

Abstract
Because our beliefs regarding our individuality, autonomy, and personhood are intimately bound up with our brains, there is a public fascination with cerebral organoids, the “mini-brain,” the “brain in a dish”. At the same time, the ethical issues around organoids are only now being explored. What are the prospects of using human cerebral organoids to better understand, treat, or prevent dementia? Will human organoids represent an improvement on the current, less-than-satisfactory, animal models? When considering these questions, two major issues arise. One is the general challenge associated with using any stem cell–generated preparation for in vitro modelling (challenges amplified when using organoids compared with simpler cell culture systems). The other relates to complexities associated with defining and understanding what we mean by the term “dementia.” We discuss 10 puzzles, issues, and stumbling blocks to watch for in the quest to model “dementia in a dish.”
Keywords: dementia, organoids, induced pluripotent stem cells, Alzheimer’s disease, neurodegeneration, cerebral, cortical, disease model
Introduction
Cerebral organoids (Fig. 1), or “mini-brains” as they are commonly referred to, are three-dimensional tissue structures that are generated in vitro (often from pluripotent stem cells) (Pașca 2018), containing different cell types of the brain, and representing the anatomical structures of the brain (Fig. 2). Organoids are used for disease modelling for dementia (summarized in Table 1); however, there are a number of important considerations. We outline the 10 big questions that impinge on the design and interpretation of experiments using cerebral organoids from dementia patients.
Figure 1.

The experimental paradigm using cerebral organoids to investigate Alzheimer’s disease. (A) Alzheimer’s disease (AD) is the most prevalent form of dementia and is characterized by neuronal cell death to the cortex and hippocampus of the brain resulting in impaired memory, cognition, and behavior. The underlying molecular mechanisms that result in neurodegeneration remain to be defined, with patient-derived induced pluripotent stem cells (iPSCs) offering a biologically relevant model to better understand the biological basis of AD. The AD brain shows a reduced volume due to the loss of synapses and neurons. (B) During normal development pluripotent stem cells differentiate into multiple cell types of the body, including cells from the three germ layers, the ectoderm, mesoderm, and endoderm. Terminally differentiated cells, such as skin cells, can be reprogrammed to generate iPSCs that have the capacity to differentiate into multiple cell types of the body. (C) To model AD in a dish, patients donate cells (such as skin cells) that are reprogrammed into iPSCs. The iPSCs can then be differentiated into neurons or other cell types. The use of AD patient–derived iPSCs allows researchers to generate cells of a specific lineage, including neurons and glial cells, to characterize disease phenotypes that may be a result of the patient’s genetic makeup and to develop new therapeutics via drug screening. (D) Cerebral organoids are three-dimensional stem cell cultures that have the advantage over traditional two-dimensional culture approaches as they allow the stem cells to self-organize into structures, form signaling networks and develop cell-cell interactions that better mimic in vivo neurodevelopment. These three-dimensional culture systems allow us to better model and further interrogate the roles that different neural and glial cell types play in neurodegeneration. The cerebral organoids contain a mixture of neural cell types that represent the anatomical structure of the brain.
Figure 2.

Generalized schematic of cerebral organoid differentiation based on Lancaster and Knoblich (2014). (A) Small molecule inhibitors are added to human pluripotent stem cells to drive differentiation toward a cortical phenotype. Scale bar = 200 µm. (B) Following neural induction, neural rossettes are harvested for three-dimensional culture as neurospheres. Scale bar = 200 µm. (C) Cortical neurospheres are cultured in suspension to allow for maturation and expansion. Following 2 weeks, neurospheres are maintained for longer times to allow for further self-organization and differentiation. Cortical neurospheres self-organize into distinct structures. Neural rosettes observed in vitro during cerebral organoid culture (highlighted in the rectangle) represent the neural tube formation in vivo during human neurodevelopment. Scale bar = 200 µm. (D) Cerebral organoids are maintained for long-term culture allowing for neural differentiation that recapitulates human brain development, and continue to grow in size. Scale bar = 200 µm. Cerebral organoids can be cryosectioned and characterized by immunocytochemistry, using antibodies directed at specific neuronal and glial markers to show the heterogeneous populations of cells contained within an individual organoid. A representative example of a cerebral organoid derived from a healthy individual shows the nuclear stain, Hoechst, blue (E); the mature neuronal marker microtubule associated protein 2 (MAP2), green (F); the astrocyte marker glial fibrillary acidic protein (GFAP), red (G); the merged overlay (H; scale bar = 25 µm); and a magnified image to show the mix of neurons and astrocytes in the organoid (I; scale bar = 50 µm).
Table 1.
Summary of Organoid Models Used in Alzheimer’s Disease Research.
| Summary | Cell Line | Days Matured | Reference |
|---|---|---|---|
| The authors successfully recapitulated Alzheimer’s disease pathology, including Aβ aggregates and hyperphosphorylated tau in cerebral organoids. Choi and colleagues (2014) showed that inhibition of Aβ generation reduced Aβ pathology and tauopathy; glycogen synthase kinase 3 regulated Aβ-mediated tau phosphorylation. | ReN cell VM overexpressing human APP and PSEN1 containing YOAD mutations | 42–84 days | Choi and others (2014) |
| The 3D neuronal model generated recapitulated both tau and amyloid pathology. β and γ-secretase inhibitors were more efficient in reducing Aβ levels in 2D than in 3D neuronal cultures and the response to drug treatment was highly variable among the different iPSC lines. | iPSCs derived from PBMCs of five LOAD patients | 42–63 days | Lee and others (2016) |
| Using iPSC-derived cerebral organoids AD phenotypes were observed, including Aβ aggregates, hyperphosphorylated tau, and endosome abnormalities. Patient-derived organoids with β- and γ-secretase inhibitors resulted in a significant reduction in Aβ and tau pathology. | iPSCs derived from four YOAD patients with APP duplication or PSEN1 mutation | 60–100 days | Raja and others (2016) |
| Cerebral organoids derived from YOAD and Down syndrome (DS) patients spontaneously developed structures reminiscent of Aβ plaques and NFTs over time. These structures were not observed in iPSCs derived from healthy controls, patients affected by Creutzfeldt-Jakob disease or mouse embryonic stem cells and iPSCs. | iPSCs derived from a YOAD patient with a mutation in PSEN1 and a DS patient | 30–110 days | Gonzalez and others (2018) |
| APOE4 cerebral organoids demonstrated accumulation of Aβ and tau hyperphosphorylation after 6 months, whereas fAD organoids used in Raja and others (2016) showed pathology after 2 months. APOE4 alone was sufficient to induce AD pathology. APOE4 in LOAD organoids had significantly higher levels of Aβ after 6 months compared to age-matched APOE3 organoids. These findings provide evidence for the role of APOE4 in sAD; gene editing to APOE3 protected against AD pathology. | iPSC line derived from LOAD patient and healthy control. Used CRISPR/Cas9 to convert APOE4 to APOE3 or vice versa. | 180 days | Lin and others (2018) |
| This study generated a novel 3D triculture system using a microfluidic platform for neurons, astrocytes, and microglia. There was increased Aβ, inflammatory cytokines, chemokines and hyperphosphorylated tau in the triculture system. The model showed microglial recruitment and neurotoxic properties, including pro-inflammatory cytokine/chemokine release, axonal damage and nitric oxide release, which was proposed to damage neurons and astrocytes in culture. Knocking down Toll-like receptor 4 (TLR4) protected against neuronal and astrocyte loss. The iPSC derived triculture replicated microglial recruitment and activation and neuronal and astrocyte loss. | ReN cell VM expressing APP containing YOAD mutations with both K670N/M671L (Swedish) and V717I (London), human microglia SV40 cell line, and SCR131 iPSC neural progenitors | 21–70 days | Park and others (2018) |
| Aβ secretion and accumulation was promoted by the chemical inducer Attin-5, which increased Aβ42 and Aβ42/Aβ40 ratio. | iPSCs reprogrammed from CRL-2522 fibroblasts | 60–225 days | Pavoni and others (2018) |
Aβ = amyloid-β; DS = Down syndrome; LOAD = late-onset Alzheimer Dementia iPSCs = induced pluripotent stem cells; NFTs = neurofibrillary tangles; PBMCs = peripheral blood mononuclear cells; YOAD = younger-onset Alzheimer Dementi.
