. The recommended dose is 200 mg BID for patients with moderate hepatic impairment, and the dose should not exceed 250 mg daily for patients with severe hepatic impairment. Adverse events appeared consistent among the hepatic impairment groups.
Keywords: Advanced cancer, Crizotinib, Hepatic impairment, Pharmacokinetics
Introduction
Crizotinib is a potent oral small-molecule inhibitor of anaplastic lymphoma kinase (ALK), mesenchymal epithelial transition factor/hepatocyte growth factor receptor (c-Met), recepteur d’origine nantais (RON), and c-Ros oncogene 1 (ROS1) receptor tyrosine kinases [1, 2]. Crizotinib was the first targeted therapy approved for the treatment of ALK-positive and/or ROS1-positive non-small cell lung cancer [3–5] and has been the standard of care for this patient population worldwide since its initial approval in the United States in 2011. The approved dosing regimen for crizotinib is 250 mg twice daily (BID).
It has been demonstrated that crizotinib undergoes extensive metabolism in both in vitro and clinical assessments. In a mass balance study in healthy subjects, only about 2% of the crizotinib dose was detected in urine as unchanged form (average of 22.2% of dose excreted in urine) [6]. Although unchanged crizotinib recovered in feces accounts for 53% of an administered dose, it is likely to represent the unabsorbed drug. In a separate study in healthy subjects, the absolute oral bioavailability of crizotinib was determined to be 43% [7]. In the human mass balance study, a number of metabolites of crizotinib were detected in plasma, urine, and feces. As metabolism of drugs predominantly occurs in the liver, liver function may have a potential impact on the elimination of a drug that undergoes extensive metabolism, such as crizotinib. Due to the importance of the liver in the elimination of crizotinib and the potential need to use crizotinib to treat patients with cancer who also have impaired liver function, it is important to determine the effect of hepatic impairment on the pharmacokinetics (PK) and safety of crizotinib and, based on this, to determine whether dose modification would be necessary in these patients.
The initial clinical trials with crizotinib limited enrollment to patients with minimal to no laboratory abnormalities that would indicate hepatic impairment, including initial serum total bilirubin (TB) > 1.5 × upper limit of normal (ULN) or transaminases > 2.5 × ULN. In clinical trials, elevations of hepatic transaminases [aspartate aminotransferase (AST) and alanine aminotransferase (ALT)] were among the most frequent adverse events (AEs) occurring in patients treated with crizotinib, and were reversible with dose reductions or treatment interruption [8]. A pooled analysis from two single-arm trials in patients with ALK-positive non-small cell lung cancer who received crizotinib 250 mg BID (n = 588) showed treatment-related ALT and AST elevations occurred in 73 (12%) and 52 (9%) patients, respectively. Grade 3 or 4 ALT and AST elevations occurred in 24 (4%) and 10 (2%) patients, respectively [9]. Transaminase elevations occurred most often within the first two cycles of treatment and were reversible with treatment interruption, with most patients resuming crizotinib treatment at the same or lower doses [9, 10]. In addition, rare (< 1%) cases of severe hepatotoxicity, including isolated fatal cases of treatment-related hepatotoxicity, have been reported in patients treated with crizotinib [11, 12]. However, knowledge of crizotinib PK and safety in patients with laboratory evidence of hepatic impairment remains incomplete, and there are no systematically collected data on which to make dosing recommendations for these patients. The current study is designed to evaluate the effect of hepatic impairment on the steady-state PK and safety of crizotinib in patients with advanced cancer and to determine dosing recommendations for patients with impaired hepatic function.
Materials and methods
Patients
This study enrolled patients with varying degrees of hepatic impairment and histologically or cytologically confirmed unresectable or metastatic solid tumors or lymphomas for whom no palliative or curative treatment options were available. Key inclusion criteria included age ≥ 18 years, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–2, absolute neutrophil count ≥ 750/µL, platelets ≥ 30,000/µL, hemoglobin ≥ 8.0 g/dL (≥ 7.0 g/dL for patients with hematologic malignancy), adequate renal function (creatinine < 1.5 × ULN or creatinine clearance > 60 mL/min/1.73 m2 for patients with serum creatinine levels > 1.5 × ULN), and completion of any prior chemotherapy treatments ≥ 30 days before enrollment. Patients were ineligible for the study if they had received prior crizotinib therapy, had untreated esophageal varices, ascites requiring therapeutic paracentesis more than four times per month despite medical management, an episode of hepatic encephalopathy within 4 weeks of study initiation, spinal cord compression, carcinomatous meningitis or leptomeningeal disease, acute coronary artery disease or cerebrovascular accident within 6 months of study initiation, symptomatic congestive heart failure, grade ≥ 2 cardiac dysrhythmias, prior gastrointestinal surgery that removed more than one-third of the colon or any other part of the gastrointestinal tract and/or the gallbladder, or use of agents that are known cytochrome P450 3A4 inhibitors, inducers, or substrates.
All patients provided written informed consent before enrollment. The institutional review board or independent ethics committee at each participating center approved the protocol, which complied with the International Conference on Harmonisation Good Clinical Practice guidelines, the Declaration of Helsinki, and local laws.
Study design and treatment
This multicenter, open-label, nonrandomized, phase 1 clinical trial (ClinicalTrials.gov identifier: NCT01576406) sequentially enrolled and assigned eligible patients to treatment groups based on hepatic function, as assessed by AST and TB according to National Cancer Institute guidance on hepatic impairment studies in patients with cancer [13] (described in Table 1). Child–Pugh scores were calculated and recorded as part of liver function but were not used to stratify patients into treatment groups.
Table 1.
