

Abstract
Post-translational modifications (PTMs) play critical roles in biological processes and have significant effects on the structures and dynamics of proteins. Top-down proteomics methods were developed for and applied to the study of intact proteins and their PTMs in human samples. However, the large dynamic range and complexity of human samples makes the study of human proteins challenging. To address these challenges, we developed a 2D pH RP/RPLC-MS/MS technique that fuses high-resolution separation and intact protein characterization to study the human proteins in HeLa cell lysate. Our results provide a deep coverage of soluble proteins in human cancer cells. Compared to 225 proteoforms from 124 proteins identified when 1D separation was used, 2778 proteoforms from 628 proteins were detected and characterized using our 2D separation method. Many proteoforms with critically functional PTMs including phosphorylation were characterized. Additionally, we present the first detection of intact human GcvH proteoforms with rare modifications such as octanoylation and lipoylation. Overall, the increase in the number of proteoforms identified using 2DLC separation is largely due to the reduction in sample complexity through improved separation resolution, which enables the detection of low abundance PTM modified proteoforms. We demonstrate here that 2D pH RP/RPLC is an effective technique to analyze complex protein samples using top-down proteomics.
Keywords: top-down proteomics, Mass Spectrometry, liquid chromatography, RPLC, intact proteoforms
Graphical Abstract
INTRODUCTION
The ability to identify functional proteins with post-translational modifications (PTMs) is a crucial element in understanding cellular function. For example, acetylation of acidic residues can impact protein function and acetylation of protein N-terminal amines can protect proteins from degradation [1]. Futhermore, the acetylation of primary amines on histones often regulates protein-DNA interactions [2–5]. Protein PTMs have emerged in the post-genomic era as critical features in regulating and diversifying the biological activity of proteins. However, identifying specific proteoforms with PTMs and understanding their function on individual proteins is currently limited by the lack of effective and accessible analytical methods. Therefore, there is a need to develop a high-throughput approach to characterize intact proteins and their modified proteoforms at the systems level.
Proteomics is a high-throughput bio-analytical approach to study proteins in a complex sample using mass spectrometry (MS) based methods. Currently, there are two fundamental approaches to MS-based protein identification and characterization: bottom-up and top-down proteomics. Recent developments in bottom-up proteomics significantly expanded the depth of proteomic analysis, both qualitatively and quantitatively [6, 7]. Two independent bottom-up based human proteomics studies confirmed ~20,000 protein-coding genes from human samples, which provided direct evidence for the actual translation of over 90% of putative protein-coding genes [8, 9]. The throughput of top-down proteomics has also improved significantly during the last decade due to the advancements in chromatographic separation and high-resolution MS instrumentation [10–17].
Reversed-phase liquid chromatography (RPLC) is the most prevalent separation technique in top-down proteomics. Long capillary column (i.e., 1 m length, 75 μm i.d.) ultra-high-pressure liquid chromatography has been coupled to high-resolution top-down mass spectrometry (UPLC-HRMS) for the analysis of intact proteins in complex mixtures [18, 19]. However, in cases of high complexity and large dynamic range, such as in human cell lysate, 1D-RPLC may not provide sufficient proteome coverage due to a limited sample loading capacity. To overcome this challenge, different LC separation formats are often combined to increase the separation power based on the orthogonality. LC techniques often used in proteomics include reversed-phase chromatography (RPLC), ion exchange chromatography (IEX), hydrophilic interaction chromatography (HILIC), hydrophobic interaction chromatography (HIC), size exclusion chromatography (SEC), etc. [20–25]. Several electrophoresis-based separation methods such as gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) [26] and solution isoelectric focusing (sIEF)[27] can be applied as pre-fractionation methods for RPLC; however, these methods cannot be directly coupled to MS for detection. Capillary electrophoresis (CE) is a very promising electrophoresis-based separation technique that can be coupled after RPLC separation due to the small sample loading amount. CE can be directly coupled with MS either by the sheathless interface [28] or by a sheath-flow based interface [29]. McCool et al. reported a three-dimensional platform, SEC-RPLC-CZE-MS/MS, that was applied to study intact proteins in E. coli lysate. A total of 5705 proteoforms from 850 proteins were identified using this method [14].
Recently, our lab developed a two-dimensional separation platform using high-pH and low-pH RPLC for top-down proteomics [23]. This method was applied to analyze intact proteins and proteoforms in E. coli cell lysate, and our results demonstrated that the platform provided high-resolution protein separation due to the comparatively good separation resolution and orthogonality between the two RPLC formats. Another advantage of this approach is the simple sample handling procedure which utilizes MS-compatible buffers. In this study, we further applied 2D pH RP/RPLC coupled with top-down MS to characterize intact proteins and proteoforms in human cell lysate (e.g., HeLa cell lysate). A fraction-to-fraction orthogonal selectivity analysis between high-pH and low-pH separations was performed. In addition, we compared different cell lysis sample preparation methods (i.e, with and without the removal of small proteins with a 10 kDa MW cutoff filter). In total, 2791 proteoforms from 626 proteins were identified and manually confirmed. A total of 20 different types of PTMs were characterized, including intact proteoforms with phosphorylation, lipoylation, and glutathionylation. Overall, our results suggested that the two-dimensional high-pH and low-pH RPLC-MS can be easily adapted to complex protein samples for deep proteoform characterization.
MATERIALS AND METHODS
Chemicals and reagents.
All chemicals including LC-MS grade water and organic solvents were purchased from Sigma-Aldrich (Milwaukee, WI) unless noted otherwise. Jupiter particles used as column packing material were purchased from Phenomenex (Torrance, CA).
HeLa cell culture and cell lysate preparation.