Question 1: How Do We Define Dementia? What Is Its True Burden?
Dementia is a syndrome that is caused by different neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease (Silverberg and others 2018). Few people in medical research underestimate the prevalence of the disease on which they work; meanwhile prevalence and incidence rate estimates are affected by comorbidity and misdiagnoses (van der Flier and Scheltens 2005). Coexisting uneasily with this is a public fear of losing one’s mind through dementia, of alarming (or alarmist) predictions of runaway personal costs due to the basic loss of control and dignity that is central to dementia, and the economic costs confronting the health and aged care systems. Accurate figures are necessary so the health care system can estimate the burden of disease and plan for the personal and institutional cost of treatment. Accurate figures are also essential to determine if population-based interventions are effective.
Dementia describes a syndrome where there is progressive loss of cognitive skills over time, sufficient to interfere with independent living. This can be due to one of several underlying causes, or to more than one cause acting together. During the normal process of ageing, many people lose cognitive skills, but for the majority this is not sufficient to cause more than inconvenience or anxiety. By contrast, by the age of 90, 28% of men and 45% of women develop dementia, according to a population study in the United States (Corrada and others 2008). Similar estimates come from other high-income countries, but it is not clear if the same applies in middle-income countries that now are achieving an ageing profile similar to the United States or Australia, such as China and India.
We also do not have accurate information on whether the age-adjusted incidence of dementia is genuinely decreasing when compared to 20 years ago (Satizabal and others 2016; Seblova and others 2018; Wu and others 2017). Addressing this problem is vitally important for obtaining accurate epidemiological estimates of incidence, progression, and the impact of care. Drift in diagnostic criteria over time (Jack and others 2018) has made such comparisons difficult. The way in which the financial burden of care is allocated by disease also will affect the diagnoses that are offered (McPhail 2016). Defining dementia and its burden is important when considering using organoids as a disease model; if we are not able to define or accurately diagnose disease we could be studying cells from people that do not represent dementia patients.
Question 2: What Is the True Etiology of Dementia?
Most research begins with a clinical assessment to determine whether a patient has dementia. Experienced geriatricians, psychiatrists, neurologists, and psychologists can distinguish dementia from other mental illness or delirium. However, differential diagnosis between the four common dementia etiologies is challenging. Alzheimer dementia (AD), vascular dementia (VD), dementia with Lewy Bodies (DLB), and frontotemporal lobar dementia (FTLD) share many clinical features, but different etiologies, and relying solely on clinical assessment has limitations if all four are lumped together as “dementia.” It is particularly important to separate VD from the other causes of dementia, as it is often preventable, has a major environmental component, and does not “progress” as Alzheimer disease does.
Furthermore, several age-related neurodegenerative diseases can coexist in the elderly brain (Boyle and others 2018) and hence use of biomarkers for phenotyping will be limited unless all four major causes of dementia are considered. This rarely happens at present. Stem cells generated from research participants (invariably labelled with a single diagnosis) could have multiple background neuropathologies, potentially compromising both “disease” and “control” cell lines.
Given that a person with dementia may have a biomarker pattern indicating more than one etiology, the challenging question then becomes how to best define the “prime mover.” For AD, combining genetics (the presence of APOE4), biomarkers (amyloid-beta fragment ratios in blood and/or cerebral spinal fluid), and imaging (amyloid as seen using positron emission tomography) is diagnostic (Jack and others 2018; Villemagne and others 2018). Similar multidisciplinary approaches are being developed to diagnose DLB and FTLD, incorporating genetic risk, biomarker assessment, and imaging (Meeter and others 2017). The diagnosis of VD relies primarily on neuroimaging (using magnetic resonance imaging) to reveal either macro- or microvascular disease. Given the advanced state of these technologies, any stem cell–related modelling approach must start by demanding a high-quality diagnostic workup of the persons providing the donor cells. For data from organoids to be meaningful, we need to be able to label each with an accurate history, including types of dementia and staging.
Question 3: Comorbidity and Why Is Age the Biggest Risk Factor?
Most people with dementia are elderly. Many elderly people have comorbidities, such as hypertension, coronary artery disease, depression, or diabetes. Vascular risk factors and related conditions that develop in later life, including cardiovascular disease, diabetes, hypertension, and stroke, are all associated with an increased risk of dementia (Breteler 2000). Furthermore, it is not clear whether different morbidities combine in an additive or synergistic way to affect brain function (Abner and others 2016). Is it valid to model dementia etiology without taking into account these major chronic diseases with known complex connections to brain function? Thoroughly characterizing donors will require considering not only comorbidity but also issues beyond dementia-related history, including non–central nervous system systemic health. To address this, an accepted framework or set of guidelines developed by the research community, funding bodies, and regulators would vastly help in standardizing the information gathered per donor for the purposes of disease modeling.
Although research attention has, understandably, focused on genetic and environmental risk, and on diagnosis, the single biggest risk factor for all forms of dementia is age. Induced pluripotent stem cells (iPSCs) are thought to be the equivalent, in stage, to fetal cells; if they are aged in the laboratory, is this the equivalent of ageing in a person? Can organoids be used to study the mechanisms by which ageing contributes to dementia?
Question 4: Just How Much of the Risk of Alzheimer Dementia Is Genetic and What Is the Role of APOE4?
There are a very small number of families that are affected by Mendelian younger-onset forms of AD (YOAD, familial AD). Symptoms of dementia in YOAD often begin between 40 and 50 years of age due to a fully penetrant mutation in one of the APP, PSEN1, or PSEN2 genes (Goate and others 1991; Levy-Lahad and others 1995; Sherrington and others 1995). For these families, the risk is genetic. However, the great majority of cases of Alzheimer dementia are late-onset (LOAD; >65 years of age), not Mendelian (although the disease “runs in families”), and influenced by more common genetic variants, notably those within the apolipoprotein E (APOE) gene (Strittmatter and others 1993) but also other genes (Fig. 3). The term “sporadic” is a misnomer; Alzheimer dementia is not sporadic, even if we do not yet understand all of the predetermining genetic and environmental risk factors.
Figure 3.

Genetic risk for Alzheimer’s disease. Mutations in APP, PSEN1, and PSEN2 that cause younger-onset AD (YOAD; also known as “familial AD”) are rare with the vast majority of genetic risk for late onset (LOAD; also known as “sporadic AD”) arising through common variants in multiple genes that each increase risk but which individually are not causative. Figure modified from Karch and Goate (2015).