Hepatic function and crizotinib dosing assignments
| Group A1 (match B) 250 mg BID | Group A2a (match C2) 200 mg BID | Group B 250 mg BID | Group C1b 250 mg QD | Group C2b 200 mg BID | Group Db 250 mg QD | |
|---|---|---|---|---|---|---|
| Liver function | Normal | Normal | Mild impairment | Moderate impairment | Moderate impairment | Severe impairment |
| TB ≤ ULN and AST ≤ ULN | TB ≤ ULN and AST ≤ ULN | TB ≤ ULN and AST > ULN; OR TB > 1.0 –≤ 1.5 × ULN and any AST | TB > 1.5–≤3 × ULN and any AST | TB > 1.5–≤3 × ULN and any AST | TB > 3 × ULN and any AST | |
| Starting dose | 250 mg BID | 200 mg BID | 250 mg BID | 250 mg QD | 200 mg BID | 250 mg QD |
AST aspartate aminotransferase, BID twice daily, PK pharmacokinetics, QD once daily, TB total bilirubin, ULN upper limit of normal
Enrollment of patients in control Group A2 was deferred until Group C1 PK results were available, so that the patients in control Group A2 and those in Group C2 would receive the same dose. Patients in control Group A2 were able to receive crizotinib 250 mg BID after PK assessments were completed
Two-stage enrollment was used in Group C; Stage 1 (Group C1) enrolled three patients at a crizotinib dose of 250 QD. Stage 2 (Group C2) was initiated once the PK analyses from Stage 1 were completed. The Stage 2 crizotinib dose in Group C and the dose for Group D were determined on the basis of Stage 1 PK results and assessment of tolerability
For each of the hepatic function groups, crizotinib was administered in 28-day cycles. Groups A1 and A2 (normal hepatic function) served as controls matched by age, weight, race, sex, and ECOG PS for Groups B (mild hepatic impairment) and C (moderate hepatic impairment), respectively. Patients assigned to Group D (severe hepatic impairment) were not matched with a control group. Patients in Groups A1 and B received crizotinib 250 mg BID. As the matched normal control for Group C, patients in Group A2 received the same dosing regimen as Group C until the completion of PK assessment on cycle 2, day 1, at which point the dose for crizotinib could be increased to the approved therapeutic dose of 250 mg BID. To protect patient safety and find an appropriate dose for patients with a higher degree of liver impairment, a two-stage design was adopted for Group C. At the first stage (C1), patients received a reduced dose of 250 mg once daily (QD) (C1). After evaluation of the preliminary PK and safety results from C1, a dose of 200 mg BID was determined to be an appropriate dose for patients with moderate hepatic impairment in Group C (C2). A dose of 250 mg QD was selected for patients in Group D.
Once assigned to a specific treatment group according to hepatic function, patients remained in that group, even in the presence of changes in hepatic function. Treatment was continued until disease progression, patient refusal, or unacceptable toxicity occurred. Crizotinib treatment interruptions and dosing adjustments were permissible for treatment-emergent toxicities described in Supplementary Tables S1, S2, and S3. With the exception of pneumonitis, which required permanent treatment discontinuation, patients were eligible to resume treatment at the previous dose once the toxicity resolved.
PK assessments
Blood samples were collected at times 0 (predose), 1, 2, 4, 6, 8, and 12 h (BID dosing) or 24 h (QD dosing) after the morning crizotinib dose on cycle 1, day 1 and cycle 2, day 1 for the determination of plasma concentrations of crizotinib and its metabolite, PF-06260182. Additional blood samples were also collected before the first crizotinib dose on cycle 1, day 1 and 4 h after the morning crizotinib dose on cycle 2, day 1 for the evaluation of plasma protein binding in each patient. Plasma samples for determination of crizotinib and PF-06260182 were analyzed using a validated, sensitive, and specific high-performance liquid chromatography–tandem mass spectrometric method (Covance Bioanyalytical Services, LLC, Indianapolis, IN, USA) in compliance with Pfizer’s standard operating procedures. Protein-binding samples were dialyzed (Covance Laboratories, Madison, WI, USA), and the extent of plasma protein binding of crizotinib and PF-06260182 in those samples was calculated (Covance Laboratories) using the plasma ultrafiltrate concentrations generated by Covance Bioanalytical Services, LLC.
Relevant PK parameters for crizotinib and PF-06260182 were calculated for each patient and treatment day using non-compartmental analysis (NCA) of plasma concentration–time curve, if data permitted, via eNCA v2.2.4 (Pfizer, Groton, CT, USA). Concentrations below the lower limit of quantification were set to 0 ng/mL for the analysis. Actual sample collection times were used during NCA. Primary PK endpoints assessed were area under the concentration–time curve as daily exposure (AUCdaily) and maximum observed plasma concentration (Cmax) at steady state (cycle 2, day 1). The secondary PK endpoints assessed were area under the concentration–time curve (AUC) from time zero to the last quantifiable plasma concentration; time to the last measurable concentration; Cmax after a single dose (cycle 1, day 1); AUC from time zero to tau hours postdose (AUCtau), where tau was 12 h for BID dosing and 24 h for QD dosing at steady state (cycle 2, day 1); average plasma concentration during one dosing interval at steady state (cycle 2, day 1); apparent oral clearance (CL/F) at steady state (cycle 2, day 1); and metabolite-to-parent ratios corrected for molecular weight for AUCtau and Cmax for PF-06260182. The PK-evaluable population included patients in the safety analysis population who completed cycle 2, day 1 PK sample collections; had no dose modifications from cycle 1, day 1 through cycle 2, day 1; received > 80% of the prescribed dose during the 14 days before cycle 2, day 1; and did not vomit crizotinib on cycle 2, day 1. AUCdaily was calculated as twice the AUCtau for BID dosing and AUCtau for QD dosing. The effect of hepatic impairment on PK parameters was assessed by constructing 90% confidence intervals (CIs) around the estimated difference between each of the hepatic impairment groups (Test) and normal hepatic function (Reference) using a one-way analysis of variance model based on natural log-transformed data.
Safety assessment
All AEs were reported regardless of treatment group or suspected causal relationship (assessed by the investigator) to crizotinib. AEs were classified by type, incidence, severity (graded by National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0), timing, seriousness, and relatedness to crizotinib. The safety population included all enrolled patients who received at least one dose of crizotinib. Only treatment-related AEs (TRAEs) are summarized and included in this report.
Antitumor activity assessment
Tumor assessments were performed at screening and subsequently every odd-numbered cycle or according to standard practice at the individual study site and also at the end of treatment or at study withdrawal. Patients who received at least one dose of crizotinib and had an adequate baseline tumor measurement were included in the efficacy analysis. Objective response rate was defined as the percentage of patients with confirmed complete response or confirmed partial response according to Response Evaluation Criteria in Solid Tumors version 1.1, based on the investigator assessment, relative to the response-evaluable population. The point estimate along with the corresponding two-sided 95% CI using the exact method was calculated. Duration of response was defined as the time (in weeks) from the first documentation of objective tumor response (complete or partial response) that was subsequently confirmed to the first documentation of objective tumor progression or death on study due to any cause, whichever occurred first.
Results
A total of 88 patients were enrolled. Of these, 26 had normal hepatic function, 20 had mild hepatic impairment, 26 had moderate hepatic impairment, and 16 met criteria for severe hepatic impairment. The median age for all patients was 60.0 years (range 31–78 years); 64.8% were male; and 18.2, 73.9, and 8.0% had an ECOG PS of 0, 1, and 2, respectively. The most common primary diagnoses for the total population were hepatocellular carcinoma (48.9%) and colorectal cancer (27.3%). Demographics and baseline characteristics were similar across groups (Table 2).
Table 2.
