

Abstract
Background
AML patients with FLT3/ITD mutations have poor response to cytarabine-based chemotherapy. FLT3 inhibitors (FLT3i) may resensitize cells to cytarabine (CYT). Improving treatment outcome of this combination may benefit from a mechanistic extrapolation approach from in vitro data.
Methods
The effects of CYT and several FLT3i on cell proliferation and cell cycle kinetics were examined in AML cell lines. The effect of FLT3i (quizartinib, midostaurin, sorafenib) on cell proliferation and cell cycle kinetics was assessed in AML cell lines with differing FLT3 status; HEL (negligible expression of wild-type FLT3), EOL1 (wild-type FLT3), MV4–11 (FLT3-ITD resulting in constitutively active isoform). Semi-mechanistic cell cycle models for CYT and FLT3i were developed. Clinical CYT and quizartinib pharmacokinetic dosage regimens were modeled. Survival of AML patients was described via a hazard model. Simulations exploring different CYT/quizartinib regimens were conducted with the goal of improving treatment outcome.
Results
FLT3 status was associated with sensitivity to CYT (HEL cells most sensitive > EOL1 > MV4–11 cells). This order of sensitivity is reversed for FLT3i. Cytarabine induced apoptosis in the S-phase while all FLT3i induced apoptosis and cell cycle arrest at G1 phase. Simulations of candidate clinical regimens predict better cell kill upon adding quizartinib simultaneously with or immediately after CYT exposure. Overall survival was predicted to be significantly better with quizartinib 200 mg administered every 48 h vs every 24 h in patients with FLT3 aberrations.
Conclusion
Simultaneous administration of quizartinib and CYT every other day is a promising combination regimen for AML patients with FLT3 mutations.
Keywords: Pharmacodynamic modeling, AML, Cell cycle, Cytotoxicity, Combination
Introduction
Despite recent progress in acute myeloid leukemia (AML) treatment, 5-year overall survival rate remains dismal at 27% [1] with cure rates for patients older than 60 years remaining less than 10% [2–4]. AML treatment typically involves induction chemotherapy for eligible patients with cytarabine (CYT) and an anthracycline [5]. Most of these aggressive malignancies are resistant to intensive CYT-based regimens and will relapse after initially achieving complete remission (CR) [6]. This poor clinical picture highlights the need for improvements at all treatment stages and lines of therapy.
Treatment failure and high relapse rates can be attributed primarily to poor cytogenetic profiles, eg, monosomal karyo-type (MK), and unfavorable genetic findings such as mutations in the FMS-like tyrosine kinase 3 (FLT3). Specifically, internal tandem duplication mutations (FLT3-ITD) are considered the most frequent somatic mutation in AML cases (~ 30% of AML cases) [7–11]. Several studies highlighted the prognostic impact of FLT3 abnormalities where AML patients with FLT3/ITD mutations have higher relapse rates, lower second CR rate, and significant survival disadvantage in both adults and pediatric patients [11–17].
Several FLT3 inhibitors (FLT3i) are currently approved or undergoing clinical testing. Midostaurin received approval by the Food and Drug Administration (FDA). Others include sorafenib, crenolinib, gilteritinib, and quizartinib. The FDA recently declined to approve quizartinib for patients with relapsed or refractory FLT3-mutated AML primarily due to concerns on lack of improvement of event-free survival and safety concerns of QTcF prolongation [18]. This suggests that despite the high potency, specificity, and sustained in vivo FLT3 inhibition of these compounds, the results have thus far been suboptimal; an expected outcome of single-agent treatment of such a resistant and aggressive malignancy [19–21]. Several mechanisms can explain the acquired resistance to FLT3i. The activation of other survival signals such as signal transducers and activators of transcription (STAT) pathway, inhibitor of apoptosis proteins (IAP), Notch, and RAS pathways, which are induced by sustained exposure to FLT3i can provide an escape mechanism from the cytostatic and cytotoxic effects of FLT3 inhibition [22–25]. Another explanation is the upregulation of FLT3 receptor expression on the blast surface to overcome FLT3 inhibition [26].
Combining conventional chemotherapeutic agents (e.g., CYT or daunorubicin) with FLT3i has the potential to resensitize cells to CYT therapy and improve treatment outcome. However, CYT and FLT3i have distinct mechanisms of action on cell cycle kinetics. CYT exerts its cytotoxic effect on the S-phase of the cell cycle specific [27, 28], while FLT3i may exhibit cytostatic as well as cytotoxic effects [29, 30]. Therefore, careful design of FLT3i/CYT dosing regimens and treatment schedules is warranted. For example, Lewis et al. [29] showed that pretreatment with FLT3 inhibitor, CEP-701, followed by CYT exposure resulted in an adverse outcome with increased FLT3/ITD AML cell proliferation suggesting an antagonistic effect with regimens involving initial FLT3i therapy followed by CYT. This could be due to FLT3i-mediated G1 cell cycle arrest and depletion of AML cells from the S-phase where CYT is believed to exert its cytotoxic effects. Thus, combining these agents requires adequate understanding of the nature of disease progression, cell cycle dynamics, and the underlying mechanisms of action of chemotherapeutic agents such as cytarabine and FLT3i.
Given the aggressive nature of AML, the complexity of treatment resistance, and the cell cycle dependency of CYT and FLT3i, semi-mechanistic models that describe the cell cycle dynamics of AML and consider the distinct effects of CYT and FLT3i on different phases of the cell cycle can provide a useful tool to guide the choice of promising dosing regimens of the CYT/FLT3i combination. Furthermore, correlating disease progression with models that predict patient survival enables early comparison of expected clinical outcomes from various dosing regimens and treatment schedules of combining CYT/FLT3i [31, 32].
In this report, an extrapolation approach based on in vitro data is proposed to improve treatment outcome with CYT/FLT3i combination regimens. We developed a cell cycle model to describe cell proliferation and cell cycle kinetics of several AML cell lines with differing FLT3 status. The model accounts for the development of resistance to FLT3i. Further, we developed a survival model that adds to the cell cycle model to compare different FLT3i regimens combined with the standard of care CYT therapy [2, 33].
Materials and methods
A flowchart of the methods employed in the current analysis is presented in Supplemental Figure 1.
Cell culture and reagents
Human AML cell lines HEL, MV4–11, and EOL-1 were purchased from DSMZ (German Collection of Microorganisms and Cell Cultures). HEL cells express little or no FLT3 with no mutation (wild-type). EOL-1 cells express wild-type FLT3 which depends on FL ligand for activation [34, 35]. MV4–11 cells are FLT3/ITD resulting in constitutively active kinase and detectable levels of activated FLT3 [34]. Cell lines were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY) and cultured in 5% CO2 at 37 °C. Sorafenib and quizartinib were purchased from LC laboratories (Woburn, MA) and midostaurin was purchased from Selleckchem (Houston, TX).
Cryopreserved and fresh low-density fraction bone mar-row samples from ten AML patients, containing >75% blasts with more than 109 cells, and cord blood (CB) mononuclear cells were obtained from the Roswell Park Cancer Institute (RPCI) Hematopoietic Procurement Facility. All patients gave informed consent according to the Institutional Review Board (IRB) approval obtained from RPCI. All 10 patients failed treatment with FLT3i. Upon use, frozen samples were thawed rapidly, incubated in RPMI 1640/10% FBS medium overnight, and then subjected to another round of density centrifugation to eliminate dead cells that had undergone apoptosis during thawing. FLT3 mutation status was determined as previously described [36].
Cell growth assay
Cells (1 × 106 cells/mL) were suspended in culture media. Then 4 mL of cell suspension was added to each well of a 6-well plate and incubated with varying concentrations of CYT or FLT3i in triplicates. The number of cells was counted at each time point (0, 24, 48, 72, and 96 h after incubation with CYT or FLT3i) using a trypan blue dye exclusion assay. Each combination of drug, concentration, and cell line was replicated 3 times.
