

Abstract
Recent advances in understanding the role of eukaryotic translation initiator factor 4E (eIF4E) in tumorigenesis and cancer progression have generated significant interest in therapeutic agents that indirectly or directly target aberrant activation of eIF4E in cancer. Here, we address the general function of eIF4E in translation initiation and cancer, present evidence supporting its role in cancer initiation and progression, and highlight emerging therapeutics that efficiently target hyperactivated eIF4E. In doing so, we also highlight the major differences between these therapeutics that may influence their mechanism of action.
Background
eIF4E and translation control
The multistep process by which ribosomes are recruited to mature mRNA sequences is a highly regulated process that requires the coordination of free 40S ribosomal subunits, mRNA, and the eukaryotic translation initiator factor 4F (eIF4F) translation initiation complex. eIF4F is a trimeric complex composed of eukaryotic translation initiator factor 4E (eIF4E), the 5′ cap mRNA-binding protein; eIF4A, an RNA helicase; and eukaryotic translation initiator factor 4G (eIF4G), a scaffolding molecule. The eIF4E protein provides the critical interface between mRNA, recruitment of eIF4A and eIF4G, and the 40S ribosomal subunit because it binds to the 7-methyl guanosine cap structure at the end of mRNAs (1, 2). eIF4E is regulated at multiple levels, including transcriptionally, by phosphorylation of serine 209, as well as through inhibitory interactions with a series of binding proteins (4EBP; Fig. 1A). The tight regulation of eIF4E availability and activity provides a rapid mechanism to modulate translation in response to hypoxia, DNA damage, nutrient availability, and oncogenic stimulation.
Fig. 1.
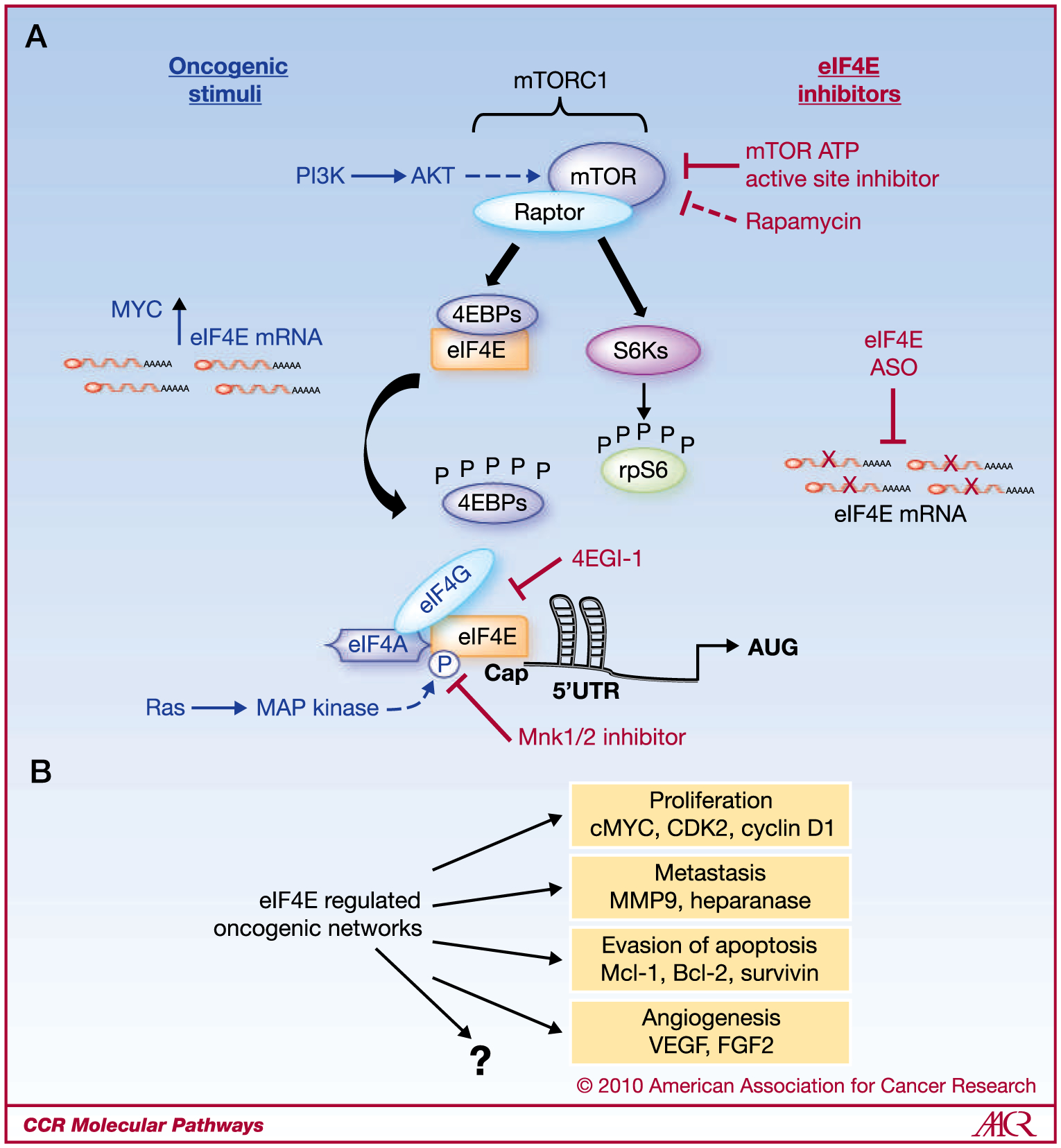
A, simplified cartoon of the 4EBP/eIF4E signaling axis. Oncogenic stimuli (blue) such as PI3K/AKT/mTOR and RAS increase eIF4E activity, whereas oncogenic MYC increases eIF4E mRNA. Hyperactivity of eIF4E can be targeted through multiple mechanisms (red), which include ATP-active site inhibitors of mTOR, MNK1/2 kinase inhibitors, 4EGI-1, and eIF4E ASOs. Rapamycin is an inefficient inhibitor of eIF4E hyperactivity. B, eIF4E regulates the translation of multiple oncogenic networks that control cell survival, proliferation, metastasis, and angiogenesis. Currently, little is known about the entire repertoire of eIF4E targets activated in cancer, their role in specific steps of eIF4E-mediated tumorigenesis and cancer progression, as well as their various effects in different tissue types.
eIF4E binding proteins 1–3 (4EBP1–3) are a family of three small acidic proteins that compete with eIF4G to bind the dorsal surface of eIF4E. In a hypophosphorylated state, 4EBPs prevent the formation of the eIF4F complex by preventing the recruitment of eIF4G to the 5′ cap of mRNA (Fig. 1A). However, upon mitogen, growth factor, and cytokine stimulation, 4EBPs are phosphorylated at serine-threonine residues leading to a conformational alteration that releases eIF4E, allowing for binding by eIF4G. The mammalian target of rapamycin (mTOR) is the primary kinase that phosphorylates 4EBPs and serves a priming function for subsequent phosphorylations that lead to the dissociation of 4EBP from eIF4E.