The HeLa cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) with 10% Fetal Bovine Serum (FBS) and 2% Penicillin-Streptomycin. The growth status of the cells was monitored using a microscope. When the cells were 80–90% confluent, they were washed with icecold PBS buffer and scratched off the petri dish. The cells were centrifuged at 2,500 × g, 4 °C for 30 min. Ten plates (10 cm diameter) of HeLa cells were resuspended to 10 mL in lysis buffer (1μM PMSF and 20mM NaF in PBS, pH=7.4). The cells were broken using a sonication homogenizer on ice for 30 minutes and insoluble matter was removed by centrifugation at 15,000 × g, 4 °C for 30 minutes. The lysate (unfiltered sample) was aliquoted and stored at −80 °C. For a portion of the lysate sample, small proteins or degradation products were removed using a 10 kDa MWCO spin filter at 4 °C (two times with 25 mM ammonium bicarbonate), referred to as “filtered sample”.
High-pH RPLC fractionation.
The intact proteins from HeLa cell lysate were separated and analyzed using the 2D pH RP/RPLC-MS/MS platform as previously described [23]. Briefly, 1 mg of proteins were fractionated by an offline high-pH RPLC separation (pH = 10) using an XBridge Protein BEH C4 column (300 Å, 3.5μm, 2.1 mm × 250 mm) from Waters, Inc. (Milford, MA). The mobile phase A (MPA) was 20 mM ammonium formate in water (pH = 10) and mobile phase B (MPB) was 20 mM ammonium formate in acetonitrile (pH = 10). The unfiltered sample was separated using a gradient from 3% to 90% MPB over 60 minutes. The eluted proteins were fractionated into 24 2.5-min fractions starting from 16 minutes after sample injection (i.e., the first detected UV peak). Based on the results from the unfiltered sample, we fractionated the filtered sample into 9 fractions (Supplementary Figure 1A). All the fractions were vacuum dried and resuspended to a final volume of 50 μL using 25 mM ammonium bicarbonate before injecting onto the second-dimension column.
Low-pH RPLC-MS analysis.
10 μg of protein (filtered, unfiltered, or fractions from high-pH RPLC separation) from the high-pH fractions was loaded onto a trapping column (150 μm i.d., 10 cm length, Jupiter particles, 5 μm diameter, 300 Å pore size) and separated using a home-packed C5 RPLC capillary column (75 μm i.d., 70 cm length, Jupiter particles, 5 μm diameter, 300 Å pore size) on a modified Thermo Scientific (Waltham, MA, USA.) Accela LC system [19]. Specifically, MPA was 0.01% TFA, 0.585% acetic acid, 2.5% isopropanol, and 5% acetonitrile in water. MPB was 0.01% TFA, 0.585% acetic acid, 45% isopropanol, and 45% acetonitrile in water [18, 30, 31]. A 200-min gradient from 10% to 65% of MPB was applied with a flow rate of 400 nL/min. The LC elution was sprayed from an etched capillary tip through a customized nano-ESI interface to an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific, Hanover Park, IL, Bremen, Germany, USA) [32]. The temperature of the inlet capillary was set to 275 °C and the spray voltage was 2.6 kV. Full MS scans used the resolving power setting of 100,000 (at m/z 400) with two micro scans. Collision-induced dissociation (CID) with a normalized collision energy of 35 eV was applied. The top 6 most abundant precursor ions in the full MS scans were selected for MS/MS fragmentation. The MS/MS data were obtained at a resolving power setting of 60,000 (at m/z 400) with two micro scans and an isolation window of 3.0 m/z. The AGC target was set as 5×105 for full mass scans and 3×105 for MS/MS scans. All the data was collected using Xcalibur 3.0 software (Thermo Fisher Scientific, Bremen, Germany).
Data analysis.
Raw data were deconvoluted and searched against the Swiss-Prot reviewed homo sapiens protein database (published on May 2018, 20355 proteins) using TopPIC Suite [33]. All parameters were set as follows: the error tolerance was 15 ppm; the maximum value for the unexpected mass shift was 500 Da; the maximum number of the unexpected mass shifts was 1. The decoy database was utilized to calculate PSM level FDRs and proteoform level FDRs in TopPIC. The E-value cutoff value was 8.81E-8 for PSM-level FDRs <1% and 5.31E-9 for proteoform-level FDRs < 1%. For each dataset, we only considered proteoforms with E-values less than the proteoform-level FDR cutoff values. Proteoform identifications in different fractions were aggregated based on their retention time (~3 min normalized retention time window) and detected masses (15 ppm). No protein level FDR was calculated. All proteins with proteoforms that passed the E-value cutoff were reported. All identified proteoforms were also manually evaluated. MASH Suite [34] and ProSight Lite [35] were used for manual interpretation and spectrum presentation.
RESULTS AND DISCUSSION
2D pH RP/RPLC top-down MS on HeLa cell lysate.
In this study, we applied the previously developed 2D pH RP/RPLC separation and top-down MS to characterize intact proteins and proteoforms in human HeLa cell lysate. A total of 24 first-dimension fractions were analyzed with long-column high-pressure RPLC with top-down MS [23] and by SDS-PAGE (Figure 1A and 1B). In total, 2778 unique proteoforms from 628 proteins were identified and manually confirmed (Supplementary Table 1). 77 % of the proteoforms were detected in 1 fraction, 14 % of the proteoforms were detected in 2 fractions, and 9 % of the proteoforms were detected in 3 or more fractions. Our results suggest that high-pH RPLC provides good separation power as the first dimensional separation.
Figure 1.

Protein and proteoform identification count of HeLa cell lysate. (A) First-dimension fractions performed by SDS-PAGE. (B) Protein and proteoform identification count in each fraction under high-pH conditions. (C) Venn diagram of proteins identified using the 1D and 2D platforms. (D) Venn diagram of proteoforms identified using the 1D and 2D platforms.