Cell cycle analysis
Cells were washed with PBS and fixed in 70% ice-cold ethanol at each time point (0, 24, 48, 72, and 96 hours after incubation with CYT or FLT3i). Cells were then stained with propidium iodide. Cell cycle analysis was performed with flow cytometer. Cell cycle distributions were used to quantify the fraction of viable cells in each phase of the cell cycle. Triplicate samples were used for every time point. The subG1 events were quantified as an indication of the number of apoptotic events. Each combination of drug, concentration, and cell line was replicated 3 times.
Cell cycle model
A compartmental model was used to describe the distribution of cells among the various phases of the cell cycle (Fig. 1). Each compartment represents a distinct phase of the cell cycle. The G2 and M phases were lumped together as both display 4 N DNA content in flow cytometry analysis and thus could not be distinguished. The assumptions employed in this model are listed in Table S1. All model parameters are listed in Table 1. First-order rate constants (k1, k2, and k3) reflect rates of transition among consecutive phases. Apoptosis was assumed to be the primary mode of cell death and cells in all phases can commit to apoptosis via a first-order rate constant, kd. The total number of viable cells determined via trypan blue exclusion experiments (Ctot) is considered the sum of the cell numbers in the G1, S, and G2∕M phases (Eq. 1). The number of viable cells in each compartment is described as the fraction of cells in each compartment as determined by flow cytometry (Eqs. 2–4). The number of cells stained with trypan blue was used to represent the death (i.e., apoptosis) compartment. The mass balance of cells in each compartment is presented in Eqs. 5–8. To reflect the reduced replication efficiency of cancer cells and the G1 cell cycle arrest observed at high cell loads, two logistic functions were introduced on k1 (the rate of transition from G1 to S phase) and k3 (the rate of transition from G2∕M to G1 phase) with Rmax being the maximum cell carrying capacity (Eqs. 5 and 7). CYT was assumed to increase cell death exclusively from the S-phase using a stimulatory Emax function on kd, STIMARA−C (Eqs. 6 and 11). The effects of FLT3 inhibitors are modeled such that they induce cell cycle arrest at the G1 phase via an inhibitory Imax function (INHFLT3) (Eqs. 6 and 9) while their cytotoxic effect are mediated by stimulating cell death from the G1 phase via a stimulatory Emax function, STIMFLT3 (Eqs. 5 and 10). The FLT3i are assumed to exhibit similar sensitivity to inducing G1 cell cycle arrest or inducing cell death from G1 phase (Eq. 11A). The development of resistance to FLT3i was assumed to be related to desensitization of FLT3 receptors upon drug exposure. The number of FLT3 receptors is assumed to be proportional to the capacity of cell cycle arrest via the FLT3i, Imax term. Thus, FLT3 exposure will lead to FLT3 receptor desensitization and consequently reduced Imax. Further, the FLT3i are assumed to exhibit full inhibition capacity at the start of treatment (i.e., Imax(0) = 1). At steady state, the rates of receptor synthesis and turnover are the same (Eq. 13). The change in Imax was modeled using a desensitization model as described in Eqs. 12–14. The full model equations are presented below:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (11A) |
Fig. 1.
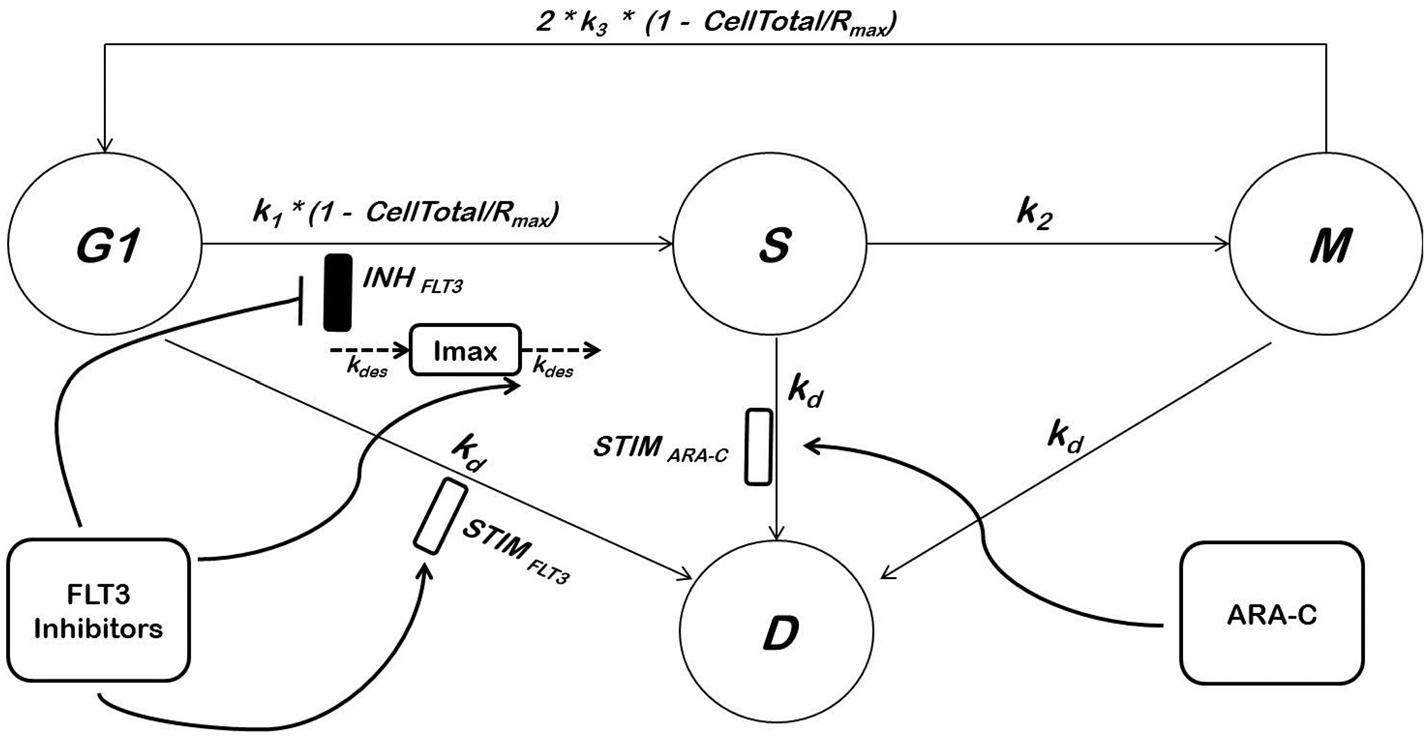
Compartmental model of FLT3 inhibitor and CYT cell cycle and apoptotic effects. Please refer to text and Table 1 for details and definitions
Table 1.
Pharmacodynamic model parameter estimates for MV4–11
| Parameter | Definition | Value | CV (%) |
|---|---|---|---|
| k1 (/h) | G1−S 1st-order rate transition | 0.037 | 4.0 |
| k2 (/h) | S−G2/M 1st-order rate transition | 0.06 | 3.1 |
| k3 (/h) | G2/M−G1 1st-order rate transition | 0.17 | 4.2 |
| kd (/h) | 1st-order death rate | 0.0012 | 16.9 |
| Rmax (cells/mL) | Maximum cell carrying capacity | 5 × 1020 | Fixed |
| Imax (no unit) | Maximum inhibition capacity of FLT3i | 1 | Fixed |
| EmaxQuizartinib (no unit) | Maximum stimulation of cell death from G1 phase by quizartinib | 8.0 | 24.2 |
| Sensitivity to quizartinib | 1.2 | 14.8 | |
| EmaxMidostaurin (no unit) | Maximum stimulation of cell death from G1 phase by midostaurin | 81.8 | 19.0 |
| Sensitivity to midostaurin | 92.3 | 14.4 | |
| EmaxSorafenib (no unit) | Maximum stimulation of cell death from G1 phase by FLT3 sorafenib | 39.0 | 18.5 |
| Sensitivity to sorafenib | 10.6 | 10.8 | |
| Emax ARA−C | Maximum stimulation of cell death by CYT | 304.8 | 21.9 |
| Sensitivity to cytarabine | 2370.8 | 20.3 | |
| kdes (no unit) | Desensitization rate | 1.4 × 10−5 | 23.5 |
If , then
| (12A) |
If , then
| (12B) |
Assuming,
| (13) |
Equations 12A and 12B can be reduced to: If , then
| (14A) |
If , then
| (14B) |
Model fittings and simulations
The cell cycle model was fitted to growth curves and cell cycle distributions of sorafenib, quizartinib, midostaurin, and CYT incubated with each cell line simultaneously to determine the transition rate constants (k1, k2, k3, and kd) that govern the growth of each cell line as well as drug-related parameters (, , , , and kdes). All model fittings were conducted using the non-linear maximum likelihood estimation algorithm in Phoenix (Version 6.4). Goodness-of-fit was judged by inspection, residuals, and the Akaike Information Criterion (AIC). Model simulations were run in Berkeley Madonna using the Stiff integration method.