eIF4E is also intrinsically regulated by phosphorylation at serine 209 by the extracellular signal-regulated kinase (ERK), and p38 MAP kinase targets MNK1 and MNK2 (3–5). The functional role of eIF4E phosphorylation is context dependent. At steady state, MNK1/MNK2 knock-out mice do not exhibit any overt defects in cell growth during development (5). However, phosphorylation of serine 209 is necessary to maintain the full transforming potential of eIF4E in vitro and to potentiate MYC driven tumorigenesis in vivo (6, 7). It should be noted that in the preceding studies mutant forms of eIF4E were overexpressed. It is uncertain if endogenous eIF4E S209 phosphorylation is required for oncogenesis. In order to address this question, an eIF4E S209 knock-in mutant murine model has been generated that seems to decrease tumor-formation downstream loss of the tumor suppressor PTEN.3 Thus at this time, eIF4E phosphorylation does not seem to have a significant function in normal tissue development, but may provide a selective advantage through improved translation efficiency, which facilitates transformation in the setting of oncogenic stress.
The exact mechanism by which eIF4E and the eIF4F complex induce oncogenic transformation is hotly debated; however, it is believed to be mediated in part through enhanced translation of a subset of mRNAs involved in critical cellular processes implicated in oncogenesis such as cell proliferation, evasion of apoptosis, angiogenesis, and metastasis (8, 9). These putative targets include cyclin D1, cyclin-dependent kinase 2 (CDK2), cMYC, Mcl-1, Bcl-2, survivin, vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2), matrix metalloproteinase 9 (MMP9), and heparanase (Fig. 1B). These transcripts have long 5′ untranslated regions (5′ UTR) and high G/C contents, which result in complex hairpin structures that require the helicase activity of eIF4A to permit scanning of the 40S ribosomal subunit. These mRNAs are termed “weak” mRNA because they are poorly translated and are thought to rely more on increased eIF4E activity for their translation, thereby providing an additional barrier for tight regulation over protein expression. In the setting of oncogenic stimuli, these “weak” mRNA are given a translational boost through the hyperactivity of eIF4E, allowing for increased eIF4F complex formation and increased eIF4A helicase activity, which results in improved translational efficiency. As a result, subsets of mRNAs that may contribute to oncogenesis are selectively activated (Fig. 1B). Many of these targets have been uncovered in cell-based eIF4E overexpression systems. However, few of these targets have been validated in more physiologically relevant in vivo cancer models, so the exact role that these putative eIF4E targets play in tumor formation and progression is uncertain. eIF4E targets may also differ between different tissue types, which adds a significant layer of complexity to our understanding of translationally controlled oncogenic networks regulated by eIF4E.