Demographics
| Pharmacokinetics-evaluable population (n = 48) | Group A1 (n = 8) 250 mg BID | Group A2 (n = 9) 200 mg QD | Group B (n = 10) 250 mg BID | Group C1 (n = 7) 250 mg QD | Group C2 (n = 8) 200 mg BID | Group D (n = 6) 250 mg QD |
|---|---|---|---|---|---|---|
| Sex, n (%) | ||||||
| Male | 4 (50.0) | 6 (66.7) | 6 (60.0) | 6 (85.7) | 6 (75.0) | 4 (66.7) |
| Female | 4 (50.0) | 3 (33.3) | 4 (40.0) | 1 (14.3) | 2 (25.0) | 2 (33.3) |
| Age (years) | ||||||
| Median (range) | 63.5 (31–78) | 65.0 (51–78) | 61.0 (42–78) | 52.0 (48–73) | 61.0 (52–75) | 57.5 (54–69) |
| Age category, n (%) | ||||||
| <65 years | 4 (50.0) | 4 (44.4) | 6 (60.0) | 5 (71.4) | 6 (75.0) | 4 (66.7) |
| ≥65 years | 4 (50.0) | 5 (55.6) | 4 (40.0) | 2 (28.6) | 2 (25.0) | 2 (33.3) |
| ECOG PS, n (%) | ||||||
| 0 | 4 (50.0) | 1 (11.1) | 3 (30.0) | 1 (14.3) | 2 (25.0) | 0 |
| 1 | 4 (50.0) | 8 (89.0) | 7 (70.0) | 6 (85.7) | 6 (75.0) | 5 (83.3) |
| 2 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) |
| Race, n (%) | ||||||
| White | 6 (75.0) | 5 (55.6) | 5 (50.0) | 5 (71.4) | 6 (75.0) | 5 (83.3) |
| Black | 0 | 1 (11.1) | 1 (10.0) | 0 | 0 | 0 |
| Asian | 1 (12.5) | 3 (33.3) | 1 (10.0) | 0 | 2 (25.0) | 0 |
| Japanese | 0 | 0 | 0 | 0 | 0 | 0 |
| Korean | 0 | 0 | 0 | 0 | 0 | 0 |
| Chinese | 0 | 2 (22.2) | 1 (10.0) | 0 | 1 (12.5) | 0 |
| Other | 1 (12.5) | 1 (11.0) | 0 | 0 | 1 (12.5) | 0 |
| Other | 1 (12.5) | 0 | 3 (30.0) | 2 (28.6) | 0 | 1 (16.7) |
| Safety population (n = 88) | Group A1 (n = 11) 250 mg BID | Group A2 (n = 15) 200 mg BID | Group B (n = 20) 250 mg BID | Group C1 (n = 10) 250 mg QD | Group C2 (n = 16) 200 mg BID | Group D (n = 16) 250 mg QD |
| Sex, n (%) | ||||||
| Male | 7 (63.6) | 9 (60.0) | 13 (65.0) | 8 (80.0) | 10 (62.5) | 10 (62.5) |
| Female | 4 (36.4) | 6 (40.0) | 7 (35.0) | 2 (20.0) | 6 (37.5) | 6 (37.5) |
| Age (years) | ||||||
| Median (range) | 63.0 (31–78) | 65.0 (42–78) | 60.5 (42–78) | 58.5 (48–73) | 60.0 (43–75) | 57.5 (47–70) |
| Age category, n (%) | ||||||
| < 65 years | 6 (54.5) | 7 (46.7) | 13 (65.0) | 7 (70.0) | 13 (81.3) | 13 (81.3) |
| ≥ 65 years | 5 (45.5) | 8 (53.3) | 7 (35.0) | 3 (30.0) | 3 (18.8) | 3 (18.8) |
| ECOG PS, n (%) | ||||||
| 0 | 5 (45.5) | 1 (6.7) | 4 (20.0) | 3 (30.0) | 2 (12.5) | 1 (6.3) |
| 1 | 6 (54.5) | 14 (93.3) | 15 (75.0) | 7 (70.0) | 12 (75.0) | 11 (68.8) |
| 2 | 0 | 0 | 1 (5.0) | 0 | 2 (12.5) | 4 (25.0) |
| Race, n (%) | ||||||
| White | 9 (81.8) | 8 (53.3) | 10 (50.0) | 7 (70.0) | 14 (87.5) | 12 (75.0) |
| Black | 0 | 3 (20.0) | 2 (10.0) | 0 | 0 | 1 (6.3) |
| Asian | 1 (9.1) | 4 (26.7) | 3 (15.0) | 0 | 2 (12.5) | 2 (12.5) |
| Japanese | 0 | 0 | 0 | 0 | 0 | 0 |
| Korean | 0 | 1 (6.7) | 0 | 0 | 0 | 2 (12.5) |
| Chinese | 0 | 2 (13.3) | 1 (5.0) | 0 | 1 (6.3) | 0 |
| Other | 1 (9.1) | 1 (6.7) | 2 (10.0) | 0 | 1 (6.3) | 0 |
| Other | 1 (9.1) | 0 | 5 (25.0) | 3 (30.0) | 0 | 1 (6.3) |
| Primary diagnosis, n (%) | ||||||
| Hepatocellular carcinomaa | 0 | 2 (13.3) | 14 (70.0) | 6 (60.0) | 11 (68.8) | 10 (62.5) |
| Colorectal cancerb | 2 (18.2) | 3 (20.0) | 4 (20.0) | 4 (40.0) | 3 (18.8) | 3 (18.8) |
| Prostate cancer | 0 | 3 (20.0) | 0 | 0 | 0 | 0 |
| Adenoid cystic carcinomac | 2 (18.2) | 1 (6.7) | 0 | 0 | 0 | 0 |
| Cholangiocarcinoma | 1 (9.1) | 0 | 0 | 0 | 1 (6.3) | 1 (6.3) |
| Acinic cell carcinoma | 0 | 0 | 1 (5.0) | 0 | 0 | 0 |
| Adenocarcinoma of unknown primary | 0 | 0 | 0 | 0 | 1 (6.3) | 0 |
| Pancreatic cancerd | 0 | 1 (6.7) | 0 | 0 | 0 | 1 (6.3) |
| Anal squamous cell carcinoma | 0 | 0 | 0 | 0 | 0 | 1 (6.3) |
| Bladder cancer | 0 | 0 | 1 (5.0) | 0 | 0 | 0 |
| Chondrosarcomae | 1 (9.1) | 1 (6.7) | 0 | 0 | 0 | 0 |
| Leiomyosarcoma | 0 | 1 (6.7) | 0 | 0 | 0 | 0 |
| Lung cancerf | 1 (9.1) | 1 (6.7) | 0 | 0 | 0 | 0 |
| Neuroblastoma | 1 (9.1) | 0 | 0 | 0 | 0 | 0 |
| Esophageal cancer | 1 (9.1) | 0 | 0 | 0 | 0 | 0 |
| Ovarian cancer | 1 (9.1) | 0 | 0 | 0 | 0 | 0 |
| Renal cell carcinoma | 0 | 1 (6.7) | 0 | 0 | 0 | 0 |
| Teratoma | 1 (9.1) | 0 | 0 | 0 | 0 | 0 |
| Thyroid cancer | 0 | 1 (6.7) | 0 | 0 | 0 | 0 |
BID twice daily, ECOG PS Eastern Cooperative Oncology Group performance status, QD once daily
Hepatocellular carcinoma includes hepatocellular cancer and hepatic cancer metastatic
Colorectal cancer includes colon cancer metastatic, colon cancer, rectal cancer, adenocarcinoma of colon, rectal adenocarcinoma and rectal cancer metastatic
Adenoid cystic carcinoma includes adenoid cystic carcinoma of salivary gland
Pancreatic cancer includes adenocarcinoma of pancreas and pancreatic carcinoma
Chondrosarcoma includes chondrosarcoma metastatic
Lung cancer includes lung adenocarcinoma and lung neoplasm malignant
Pharmacokinetics
Overall, 48 patients met the criteria for PK evaluation and were included in the PK-evaluable population. Among those patients who were not PK evaluable, the majority did not have PK samples collected on cycle 2, day 1. Preliminary PK data for patients treated with crizotinib 250 mg QD during Stage 1 of Group C showed that total systemic exposure was approximately one-half the exposure in normal controls (Group A1) (Table 3). Relevant PK parameters for crizotinib and PF-06260182 are summarized by hepatic function and starting dose in Table 3.