The importance of APOE4 has been known for more than 25 years (Qian and others 2017; Spinney 2014). Approximately 25% of the population has one copy of APOE4, and approximately 2% is homozygous for this variant, but both APOE4 prevalence and its impact vary somewhat for different ethnic groups (Rajabli and others 2018). Possession of one allele of APOE4 increases risk of developing AD threefold, while two copies of APOE4 increases risk 15-fold, matching a co-dominant model. Healthy, non-AD adults with high levels of amyloid show higher levels of memory decline if they are APOE4 carriers (Lim and others 2016; Thai and others 2015). Although APOE4 is the major genetic determinant for developing LOAD, around half of those with an APOE4 allele will not develop dementia (Qian and others 2017). The impact of risk factor genes beyond APOE therefore needs to be considered. This has led to development of a polygenic risk score, which takes into account the contributions of alleles of many genes, each with a small effect contributing to the risk of developing LOAD (Escott-Price and others 2015). Some of the “minor risk genes” describe functions specific to neurons, such as synaptic processes and axonal transport, while others affect pathways in lipid metabolism and inflammation (Lambert and others 2013).
It is important to note that, unlike the Mendelian autosomal dominant mutations, APOE4 is a risk factor and is not fully penetrant. While it is usually assumed that this involves interactions with other genetic variants, it is still possible that genetics and lifestyle interact to lower or increase AD risk. The ethical issues regarding privacy, consent, data sharing, and risk of re-identification associated with acquiring a full genetic profile from individuals can be a barrier to stem cell–based studies. However, stem cell studies of dementia would be improved by knowledge of the full genetic profile of each donor; advanced molecular technologies, such as genome editing and single-cell RNA sequencing, should assist in assigning specific cell functions to particular combinations of risk variants.
Question 5: Are Protein Aggregates the Main Culprit?
The amyloid hypothesis (Hardy and Higgins 1992) was the first attempt to explain the relationship between the key pathological features of AD (beta-amyloid [Aβ] plaques and tau neurofibrillary tangles) and the development of dementia. It argues for a central role for Aβ as the trigger for the key features of AD. It is based on three major findings: (1) that mutations in the amyloid precursor protein (APP) gene itself, and in pathways associated with Aβ production, cause familial YOAD; (2) that there is a large amount of Aβ found in the brains of affected individuals; and (3) that persons with Down syndrome, who synthesize higher amounts of Aβ as the gene for Aβ is on chromosome 21, develop YOAD.
Although amyloid is important in AD, there is a growing view that the amyloid hypothesis is not complete (Makin 2018). Amyloid load does not correlate closely with cognitive loss (Hedden and others 2013), and recent trials that successfully removed amyloid from the brains of those with Alzheimer dementia did not have an impact on clinical progression (Wang and others 2017).
Two alternative explanations for the cause of AD focus, respectively, on the role of tau, or inflammation (Figure 4). Tau pathology occurs during ageing, starting in the lateral entorhinal cortex (Braak and others 2011). It is possible that amyloid catalyzes the spread of tau rather than directly inducing tau pathology. This model could potentially explain the “no going back” theory: once tau deposition is initiated, removing amyloid would not be effective at stopping AD progression (Jacobs and others 2018; Price and Morris 1999). A range of methods have been used to assess Aβ and tau “pathology” in cerebral organoids from AD patients and controls (Figure 5).
Figure 4.

Summary of the amyloid, tau, and inflammation hypotheses for Alzheimer’s disease. Amyloid precursor protein (APP) is a membrane protein that is proteolyticaly cleaved by multiple enzymes. The non-amyloidgenic pathway proceeds when APP is cleaved by the activity of α-secretase (α-sec) to soluble APP α (sAPPα) and APP carboxy terminal fragment α (APP-CTFα), these products are cleaved by γ-secretase (γ-sec) to produce truncated Aβ (p3) and the cytoplasmic polypeptide named AICD. The amyloidgenic pathway occurs when APP undergoes cleavage by β-secretase (β-sec) to form sAPPβ and APP-CTFβ. These proteins are cleaved by γ-sec to form AICD and amyloid-β (Aβ). In YOAD, mutations in APP, PSEN1, and PSEN2 there is an increase in the 42-residue Aβ42, relative to Aβ40, which leads to an increase in Aβ oligomers and amyloid plaque formation (Thinakaran and Koo 2008). Tau binds to microtubules and is important in cytoskeletal function. In AD, tau is hyperphosphorylated resulting in cytoskeletal dysfunction. Hyperphosphorylated tau detaches from microtubules and forms paired helical filaments that aggregate and form neurofibrillary tangles (NFTs). Inflammation is hypothesized to contribute to cognitive loss in AD. Astrocytes and microglia are activated in the AD brain, releasing pro-inflammatory cytokines (e.g., tumor necrosis factor-α [TNFα], interleukin [IL]-1β, IL-6, interferon-γ [IFNγ]) and chemokines (e.g., MIP-1α and MIP-1β) resulting in neuronal death, either by directly damaging neurons or by failing in their normal function to clear aggregates from the brain (Azizi and others 2015). There are multiple underlying molecular pathways leading to AD; intersecting pathways between amyloid, tau, inflammation, and other processes contribute to a complex mechanism that drives neurodegeneration.
Figure 5.

Assessing Aβ and tau pathology in human brain organoids. (A) The localization of Aβ and phosphorylated tau (p-tau) has been visualized in organoids by immunocytochemistry (Choi and others 2014; Gonzalez and others 2018; Lee and others 2016; Lin and others 2018; Park and others 2018; Pavoni and others 2018; Raja and others 2016). (B) β-sheet aggregates have been stained with the fluorescent Thioflavin-S (Thio S) dye. Thio S staining was proposed to identify tau pathology in AD organoids (Raja and others 2016), though the precise molecular identity of the aggregates needs to be confirmed. (C) Enzyme-linked immunosorbent assays (ELISAs) have been used to quantify secreted Aβ in the organoid medium (Choi and others 2014; Gonzalez and others 2018; Lee and others 2016; Lin and others 2018; Park and others 2018; Pavoni and others 2018; Raja and others 2016). (D) Protein levels of Aβ and p-tau have been compared in AD and control organoids by western blotting semiquantitative analysis (Choi and others 2014; Gonzalez and others 2018; Lin and others 2018; Park and others 2018; Raja and others 2016). See also Table 1.
The suggestion that inflammation is a driver of cognitive loss is supported by data demonstrating key roles for immune cells in controlling relevant normal brain function (Szepesi and others 2018). Many of the minor genes implicated in AD are associated with microglial or immune cell function, whether through modulation of inflammation or regulating non-inflammatory processes, such as synapse loss (Hong and others 2016a; Hong and others 2016b; Kinney and others 2018).
Twenty-five years after it was first proposed, the amyloid hypothesis remains hotly debated, with many voices arguing for the need for new ideas (Makin 2018). However, there is no need to assume that there is only one player in the underlying molecular pathways leading to AD. Amyloid, tau, and inflammation (and other processes) are best seen as interacting parts in a complex process that leads to dementia.
Rarer diseases than AD that are characterized by aggregates of proteins other than amyloid can lead to neurodegeneration in the absence of amyloid plaques (non-Alzheimer dementias), such as tau aggregation in age-related tauopathy, progressive supranuclear palsy, or dementia with Lewy bodies and TDP-43 aggregation in hippocampal sclerosis or frontotemporal lobar dementia (Nelson and others 2016). TDP-43 pathology biomarkers are unreliable (Steinacker and others 2019), while tau imaging tools remain problematic in terms of off-target (non-tau) binding and a reduced ability to identify non-AD tau aggregation (Leuzy and others 2019). Developing reliable biomarkers is important for accurate disease models and to understand the link between aggregation of certain proteins and dementia.
Question 6: How Reproducible Are the Data Generated from iPSCs?