Simulations of clinical response to therapy
To predict the dynamics of leukemia upon treating AML patients with various dosing regimens of FLT3i and CYT, the integrated PK/disease progression model was used to perform clinical trial simulations using different dosing regimens and treatment schedules. Plasma concentration versus time profiles for midostaurin and CYT were digitized from the literature using Graph Digitizer 2.1 × software [37, 38]. PK models were fit to midostaurin and CYT PK data using maximum likelihood estimation algorithm in Phoenix (Version 6.4). Estimated PK parameters and the associated inter-individual variability [39, 40] were fixed and used as input in the pharmacodynamic model. Cell cycle-related parameters (k1, k2, k3, and kd) and drug-related parameters (, , , , and kdes) were fixed to the estimates obtained from the cell cycle model. Plasma concentrations were corrected for protein binding as obtained from the literature [38, 41]. Initial leukemic cell counts in AML patients were fixed to clinically plausible value (6 × 1010 cells/L) [42]. Distribution of cells among different cell cycle phases (G1F, SF, and G2∕MF) was assumed to follow the same distribution as observed in the in vitro cell line data. The effects of different dose levels, dosing interval, and sequence of administration of CYT and midostaurin in combinations were explored via simulations in Berkeley Madonna.
Survival model
We mathematically described the effects of different covariates such as genetic representation on survival. Survival data from [33] was digitized using Graph Digitizer 2.1× (Supplemental Figure 2). The rate of patient deaths was described by the hazard function:
| (15) |
where HR is the hazard rate related to patient’s molecular information (GEN) with the coefficient α1, and the intercept of the hazard function, α0. Patients were stratified according to their cytogenetic and molecular profiles into two prognostic groups as previously described [33] with the favorable genetic profile (patients with mutant CEBPA or mutant NPM1 without FLT3-ITD) assigned a value of 0 while patients with unfavorable genetic profile, i.e., other genotypes, were assigned a value of 1 for GEN. ‘Other genotypes’ were defined as in [33], i.e., the FLT3-ITD genotype and the triple-negative genotype consisting of wild-type NPM1 and CEBPA without FLT3-ITD (Supplemental Fig. 2). Survival of AML patients was described as:
| (16) |
Linking disease progression and drug exposure to patient survival
To simulate the clinical impact of different quizartinib treatment regimens on patient survival, we linked the cell cycle model, which describes AML disease progression, drug exposure, sensitivity, and resistance to FLT3 inhibition to overall survival. Thus, for each quizartinib proposed regimen, a Monte Carlo simulation of a clinical trial of 100 patients was conducted using the parametric simulation method in Berkeley Madonna version 9.1.19 [43]. This simulation included variability on both cell cycle parameters as well as inter-individual variability on PK exposure for both CYT and quizartinib [39, 40]. Two quizartinib dosing regimens were simulated (either 200 mg once daily or 200 mg once every two days) in combination with cytarabine according to the following regimen (Cycle 1: 1 gm/m2 every 12 h for the 6 days, i.e., 2.3 mg for a 70 kg patient, followed by 2 weeks cytarabine-free period, followed by Cycle 2: 2 gm/m2 infusion for 6 h twice daily on days 1, 2, 4, and 6). All parameter values were fixed to that obtained from the model fitting for the in vitro data using MV4–11 cell line parameter values with inter-individual variability parameters assumed to be equal to inter-flask variability. Desensitization of quizartinib Imax was linked to free plasma concentrations (assumed to be 0.5%). Patient survival was assumed to be related to leukemic cell load [42]. Survival was assumed to be correlated to the time taken by the leukemic cell load to reach the maximum cell number in a patient (Time to Rmax, TTR). The value for Rmax was set to a clinically plausible value (1 × 1014 cells/L) [42]. The values for TTR were assigned to the survival data such that patients with lower survival have shorter TTR.
Results
Response of AML cell lines to CYT and FLT3 Inhibitors
Three cell lines (MV4–11, EOL-1, and HEL) were exposed to a wide range of concentrations of CYT and FLT3i. The sensitivity estimates for each of the cell lines to different treatments (i.e., KC50) estimated via the cell cycle model are presented in Supplemental Table 2. The sensitivity of FLT3/ITD mutant cell line MV4–11 to FLT3i was similar to that in the wild-type FLT3 expressing cell line EOL-1 as demonstrated by similar KC50 values (Supplemental Table 2). Quizartinib, midostaurin, and sorafenib showed no effects on viable cell counts or cell cycle distribution of the HEL cell line that lacks FLT3 (Supplemental Table 2, Supplemental Fig. 4). The MV4–11 and EOL-1 cells lines were less sensitive to CYT than HEL cell line (Supplemental Table 2). The EOL-1 cell line was more sensitive to sorafenib (KC50 = 1 nM) than the MV4–11 cell lines (KC50 = 81.1 nM) (Supplemental Table 2). FLT3i induced cell cycle arrest at the G1 phase of the FLT3/ITD mutant cell line MV4–11 and the wild-type FLT3 expressing cell line EOL-1 (Fig. 2). The time course for the viable cell count for the MV4–11 cell line in response to cytarabine and FLT3i is presented in Supplemental Fig. 3. Given that quizartinib was the most potent FLT3i in the MV4–11 cell line (Supplemental Table 2) and given the similar sensitivity of the MV4–11 and EOL-1 cell lines to the FLT3i tested, further experiments were mainly focused on quizartinib and the FLT3/ITD mutant cell line MV4–11.
Fig. 2.