The same sample was also analyzed using 1D low-pH RPLC top-down MS without pre-fractionation; 124 intact proteins and 225 intact proteoforms were identified. (Supplementary Table 2). We compared the results of 1D and 2D separation of the same sample (Figure 1C and 1D). We observed an increase in the number of proteoform identifications when 2D separation was used and there are two possible reasons for this increase: (1) there is a higher chance that MS/MS analysis will be performed on low abundance proteoforms in 2D analysis due to extended MS analysis time; (2) improved separation performance in 2D analysis reduces sample complexity and allows for the detection of proteoforms that cannot be detected in 1D analysis because many co-eluted species split the ion current. To evaluate the effects of improved separation, we performed two 1D RPLC experiments using E. coli cell lysate: one with a 70-minute gradient (lower peak capacity) and one with a 200-minute gradient (higher peak capacity). All the detected mass features were deconvoluted with a 1-minute moving average window (i.e, all spectra in a 1-minute window were summed and peak deconvolution was performed). We compared the detected mass features in different MW ranges (Supplementary Figure 1). In total, we detected 137 mass features > 5 kDa from the 70-minute run, and 367 mass features > 5 kDa from the 200-minute run. Our results suggested better separation facilitated the detection of more species and provided higher chances to identify them using MS/MS analysis. Previously, we demonstrated that 2D pH RP/RPLC separation of E. coli cell lysate has high peak capacity due to good separation power on both high-pH RPLC and low-pH RPLC. Here, we applied 2D pH RP/RPLC to human cell lysate resulting in significantly improved detection of intact mass features (2637 mass features > 5 kDa) compared to the 1D 200-min RPLC run.
We further evaluated two cell lysate preparation approaches for 2D pH RP/RPLC top-down MS analysis (i.e., with and without the removal of small proteins using a 10 kDa MW cutoff filter). Based on the 24-fraction identification results from the unfiltered sample, the filtered samples were fractionated into 9 fractions (Supplementary Figure 2A) to reduce the MS operation time. A total of 1710 unique mass features > 5 kDa were detected in 9 fractions vs. 2637 unique mass features > 5 kDa in 24 fractions. Using the same MS conditions, fewer mass features were detected when 9 fractions were collected, but much less MS analysis time was required. We also compared the masses of detected proteoforms in different MW ranges (Supplementary Figure 2B). The MW distribution is similar between these two fractionation schemes and both approaches have similar percentages of detected mass features less than 10 kDa. One possible reason for the discrepancy in percentage of proteoforms is that some proteins may degrade during the concentration process (i.e., speedVac) before MS analysis. If protein degradation during concentration is an issue, filter-based concentrators or an online 2D system can be adapted in the future to reduce sample degradation.
In general, current commercially available MS instruments are limited in their ability to detect large proteins (MW > 30 kDa) with low abundance due to the low S/N ratios inherent to mass measurement. In our study, only a very small portion of proteins > 20 kDa were detected even though many large proteins were observed in the same fractions by SDS-PAGE. Gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) has been coupled with RPLC for deep intact human proteoform characterization[27, 37], and different fragmentation approaches such as UPVD[39] and Ai-ETD[13] have been coupled with GELFrEE for better sequence coverage of PTM localization in human samples. In addition, many proteoforms > 30 kDa were successfully detected in GELFrEE fractions that contained enriched large proteoforms using a 21T FTICR-MS [15]. By removing co-eluting small proteoforms, GELFrEE allows for the detection of large proteoforms with low S/N ratios in complex samples. However, GELFrEE often requires MS-incompatible detergents such as SDS as well as specific instrumentation. Therefore, GELFrEE is challenging to use online with MS analysis. SEC is another size-based separation that may enhance MS detection of large proteoforms and has been coupled online with MS analysis [36, 40]. However, SEC in general does not provide good separation resolution in the full MW range because the separation selectivity of SEC depends on the resin pore size. Our results suggested that both high-pH RPLC and low-pH RPLC provide good separation power, which allows for deep proteoform detection in complex samples. With an additional dilution step, our approach can be easily adapted into an online platform to improve the robustness and sensitivity of the 2D pH RP/RPLC. Additionally, larger proteins can be enriched prior to 2D pH RP/RPLC separation using size-based approaches such as GELFrEE or SEC for better detection of larger proteins.
Orthogonality between high-pH RPLC and low-pH RPLC for intact protein separation.
In bottom-up proteomics, it has been reported that high-pH RPLC only has semi-orthogonality with low-pH RPLC for peptide analysis [41, 42]. For example, more hydrophobic peptides tend to elute late regardless of high or low pH. Therefore, the separation window of the secondary dimension cannot be fully utilized, which limits the usage of online 2D pH RP/RPLC analysis in bottom-up proteomics. Our previous study in top-down proteomics suggested that high-pH RPLC had good orthogonality against low-pH RPLC [23]. To further assess if these two separation forms have semi-orthogonality or full orthogonality for intact proteoform analysis, we evaluated proteoform elution patterns in high-pH fractions (Figure 2). For each fraction (each column on Figure 2), a heatmap was generated using the relative number of uniquely identified proteoforms in each bin (10-minute windows). It has been demonstrated that in an ideally orthogonal system, area coverage > 63% represents full orthogonality [41]. Our results suggested that proteoforms were distributed across the entire efficient elution window of the secondary dimension in most of the fractions (81% bins containing 5 or more mass features > 2.5 kDa). The good orthogonality between RPLC performed under different pH conditions can be explained by the change in charge distribution caused by the change in pH of the mobile phase [41, 43, 44]. Compared to peptide chains, intact proteins are more complicated with more dynamic and versatile changes in charge distribution under different pH conditions. These changes in charge distribution can significantly affect protein retention and elution orders [23, 45].
Figure 2.

The proteoform elution patterns in high-pH fractions. Each bin in the heatmap represents the number of detected mass features > 2.5 kDa in each 10-min elution window in the low-pH RPLC separation.