If cerebral organoids are to be used as research workhorses in the study of dementia, there is an implicit assumption that they can be prepared with a high degree of replicability. However, there are still many questions about whether different stem cell lines, and the organoids made from them, are sufficiently identical to allow comparisons to be made between them.
Many groups have generated patient-specific iPSC lines carrying mutations in the genes PSEN1, PSEN2, and APP (Arber and others 2017). While iPSC lines show some phenotype expression associated with AD after differentiation to brain organoids, iPSC line variability is an issue when identifying potential phenotypes. Functional variation between iPSC lines and clones relate to their capacity to differentiate to a particular germ layer or cell identity. These functional differences are underpinned by variation at the transcriptional and epigenetic levels, which may be due to genetic variation, level of reprogramming, copy number changes during reprogramming, parental cell origin, and/or culture conditions (Ortmann and Vallier 2017; Polo and others 2010; Popp and others 2018; Schwartzentruber and others 2018; Stadtfeld and others 2010).
Genome editing using homology-directed repair techniques, such as CRISPR/Cas9 is a powerful tool for disease modelling. The use of precise genome editing can provide parallel cell lines that are genetically identical across the genome and differ only for a single disease-causing or disease-relevant mutation. The ability to compare gene-edited isogenic iPSC lines enables the investigation of molecular and cellular differences and strengthens tools for reverse genetic screening in iPSC disease models, increasing the likelihood that definitive disease profiles can be uncovered.
Because variation between and within iPSC lines is exceptionally important, and indeed imposes a limit on the generalizability of findings, there is great value in groups sharing the same lines experimentally, and comparing data for these lines both for similar and different experiments. In essence, this is a test of our commitment to open science and collaboration. It is promising that initiatives are proposed to facilitate sharing iPSC lines from patients with different Mendelian forms of YOAD (Karch and others 2018).
It is also important to consider the extent to which cellular heterogeneity occurs during and after iPSC differentiation, even when using “robust” differentiation protocols. Variation is observed both within one laboratory and between laboratories while using the same iPSC lines and following precisely the same differentiation protocols (Schwartzentruber and others 2018; Volpato and others 2018). This highlights the need for comprehensive genetic and phenotypic analyses of differentiated iPSC-derived cell types to compare results obtained between laboratories. When there is a conflict between data, whether within a laboratory or between laboratories, this may reflect authentic heterogeneity at the cellular level. Variability between iPSC lines, iPSC clones, and their derivatives shows that we need to treat data obtained with caution, and go back to the “whole person” for true validation of findings. This is not a simple task but it may be critical to our understanding of disease; for example, for AD it is not possible to truly confirm this diagnosis until the post mortem brain pathology can be confirmed (Perl 2010). In living patients amyloid imaging can be used as a biomarker for amyloid levels (Klunk 2011) but this is costly and not widely available (Vandenberghe and others 2013) and, as a result, is often not performed prior to inclusion of patient samples in stem cell studies.
Question 7: What about the Environment? Can We Model Modifiable Risk and Protective Factors for AD?
We frequently see the statement that about a third of the worldwide burden of dementia is attributable to modifiable risk factors in the environment (Livingston and others 2017; Norton and others 2014). It is therefore interesting to consider whether organoids might offer a model to investigate these phenomena. This would be of value for it is not clear whether such epidemiologic estimates arise from mixing different forms of dementia together (in particular, VD and AD), or are due to the inherent promiscuity of the aged brain, pathologically speaking.
To model a complex protective factor would require that: (1) organoids develop a bona fide pathological phenotype (i.e., for AD, amyloidosis + tauopathy + neurodegeneration) and (2) a readout of global organoid “neuronal functionality” is possible.
To take a popular example, several studies suggest exercise may attenuate the rate of cognitive decline in at risk elders. Processes implicated include structural plasticity in the hippocampus, upregulation of systemic brain-derived neurotrophic factor, functional brain network change, and peripheral changes in immune-related cytokines and myokines (Kivipelto and others 2018; Larson and others 2006; McEwen and others 2018; Muller and others 2017). Other studies dispute the beneficial effect of exercise on cognition (Young and others 2015). Disentangling the impact of exercise on dementia using human organoids, combining mechanical, biochemical, and genomic approaches, represents the precision medical science we hope to achieve.
More ambitiously, to address the role of education as a protective factor for dementia, the “cognitive capacity” of an organoid could be estimated by its ability to habituate to a stimulus or even encode a stimulus pattern (i.e., learn) and then retain it over an interstimulus interval (i.e., memory)—ideas elaborated on in the next section. This represents a transition from the study of organoid structure to the study of organoid function.
Perhaps more attainable are models related to high prevalence risk factors. Smoking is the strongest environmental toxin for dementia, responsible for an estimated 4.7 million cases in 2010 (Norton and others 2014). As a first exploratory step, it would be interesting to model different levels of tobacco smoke concentration in an incubator, studying the effect on organoids for each type of dementia (AD, VD, DLB, and FTLD). Another group of candidate risk factors are metabolic: hypertension, type 2 diabetes, and obesity (Clark and others 2018; Gabin and others 2017; Kivipelto and others 2018; Klimova and others 2018; Pegueroles and others 2018). It may be possible to study neuronal and glial responses to aberrant metabolism by exposing organoids to varying levels of glucose and/or lipids. Other proposed environmental risk factors for developing AD include microbial infections, such as herpes simplex (Fulöp and others 2018; Lin and others 1996). Introducing an immune-related environment in organoids, by the use either of microglia or of macrophages, together with viral or bacterial pathogens, may allow the study of whether herpes simplex contributes to AD pathology.
Another potential insult to the brain is ischemia and hypoxia. There is a rich literature linking ischemia, hypoxia, and sleep disorders to AD pathology, offering circumstantial evidence (Valenzuela and others 2012). While the long-term objective is to create vascularized organoids perfusable with oxygen at different dissolved tensions, hypoxia could be modelled in a simple fashion by controlling atmospheric oxygen fraction over cultures. Low oxygen environments are known to affect neural stem cell proliferation, differentiation, and maturation (Xie and Lowry 2018). Whether the same holds for complex multicellular organoids is a novel question, and any impact on the development of AD pathology is unknown.
Question 8: Can We Measure Cognition, and “Cognitive Reserve,” in Organoids?
Memory loss and cognitive decline are the mainstays of a clinical dementia diagnosis, often without data on the presence of neurodegenerative pathology. Studies on cognitive abilities show that early deposition of amyloid is related to worse cognitive and memory performance, even before individuals meet the clinical criteria for dementia (Rodrigue and others 2012). Given that amyloid load does not directly correlate with symptoms (Hedden and others 2013) it is not clear whether this is the direct result of amyloid itself or an indirect effect of other cell biological and degenerative processes. The biological processes underlying such cognitive impairment are manifold, and most often include loss of connections between neurons (Dorostkar and others 2015). Such disarrayed communication breaks down the synchronized activity of entire areas, with working memory function quickly lost (Morrison and Baxter 2012). Being able to study the biological mechanisms behind cognitive decline is therefore essential for an in vitro dementia model. While histological tools exist to assess neurodegeneration, synaptic dysfunction, or alterations in neuronal/glial interactions, real value would come from gaining this information from living tissue, with the advantages of longitudinal interrogation.