eIF4E in cancer initiation and progression
The oncogenic potential of eIF4E hyperactivity has been well described both in vitro and in vivo. Overexpression of eIF4E is sufficient to induce transformation of fibroblasts and primary epithelial cells (10, 11). eIF4E overexpression in vivo leads to increased cancer susceptibility; eIF4E transgenic mice develop lymphomas, angiosarcomas, lung carcinomas, and hepatomas (12). Additionally, eIF4E overexpression can overcome MYC-induced apoptosis, which shows that eIF4E possesses intrinsic oncogenic activity that can overcome the cellular barrier of apoptosis resulting from MYC-induced oncogenic stress (12, 13). eIF4E gene amplification has been reported in human breast cancer and in head and neck cancer specimens compared with noncancerous control samples (14, 15).
Although an increase in cellular eIF4E activity is in itself oncogenic, common signaling pathways that are heavily mutated or amplified in human cancers directly impact eIF4E activity as well. For instance, the eIF4E promoter contains two E box domains that are direct targets of cMYC. cMYC overexpression drives eIF4E transcription, whereas dominant negative MYC represses eIF4E transcription (16). Our laboratory recently showed that, in the setting of oncogenic activation of the phosphoinositide 3-kinase (PI3K)/AKT/mTOR signaling pathway, phosphorylation of 4EBP1 resulting in hyperactivation of eIF4E is required for lymphomagenesis, as well as cancer progression in vivo. Interestingly, inhibition of phosphorylation of ribosomal protein S6 (rpS6; another downstream arm of mTOR) by mTOR does not decrease the rate of tumorigenesis and, thus, supports the primary role of the 4EBP1/eIF4E axis in cancer formation downstream of oncogenic mTOR (17). Activation of the oncogenic RAS- (an upstream regulator of the MNK kinase) and AKT-signaling pathways induces the formation of glioblastoma multiforme (GBM) in neural progenitor cells, which is accompanied by a dramatic upregulation in translational regulation of oncogenic networks (18). However, a critically unresolved question is whether eIF4E hyperactivation plays a direct role downstream of oncogenic MYC and RAS toward cancer progression. Downstream of oncogenic AKT, we have uncovered that blocking eIF4E hyperactivity after tumor formation in vivo results in inhibition of tumor growth (17). Taken together, eIF4E is an oncogene, as well as an important target for oncogenic translational deregulation in the context of MYC, PI3K/AKT/mTOR, and RAS overexpression (Fig. 1A). Therefore, eIF4E represents an attractive therapeutic target in human cancers because it embodies an integrating focal point downstream of these bona fide oncogenic pathways.
Clinical relevance of eIF4E in human cancers
eIF4E overexpression is common in multiple cancer types, including malignancies of the prostate, breast, stomach, colon, lung, skin, and the hematopoietic system. Increased expression of eIF4E is associated with increasing grade of disease (19–26). Furthermore, elevated total eIF4E levels along with 4EBP1 hyperphosphorylation, which increases the availability of eIF4E to bind with eIF4G and enhance cap-dependent translation, have been observed in both breast cancer and prostate cancer and correlate with both decreased progression-free and overall survival (19, 20). Deregulation of eIF4E activity is present in a large spectrum of human malignancies. As such, elevated eIF4E levels may serve as a biomarker that predicts disease progression, overall survival, or relapse after definitive therapy. This finding has been most specifically delineated in breast cancer; patients with lower eIF4E levels post neoadjuvant therapy have a significant improvement in disease-free survival (27). In node-negative breast cancer, patients who express high levels of eIF4E have a twofold increase in relative risk for recurrence and a four-fold increase in relative risk for death (21). High eIF4E levels in patients with triple-negative [estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (Her2)/neu–negative] breast cancer have an increased incidence in cancer recurrence, and elevated levels of 4EBP1 phosphorylation are associated with high-grade disease and higher incidences of recurrence in breast cancer (28, 29). In postradical prostatectomy prostate cancer patients, elevated eIF4E expression as well as increased 4EBP1 phosphorylation is predictive of worse overall survival (19). These studies show that eIF4E levels, as well as indirect readouts for eIF4E activation such as 4EBP1 phosphorylation, have predictive power in clinical cancer care. Furthermore, we hypothesize that these biomarkers may also predict for patient response to eIF4E-targeted therapies. An area of clinical eIF4E research that has not been thor-oughly studied because of the paucity of adequate matched tissue samples is eIF4E status during metastasis. Although some evidence suggests that eIF4E levels remain high in metastatic samples, serial measurements of the same patients from diagnosis to metastasis of eIF4E levels have not been undertaken. This issue is important both for the understanding of the role of translational deregulation in metastasis as well as confirming the utility of targeting eIF4E in the setting of lethal metastatic disease.