To further evaluate the effects of pI values on intact protein retention behavior in high-pH RPLC separation, we filtered all identified intact proteoforms from the 9-fraction sample. The pI values were calculated for each filtered proteoform and the average pI values and their standard deviations were plotted in each fraction (Supplementary Figure 2C). The average pI values were between 4 and 7 for most fractions, which suggests that the pI value alone does not alter the retention behavior. Still, the average pI value (4.48) in fraction 1 was significantly lower than other fractions indicating that pI may have some effect on separation. At pH 10, most proteins with pI values around 4 carry multiple negative charges, which reduces their retention times. In general, we didn’t detect many proteins with pI values larger than 8. One possible reason is that proteins with high pI values do not carry many charges and may not be easily eluted from the high-pH RPLC column.
Identification and characterization of intact proteins.
Overall, a total of 20 different types of PTMs were characterized in the proteoforms detected using the 2D analysis (Supplementary Table 3). Only 6 types of PTMs were detected using 1D analysis. Several commonly observed PTMs were detected including phosphorylation, acetylation, and glutathionylation. We were able to identify 96 intact proteoforms with phosphorylation and 771 intact proteoforms with acetylation. Several less common functional PTMs were also detected using the 2D platform including lipoylation and octanoylation. To increase the number of proteoforms detected using 1D RPLC separation, a combined top-down and intact mass analysis, such as that developed by Smith’s group, may be utilized [38]. The Smith group proposed a strategy to combine both top-down MS and intact mass determination, which addresses the issue that some proteoforms may be detected, but not identified, using MS/MS. The mass shift between experimentally detected species in the same run was calculated and experimental delta mass histograms were plotted (Supplementary Figure 3). Mass shifts (16 Da and 80 Da) were often detected in pairs in both 9-fraction analysis and 24-fraction analysis, which may correspond to oxidation and phosphorylation. Interestingly, although we identified 761 intact proteoforms with acetylation, we did not observe many proteoform pairs with 42 Da shifts. This may due to the fact that many acetylated species we detected have static N-term acetylation.
We also conducted a comparison of the proteoforms detected using both the 1D and 2D platforms (Figure 3). We observed significant improvements of the S/N ratios with higher signal and lower background noise when 2DLC separation methods were used, which is consistent with previous literature [46]. For example, calmodulin-1 (Swiss-Prot, P0DP23) was detected in both 1D and 2D results, but there was a much higher S/N ratio in 2D (S/N=647) than in 1D (S/N=37) (Figure 3). Additionally, a proteoform of calmodulin-1 which lost 3 amino acids (i.e., TyrAlaLys) at the C-terminus was detected using 2D (S/N=23) but not 1D separation. The C-terminus deletion mutants of calmodulin-1 were reported to reduce the Ca2+ binding ability because it had minor conformers [47]. This suggest that the C-terminus of calmodulin-1 is active in inter-domain communication while Ca2+ induces structural transition so the truncated proteoform detected is functionally relevant. To summarize, our results suggest that the 2D pH RP/RPLC separation and top-down MS can be applied to complex protein samples for deep proteoform characterization, especially to detect and characterize low abundance proteoforms and PTMs. Additionally, the application of 2D LC separation improves S/N ratio, which increases the limit of detection of the analytes [46].
Figure 3.

MS detection of calmodulin-1 proteoforms. (A) Full MS spectrum and extracted ion chromatograms (EICs) of proteoforms after 2D separation. (B) Full MS spectrum and extracted ion chromatograms (EICs) of proteoforms after 1D separation.
Characterization of intact proteoforms with phosphorylation.
The 2D LC technique applied in our study increases the detection of phosphorylated proteoforms. Many low abundance PTM-modified proteoforms were detected using the 2D platform with good S/N ratios, but were not observed in the 1D analysis. Two proteoforms of parathymosin (Swiss-Prot, P20962), with and without phosphorylation, were characterized in fraction 5 during the 2D analysis (Figure 4A). The MS/MS spectra elucidated mass differences between b51 and b53 ions indicating the possible locations of the phosphorylation (Figure 4B). We observed a decrease in the elution time of the phosphorylated proteoforms in both 1D and 2D results because the phosphorylation added an additional charge to the protein that may have decreased the hydrophobicity. Decreased hydrophobicity of the phosphorylated proteins weakened the binding of these proteins to the stationary phase in RPLC, so they often eluted earlier compared to non-phosphorylated proteins [48]. Additionally, some Fe[III] adducts were detected from phosphorylated parathymosin proteins (* in Figure 4A). The Fe[III] adducts may come from the stainless-steel parts of the high-pressure system, such as the frit and the union. To avoid the production of Fe[III] adducts and increase the sensitivity for phosphoprotein detection, a “metal-free” RPLC-ESI-MS platform could be adapted to our 2D pH RP/RPLC platform [18].
Figure 4.

MS detection and MS/MS identification of parathymosin proteoforms. (A) Full MS spectrum of proteoforms. The asterisk (*) shows the Fe[III] adduct. The EIC of each proteoform is shown. (B) MS/MS identification of proteoforms. The orange serine residue on the N-terminus is acetylated. The tyrosine residue highlighted in blue is phosphorylated.
Ribosomal proteins are involved in protein synthesis by providing control mechanisms for transcription and translation [49]. The 60S acidic ribosomal protein P2 (Swiss-prot, P05387, RPLP2) is coded by the gene RPLP2 and participates in the elongation step in protein synthesis and GTPase activation [50, 51]. Phosphorylation has been proven to stimulate interaction with eukaryotic translation elongation factor 2 (eEF-2) [51]. Phosphorylation of serine residues located near the C-terminus increases the affinity of RPLP2 for eEF-2. The phosphorylation of these residues also cause changes in eEF-2 activity, which suggests the C-terminus is involved in this functionality. The phosphorylation on the C-terminus of RPLP2 is also related to autoimmune disease. Autoantibodies against the C-terminus peptide of 60S acidic ribosomal proteins exist in 15% of systemic lupus erythematosus patients [52].