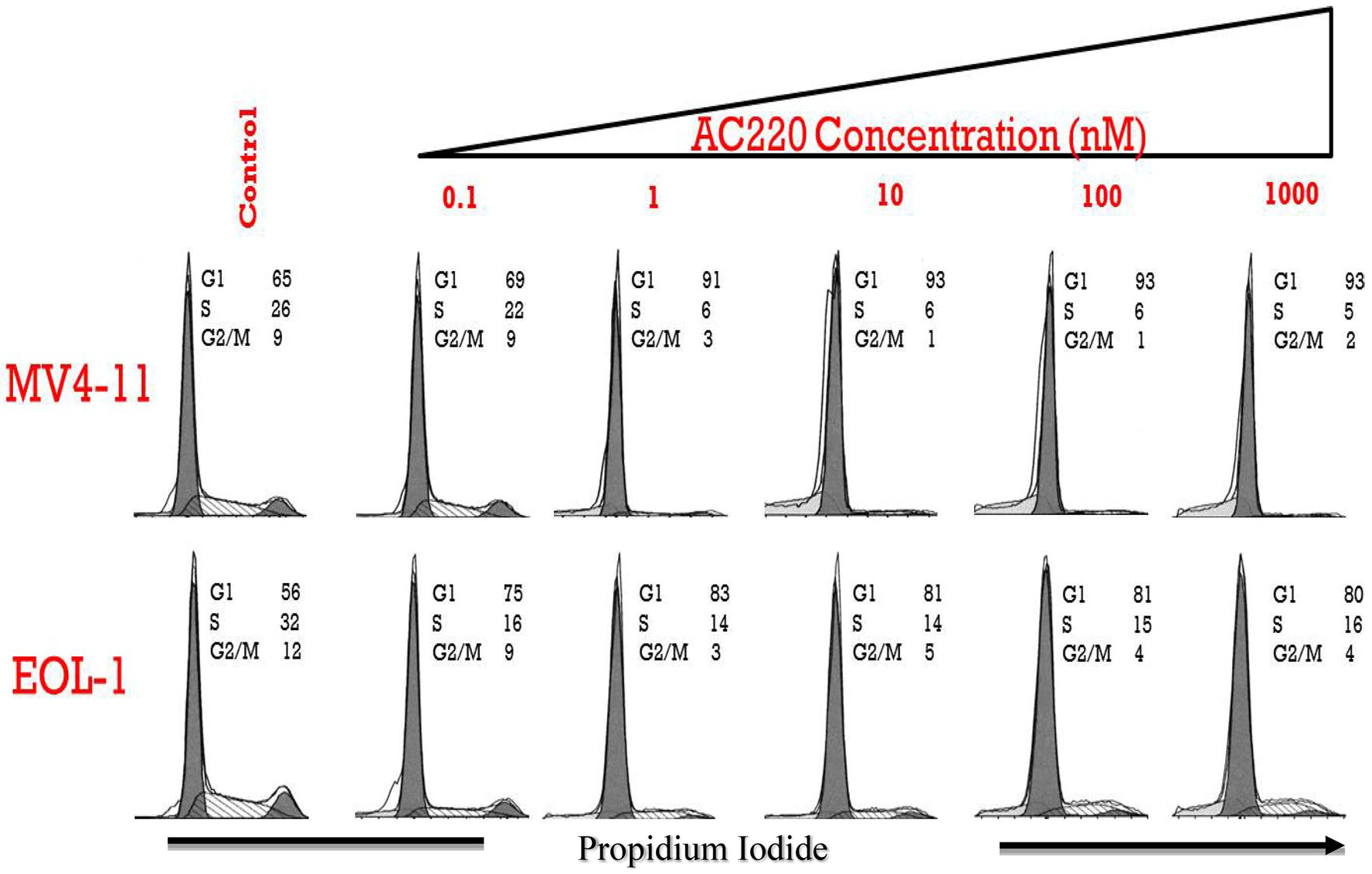
Cell cycle profile of cells treated with quizartinib. AC220 = quizartinib
Cell cycle model
The cell cycle model (Fig. 1) was fit to cell cycle/cell proliferation data from different drugs (CYT, quizartinib, midostaurin, sorafenib) for the MV4–11 cell line simultaneously to obtain cell line-specific cell cycle-related parameters (k1, k2, k3, and kd). Parameter estimates for MV4–11 cells are provided in Table 1. Model parameters were estimated with reasonable precision. Since several FLT3i are currently undergoing clinical trials, comparing their cytostatic and cytotoxic effects can be beneficial. For MV4–11 cell line, quizartinib was the most potent FLT3i (KC50=1.2 ng/mL, 95% CI 0.91–1.66 ng/mL), followed by sorafenib (KC50 = 10.6 ng/mL, 95% CI 8.4–12.9 ng/mL), and midostaurin (KC50 = 92.3 ng/mL, 95% CI 66.3–118.3 ng/mL).
Predicting optimal dose and dosing frequency for FLT3i in combination with CYT
To test the effects of different quizartinib dosing regimens on the leukemic cell load, we digitized plasma concentration profiles for quizartinib and CYT [37, 44]. A one-compartment model with first-order absorption and elimination rates well described quizartinib PK; while a two-compartment model with linear elimination was used to describe CYT PK. Estimated PK parameters (Supplemental Table 3) were fixed and used to drive the pharmacodynamic model. All PD parameters were fixed to estimates obtained from model fitting of in vitro data.
In order to investigate how the dosing interval and the dose level affect cell proliferation and cell cycle redistribution, simulations using the cell cycle model were conducted with varying the magnitude of each of these variables individually while setting the other to a constant value. First, we tested the optimal dosing interval for the selected dose level (200 mg). Dosing intervals of daily, every 2, 3, and 4 days were tested. Model simulations showed that a dosing interval of every 2 days prolonged TTR by almost one year compared to once-daily dosing and maintained relatively low AML cell counts for a longer duration (Fig. 3a). All dosing regimens including the every 48-h interval resulted in decline in Imax suggesting that development of resistance to FLT3i might be inevitable, however, a once-daily dosing resulted in more potent initial decline in leukemic cell counts (Fig. 3a) as well as faster decline in Imax and more pronounced resistance to FLT3i (Fig. 3b) vs every 48-h. Every 3- and every 4-day dosing of quizartinib resulted in faster disease progression versus every 2 days.
Fig. 3.
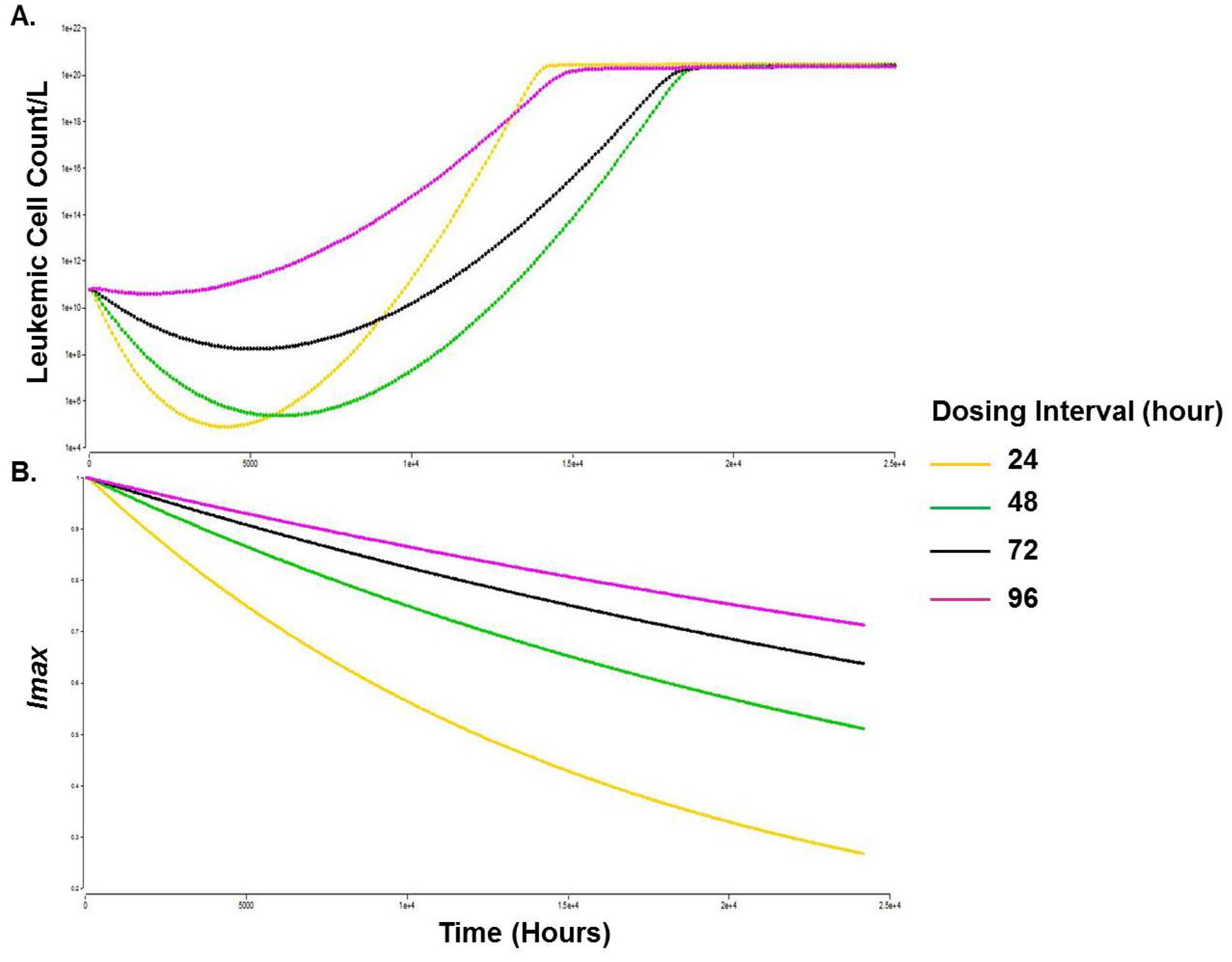
Simulations of time course of leukemic cell counts, (a) and the decline in Imax, (b) with different dosing intervals. Time scale (0–25,000 h, i.e., ~ 34 months). Leukemic cell count/L, Y-axis (1 × 104 to 1 × 1022). Imax, Y axis (0.2–1)
Further, we tested whether the clinically tested 200 mg dose of quizartinib is optimal for efficacy while fixing the dosing interval as once every 48 h. Model simulations indicate that the 200 mg dose level achieved most cell kill and prolonged TTR. Dose escalation to 300 mg or 400 mg resulted in faster time to nadir and slightly more temporal reductions in AML cell counts, but also faster rates of resistance developed from exposure to high quizartinib concentrations that led to faster growth of AML cells and shorter TTR (Fig. 4). Doses less than 200 mg were not effective in reducing the leukemic cell count (Fig. 4). Introducing a quizartinib holiday of up to 1 week resulted in a worse AML cell load and did not prolong TTR (data not shown).