There is clearly a growing need to develop technologies that can reliably measure neuronal functionality within organoids. Functional assays, such as calcium imaging (Fig. 6) or microelectrode arrays (Fig. 7) can be used to measure neuronal activity and formation of synchronous neuronal networks, and have already been successfully applied to iPSC-derived organoids (Quadrato and others 2017). An important challenging factor to consider is the time needed for neurons to mature and form networks within the organoids, which may take several months (Quadrato and others 2017). It is not currently clear how the organoids “age” or “mature” over time, in terms of their functional activity. This process needs to be characterized so that we can understand whether changes in functional outputs are disease related or an artefact of chronic long-term culture conditions. Other complimentary approaches may be to optically measure calcium transients or changes in membrane potentials within a multitude of neurons in real time and across the whole three-dimensional structure, or measuring neurotransmitter levels, such as glutamate, generated within the organoids (Nasr and others 2018; Sloan and others 2018). A major challenge is to develop live imaging tools and electrical recording techniques that overcome the z-plane depth limitation of organoid models. Another challenge is to develop organoid-specific data analysis tools that accurately model the structural and functional information that can be matched with clinical data.
Figure 6.

Functional assessment of cerebral organoids by calcium imaging. (A) A representative image of a culture derived from a YOAD cerebral organoid loaded with the ratiometric calcium indicator Fura2-AM. A 9-month-old organoid was seeded onto a glass coverslip for Ca2+ imaging. The image shows the overlay of 340 nm and 380 nm channels; each colored circle corresponds to a region of interest (ROI) represented in (B). When Ca2+ binds to the indicator, fluorescence at 340 nm increases, while 380 nm fluorescence decreases. The 340/380 ratio is thus used as a measurement of Ca2+ responses to drugs or agonists or to assess spontaneous activity. (B) Relative change in fluorescence intensity over time of specific ROIs from (A) using Fura2-AM. Neurons can be stimulated with chemicals that are perfused into the bath chamber and the Ca2+ responses recorded. The culture was exposed to the excitatory neurotransmitter glutamate (20 µM; perfusion indicated by the horizontal black bar) to elicit a Ca2+ response, followed by high K+ (60 mM; perfusion indicated by the horizontal black bar) to mimic membrane depolarization. Responses may be fast or slow transient increases or prolonged increases that do no return to baseline within the timeframe of the experiment.
Figure 7.
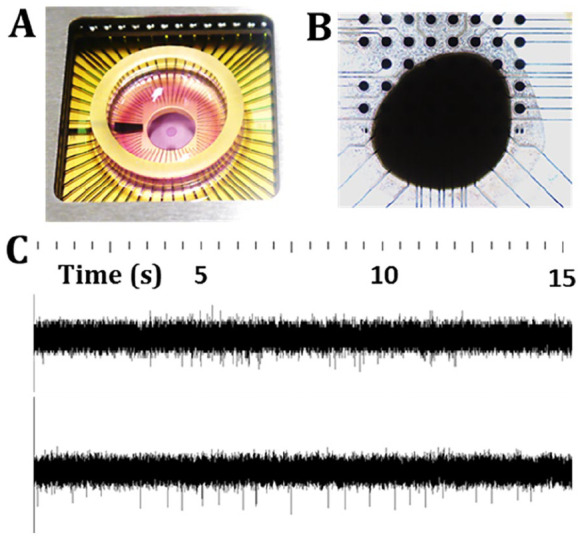
Electrophysiological assessment of cerebral organoids by microelectrode arrays. (A) Organoids can be seeded in well chambers (B) on top of regularly spaced arrays of electrodes. (C) Organoids are electrically active; shown are recordings from two different organoids at 25 kHz, whereby spikes identify electrophysiological activity. The recordings show a difference in spontaneous activity from the two organoids, which can be quantified via various parameters, such as spike amplitude and firing rate. Experiments can be designed to compare organoid responses to chemical or electrical stimulation and can provide information on neuronal network dynamics and the formation of functional circuits.
On the flip side to memory loss and cognitive decline, it would also be valuable to use organoids to model and measure cognitive reserve—a concept often invoked to explain the disconnect observed in ~30% of older persons who express suprathreshold levels of AD pathology in terms of amyloid deposition (whether at post mortem or by in vivo imaging) but do not exhibit dementia (Stern and others 2018). In essence, cognitive reserve refers to the adaptability, efficiency, and flexibility of the brain and its cognitive processes in the face of stressors such that overall function is maintained and protected (Stern and others 2018). Determinants of greater cognitive reserve include lifelong mental stimulation and challenge, including education, occupational complexity, and intellectual leisure pursuits. Organoids may be useful in modelling such determinants by measuring neuronal firing activity, particularly in synchronous neuronal networks, in response to different levels of stimulation. Currently, it is difficult to assess how measurements of function in organoids scale up to measurements in whole brains. A theoretical approach that could be used is the Perturbational Complexity Index that was developed and tested in healthy subjects and coma patients (Casali and others 2013). The technique uses transcranial stimulation to assess consciousness that is independent of sensory processing. However, the issue of consciousness in brain organoids leads to significant ethical implications (see Question 10). At present there is no evidence from published studies that brain organoids exhibit consciousness, and in fact, single cell analyses suggest brain organoids show a lack of maturity (Bhaduri and others 2020).
Question 9: Can Organoids Be Used for Drug Discovery?
There is a strong hope that organoids will be of immediate value for use in drug screening and development. However, while human brain organoids have potential to model AD and other dementias, it is not yet clear whether they can be adapted for applications in drug discovery.
The promise of organoids lies in their ability to model complex processes and it is precisely this complexity that is at odds with the industrial practice of targeted drug discovery. Typically, drug screens adopt a reductionist approach, making use of simple, direct molecular assays in the initial chemical screening phase to identify active “hits” from libraries of chemical scaffolds. Organoids are therefore unlikely to be useful at the initial chemical library screening stage. Rather, they may come into their own for identification of biochemical pathways or specific molecules that modulate the course of disease, a research process often termed “target identification,” which precedes the commitment to a targeted drug discovery campaign (Fig. 8).
Figure 8.

Schematic showing opportunities for the use of brain organoids in the drug development pipeline. In particular, stem cell–derived culture models have the potential to improve lead optimization for “formal preclinical” and clinical development.
Organoids will also have important roles to play in selection of the most biologically active compounds in the later stages of lead development, when more costly, complex, and informative biological models are justifiable and valuable. Such models also have the potential to identify opportunities for repurposing approved drugs, and ultimately provide the means to personalize therapy by identifying drugs that are most effective at reversing the pathology in organoids produced from patient-derived iPSCs.
It is tempting to suggest that a complex disease requires a complex model to identify useful hits, even during the early stages of hit identification. Can the potential of organoids be harnessed at this early stage, prior to target identification, using a “phenotypic” screening process? This will depend on whether a disease-relevant assay (or combination of assays) is replicable (as discussed above) and is sufficiently sensitive to measure drug activity. Quantitative phenotypic screening assays need to be devised that are specifically designed for organoid cultures and address these issues, particularly in relation to reproducibility.
Question 10: What Are the Ethical Issues and Community Expectations Around Brain Organoids?
When stem cells were first derived from human embryos that had been stored for in vitro fertilization but were no longer wanted for reproduction, there was a great deal of ethical debate; some people with religious objections to the destruction of embryos opposed the generation of embryonic stem (ES) cells. However, most stem cell research today is performed using iPSCs, which do not raise the same level of ethical concern. Nevertheless, human ES cells are still regarded as being the “gold standard” for potency, and the International Society for Stem Cell Research has established guidelines for researchers, clinicians, and funding agencies in this regard (Daley and others 2016).