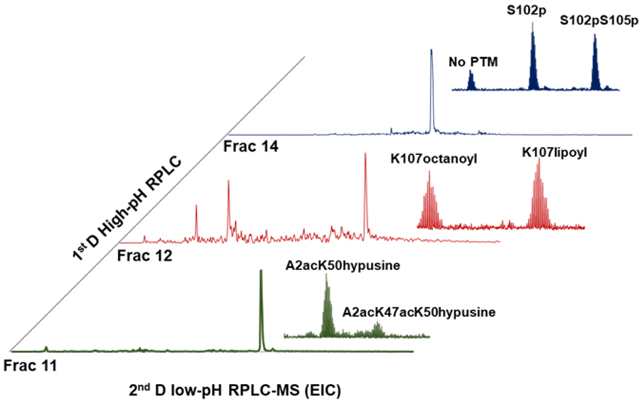
We observed a total of 10 proteoforms of RPLP2 using 2D methods with modifications including phosphorylations, oxidations, and a sequence variation, Tyr->Gln (Supplementary Figure S4). Figure 5A shows the MS spectrum of 60S acidic ribosomal protein P2 proteoforms in fraction 14 from the 2D separation. The 60S acidic ribosomal protein P2 variations include the mature proteoform RPLP2_P0, and the proteoforms RPLP2_P1 and RPLP2_P2, which have 1 and 2 phosphorylations, respectively. Figure 5B illustrates the identification and PTM characterization of RPLP2_P0, P1, and P2. The MS/MS spectra elucidated the mass differences between b96 and b107 ions of the 3 proteoforms indicating the possible locations of the phosphorylations. However, there was no fragmentation between Ser 102 and Ser 105, so the location of the phosphorylation of RPLP2_P1 could not be determined. The NET of proteoforms shifted from 119 min to 126 min and the extracted ion chromatogram (EIC) of RPLP2_P1 was split into two peaks (i.e., 120.42 and 120.87). This splitting might suggest the existence of two proteoforms with one phosphorylation on Ser102 or Ser105. To determine the location of the phosphorylation, the fragmentation method should be optimized to enhance the sequence coverage using electron transfer dissociation (ETD) and/or UVPD in the future [53]. We also found that phosphorylated proteoforms were eluted later than non-phosphorylated proteoforms, which disagrees with previous observations [48]. This may be due to the fact that both phosphorylated sites were located on the highly flexible and acidic C-terminus of the protein.
Figure 5.

MS detection and MS/MS identification of RPLP2 proteoforms. (A) Full MS spectrum of proteoforms: RPLP_P0, P1, P2. The asterisk (*) indicates the Fe[III] adduct. The EIC of each proteoform is shown. (B) MS/MS identification of RPLP_P0, P1, P2. The serine residues highlighted in blue are phosphorylated.
Identification of intact proteoforms with functional modifications.
The 2DLC technique studied here allowed for the identification of functional PTMs that have not been commonly studied. For example, octanoylation and lipoylation were characterized on the glycine cleavage system H protein (Swiss-Prot, P23434, GcvH). The GcvH protein was reported as a subunit of the glycine cleavage system (GCV). GCV is active in glycine and serine catabolism [54]. The GcvH protein shuttles the methylamine group of glycine from the P protein to the T protein [55]. We confidently identified two GcvH proteoforms with octanoylation and lipoylation (Figure 6), which have never been reported in previous top-down studies. The transit peptides of both proteoforms were cleaved suggesting that these proteoforms were mature and transferred to the mitochondrion. The difference between the y66 and y67 ions, observed via MS/MS fragmentation, suggests two different PTMs (octanoylation and lipoylation) are located at the same amino acid, Lys50, of the proteoforms. Octanoylation and lipoylation are rare modifications characterized by the addition of a heptane chain and a heptane chain ending in a 5-membered ring containing two sulfur atoms, respectively. Lipoylation plays a critical role in the function of GcvH. The oxidized form of the lipoylation serves as the acceptor of the intermediary methylamine group derived from glycine as the two sulfur atoms bind to the glycine residue [55, 56]. Lipoylation was also reported to bind with the substrates of other enzymes (e.g., pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase) [57]. Octanoylation is an intermediate of lipoylation. Jordan et al. published a mechanism proposing that octanoyl-ACP is converted to lipoyl-ACP in E. coli. In this mechanism, LipA lipoate synthase inserts two sulfur atoms at C6 and C8 on the octanyl chain [58, 59]. Our results demonstrated the first detection of octanoylation on GcvH on human proteins, which suggests that the lipoyl-GcvH was synthesized through a similar pathway in human biological systems.
Figure 6.

MS detection and MS/MS identification of GcvH proteoforms. (A) Full MS spectrum of GcvH proteoforms with octanoylation and lipoylation. (B) MS/MS identification of the GcvH with octanoylation and lipoylation. The lysine residue highlighted in orange is lipoylated. The yellow cysteine residues do not participate in disulfide bonds.
Another example of a rare PTM identified by our method is hypusine on the eukaryotic translation initiation factor 5A-1 (Swiss-Prot, P63241, eIF5A-1). eIF5A-1 is an mRNA-binding protein and is involved in translation elongation. It plays such an essential role in eukaryotic cell growth and animal development that its inactivation causes death in yeast [60, 61]. We characterized two proteoforms of eIF5A-1 containing a hypusine residue and acetylation at Lys47 (Figure 7). The hypusine was found on both proteoforms using a y110 and y98 ion with +87.08 Da. Hypusine, a unique PTM only reported on eIF5A, is derived from polyamine spermidine [60]. Interestingly, the hypusine residue is highly conserved from yeast to mammals [62]. Hypusine has a longer side chain than lysine and promotes the activity of eIF5A by enhancing translation termination through stimulating peptide release from peptide bond formation at polyproline stretches and ribosome pausing sites [60]. It was reported that the basic charge from hypusine at Lys50 is important for acetylation at Lys47 [61]. The acetylation on Lys 47 was characterized by the mass difference of a y110 and y98 ion (+42.01 Da) from two proteoforms and has been previously reported in the literature [62]. The acetylation at Lys47 also regulates the activity of eIF5A. Previous research suggests a basic charge at Lys47 is critical for eIF5A activity [62]. Overall, the deep proteoform characterization made possible by 2DLC separation allows for the detection and quantification of functional PTMs and proteoforms, which are not commonly detected using other methods.