Fig. 4.
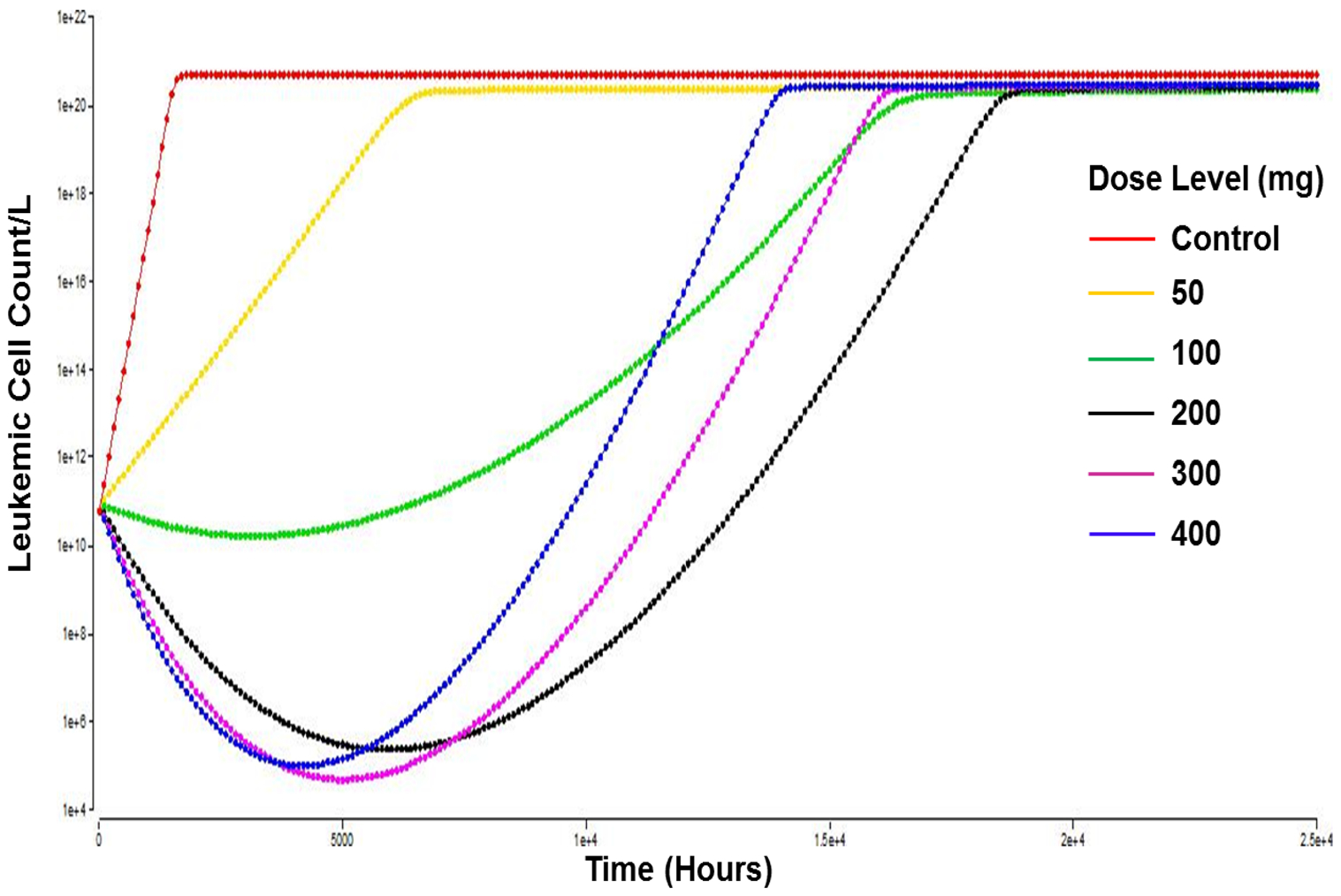
Simulations of time course of leukemic cell counts with different indicated quizartinib dose levels. The dosing frequency was selected as every 48 h. The simulations show a better response with the 200 mg regimen. Time scale, X axis, (0–25,000 h, i.e., ~ 34 months). Leukemic cell count/L, Y axis (1 × 104 to 1 × 1022)
Survival function adequately described AML patient overall survival
Important prognostic covariates such as patient cytogenetic and molecular profiles were digitized from the literature and a survival function was developed to describe survival profile of different groups of AML patients as digitized and presented in Fig. 2 [33]. Estimates of the α0 (the slope of the hazard function) and α1 (coefficient for genetic representation) were 1.13 and 0.1. The positive value for α1 indicates that an unfavorable genetic profile is associated with larger hazard ratio and consequently shorter survival. The survival function was then used to predict patient survivals based on the genetic profile, which can guide treatment options.
Linking disease progression and drug exposure to patient survival
For each quizartinib regimen (once-daily and once every other day), a clinical trial of 100 patients were simulated using the parametric simulation method [43]. All parameter values were fixed to that obtained from the in vitro experiments using MV4–11 cell line parameter values with the assumption that patient survival is associated with the time for leukemic cell burden to reach a plausible threshold count, i.e., TTR. Once the leukemic cell burden reaches the threshold count, the patient is assumed to succumb to the disease. Results of the clinical trial simulations showed a better outcome for the once every other day quizartinib regimen with median overall survival of 12.6 and 16.7 months for the every 24-h and every 48-h regimens, respectively (Fig. 5).
Fig. 5.