The fact that the specific issues around embryo destruction have been solved by scientific advances does not mean that there are no remaining ethical concerns voiced by the community (Allum and others 2017; Bredenoord and others 2017; Chalmers and others 2017; Stadelmann and Torgler 2017; Shepherd 2018). Recent discussions around the ethical issues of brain organoids describe consciousness as a moral limit of brain organoid research (Bayne and others 2020; Koplin and Savulescu 2019; Sawai and others 2019). While most people favor the use of stem cells for medical research and clinical care, they add a strong proviso: the public must be involved in open and frank discussions around ethical issues, such as privacy and consent, and the way organoids are derived and treated (Farahany and others 2018).
It is difficult to obtain informed consent to collect, study, and store biological material from people with dementia, although a common sense approach combined with information from people with early stages of dementia indicate that many of them would wish to participate (Howe 2012; Slaughter and others 2007). Because stem cell and organoid research is advancing rapidly, it is not possible to predict all of the ways in which cells might be used in future. The coalescence of these two concerns raises complications for those using organoids for dementia research, and it is imperative that researchers engage with those living with dementia and their families and carers (Pachana and others 2015; Ries and others 2017). It may be possible to use a research equivalent of an “advanced care directive,” where a person consents to future research relevant to dementia, prior to cognitive decline.
Since family history is a well-known risk factor, children of persons with AD will be a group of highly motivated individuals who wish to participate in research, just as soon as there is hope of an effective intervention. Because of this, and unlike many situations in medical research, cell donors should be reidentifiable, particularly if they carry the major risk allele APOE4. This leads to a range of ethical (and legal) issues around de-identifying data, sharing of data between research groups (Isasi and others 2014), and whether there is a responsibility to inform participants of their genetic risk status (Milne and others 2018; Timmermans and Buchbinder 2010). It is important to encourage research into the effects of revealing high-risk genotypes to asymptomatic adult children of people with AD, in the context of availability of direct-to-consumer genotyping (Green and others 2009).
The expectation of the community that they will be consulted about research using stem cells also involves a major commitment to education, both about dementia and about stem cell science (King and others 2014). On an issue as critical and costly as this, it may be appropriate for national research bodies, such as the National Institutes of Health in the United States, the Medical Research Council in the United Kingdom and the National Health and Medical Research Council of Australia to play a major role in initiating both community education and public discussion on dementia.
Final Thoughts
Caveats and Challenges: Whilst organoids provide opportunities for disease modelling, the current literature suggests cell maturation remains limited due to high levels of cellular stress (Bhaduri and others 2020). Future work needs to consider how to progress model systems to better recapitulate the spatiotemporal dynamics of differentiation and how to deal with the impact of cellular stress during long term cell culture. Addressing these difficulties is essential to improve our understanding of the model systems and the diseases they represent.
Opportunities: Stem cells and organoid technology will enable us to ask, and maybe even answer, some of the fundamental questions about pathological processes and risk associations for dementia, as well as neurodegenerative diseases more broadly (Lambert and others 2013). The most direct route is for stem cell scientists and clinicians to work closely together, in a manner where the technological advances and knowhow of scientists are guided by the most perceptive clinical insights and the most pressing health needs.
This approach has led to successes for other diseases. Some serious diseases (such as measles and polio) have been virtually eliminated, others (such as breast cancer or HIV in many countries) are much less likely to kill and are amenable to treatment, while conditions such as cardiovascular diseases (myocardial infarcts and strokes) still are common, but have been displaced to occur more often in the elderly. These transformational medical advances, be they environmental, behavioral, or pharmacological, are based on research.
Many who have worked on diseases outside the brain are surprised at how little we know about the fundamentals of Alzheimer disease and other causes of dementia. We eagerly await data showing whether pre-symptomatic removal of amyloid from the brain delays the onset of Alzheimer dementia, particularly if initiated prior to symptoms. At this stage, iPSC lines and cerebral organoids may reflect some of the clinical features of the patients from whom they were obtained, particularly with respect to pathology and etiology. If this proves to be true and technical variation can be controlled, then cerebral organoids may become a very powerful research tool for the study of dementia. There is room for optimism, but also a need for caution, with an assurance that rigorous and replicable hypothesis-driven experiments can translate back to real people with real-world clinical problems.
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MV is the scientific founder of Skin2NeuronPty Ltd and has a financial interest in the company.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Australian Dementia Stem Cell Consortium has received generous start-up travel grants from the Australian NHMRC National Institute for Dementia Research. Authors have been supported by Dementia Australia Research Foundation, Yulgilbar Alzheimer’s Research Program, DHB Foundation (AP), Brain Foundation (DH, AP), the C.F. Leung Memorial Trust (AP), the University of Melbourne (AP) and Operational Infrastructure Support from the Victorian Government (DH, AP), Monash University (AG), JO and JR Wicking Trust (Equity Trustees) (ALC and AEK), University of Sydney (MV), and generous gifts from the Sinclair, Smith and Jolly families (MV). AEK is supported by a National Health and Medical Research Council (NHMRC) of Australia Boosting Dementia Research Leadership Fellowship (APP1136913). AG is supported by a NHMRC-ARC Dementia Research Development Fellowship (GNT1097461). AP is supported by an ARC Future Fellowship (FT140100047) and a NHMRC Senior Research Fellowship (1154389). LO is supported by a NHMRC of Australia Boosting Dementia Research Leadership Fellowship (APP1135720). MV is supported by a NHMRC Career Development Fellowship (APP1112813). VG is supported by Australian Research Council’s Discovery Early Career Researcher Award (DE180100775).