Table 3.
Summary of crizotinib and PF-06260182 PK parameters following single and continuous oral doses
| Group A1 (n = 8) 250 mg BID | Group A2 (n = 9) 200 mg BID | Group B (n = 9)a 250 mg BID | Group C1 (n = 7) 250 mg BID | Group C2 (n = 8) 200 mg BID | Group D (n = 6) 250 mg QD | |
|---|---|---|---|---|---|---|
| Crizotinib | ||||||
| Cycle 1, day 1 (single dose) | ||||||
| AUClast (ng h/mL) | 732.30 (84) | 520.80 (49) | 557.50 (78) | 610.40 (128) | 684.90 (89) | 856.70 (57) |
| Tlast (h) | 12.00 (10.90–12.90) | 11.30 (10.80–12.00) | 12.00 (9.65–12.00) | 24.00 (23.60–24.00) | 12.00 (12.00–12.00) | 24.00 (23.90–24.00) |
| Cmax (ng/mL) | 101.90 (92) | 84.52 (67) | 102.30 (66) | 57.74 (112) | 99.59 (88) | 90.69 (63) |
| Tmax (h) | 4.00 (1.00–6.00) | 4.00 (1.00–6.00) | 4.00 (1.92–10.90) | 2.02 (1.00–4.05) | 4.00 (1.00–8.00) | 3.00 (1.00–6.00) |
| Cycle 2, day 1 (multiple dose) | ||||||
| AUCdaily (ng h/mL) | 7107 (48) | 5422 (66) | 6476 (73) | 2305 (83) | 8108 (58) | 4596 (63) |
| Cmin (ng/mL) | 238.60 (52) | 170.90 (75) | 179.10 (101) | 47.44 (376) | 287.00 (58) | 135.50 (102) |
| Cmax (ng/mL) | 375.10 (50) | 283.90 (65) | 342.10 (68) | 152.90 (58) | 408.30 (56) | 272.40 (29) |
| AUCtau (ng h/mL) | 3552 (48) | 2712 (66) | 3238 (73) | 2305 (83) | 4057 (58) | 4596 (63) |
| Cavg (ng/mL) | 295.80 (48) | 225.90 (66) | 269.70 (73) | 96.10 (83) | 338.10 (58) | 191.70 (63) |
| CL/F (L/h) | 70.39 (48) | 73.79 (66) | 77.21 (73) | 108.50 (83) | 49.26 (58) | 54.36 (63) |
| Tmax (h) | 4.00 (0.98–4.02) | 3.98 (1.00–6.00) | 4.00 (1.67–11.20) | 2.00 (1.05–4.05) | 3.00 (2.00–6.00) | 3.99 (2.00–6.05) |
| Fu | 0.03624 (26) | 0.03066 (27) | 0.04315 (31) | 0.05406 (20) | 0.04152 (34) | 0.03523 (46) |
| PF-06260182 | ||||||
| Cycle 1, day 1 (single dose) | ||||||
| AUClast (ng h/mL) | 272.4 (80) | 166.5 (31) | 90.71 (153) | 64.90 (103) | 53.36 (139) | 59.61 (81) |
| Tlast (h) | 12.0 (10.9–12.9) | 11.3 (10.8–12.0) | 12.0 (9.65–12.0) | 24.0 (23.6–24.0) | 12.0 (12.0–12.0) | 24.0 (23.9–24.0) |
| Cmax (ng/mL) | 35.48 (86) | 24.12 (33) | 13.54 (153) | 4.87 (95) | 6.995 (152) | 4.09 (88) |
| Tmax (h) | 6.04 (4.00–8.00) | 4.00 (2.00–8.00) | 4.00 (4.00–10.90) | 6.08 (5.93–8.00) | 8.00 (4.00–12.00) | 7.00 (4.00–8.00) |
| Cycle 2, day 1 (multiple dose) | ||||||
| AUCdaily (ng h/mL) | 2173 (43) | 1435 (81) | 961 (168) | 200 (48) | 784 (117) | 434 (96) |
| Cmin (ng/mL) | 69.40 (53) | 42.02 (104) | 21.95 (209) | 4.71 (55) | 27.41 (123) | 10.24 (134) |
| Cmax (ng/mL) | 108.50 (43) | 73.47 (76) | 50.34 (164) | 12.43 (55) | 39.32 (120) | 25.15 (110) |
| AUCtau (ng h/mL) | 1087 (43) | 717.4 (81) | 480.3 (168) | 200.0 (48) | 391.8 (118) | 434.0 (96) |
| MRAUCtau | 0.2968 (17) | 0.2569 (31) | 0.1439 (74) | 0.08402 (65) | 0.09360 (47) | 0.09162 (47) |
| MRCmax | 0.2804 (19) | 0.2511 (31) | 0.1428 (73) | 0.07881 (58) | 0.09337 (49) | 0.08954 (90) |
| Tmax (h) | 6.00 (0.983–10.00) | 4.03 (1.00–7.45) | 4.00 (0.00–10.80) | 6.00 (4.05–7.30) | 0.00 (0.00–6.00) | 6.69 (0.00–8.00) |
| Fu | 0.03797 (11) | 0.03822 (16) | 0.04857 (16) | 0.05788 (22) | 0.05031 (10) | 0.05177 (13) |
Data are presented as geometric mean (% coefficient of variation) or median (range)