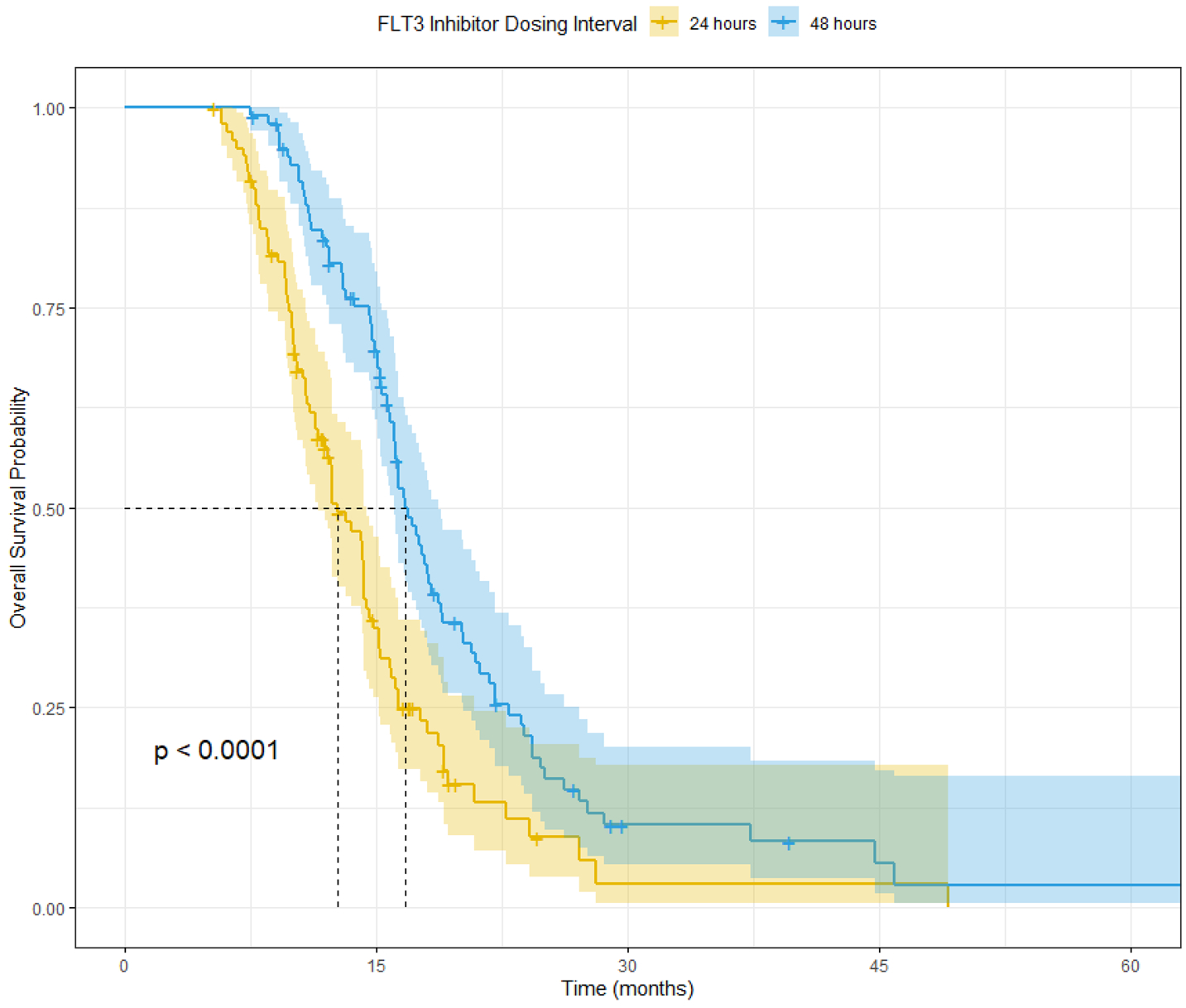
Clinical trial simulation for time course of overall survival based on in vitro data of quizartinib once-daily and once every 2 days quizartinib dosing regimen simultaneously administered with cytarabine as described in [54]. Median simulated OS (95% confidence interval) for the once daily are 12.6 (11.5, 14.4) months and the once every 2 days regimen are 16.7 (16.0, 18.7) months
Discussion
AML patients with FLT3/ITD mutations have poor response to cytarabine-based intensive chemotherapy. FLT3i may resensitize cells to CYT and improve treatment outcomes. FLT3i and CYT exert distinct mechanisms of action on the cell cycle. Attempts at improving the clinical outcome of AML patients should adopt translational models that account for the underlying genetic makeup of the cancerous cells, the cell cycle effects of standard treatments such as CYT, and the potential benefit from adding FLT3i to the treatment paradigm. In this report, we investigated the effect of CYT and several FLT3i on cell proliferation and cell cycle kinetics in AML cell lines. A mechanistic cell cycle model was developed to describe the distribution of cells among the various phases of the cell cycle, predict clinical outcome, and explore different CYT/quizartinib combination regimens.
Quizartinib, midostaurin, and sorafenib showed no effect on the viable cell count or cell cycle distribution of the HEL cell line that lacks FLT3. This indicates a highly specific mechanism of action of these agents. The MV4–11 and EOL-1 cells lines were less sensitive to CYT than HEL cell line, which is consistent with FLT3 conferring resistance to CYT therapy. The EOL-1 cell line was sensitive to sorafenib, which is consistent with previous reports on the sensitivity of this cell line to sorafenib potentially due to the inhibition of other oncogenes namely FIP1L1-PDGFRA [45].
In vitro drug exposure response curves are used to understand cell line sensitivity to drug candidates. Terms like IC50 or IC90 are often used as potency parameters without consideration of the dynamic nature of cancer cells, the shape (i.e., steepness) of the curve, and/or the probability of resistance mechanisms counteracting the drug effect. Cell cycle models have been previously used to describe the dynamic nature of cancer cells and distinguish the effects of chemotherapeutic agents on cancer cell lines [46–48]. In this report, we developed a pharmacodynamic model that mechanistically describes the effects of CYT and quizartinib on cell proliferation and cell cycle distribution. The model accounts for the dynamics of resistance to quizartinib. The semi-mechanistic nature of the model allows for exploring different combination regimens and making informed predictions on treatment outcomes.
Progression of cancer cells from the G0 quiescent phase to G1 and subsequently to S-phase is mediated via a growth factor-rich environment [49]. At high cell densities, the harsh microenvironment leads to depletion of growth factors. This can be mimicked in vitro by serum depletion or culturing cells at extremely high cell densities leading to synchronizing cells at the G0 phase [50]. Replication efficiency (i.e., duplication of number of cells via transitioning from M to G1 phase) is considerably reduced at high cell loads. To mimic these observations, a logistic function on the G1 to S transition rate, k1, and another logistic function on the M to G1 transition rate, k3, were included. At high cell densities, the distribution of cells between phases will depend on the values of k1 and k3. If k1 <<< k3, cells will accumulate at the G1 phase (Supplemental Fig. 5). Since the M to G1 replication rate is usually fast and almost invariable in cancer cells (~ 1 h), thus, at high cell densities, cells will accumulate in the G1 phase.
FLT3i are known to exert cytotoxic effects as seen on Annexin V staining [29, 30]. Since FLT3i also induced G1 cell cycle arrest (Fig. 2), we assumed that their cytotoxic effect is induced mainly from the G1 phase. In this model to allow for parameter identifiability, FLT3i were assumed to exhibit similar sensitivity to inducing G1 cell cycle arrest or inducing cell death from G1 phase. This assumption may be supported by the high residence time of cells in the G1 phase in presence of a FLT3i and therefore, cytotoxic effects of these drugs could be exerted on the G1 phase. Quizartinib-induced cell cycle arrest at the G1 phase is concentration-dependent. Interestingly, the percentage of cells in G1 phase upon quizartinib exposure is 80–93% in the MV4–11 and EOL-1 cell lines. This indicates that FLT3 inhibition is associated with almost complete blockage of the cell cycle at the G1 phase. Thus, we employed this assumption and set the initial conditions of Imax to 1.
AML cells are known to develop resistance either by prolonged incubation with FLT3i or by exposing the cells to high concentrations of FLT3i. For example, Weisberg et al. [24] generated FLT3-resistant cell lines by incubating MOLM13 AML cell line with high midostaurin concentrations for 21 days. To reflect the development of resistance to FLT3i, we assumed that Imax decreases upon exposure to FLT3i. The development of resistance is thought to be related to the extent of FLT3 inhibition and thus activation of alternative signaling pathways that overcome the FLT3 inhibitor-induced cell cycle arrest [22–25]. Therefore, the reduction of Imax was correlated to inhibition sensitivity parameters (i.e., Imax decreases if FLT3i concentrations are higher than . Similarly, restoration of FLT3i sensitivity is reflected by an increase in Imax when FLT3i concentrations fall below . This structure suggests that initial potent FLT3i is likely to develop resistance faster (i.e., when FLT3i concentrations >>> ). This assumption is supported by AML cell lines restoring their sensitivity to FLT3i upon withdrawal of FLT3i for several days or weeks [24].