ORCID iDs: Lezanne Ooi  https://orcid.org/0000-0001-9241-8268
https://orcid.org/0000-0001-9241-8268
Anthony L. Cook
https://orcid.org/0000-0003-1770-7910
Martin Engel
https://orcid.org/0000-0001-7602-1340
Anna E. King
https://orcid.org/0000-0003-1792-0965
References
- Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, and others. 2016. Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12(8):882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allum N, Allansdottir A, Gaskell G, Hampel J, Jackson J, Moldovan A, and others. 2017. Religion and the public ethics of stem-cell research: attitudes in Europe, Canada and the United States. PLoS One 12(4):e0176274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber C, Lovejoy C, Wray S. 2017. Stem cell models of Alzheimer’s disease: progress and challenges. Alzheimers Res Ther 9(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi G, Navabi SS, Al-Shukaili A, Seyedzadeh MH, Yazdani R, Mirshafiey A. 2015. The role of inflammatory mediators in the pathogenesis of Alzheimer’s disease. Sultan Qaboos Univ Med J 15(3):e305–e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne T, Seth AK, Massimini M. 2020. Are there islands of awareness? Trends Neurosci 43(1):6–16. [DOI] [PubMed] [Google Scholar]
- Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, and others. 2020. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578(7793):142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. 2018. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83(1):74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. 2011. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70(11):960–9. [DOI] [PubMed] [Google Scholar]
- Bredenoord AL, Clevers H, Knoblich JA. 2017. Human tissues in a dish: the research and ethical implications of organoid technology. Science 355(6322). 10.1126/science.aaf9414 [DOI] [PubMed] [Google Scholar]
- Breteler MMB. 2000. Vascular risk factors for Alzheimer’s disease: an epidemiologic perspective. Neurobiol Aging 21(2):153–60. [DOI] [PubMed] [Google Scholar]
- Casali AG, Gosseries O, Rosanova M, Boly M, Sarasso S, Casali KR, and others. 2013. A theoretically based index of consciousness independent of sensory processing and behavior. Sci Transl Med 5(198):198ra105. [DOI] [PubMed] [Google Scholar]
- Chalmers D, Rathjen P, Rathjen J, Nicol D. 2017. Ethics and governance of stem cell banks. Methods Mol Biol 1590:99–112. [DOI] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, and others. 2014. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515(7526):274–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LR, Koscik RL, Allison SL, Berman SE, Norton D, Carlsson CM, and others. 2018. Hypertension and obesity moderate the relationship between beta-amyloid and cognitive decline in midlife. Alzheimers Dement 15(3):418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrada MM, Brookmeyer R, Berlau D, Paganini-Hill A, Kawas CH. 2008. Prevalence of dementia after age 90: results from the 90+ study. Neurology 71(5):337–43. [DOI] [PubMed] [Google Scholar]
- Daley GQ, Hyun I, Apperley JF, Barker RA, Benvenisty N, Bredenoord AL, and others. 2016. Setting global standards for stem cell research and clinical translation: the 2016 ISSCR guidelines. Stem Cell Reports 6(6):787–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar MM, Zou C, Blazquez-Llorca L, Herms J. 2015. Analyzing dendritic spine pathology in Alzheimer’s disease: problems and opportunities. Acta Neuropathol 130(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, and others. 2015. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138(12):3673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farahany NA, Greely HT, Hyman S, Koch C, Grady C, Pasca SP, and others. 2018. The ethics of experimenting with human brain tissue. Nature 556(7702):429–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulöp T, Itzhaki RF, Balin BJ, Miklossy J, Barron AE. 2018. Role of microbes in the development of Alzheimer’s disease: state of the art—an international symposium presented at the 2017 IAGG Congress in San Francisco. Front Genet 9:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabin JM, Tambs K, Saltvedt I, Sund E, Holmen J. 2017. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: the HUNT Study. Alzheimers Res Ther 9(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, and others. 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349(6311):704–6. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Armijo E, Bravo-Alegria J, Becerra-Calixto A, Mays CE, Soto C. 2018. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry 23(12):2363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, Brown T, and others. 2009. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med 361(3):245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. 1992. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256(5054):184–5. [DOI] [PubMed] [Google Scholar]
- Hedden T, Oh H, Younger AP, Patel TA. 2013. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 80(14):1341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, and others. 2016. a. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352(6286):712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L, Stevens B. 2016. b. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe E. 2012. Informed consent, participation in research, and the Alzheimer’s patient. Innov Clin Neurosci 9(5–6):47–51. [PMC free article] [PubMed] [Google Scholar]
- Isasi R, Andrews PW, Baltz JM, Bredenoord AL, Burton P, Chiu IM, and others. 2014. Identifiability and privacy in pluripotent stem cell research. Cell Stem Cell 14(4):427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, and others. 2018. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14(4):535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs HIL, Hedden T, Schultz AP, Sepulcre J, Perea RD, Amariglio RE, and others. 2018. Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat Neurosci 21(3):424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Goate AM. 2015. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Hernandez D, Wang JC, Marsh J, Hewitt AW, Hsu S, and others. 2018. Human fibroblast and stem cell resource from the Dominantly Inherited Alzheimer Network. Alzheimers Res Ther 10(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Robinson A, Vickers J. 2014. Online education: targeted MOOC captivates students. Nature 505(7481):26. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. 2018. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4:575–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivipelto M, Mangialasche F, Ngandu T. 2018. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol 14(11):653–66. [DOI] [PubMed] [Google Scholar]
- Klimova B, Kuca K, Maresova P. 2018. Global view on Alzheimer’s disease and diabetes mellitus: threats, risks and treatment Alzheimer’s disease and diabetes mellitus. Curr Alzheimer Res 15(14):1277–82. [DOI] [PubMed] [Google Scholar]
- Klunk WE. 2011. Amyloid imaging as a biomarker for cerebral β-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol Aging (32 suppl 1): S20–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koplin JJ, Savulescu J. 2019. Moral limits of brain organoid research. J Law Med Ethics 47(4):760–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, and others. 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45(12):1452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA. 2014. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc 9(10):2329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EB, Wang L, Bowen JD, McCormick WC, Teri L, Crane P, and others. 2006. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 144(2):73–81. [DOI] [PubMed] [Google Scholar]
- Lee H-K, Velazquez Sanchez C, Chen M, Morin PJ, Wells JM, Hanlon EB, and others. 2016. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 11(9): e0163072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, Rodriguez-Vieitez E, and others. 2019. Tau PET imaging in neurodegenerative tauopathies—still a challenge. Mol Psychiatry 24(8):1112–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, and others. 1995. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269(5226):973–7. [DOI] [PubMed] [Google Scholar]
- Lim YY, Laws SM, Villemagne VL, Pietrzak RH, Porter T, Ames D, and others. 2016. A beta-related memory decline in APOE epsilon4 noncarriers: implications for Alzheimer disease. Neurology 86(17):1635–42. [DOI] [PubMed] [Google Scholar]
- Lin WR, Shang D, Itzhaki RF. 1996. Neurotropic viruses and Alzheimer disease. Interaction of herpes simplex type 1 virus and apolipoprotein E in the etiology of the disease. Mol Chem Neuropathol 28(1–3): 135–41. [DOI] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, and others. 2018. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98(6):1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, and others. 2017. Dementia prevention, intervention, and care. Lancet 390(10113):2673–734. [DOI] [PubMed] [Google Scholar]
- Makin S. 2018. The amyloid hypothesis on trial. Nature 559(7715):S4–7. [DOI] [PubMed] [Google Scholar]
- McEwen SC, Siddarth P, Abedelsater B, Kim Y, Mui W, Wu P, and others. 2018. Simultaneous aerobic exercise and memory training program in older adults with subjective memory impairments. J Alzheimers Dis 62(2):795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail SM. 2016. Multimorbidity in chronic disease: impact on health care resources and costs. Risk Manag Healthc Policy 9:143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeter LH, Kaat LD, Rohrer JD, van Swieten JC. 2017. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol 13(7):406–19. [DOI] [PubMed] [Google Scholar]