n = number of patients contributing to the summary statistics
AUCdaily area under the plasma concentration–time curve as daily exposure (AUCtau*2 for BID and AUCtau for QD), AUClast area under the plasma concentration–time curve from time zero to the last measurable concentration, AUCtau area under the plasma concentration–time curve during one dosing interval at steady state (cycle 2, day 1 only), BID twice daily, Cavg average plasma concentration during one dosing interval at steady state (cycle 2, day 1 only) (AUCtau/tau), CL/F apparent oral clearance (cycle 2, day 1 only), Cmax maximum observed plasma concentration, Cmin minimum observed plasma concentration, Fu fraction of unbound drug in plasma, MRAUCtau metabolite-to-parent ratio for AUCtau, MRCmax metabolite-to-parent ratio for Cmax, PK pharmacokinetics, QD once daily, Tmax time to Cmax, Tlast time to the last measurable plasma concentration
One patient from the PK-evaluable set vomited on cycle 1, day 1; as such, no concentration data were obtained
As shown in Fig. 1, at steady state (cycle 2, day 1), between-group comparisons of the adjusted mean ratios for the exposure parameters AUCdaily and Cmax were similar for Groups A1 and B, with geometric mean ratios for AUCdaily and Cmax of 91.12% (90% CI 56.56–146.79) and 91.20% (90% CI 57.47–144.72), respectively. Geometric mean ratios for AUCdaily and Cmax were relatively higher in patients in Group C2 receiving crizotinib 200 mg BID [149.54% (90% CI 91.85–243.46) and 143.82% (90% CI 89.11–232.12), respectively], compared to patients in Group A2. For patients in Group C2, geometric mean ratios were 114.08% (90% CI 73.57–176.89) for AUC daily and 108.87% (90% CI 70.13–168.99) for Cmax compared to patients in Group A1. Exposures were lowest in patients in Group C1 receiving crizotinib 250 mg QD. AUCdaily and Cmax in patients in Group D receiving crizotinib 250 mg QD were relatively lower compared to patients in control Groups A1 and A2, with geometric mean ratios for AUCdaily and Cmax in Group D patients approximately 64.67% (90% CI 39.50–105.89) and 72.63% (90% CI 49.07–107.50), respectively, of those in Group A1.
Fig. 1.
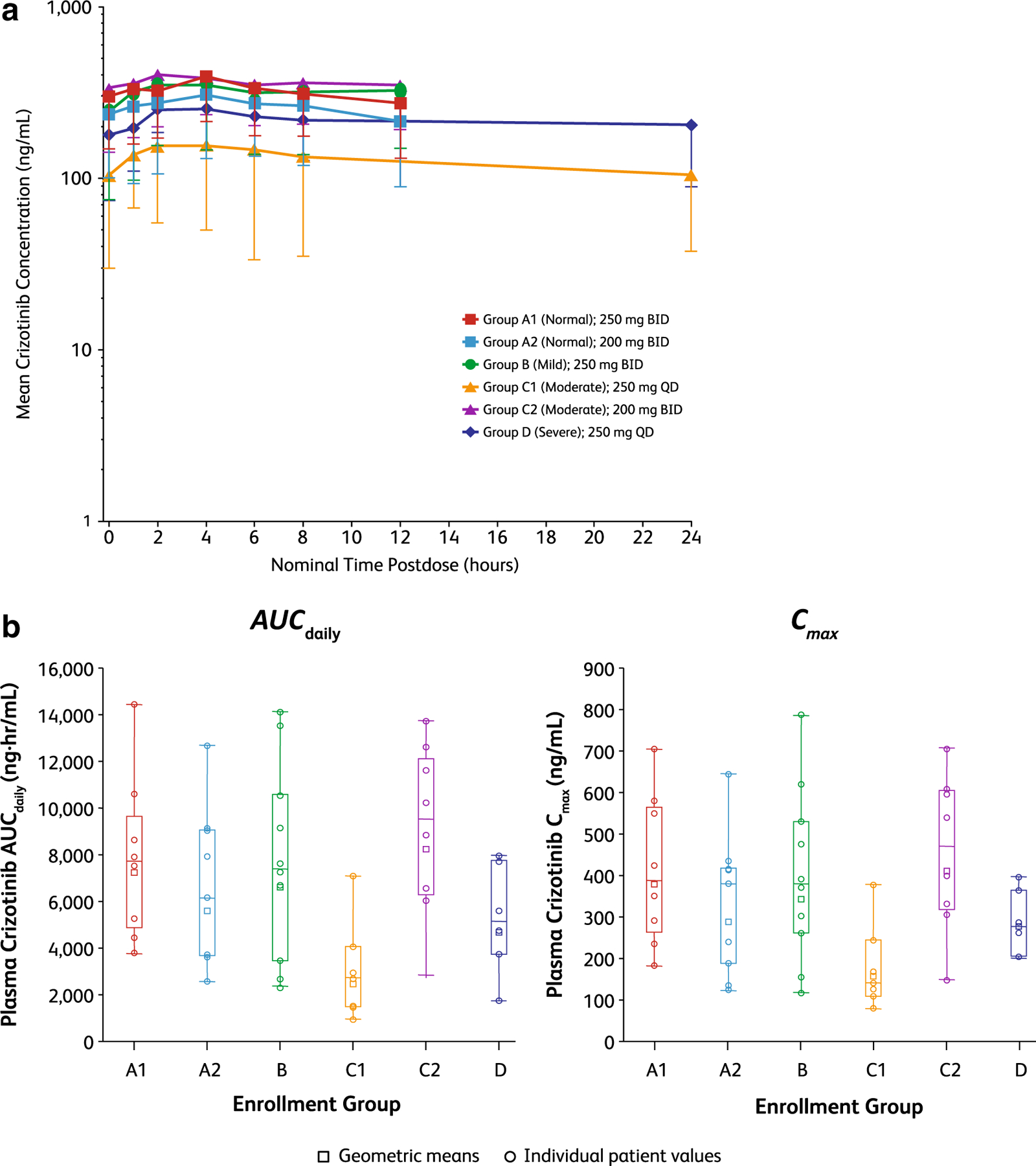
Between-group comparisons of crizotinib exposure parameters at steady state (cycle 2, day 1). a Mean crizotinib concentration versus time curves for Groups A1 through D. b Individual values and geometric mean AUCdaily and Cmax values for Groups A1 through D. Median is indicated by the line in the boxes. Box plot, 25%/75% quartiles with whiskers to the last point within 1.5 times the interquartile range. AUCdaily area under the plasma concentration–time curve as daily exposure, BID twice daily, Cmax maximum observed plasma concentration, QD once daily
The quantity of unbound crizotinib and its metabolite, PF-06260182, in plasma for each of the hepatic impairment groups is shown in Table 3. Mean unbound fractions of crizotinib and its metabolite were higher in patients with hepatic impairment compared to patients with normal hepatic function, with the exception of patients with severe hepatic impairment (Group D) for crizotinib only.