According to our cell cycle model parameterization, higher drug concentrations may adversely affect treatment response in long-term clinical trials as resistance can develop faster with prolonged exposure to FLT3 inhibitor concentrations higher than . Therefore, different FLT3i dosing regimens can be explored to maximize the clinical benefit from these agents while reducing the development of resistance. To describe the PK exposure of quizartinib and CYT, we digitized plasma concentration profiles and developed separate PK models to describe their exposure. We acknowledge that data digitization may result in imperfect number values. However, the PK profiles are only used as an approximation of the average PK in human subjects, but demonstrate key principles.
Model simulations showed that a once-daily quizartinib dosing regimen resulted in more potent initial decline in leukemic cell count compared to once every 48-h dosing. However, the once-daily regimen was associated with more pronounced resistance to quizartinib as demonstrated via faster decline in Imax. Every 3- and every 4-day dosing of quizartinib resulted in faster disease progression versus every 2 days suggesting that every 2 days dosing provides a reasonable balance between maintaining efficacious exposure and slowing the rate of resistance development. The quizartinib dose of 200 mg dose was shown to be adequate for efficacy. Model simulations indicated that the 200 mg dose achieved most cell kill and prolonged TTR. Dose escalations to 300 mg or 400 mg resulted in faster time to nadir and slightly more temporal reduction in AML cell count but faster rates of resistance developed from exposure to high quizartinib concentrations that led to faster growth of AML cells and shorter TTR. Interestingly, doses less than 200 mg were not effective in reducing the leukemic cell count (Fig. 4). These findings might explain the modest efficacy observed in a pivotal study in patients with FLT3 mutant AML where quizartinib was given at doses ≤ 60 mg once daily; which led to FDA rejection for quizartinib for this indication over concerns of lack of survival benefit as well as increased QTc prolongation [18].
Finally, we tested whether a drug holiday of 1 week will help restore sensitivity to quizartinib. Introducing quizartinib holidays of up to 1 week resulted in a worse AML cell load and did not prolong TTR (data not shown). This indicates that a drug holiday cannot be afforded as the cost of loss in efficacy outweighs the benefit gained by restoring drug sensitivity. Based on the current model, the development of resistance to quizartinib will eventually take place since the rate of restoration of drug sensitivity is slower than that of developing the resistance.
The premise of treating AML patients is reducing the cancer burden and improving overall survival. In different subsets of AML patients, high white blood cell counts at presentation are associated with poor prognosis and response to treatment [5, 51, 52]. Furthermore, patients harboring FLT3/ITD mutations are presented with high white blood cell counts [10, 53]. Thus, we assumed that patient survival is associated with time for leukemic cell burden to reach a plausible threshold count, i.e., TTR. With this assumption, patients with higher leukemic cell counts will have shorter TTR and lower survival probability. The assumption links the in vitro drug sensitivity results, which affects the leukemic cell counts, with treatment outcome, and allows testing the effects of different treatment regimens on patient survival. Thus, for each quizartinib regimen (once-daily and once every other day), a clinical trial of 100 patients was simulated using the parametric simulation method [43]. These simulations should be used with caution as there are several assumptions built in them including the use of in vitro estimates without scaling, as they do not account for differences in protein binding or inter-individual variability in PK exposure. Furthermore, the inter-individual variability in treatment response may be underpredicted in the Monte Carlo simulations as it is assumed to be equal to in vitro variability. That said, interestingly, the median overall survival in the simulations (12.6 and 16.7 months) (Fig. 5) was similar to that observed clinically in the same patient population [33]. This finding is also consistent with the simulations suggesting that every other day dosing might provide additional benefit for patients compared to once-daily regimen.
In conclusion, we developed a cell cycle model that represents a mechanistic interpretation for cytarabine and quizartinib effects in AML cell lines. Further work is warranted to verify the assumptions included in this exercise such as: (a) the similar sensitivity of FLT3i to inducing G1 cell cycle arrest or inducing cell death from G1 phase; (b) the direct application of the PD model developed using in vitro experiments to clinical outcome; (c) patient survival being directly linked to leukemic cell load, and (d) to evaluate the effects of inter-individual variability in PK and response on treatment outcomes. While acknowledging that these assumptions included in the modeling approach might affect the utility of this translational exercise, this model is intended to provide a tool to explore different FLT3i treatment regimens with standard cytotoxic therapies, both from sequencing and dosing perspective. Our results suggest that 200 mg quizartinib every other day simultaneously administered with CYT might improve treatment outcome for AML patients. These results bear consideration in light of the recent FDA rejection of quizartinib in relapsed or refractory FLT3-mutated AML primarily due to concerns on lack of improvement of event-free survival and safety concerns [18].
Supplementary Material
Acknowledgements
We dedicate this article to the memory of Dr. Meir Wetzler, an outstanding person, mentor, clinician, and scientist.
Funding This work was supported by NIH Grants GM 24211, GM 131800 and CA 16056 and the University at Buffalo-Novartis Fellowship.
Footnotes
Conflicts of interest The authors declare no potential conflicts of interest.
Ethical approval All samples obtained from patients were done in accordance with approval by the Investigational Review Board of Roswell Park Comprehensive Cancer Center. Informed consent. Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00280-020-04114-z) contains supplementary material, which is available to authorized users.