- Milne R, Diaz A, Badger S, Bunnik E, Fauria K, Wells K. 2018. At, with and beyond risk: expectations of living with the possibility of future dementia. Sociol Health Illn 40(6):969–87. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. 2012. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 13(4):240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Chan K, Myers JN. 2017. Association between exercise capacity and late onset of dementia, Alzheimer disease, and cognitive impairment. Mayo Clin Proc 92(2):211–7. [DOI] [PubMed] [Google Scholar]
- Nasr B, Chatterton R, Yong JHM, Jamshidi P, D’Abaco GM, Bjorksten AR, and others. 2018. Self-organized nanostructure modified microelectrode for sensitive electrochemical glutamate detection in stem cells-derived brain organoids. Biosensors (Basel) 8(1). 10.3390/bios8010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Trojanowski JQ, Abner EL, Al-Janabi OM, Jicha GA, Schmitt FA, and others. 2016. “New Old Pathologies”: AD, PART, and cerebral age-related TDP-43 with clerosis (CARTS). J Neuropathol Exp Neurol 75(6):482–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. 2014. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol 13(8):788–94. [DOI] [PubMed] [Google Scholar]
- Ortmann D, Vallier L. 2017. Variability of human pluripotent stem cell lines. Curr Opin Genet Dev 46:179–85. [DOI] [PubMed] [Google Scholar]
- Pachana NA, Liddle J, Peel NM, Beattie E, Juang C, Knight BG. 2015. Can we do better? Researchers’ experiences with ethical review boards on projects with later life as a focus. J Alzheimers Dis 43(3):701–7. [DOI] [PubMed] [Google Scholar]
- Park J, Wetzel I, Marriott I, Dréau D, D’Avanzo C, Kim DY, and others. 2018. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat Neurosci 21(7):941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pașca SP. 2018. The rise of three-dimensional human brain cultures. Nature 553(7689):437–45. [DOI] [PubMed] [Google Scholar]
- Pavoni S, Jarray R, Nassor F, Guyot AC, Cottin S, Rontard J, and others. 2018. Small-molecule induction of Aβ-42 peptide production in human cerebral organoids to model Alzheimer’s disease associated phenotypes. PLoS One 13(12):e0209150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegueroles J, Jimenez A, Vilaplana E, Montal V, Carmona-Iragui M, Pane A, and others. 2018. Obesity and Alzheimer’s disease, does the obesity paradox really exist? A magnetic resonance imaging study. Oncotarget 9(78):34691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl DP. 2010. Neuropathology of Alzheimer’s disease. Mt Sinai J Med 77(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, and others. 2010. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 28(8):848–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp B, Krumbiegel M, Grosch J, Sommer A, Uebe S, Kohl Z, and others. 2018. Need for high-resolution genetic analysis in iPSC: results and lessons from the ForIPS Consortium. Sci Rep 8(1):17201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC. 1999. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45(3):358–68. [DOI] [PubMed] [Google Scholar]
- Qian J, Wolters FJ, Beiser A, Haan M, Ikram MA, Karlawish J, and others. 2017. APOE-related risk of mild cognitive impairment and dementia for prevention trials: an analysis of four cohorts. PLoS Med 14(3):e1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, and others. 2017. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545(7652):48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja WK, Mungenast AE, Lin YT, Ko T, Abdurrob F, Seo J, and others. 2016. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One 11(9):e0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabli F, Feliciano BE, Celis K, Hamilton-Nelson KL, Whitehead PL, Adams LD, and others. 2018. Ancestral origin of ApoE epsilon4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet 14(12):e1007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries NM, Thompson KA, Lowe M. 2017. Including people with Dementia in research: an analysis of Australian ethical and legal rules and recommendations for reform. J Bioeth Inq 14(3):359–74. [DOI] [PubMed] [Google Scholar]
- Rodrigue KM, Kennedy KM, Devous MD, Sr, Rieck JR, Hebrank AC, Diaz-Arrastia R, and others. 2012. Beta-amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 78(6):387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satizabal CL, Beiser AS, Chouraki V, Chêne G, Dufouil C, Seshadri S. 2016. Incidence of dementia over three decades in the Framingham heart study. N Engl J Med 374(6):523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai T, Sakaguchi H, Thomas E, Takahashi J, Fujita M. 2019. The ethics of cerebral organoid research: being conscious of consciousness. Stem Cell Reports 13(3):440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J, Foskolou S, Kilpinen H, Rodrigues J, Alasoo K, Knights AJ, and others. 2018. Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet 50(1):54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seblova D, Quiroga ML, Fors S, Johnell K, Lovden M, de Leon AP, and others. 2018. Thirty-year trends in dementia: a nationwide population study of Swedish inpatient records. Clin Epidemiol 10:1679–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd J. 2018. Ethical (and epistemological) issues regarding consciousness in cerebral organoids. J Med Ethics 44(9):611–2. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, and others. 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375:754–60. [DOI] [PubMed] [Google Scholar]
- Silverberg N, Elliott C, Ryan L, Masliah E, Hodes R. 2018. NIA commentary on the NIA-AA research framework: towards a biological definition of Alzheimer’s disease. Alzheimers Dement 14(4):576–8. [DOI] [PubMed] [Google Scholar]
- Slaughter S, Cole D, Jennings E, Reimer MA. 2007. Consent and assent to participate in research from people with dementia. Nurs Ethics 14(1):27–40. [DOI] [PubMed] [Google Scholar]
- Sloan SA, Andersen J, Pasca AM, Birey F, Pasca SP. 2018. Generation and assembly of human brain region-specific three-dimensional cultures. Nat Protoc 13(9):2062–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinney L. 2014. Alzheimer’s disease: the forgetting gene. Nature 510(7503):26–8. [DOI] [PubMed] [Google Scholar]
- Stadelmann D, Torgler B. 2017. Voting on embryonic stem cell research: citizens more supportive than politicians. PLoS One 12(1):e0170656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, and others. 2010. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 465(7295):175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinacker P, Barschke P, Otto M. 2019. Biomarkers for diseases with TDP-43 pathology. Mol Cell Neurosci 97:43–59. [DOI] [PubMed] [Google Scholar]
- Stern Y, Arenaza-Urquijo EM, Bartres-Faz D, Belleville S, Cantilon M, Chetelat G, and others. 2018. Whitepaper: defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement. Epub Sep 14. 10.1016/j.jalz.2018.07.219 [DOI] [PMC free article] [PubMed]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, and others. 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90(5):1977–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szepesi Z, Manouchehrian O, Bachiller S, Deierborg T. 2018. Bidirectional microglia-neuron communication in health and disease. Front Cell Neurosci 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai C, Lim YY, Villemagne VL, Laws SM, Ames D, Ellis KA, and others. 2015. Amyloid-related memory decline in preclinical Alzheimer’s disease is dependent on APOE epsilon4 and is detectable over 18-Months. PLoS One 10(10):e0139082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. 2008. Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283(44):29615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans S, Buchbinder M. 2010. Patients-in-waiting: living between sickness and health in the genomics era. J Health Soc Behav 51(4):408–23. [DOI] [PubMed] [Google Scholar]
- Valenzuela M, Esler M, Ritchie K, Brodaty H. 2012. Antihypertensives for combating dementia? A perspective on candidate molecular mechanisms and population-based prevention. Transl Psychiatry 2:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier WM, Scheltens P. 2005. Epidemiology and risk factors of dementia. J Neurol Neurosurg Psychiatry 76(suppl 5):v2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe R, Adamczuk K, Dupont P, Laere KV, Chételat G. 2013. Amyloid PET in clinical practice: its place in the multidimensional space of Alzheimer’s disease. Neuroimage Clin 2:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villemagne VL, Doré V, Burnham SC, Masters CL, Rowe CC. 2018. Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nature Rev Neurol 14(4):225–36. [DOI] [PubMed] [Google Scholar]
- Volpato V, Smith J, Sandor C, Ried JS, Baud A, Handel A, and others. 2018. Reproducibility of molecular phenotypes after long-term differentiation to human iPSC-derived neurons: a multi-site omics study. Stem Cell Reports 11(4):897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yan T, Lu H, Yin W, Lin B, Fan W, and others. 2017. Lessons from anti-amyloid-beta immunotherapies in Alzheimer disease: aiming at a moving target. Neurodegener Dis 17(6):242–50. [DOI] [PubMed] [Google Scholar]
- Wu YT, Beiser AS, Breteler MMB, Fratiglioni L, Helmer C, Hendrie HC, and others. 2017. The changing prevalence and incidence of dementia over time—current evidence. Nat Rev Neurol 13:327–39. [DOI] [PubMed] [Google Scholar]
- Xie Y, Lowry WE. 2018. Manipulation of neural progenitor fate through the oxygen sensing pathway. Methods 133:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J, Angevaren M, Rusted J, Tabet N. 2015. Aerobic exercise to improve cognitive function in older people without known cognitive impairment. Cochrane Database Syst Rev (4):CD005381. [DOI] [PMC free article] [PubMed] [Google Scholar]