Figure 7.

MS detection and MS/MS identification of eIF5A-1 proteoforms. (A) Full MS spectrum of eIF5A-1 proteoforms with hypusine and 1 or 2 acetylations. (B) MS/MS identification of the eIF5A-1 proteoform with 1 acetylation on N-terminus. The cysteine residues highlighted in grey participate in a disulfide bond. The methionine residue highlighted in grey is removed. The yellow cysteine residues do not participate in disulfide bonds. The orange alanine residue on the N-terminus is acetylated. The Lys50 highlighted in blue is modified with a hypusine.
CONCLUSION
In our study, the 2D pH RP/RPLC-MS/MS platform was applied to study the human proteins and PTMs in HeLa cell lysate. The protein, proteoform, and PTM identifications were enhanced using the 2D platform compared to 1D. The 2D separation implemented here made possible the characterization of proteoforms with complicated PTM distribution, such as multiple phosphorylations. In addition, we presented the first detection of intact human GcvH proteoforms with rare modifications such as octanoylation and lipoylation. The 2D platform is “salt-free” with good orthogonality, making the platform easily optimized to online 2D methods. Size-based separation approaches such as GELFrEE or SEC can be used before the 2D pH RP/RPLC for large protein fractionation and characterization. Different buffer conditions and column materials can be evaluated to improve the elution of hydrophobic proteins with high pI values. To characterize specific proteins, the 2D platform may be coupled with other sample preparation methods, such as immobilized metal affinity chromatography for phosphoprotein study [63]. In summary, our application provides a comprehensive aerial view of proteins in human cancer cells. This technique may also be used with isobaric-labeling based quantification (TMT or iTRAQ) and applied to other human proteomics studies.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Anthony Burgett for providing the Hela cells. This work was partly supported by grants from NIH NIAID R01AI141625, NIAID CSGADP Pilot project (NIH 5U01AI101990-04, BRI no. FY15109843), NIH NIGMS R01GM118470, OCAST HR16-125, and OU FIP program.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCE:
- 1.Hershko A, Heller H, Eytan E, Kaklij G, Rose IA: Role of the alpha-amino group of protein in ubiquitin-mediated protein breakdown. Proceedings of the National Academy of Sciences. 81, 7021–7025 (1984) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Recht J, Tsubota T, Tanny J, Diaz R, Berger JM, Zhang X, Garcia B, Shabanowitz J, Burlingame A, Hunt D: Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proceedings of the National Academy of Sciences. 103, 6988–6993 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grunstein M: Histone acetylation in chromatin structure and transcription. Nature. 389, 349 (1997) [DOI] [PubMed] [Google Scholar]
- 4.Miao J, Lawrence M, Jeffers V, Zhao F, Parker D, Ge Y, Sullivan WJ Jr, Cui L: Extensive lysine acetylation occurs in evolutionarily conserved metabolic pathways and parasite-specific functions during P lasmodium falciparum intraerythrocytic development. Molecular microbiology. 89, 660–675 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ: Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proceedings of the National Academy of Sciences. 105, 4093–4098 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen X, Sun L: Systematic Evaluation of Immobilized Trypsin-Based Fast Protein Digestion for Deep and High-Throughput Bottom-Up Proteomics. Proteomics. 18, 1700432 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gautier V, Boumeester AJ, Lössl P, Heck AJ: Lysine conjugation properties in human IgGs studied by integrating high-resolution native mass spectrometry and bottom-up proteomics. Proteomics. 15, 2756–2765 (2015) [DOI] [PubMed] [Google Scholar]
- 8.Pruitt KD, Tatusova T, Maglott DR: NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic acids research. 35, D61–D65 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hubbard TJ, Aken BL, Beal K, Ballester B, Cáccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T: Ensembl 2007. Nucleic acids research. 35, D610–D617 (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melani RD, Skinner OS, Fornelli L, Domont GB, Compton PD, Kelleher NL: Mapping proteoforms and protein complexes from king cobra venom using both denaturing and native top-down proteomics. Molecular & Cellular Proteomics. mcp. M115. 056523 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhoads TW, Rose CM, Bailey DJ, Riley NM, Molden RC, Nestler AJ, Merrill AE, Smith LM, Hebert AS, Westphall MS: Neutron-encoded mass signatures for quantitative top-down proteomics. Analytical chemistry. 86, 2314–2319 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng Y, Gregorich ZR, Valeja SG, Zhang H, Cai W, Chen Y-C, Guner H, Chen AJ, Schwahn DJ, Hacker TA: Top-down proteomics reveals concerted reductions in myofilament and Z-disc protein phosphorylation after acute myocardial infarction. Molecular & Cellular Proteomics. mcp. M114. 040675 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riley NM, Sikora JW, Seckler HS, Greer JB, Fellers RT, LeDuc RD, Westphall MS, Thomas PM, Kelleher NL, Coon JJ: The Value of Activated Ion Electron Transfer Dissociation for High-Throughput Top-Down Characterization of Intact Proteins. Analytical Chemistry. (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCool EN, Lubeckyj RA, Shen X, Chen D, Kou Q, Liu X, Sun L: Deep Top-Down Proteomics Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry: Identification of 5700 Proteoforms from the Escherichia coli Proteome. Analytical chemistry. 90, 5529–5533 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson LC, DeHart CJ, Kaiser NK, Fellers RT, Smith DF, Greer JB, LeDuc RD, Blakney GT, Thomas PM, Kelleher NL: Identification and characterization of human proteoforms by top-down LC-21 tesla FT-ICR mass spectrometry. Journal of proteome research. 