Safety
All enrolled patients (N = 88) were included in the safety analysis. The median duration of treatment in groups A1, A2, B, C1, C2, and D was 11.0 (range 1.7–103.9) weeks, 6.4 (range 1.3–15.9) weeks, 6.4 (range 0.3–45.3) weeks, 10.0 (range 2.0–18.3) weeks, 5.7 (range 1.0–24.6) weeks, and 3.1 (range 1.1–18.1) weeks, respectively. In this study, the most frequently reported TRAEs occurring in at least 20% of the total population were nausea, vomiting, fatigue, and vision disorders (comprising blurred vision and vitreous floaters) (Table 4).
Table 4.
Most common (≥ 10%) treatment-related adverse events of any grade
| Event, n (%) | Group A1 (n = 11) 250 mg BID | Group A2 (n =1 5) 200 mg BID | Group B (n =2 0) 250 mg BID | Group C1 (n = 10) 250 mg QD | Group C2 (n = 16) 200 mg BID | Group D (n = 16) 250 mg QD | Total (N = 88) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | Any grade | Grade 3 or 4 | |
| Any event | 9 (82) | 3 (27) | 14 (93) | 3 (20) | 15 (75) | 3 (15) | 6 (60) | 1 (10) | 11 (69) | 7 (44) | 11 (69) | 5 (31) | 66 (75) | 22 (25) |
| Blood and lymph disorders | ||||||||||||||
| Anemia | 1 (9) | 1 (9) | 2 (13) | 0 | 0 | 0 | 1 (10) | 0 | 1 (6) | 1 (6) | 0 | 0 | 5 (6) | 2 (2) |
| Neutropenia | 0 | 0 | 0 | 0 | 00 | 0 | 0 | 0 | 1 (6) | 1 (6) | 1 (6) | 1 (6) | 2 (2) | 2 (2) |
| Thrombocytopenia | 0 | 0 | 0 | 0 | 1 (5) | 1 (5) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) | 1 (1) |
| Eye disorders | ||||||||||||||
| Vision blurred | 3 (27) | 0 | 1 (7) | 0 | 2 (10) | 0 | 2 (20) | 0 | 2 (13) | 0 | 0 | 0 | 10 (11) | 0 |
| Vitreous floaters | 2 (18) | 0 | 0 | 0 | 1 (5) | 0 | 3 (30) | 0 | 2 (13) | 0 | 1 (6) | 0 | 9 (10) | 0 |
| Photophobia | 0 | 0 | 2 (13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 |
| Gastrointestinal disorders | ||||||||||||||
| Diarrhea | 6 (55) | 0 | 3 (20) | 1 (7) | 3 (15) | 0 | 1 (10) | 0 | 1 (6) | 0 | 1 (6) | 0 | 15 (17) | 1 (1) |
| Nausea | 5 (45) | 0 | 7 (47) | 0 | 11 (55) | 0 | 1 (10) | 0 | 6 (38) | 0 | 4 (25) | 0 | 34 (39) | 0 |
| Constipation | 2 (18) | 0 | 1 (7) | 0 | 2 (10) | 1 (5) | 1 (10) | 0 | 1 (6) | 0 | 0 | 0 | 7 (8) | 1 (1) |
| Vomiting | 2 (18) | 0 | 7 (47) | 1 (7) | 6 (30) | 0 | 1 (10) | 0 | 6 (38) | 1 (6) | 3 (19) | 0 | 25 (28) | 2 (2) |
| Dry mouth | 1 (9) | 0 | 0 | 0 | 1 (5) | 0 | 0 | 0 | 2 (13) | 0 | 0 | 0 | 4 (5) | 0 |
| Dyspepsia | 1 (9) | 0 | 2 (13) | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 | 1 (6) | 0 | 5 (6) | 0 |
| GERD | 1 (9) | 0 | 2 (13) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (3) | 0 |
| General disorders and administration-site conditions | ||||||||||||||
| Edema peripheral | 3 (27) | 0 | 2 (13) | 0 | 2 (10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 (8) | 0 |
| Fatigue | 2 (18) | 0 | 4 (27) | 0 | 2 (10) | 0 | 2 (20) | 1 (10) | 5 (31) | 3 (19) | 4 (25) | 1 (6) | 19 (22) | 5 (6) |
| Face edema | 0 | 0 | 0 | 0 | 0 | 0 | 1 (10) | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 |
| Pyrexia | 0 | 0 | 2 (13) | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 | 0 | 0 | 3 (3) | 0 |
| Hepatobiliary disorders | ||||||||||||||
| Hyperbilirubinemia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (19) | 3 (19) | 0 | 0 | 3 (3) | 3 (3) |
| Investigations | ||||||||||||||
| ALT increased | 2 (18) | 0 | 0 | 0 | 4 (20) | 0 | 0 | 0 | 2 (13) | 0 | 1 (6) | 0 | 9 (10) | 0 |
| AST increased | 3 (27) | 0 | 0 | 0 | 4 (20) | 1 (5) | 0 | 0 | 1 (6) | 0 | 1 (6) | 1 (6) | 9 (10) | 2 (2) |
| Blood creatinine increased | 1 (9) | 0 | 1 (7) | 0 | 2 (10) | 0 | 1 (10) | 0 | 0 | 0 | 0 | 0 | 5 (6) | 0 |
| Activated PTT prolonged | 1 (9) | 1 (9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) | 1 (1) |
| Neutrophil count decreased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (19) | 2 (13) | 3 (3) | 2 (2) |
| Weight increased | 0 | 0 | 1 (7) | 0 | 0 | 0 | 1 (10) | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 |
| White blood cell count decreased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (13) | 2 (13) | 2 (2) | 2 (2) |
| Metabolic and nutritional disorders | ||||||||||||||
| Decreased appetite | 1 (9) | 0 | 3 (20) | 0 | 2 (10) | 0 | 1 (10) | 0 | 3 (19) | 0 | 0 | 0 | 10 (11) | 0 |
| Hyponatremia | 0 | 0 | 3 (20) | 2 (13) | 0 | 0 | 0 | 0 | 1 (6) | 1 (6) | 1 (6) | 1 (6) | 5 (6) | 4 (5) |
| Hypophosphatemia | 1 (9) | 1 (9) | 0 | 0 | 0 | 0 | 3 (30) | 0 | 1 (6) | 0 | 0 | 0 | 5 (6) | 1 (1) |
| Hyperglycemia | 0 | 0 | 0 | 0 | 0 | 0 | 1 (10) | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 |
| Musculoskeletal and connective tissue disorders | ||||||||||||||
| Arthralgia | 2 (18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 |
| Pain in extremity | 2 (18) | 1 (9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (2) | 1 (1) |
| Nervous system disorders | ||||||||||||||
| Dysgeusia | 2 (18) | 0 | 1 (7) | 0 | 3 (15) | 0 | 1 (10) | 0 | 1 (6) | 0 | 1 (6) | 0 | 9 (10) | 0 |
| Headache | 2 (18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 | 3 (3) | 0 |
| Respiratory disorders | ||||||||||||||
| Respiratory failure | 0 | 0 | 1 (7) | 1 (7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1) | 1 (1) |
| Skin disorders | ||||||||||||||
| Dry skin | 0 | 0 | 0 | 0 | 0 | 0 | 1 (10) | 0 | 0 | 0 | 1 (6) | 0 | 2 (2) | 0 |
| Vascular disorders | ||||||||||||||
| Flushing | 0 | 0 | 0 | 0 | 0 | 0 | 2 (20) | 0 | 0 | 0 | 0 | 0 | 2 (2) | 0 |
ALT alanine aminotransferase, AST aspartate aminotransferase, BID twice daily, GERD gastroesophageal reflux disease, PTT partial thromboplastin time, QD once daily