References
- 1.Institute NC (2018) Surveillance, Epidemiology, and End Results (SEER) Program
- 2.Buchner T, Berdel WE, Haferlach C, Haferlach T, Schnittger S, Muller-Tidow C et al. (2009) Age-related risk profile and chemotherapy dose response in acute myeloid leukemia: a study by the german acute myeloid leukemia cooperative group. J Clin Oncol 27(1):61–69 [DOI] [PubMed] [Google Scholar]
- 3.Juliusson G, Antunovic P, Derolf A, Lehmann S, Mollgard L, Stockelberg D et al. (2009) Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood 113(18):4179–4187 [DOI] [PubMed] [Google Scholar]
- 4.Rees JK, Gray RG, Swirsky D, Hayhoe FG (1986) Principal results of the Medical Research Council’s 8th acute myeloid leukaemia trial. Lancet 2(8518):1236–1241 [DOI] [PubMed] [Google Scholar]
- 5.Estey E, Smith TL, Keating MJ, McCredie KB, Gehan EA, Freireich EJ (1989) Prediction of survival during induction therapy in patients with newly diagnosed acute myeloblastic leukemia. Leukemia 3(4):257–263 [PubMed] [Google Scholar]
- 6.Keating MJ, Kantarjian H, Smith TL, Estey E, Walters R, Andersson B et al. (1989) Response to salvage therapy and survival after relapse in acute myelogenous leukemia. J Clin Oncol 7(8):1071–1080 [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S et al. (2001) Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97(8):2434–2439 [DOI] [PubMed] [Google Scholar]
- 8.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K et al. (1996) Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 10(12):1911–1918 [PubMed] [Google Scholar]
- 9.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F et al. (2004) The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell 13(2):169–178 [DOI] [PubMed] [Google Scholar]
- 10.Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S et al. (1999) Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood 93(9):3074–3080 [PubMed] [Google Scholar]
- 11.Rombouts WJ, Blokland I, Lowenberg B, Ploemacher RE (2000) Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia 14(4):675–683 [DOI] [PubMed] [Google Scholar]
- 12.Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT (2001) Genomic structure of human FLT3: implications for mutational analysis. Br J Haematol 113(4):1076–1077 [DOI] [PubMed] [Google Scholar]
- 13.Abu-Duhier FM, Goodeve AC, Wilson GA, Gari MA, Peake IR, Rees DC et al. (2000) FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br J Haematol 111(1):190–195 [DOI] [PubMed] [Google Scholar]
- 14.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA et al. (2001) The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 98(6):1752–1759 [DOI] [PubMed] [Google Scholar]
- 15.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD et al. (2001) Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res 61(19):7233–7239 [PubMed] [Google Scholar]
- 16.Knapper S (2011) The clinical development of FLT3 inhibitors in acute myeloid leukemia. Expert Opin Investig Drugs 20(10):1377–1395 [DOI] [PubMed] [Google Scholar]
- 17.Ravandi F, Kantarjian H, Faderl S, Garcia-Manero G, O’Brien S, Koller C et al. (2010) Outcome of patients with FLT3-mutated acute myeloid leukemia in first relapse. Leuk Res 34(6):752–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.FDA (2019) Oncologic Drugs Advisory Committee (ODAC): Quizartinib in AML. https://www.fdagov/media/124896/download. Accessed 18 Mar 2020
- 19.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K et al. (2004) Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 103(10):3669–3676 [DOI] [PubMed] [Google Scholar]
- 20.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD et al. (2005) Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 105(1):54–60 [DOI] [PubMed] [Google Scholar]
- 21.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C et al. (2010) Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 28(28):4339–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG et al. (2009) Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood 113(17):4052–4062 [DOI] [PubMed] [Google Scholar]
- 23.Stolzel F, Steudel C, Oelschlagel U, Mohr B, Koch S, Ehninger G et al. (2010) Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann Hematol 89(7):653–662 [DOI] [PubMed] [Google Scholar]
- 24.Weisberg E, Ray A, Nelson E, Adamia S, Barrett R, Sattler M et al. (2011) Reversible resistance induced by FLT3 inhibition: a novel resistance mechanism in mutant FLT3-expressing cells. PLoS ONE 6(9):e25351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruggeri B, Singh J, Gingrich D, Angeles T, Albom M, Yang S et al. (2003) CEP-7055: a novel, orally active pan inhibitor of vascular endothelial growth factor receptor tyrosine kinases with potent antiangiogenic activity and antitumor efficacy in preclinical models. Cancer Res 63(18):5978–5991 [PubMed] [Google Scholar]
- 26.Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, Chopra R et al. (2006) A phase 2 trial of the FLT3 inhibitor lest-aurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 108(10):3262–3270 [DOI] [PubMed] [Google Scholar]
- 27.Momparler RL (1974) A model for the chemotherapy of acute leukemia with 1-beta-D-arabinofuranosylcytosine. Cancer Res 34(8):1775–1787 [PubMed] [Google Scholar]
- 28.Leclerc JM, Momparler RL (1984) Effect of the interval between exposures to cytarabine on its cytotoxic action on HL-60 myeloid leukemic cells. Cancer Treat Rep 68(9):1143–1148 [PubMed] [Google Scholar]
- 29.Levis M, Pham R, Smith BD, Small D (2004) In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood 104(4):1145–1150 [DOI] [PubMed] [Google Scholar]
- 30.Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD et al. (2002) A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 99(11):3885–3891 [DOI] [PubMed] [Google Scholar]
- 31.Claret L, Girard P, Hoff PM, Van Cutsem E, Zuideveld KP, Jorga K et al. (2009) Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol 27(25):4103–4108 [DOI] [PubMed] [Google Scholar]
- 32.Mould DR (2012) Models for disease progression: new approaches and uses. Clin Pharmacol Ther 92(1):125–131 [DOI] [PubMed] [Google Scholar]
- 33.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L et al. (2008) Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358(18):1909–1918 [DOI] [PubMed] [Google Scholar]
- 34.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG (2003) FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 17(1):120–124 [DOI] [PubMed] [Google Scholar]
- 35.Ampasavate C, Jutapakdee W, Phongpradist R, Tima S, Tanti-worawit A, Charoenkwan P et al. (2019) FLT3, a prognostic bio-marker for acute myeloid leukemia (AML): quantitative monitoring with a simple anti-FLT3 interaction and flow cytometric method. J Clin Lab Anal 33(4):e22859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD et al. (2003) Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn 5(2):96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan SH, Huffman DH, Azarnoff DL, Hoogstraten B, Larsen WE (1974) Pharmacokinetics of 1-beta-D-arabinofuranosylcytosine in humans. Cancer Res 34(2):392–397 [PubMed] [Google Scholar]
- 38.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B et al. (2009) AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 114(14):2984–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krogh-Madsen M, Bender B, Jensen MK, Nielsen OJ, Friberg LE, Honore PH (2012) Population pharmacokinetics of cytarabine, etoposide, and daunorubicin in the treatment for acute myeloid leukemia. Cancer Chemother Pharmacol 69(5):1155–1163 [DOI] [PubMed] [Google Scholar]
- 40.Yin OQ, Wang Y, Schran H (2008) A mechanism-based population pharmacokinetic model for characterizing time-dependent pharmacokinetics of midostaurin and its metabolites in human subjects. Clin Pharmacokinet 47(12):807–816 [DOI] [PubMed] [Google Scholar]
- 41.Slevin ML (1984) Relationship between protein binding and extravascular drug concentrations of a water-soluble drug, cytosine arabinoside. J R Soc Med 76(5):365–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Jonge HJ, Valk PJ, de Bont ES, Schuringa JJ, Ossenkoppele G, Vellenga E et al. (2011) Prognostic impact of white blood cell count in intermediate risk acute myeloid leukemia: relevance of mutated NPM1 and FLT3-ITD. Haematologica 96(9):1310–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Y, Kosorok MR, Zeng D (2009) Reinforcement learning design for cancer clinical trials. Stat Med 28(26):3294–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.James J et al. (2008) Clinical pharmacokinetics and FLT3 phosphorylation of AC220, a highly potent and selective inhibitor of FLT3. Am Soc Hematol 2008:2637 [Google Scholar]
- 45.Lierman E, Folens C, Stover EH, Mentens N, Van Miegroet H, Scheers W et al. (2006) Sorafenib is a potent inhibitor of FIP1L1-PDGFRalpha and the imatinib-resistant FIP1L1-PDGFRalpha T674I mutant. Blood 108(4):1374–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Florian JA Jr, Eiseman JL, Parker RS (2005) Accounting for quiescent cells in tumour growth and cancer treatment. Syst Biol (Stevenage) 152(4):185–192 [DOI] [PubMed] [Google Scholar]
- 47.Sherer E, Hannemann RE, Rundell A, Ramkrishna D (2006) Analysis of resonance chemotherapy in leukemia treatment via multistaged population balance models. J Theor Biol 240(4):648–661 [DOI] [PubMed] [Google Scholar]
- 48.Hamed SS, Roth CM (2011) Mathematical modeling to distinguish cell cycle arrest and cell killing in chemotherapeutic concentration response curves. J Pharmacokinet Pharmacodyn 38(3):385–403 [DOI] [PubMed] [Google Scholar]
- 49.Sherr CJ (1996) Cancer cell cycles. Science 274(5293):1672–1677 [DOI] [PubMed] [Google Scholar]
- 50.Davis PK, Ho A, Dowdy SF (2001) Biological methods for cell-cycle synchronization of mammalian cells. Biotechniques 30(6):1322–1326 [DOI] [PubMed] [Google Scholar]
- 51.Dutcher JP, Schiffer CA, Wiernik PH (1987) Hyperleukocytosis in adult acute nonlymphocytic leukemia: impact on remission rate and duration, and survival. J Clin Oncol 5(9):1364–1372 [DOI] [PubMed] [Google Scholar]
- 52.Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M et al. (2006) Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 107(10):4011–4020 [DOI] [PubMed] [Google Scholar]
- 53.Frohling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K et al. (2002) Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood 100(13):4372–4380 [DOI] [PubMed] [Google Scholar]
- 54.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK et al. (2010) Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115(3):453–474 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.