16, 1087–1096 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karch KR, Coradin M, Zandarashvili L, Kan Z-Y, Gerace M, Englander SW, Black BE, Garcia BA: Hydrogen-deuterium exchange coupled to top-and middle-down mass spectrometry reveals histone tail dynamics before and after nucleosome assembly. Structure. 26, 1651–1663. e1653 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dang X, Singh A, Spetman BD, Nolan KD, Isaacs JS, Dennis JH, Dalton S, Marshall AG, Young NL: Label-free relative quantitation of isobaric and isomeric human histone H2A and H2B variants by Fourier transform ion cyclotron resonance top-down MS/MS. Journal of proteome research. 15, 3196–3203 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, Yang F, Zhao R, Tolic N, Robinson EW, Camp DG, Smith RD, Pasa-Tolic L: Integrated workflow for characterizing intact phosphoproteins from complex mixtures. Analytical chemistry. 81, 4210–4219 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, Liu X, Muther J, James AJ, Smith K, Wu S: Top-down Mass Spectrometry Analysis of Human Serum Autoantibody Antigen-Binding Fragments. Scientific Report, (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bloh AM, Campos JM, Alpert G, Plotkin SA: Determination of N-formimidoylthienamycin concentration in sera from pediatric patients by high-performance liquid chromatography. Journal of Chromatography B: Biomedical Sciences and Applications. 375, 444–450 (1986) [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Gu D, Aisa HA, Ito Y: Studies on the effect of column angle in figure-8 centrifugal counter-current chromatography. Journal of Chromatography A. 1218, 6128–6134 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen B, Peng Y, Valeja SG, Xiu L, Alpert AJ, Ge Y: Online hydrophobic interaction chromatography–mass spectrometry for top-down proteomics. Analytical chemistry. 88, 1885–1891 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Ma H, Smith K, Wu S: Two-dimensional separation using high-pH and low-pH reversed phase liquid chromatography for top-down proteomics. International Journal of Mass Spectrometry. 427, 43–51 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiu L, Valeja SG, Alpert AJ, Jin S, Ge Y: Effective protein separation by coupling hydrophobic interaction and reverse phase chromatography for top-down proteomics. Analytical chemistry. 86, 7899–7906 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M, Wu S, Stenoien DL, Zhang Z, Connolly L, Freitag M, Paša-Tolić L: Profiling Changes in Histone Post-translational Modifications by Top-Down Mass Spectrometry. Springer, (2017) [DOI] [PubMed] [Google Scholar]
- 26.Vellaichamy A, Tran JC, Catherman AD, Lee JE, Kellie JF, Sweet SM, Zamdborg L, Thomas PM, Ahlf DR, Durbin KR: Size-sorting combined with improved nanocapillary liquid chromatography− mass spectrometry for identification of intact proteins up to 80 kDa. Analytical chemistry. 82, 1234–1244 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M: Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 480, 254–258 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haselberg R, de Jong GJ, Somsen GW: Low-flow sheathless capillary electrophoresis–mass spectrometry for sensitive glycoform profiling of intact pharmaceutical proteins. Analytical chemistry. 85, 2289–2296 (2013) [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Sun L, Knierman MD, Dovichi NJ: Fast separation and analysis of reduced monoclonal antibodies with capillary zone electrophoresis coupled to mass spectrometry. Talanta. 148, 529–533 (2016) [DOI] [PubMed] [Google Scholar]
- 30.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Kaiser BLD, Nakayasu ES, Cort JR, Pevzner P, Smith RD: Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proceedings of the National Academy of Sciences. 110, 10153–10158 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu S, Brown RN, Payne SH, Meng D, Zhao R, Tolić N, Cao L, Shukla A, Monroe ME, Moore RJ: Top-down characterization of the post-translationally modified intact periplasmic proteome from the bacterium Novosphingobium aromaticivorans. International journal of proteomics. 2013, (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly RT, Page JS, Tang K, Smith RD: Array of chemically etched fused-silica emitters for improving the sensitivity and quantitation of electrospray ionization mass spectrometry. Analytical chemistry. 79, 4192–4198 (2007) [DOI] [PubMed] [Google Scholar]
- 33.Kou Q, Xun L, Liu X: TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics. 32, 3495–3497 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guner H, Close PL, Cai W, Zhang H, Peng Y, Gregorich ZR, Ge Y: MASH Suite: a user-friendly and versatile software interface for high-resolution mass spectrometry data interpretation and visualization. Journal of The American Society for Mass Spectrometry. 25, 464–470 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, Thomas PM: ProSight Lite: graphical software to analyze top down mass spectrometry data. Proteomics. 15, 1235–1238 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valeja SG, Xiu L, Gregorich ZR, Guner H, Jin S, Ge Y: Three Dimensional Liquid Chromatography Coupling IEC/HIC/RPC for Effective Protein Separation in Top-Down Proteomics. Analytical chemistry. 87, 5363 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL: Quantitation and identification of thousands of human proteoforms below 30 kDa. Journal of proteome research. 15, 976–982 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaffer LV, Rensvold JW, Shortreed MR, Cesnik AJ, Jochem A, Scalf M, Frey BL, Pagliarini DJ, Smith LM: Identification and Quantification of Murine Mitochondrial Proteoforms Using an Integrated Top-Down and Intact-Mass Strategy. Journal of proteome research. 17, 3526–3536 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cleland TP, DeHart CJ, Fellers RT, VanNispen AJ, Greer JB, LeDuc RD, Parker WR, Thomas PM, Kelleher NL, Brodbelt JS: High-throughput analysis of intact human proteins using UVPD and HCD on an orbitrap mass spectrometer. Journal of proteome research. 16, 2072–2079 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai W, Tucholski T, Chen B, Alpert AJ, McIlwain S, Kohmoto T, Jin S, Ge Y: Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Analytical Chemistry. 89, 5467–5475 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilar M, Olivova P, Daly AE, Gebler JC: Orthogonality of separation in two-dimensional liquid chromatography. Analytical chemistry. 77, 6426–6434 (2005) [DOI] [PubMed] [Google Scholar]