The majority of TRAEs were grade 1 or 2 in severity. Of 66 patients with TRAEs, maximum grade 1 and 2 TRAEs occurred in 19 patients (21.6%) and 25 patients (28.4%), respectively. No single grade 3 TRAE occurred in more than five patients (5.7%), and grade 4 TRAEs were reported in three patients (3.4%). TRAEs occurring in ≥ 10% of each group and all grade 3 or 4 TRAEs are displayed in Table 4. Overall, 20 patients (22.7%) had temporary treatment discontinuations associated with TRAEs (Group A1, n = 3; Group A2, n = 6; Group B, n = 4; Group C1, n = 0; Group C2, n = 3; and Group D, n = 4). The most frequently occurring grade 2 or 3 TRAEs leading to temporary discontinuations were neutropenia (n = 4), vomiting (n = 4), fatigue (n = 3), and nausea (n = 3). Four of the grade 3 events resulted in permanent discontinuation of therapy: activated partial thromboplastin time prolongation in Group A1, hyponatremia in Group A2, and fatigue in one patient in Group C2 and one patient in Group D. TRAE-associated dose reductions occurred in 0, 0, 2 (10%), 0, 1 (6%), and 0 patients in Groups A1, A2, B, C1, C2, and D, respectively.
Deaths occurred in 36 (40.9%) of 88 patients overall, with 23 of the deaths (26.1%) occurring within 28 days of the last crizotinib dose and the remaining 13 (14.8%) occurring more than 28 days following the last dose of crizotinib. No deaths were a result of crizotinib toxicity, and 35 of the 36 (97%) deaths were a result of disease progression.
Antitumor activity
Among patients that were not selected based on ALK or ROS1 positivity, the objective response rate was 3.4% (95% CI 0.7–9.6) consisting of three confirmed partial responses in patients with adenocarcinoma of the lung (n = 1), hepatocellular cancer (n = 1), and cholangiocarcinoma (n = 1) in Groups A1, B, and C2, respectively. Response durations in these three patients were 96.0, 17.4, and 17.3 weeks, respectively. Before starting crizotinib treatment, the patient with lung cancer had no prior therapy; the patient with hepatocellular cancer had one prior systemic therapy (sorafenib in the adjuvant setting) and radiofrequency ablation; and the patient with cholangiocarcinoma had systemic therapy with cisplatin and external beam radiation therapy.
Of the 88 patients, 25 (28.4%) had stable disease, four of whom had stable disease for 6 months or longer. Among those four patients with stable disease for ≥ 6 months, two had hepatocellular carcinoma, one had adenoid cystic carcinoma, and one had neuroblastoma.
Discussion
The primary objectives of this study were to evaluate the effect of hepatic impairment on the steady-state PK and safety of crizotinib in patients with advanced cancer. The steady-state CL/F in patients with normal hepatic function (Groups A1 and A2) receiving crizotinib 200 mg BID and 250 mg BID doses, respectively, was similar, indicating linear PK between 200 and 250 mg BID in patients with normal hepatic function. The PK of crizotinib in patients with mild hepatic impairment (Group B) was comparable to that of matched control patients with normal hepatic function (Group A1) with similar CL/F observed at steady state, indicating that mild hepatic impairment has no or minimal effects on the PK of crizotinib. Preliminary PK data for patients treated with crizotinib 250 mg QD during Stage 1 of Group C showed that total systemic exposure was approximately one-half the exposure in normal controls (Group A1). This indicated that the elimination of crizotinib after an oral dose in patients with moderate hepatic impairment who received 250 mg QD dosing was comparable to that of patients with normal hepatic function who received 250 mg BID dosing. Thus, a crizotinib dose of 250 mg BID would be considered to be an appropriate dose for patients with moderate hepatic impairment based on these PK results. However, it had not been established whether a linear PK relationship would be expected for patients with moderate hepatic impairment when the dose is increased from 250 mg QD to 250 mg BID. Therefore, it was decided to use a more conservative approach in selecting the dose for Stage 2 of Group C (Group C2) and for Group D. The final doses for Groups C2 and D were 200 mg BID and 250 mg QD, respectively, based on clinical and PK data.
When the dose increased from 250 mg QD (Group C1) to 200 mg BID (Group C2) for patients with moderate hepatic impairment, the total exposure of crizotinib increased in a more than dose-proportional manner (mean AUCdaily increased from 2305 to 8108 ng h/mL), indicating nonlinear PK of crizotinib in patients with moderate hepatic impairment tested between 250 mg QD and 200 mg BID.
When receiving the same crizotinib dose of 200 mg BID, patients with moderate hepatic impairment (Group C2) showed higher systemic exposure and lower clearance compared to the matched control group (Group A2) with normal hepatic function, suggesting decreased overall elimination of crizotinib in patients with moderate hepatic impairment. In addition, the total systemic exposure of crizotinib in patients with moderate hepatic impairment receiving crizotinib 200 mg BID (Group C2) was comparable to that observed in patients with normal hepatic function receiving a dose of 250 mg BID (Group A1).
The total systemic exposure of crizotinib in patients with severe hepatic impairment receiving crizotinib 250 mg QD (Group D) was lower than that observed in patients with normal hepatic impairment receiving a dose of 250 mg BID (Group A1) but higher than that observed in patients with moderate hepatic impairment receiving 250 mg QD (Group C1). Based on this observation and the above-described experience increasing the dose from 250 mg QD to the next dose level of 200 mg BID in patients with moderate hepatic impairment, crizotinib doses higher than 250 mg QD are not recommended and have not been tested in patients with severe hepatic impairment.
The results from this study showed that the metabolism of crizotinib to PF-06260182 was lower in patients with hepatic impairment than in patients with normal hepatic function, as indicated by the decrease in the metabolite-to-parent ratios in patients with hepatic impairment (mild, moderate, or severe).
The mean value of unbound fraction in Group D for crizotinib (0.03523) was similar to that in patients with normal hepatic function (0.03624 and 0.03066 for Groups A1 and A2, respectively). However, it should be noted that the variability of protein binding in Group D (46%) was higher than in the other hepatic function groups (34% or lower) and the number of PK-evaluable patients in Group D was the smallest (n = 6). Thus, the results of Group D should be interpreted with caution. In addition, it is unlikely that the protein-binding change was associated with the degree of hepatic impairment, as the mean unbound fraction was similar for Groups B and C2.
Crizotinib was generally tolerable, and TRAEs were manageable with dosing interruption, dose reduction, and/or standard medical therapy. Most TRAEs were grade 1 or 2. The frequency of permanent treatment discontinuations associated with TRAEs was low (< 5%), and the safety profile observed in this study was generally consistent with the known safety profile for crizotinib. Moreover, there were no differences in the frequency of AEs based on the degree of hepatic impairment.
The results from this study, based on the observed plasma exposures in patients with varying degrees of hepatic impairment, suggest that crizotinib can be administered to patients with hepatic impairment with appropriate dose adjustment. No dosing adjustments are recommended for patients with mild hepatic impairment. Patients with moderate hepatic impairment are recommended to start crizotinib at 200 mg BID. The crizotinib dose for patients with severe hepatic impairment should not exceed 250 mg QD.
Supplementary Material
Acknowledgements
The authors wish to thank all the participating patients and their families, as well as the investigators, research nurses, study coordinators, and operations staff who contributed to this study.
Funding This study was sponsored by Pfizer Inc. and was supported by Cancer Center Support Grant P30CA054174 from the Institute for Drug Development, Cancer Therapy and Research Center at University of Texas Health Science Center San Antonio, San Antonio, TX. Editorial/medical writing support was provided by Michelle Daniels of inScience Communications, Springer Healthcare (Philadelphia, PA, USA), and was funded by Pfizer Inc.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00280-018-3517-8) contains supplementary material, which is available to authorized users.
Clinical trial registration no NCT01576406.
Conflict of interest ABE-K has served as a consultant, advisor, or speaker for AstraZeneca, Bayer, Bristol-Myers Squibb, Celgene, CytomX, Merrimack Pharmaceutical, and Roche-Genentech. CLO has served as a consultant, advisor, or speaker for Amgen, Bristol-Myers Squibb, and Heron Therapeutics and has received research funding from AstraZeneca and Bayer. KKC has received research funding from Bayer, Boston Biomedical, Incyte, MedImmune, Merck, Novartis, Pfizer, and Sanofi. HX is an employee of and owns stock in Pfizer and holds a patent or receives royalties from GTx Inc. MO is an employee of Pfizer. JC and TU are employees of and own stock in Pfizer. BFE-R has served as a consultant or advisor for BTG International, Lexicon, and Merrimack Pharmaceutical, and has received research funding from AVEO Pharmaceuticals, Boston Biomedical, Bristol-Myers Squibb, Cleave Biosciences, Inc., Genentech, Hoosier Cancer Research Network, Inc., Merck, Novartis, and TAIHO Oncology.
Compliance with ethical standards
Ethical approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent Informed consent was obtained from all individual participants included in the study.
References
- 1.Yasuda H, de Figueiredo-Pontes LL, Kobayashi S, Costa DB (2012) Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol 7:1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, Nambu MD, Los G, Bender SL, Mroczkowski B, Christensen JG (2007) An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res 67:4408–4417 [DOI] [PubMed] [Google Scholar]
- 3.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, Zheng Z, Tan W, Stephenson P, Shreeve SM, Tye LM, Christensen JG, Wilner KD, Clark JW, Iafrate AJ (2014) Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 371:1963–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Iyer S, Reisman A, Wilner KD, Tursi J, Blackhall F (2014) First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371:2167–2177 [DOI] [PubMed] [Google Scholar]
- 5.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, Moro-Sibilot D, Camidge DR, Mok T, Hirsh V, Riely GJ, Iyer S, Tassell V, Polli A, Wilner KD, Janne PA (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 368:2385–2394 [DOI] [PubMed] [Google Scholar]
- 6.Johnson TR, Tan W, Goulet L, Smith EB, Yamazaki S, Walker GS, O’Gorman MT, Bedarida G, Zou HY, Christensen JG, Nguyen LN, Shen Z, Dalvie D, Bello A, Smith BJ (2015) Metabolism, excretion and pharmacokinetics of [14C]crizotinib following oral administration to healthy subjects. Xenobiotica 45:45–59 [DOI] [PubMed] [Google Scholar]
- 7.Xu H, O’Gorman M, Boutros T, Brega N, Kantaridis C, Tan W, Bello A (2015) Evaluation of crizotinib absolute bioavailability, the bioequivalence of three oral formulations, and the effect of food on crizotinib pharmacokinetics in healthy subjects. J Clin Pharmacol 55:104–113 [DOI] [PubMed] [Google Scholar]
- 8.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, Salgia R, Fidias P, Engelman JA, Gandhi L, Janne PA, Costa DB, Shapiro GI, Lorusso P, Ruffner K, Stephenson P, Tang Y, Wilner K, Clark JW, Shaw AT (2012) Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol 13:1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnell P, Safferman AZ, Bartlett CH, Tang Y, Wilner K (2012) Clinical presentation of hepatotoxicity-associated crizotinib in ALK-positive (ALK+) advanced non-small cell lung cancer (NSCLC) [abstract]. J Clin Oncol 30:7598 [Google Scholar]
- 10.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363:1693–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Geel RM, Hendrikx JJ, Vahl JE, van Leerdam ME, van den Broek D, Huitema AD, Beijnen JH, Schellens JH, Burgers SA (2016) Crizotinib-induced fatal fulminant liver failure. Lung Cancer 93:17–19 [DOI] [PubMed] [Google Scholar]
- 12.Sato Y, Fujimoto D, Shibata Y, Seo R, Suginoshita Y, Imai Y, Tomii K (2014) Fulminant hepatitis following crizotinib administration for ALK-positive non-small-cell lung carcinoma. Jpn J Clin Oncol 44:872–875 [DOI] [PubMed] [Google Scholar]
- 13.Cancer Therapy Evaluation Program (CTEP) Organ Dysfunction Working Group Protocol Template (2015) Specific instructions for the use of protocol templates for organ dysfunction studies. https://ctep.cancer.gov/protocolDevelopment/docs/CTEP_Organ_Dysfunction_Protocol_Template.docx. Accessed 7 Sept 2017
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.