- 42.Dwivedi RC, Spicer V, Harder M, Antonovici M, Ens W, Standing KG, Wilkins JA, Krokhin OV: Practical implementation of 2D HPLC scheme with accurate peptide retention prediction in both dimensions for high-throughput bottom-up proteomics. Analytical chemistry. 80, 7036–7042 (2008) [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T, Shen Y, Monroe ME, Lopez-Ferrer D, Reno T: Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 11, 2019–2026 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delmotte N, Lasaosa M, Tholey A, Heinzle E, Huber CG: Two-dimensional reversed-phase x ion-pair reversed-phase HPLC: an alternative approach to high-resolution peptide separation for shotgun proteome analysis. J Proteome Res. 6, 4363–4373 (2007) [DOI] [PubMed] [Google Scholar]
- 45.Baghdady YZ, Schug KA: Qualitative evaluation of high pH mass spectrometry-compatible reversed phase liquid chromatography for altered selectivity in separations of intact proteins. Journal of Chromatography A. (2019) [DOI] [PubMed] [Google Scholar]
- 46.Choi B, Hercules D, Gusev A: LC-MS/MS signal suppression effects in the analysis of pesticides in complex environmental matrices. Fresenius’ journal of analytical chemistry. 369, 370–377 (2001) [DOI] [PubMed] [Google Scholar]
- 47.Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, Ristig D, Schuler V, Meigel I, Lampert C: C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. Journal of Neuroscience. 21, 1189–1202 (2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim J, Petritis K, Shen Y, Camp II DG, Moore RJ, Smith RD: Phosphopeptide elution times in reversed-phase liquid chromatography. Journal of Chromatography A. 1172, 9–18 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uechi T, Tanaka T, Kenmochi N: A complete map of the human ribosomal protein genes: assignment of 80 genes to the cytogenetic map and implications for human disorders. Genomics. 72, 223–230 (2001) [DOI] [PubMed] [Google Scholar]
- 50.Rich BE, Steitz JA: Human acidic ribosomal phosphoproteins P0, P1, and P2: analysis of cDNA clones, in vitro synthesis, and assembly. Molecular and cellular biology. 7, 4065–4074 (1987) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bargis-Surgey P, Lavergne JP, Gonzalo P, Vard C, Filhol-Cochet O, Reboud JP: Interaction of elongation factor eEF-2 with ribosomal P proteins. European journal of biochemistry. 262, 606–611 (1999) [DOI] [PubMed] [Google Scholar]
- 52.Artero-Castro A, Perez-Alea M, Feliciano A, Leal JA, Genestar M, Castellvi J, Peg V, Ramon y Cajal S, LLeonart ME: Disruption of the ribosomal P complex leads to stress-induced autophagy. Autophagy. 11, 1499–1519 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sidoli S, Lin S, Xiong L, Bhanu NV, Karch KR, Johansen E, Hunter C, Mollah S, Garcia BA: Sequential window acquisition of all theoretical mass spectra (SWATH) analysis for characterization and quantification of histone post-translational modifications. Molecular & Cellular Proteomics. 14, 2420–2428 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kikuchi G: The glycine cleavage system: composition, reaction mechanism, and physiological significance. Molecular and cellular biochemistry. 1, 169–187 (1973) [DOI] [PubMed] [Google Scholar]
- 55.Koyata H, Hiraga K: The glycine cleavage system: structure of a cDNA encoding human H-protein, and partial characterization of its gene in patients with hyperglycinemias. American journal of human genetics. 48, 351 (1991) [PMC free article] [PubMed] [Google Scholar]
- 56.Fujiwara K, Okamura-Ikeda K, Motokawa Y: Chicken liver H-protein, a component of the glycine cleavage system. Amino acid sequence and identification of the N epsilon-lipoyllysine residue. Journal of Biological Chemistry. 261, 8836–8841 (1986) [PubMed] [Google Scholar]
- 57.Zhou ZH, McCarthy DB, O’Connor CM, Reed LJ, Stoops JK: The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proceedings of the National Academy of Sciences. 98, 14802–14807 (2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hermes FA, Cronan JE: Scavenging of cytosolic octanoic acid by mutant LplA lipoate ligases allows growth of Escherichia coli strains lacking the LipB octanoyltransferase of lipoic acid synthesis. Journal of bacteriology. 191, 6796–6803 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jordan SW, Cronan JE Jr: The Escherichia coli lipB gene encodes lipoyl (octanoyl)-acyl carrier protein: protein transferase. Journal of bacteriology. 185, 1582–1589 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park MH, Wolff EC: Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. Journal of Biological Chemistry. 293, 18710–18718 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saini P, Eyler DE, Green R, Dever TE: Hypusine-containing protein eIF5A promotes translation elongation. Nature. 459, 118 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cano VS, Jeon GA, Johansson HE, Henderson CA, Park JH, Valentini SR, Hershey JW, Park MH: Mutational analyses of human eIF5A-1–identification of amino acid residues critical for eIF5A activity and hypusine modification. The FEBS journal. 275, 44–58 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qu Y, Wu S, Zhao R, Zink E, Orton DJ, Moore RJ, Meng D, Clauss TR, Aldrich JT, Lipton MS: Automated immobilized metal affinity chromatography system for enrichment of Escherichia coli phosphoproteome. Electrophoresis. 34, 1619–1626 (2013) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.