Clinical-Translational Advances
mTOR ATP-active site inhibitors
Targeting mTOR is a therapeutically attractive option for inhibiting eIF4E hyperactivity because 4EBPs are direct substrates of the mTOR kinase (Fig. 1A). mTOR is a protein kinase that, when activated, forms two distinct multi-meric kinases: mTORC1 and mTORC2. mTORC1 substrates are 4EBPs and S6 kinases (S6K), whereas mTORC2 predominantly phosphorylates AKT at the S473 site. First generation allosteric mTOR inhibitors such as rapamycin, RAD001, and CCI-779 have done poorly in clinical trials against many human cancers, despite the evidence from preclinical data supporting the potential for efficacy. The discrepancy can be primarily attributed to the historically accepted convention of using the phosphorylated activation of S6Ks and ribosomal S6 (rpS6), one of their targets, as the predominant readout for mTOR inhibition. Rapamycin is a potent inhibitor of S6Ks phosphorylation in nearly all cell types. As such, it has been assumed that inhibition of S6 kinases and rpS6 phosphorylation by first generation mTOR inhibitors reflects inhibition of all mTOR kinase function. However, recently it has been shown that rapamycin and associated analogs inconsistently inhibit the phosphorylation of downstream targets of mTOR, particularly 4EBP1 (17, 30). This inconsistent inhibition is problematic because eIF4E hyperactivation is left unchecked upon treatment with rapalogs. Our laboratory recently showed that phosphorylation of 4EBP1 is necessary for oncogenesis and tumor maintenance in AKT-driven lymphomas, whereas phosphorylation of rpS6 is dispensable (17). Thus, continued activation of the 4EBP1/eIF4E axis downstream of oncogenic mTOR is the likely culprit behind the clinical failure of rapamycin and associated analogs.
In light of the clear need for more potent inhibitors of mTOR, a series of ATP-active site inhibitors of mTORC1 and mTORC2, such as PP242, Torin1, WYE-132, Ku-0063794, Palomid 529, and AZD8055, were recently reported by several groups (31–36). All of these compounds target the catalytic cleft of mTOR and reversibly compete against ATP to bind the mTOR catalytic domain. Unlike allosteric inhibitors of mTOR such as rapamycin, which predominantly inhibit S6Ks phosphorylation, PP242 effectively blocks the phosphorylation of all three mTOR targets, which include 4EBP1, rpS6, and AKT. This results in a more significant antiproliferative effect in cultured murine and human cell lines compared with rapamycin (31). More recently, our laboratory showed, in tumors from lymphoma-prone AKT transgenic mice, that, by mutating 4EBP1 and rendering it insensitive to mTOR phosphorylation, PP242 was no longer able to induce apoptosis (17). Furthermore, it has been shown that both PP242 and Torin1 retain their antiproliferative effect in mouse embryonic fibroblasts that are unable to phosphorylate AKT at the S473 site through destabilization of the mTORC2 complex (31, 32). Thus, the antitumor effect of ATP-active site inhibitors of mTOR is mediated through inhibition of the 4EBP1/eIF4E axis, resulting in a decrease in cap-dependent translation. These findings, however, do not exclude the possibility that inhibition of S6Ks and AKT phosphorylation or other targets may contribute to the antitumor tumor efficacy of mTOR ATPactive site inhibitors in other cellular contexts.
Multiple pharmaceutical companies have ATP-active site inhibitors in their pipelines, and we suspect that many more will be tested in phase I trials in the coming years. A more potent form of PP242, INK128, is currently in phase I clinical trials in patients with advanced solid tumors as well as multiple myeloma (NIH sponsored and/or endorsed clinical trials: NCT01058707, NCT01118689). AZD2014 is also being studied for safety, tolerability, and pharmacokinetics in late-stage cancers (NCT01026402). Palomid 529 is being tested in patients with advanced neo-vascular age-related macular degeneration (NCT01033721). Dual PI3K/mTOR ATP-analog inhibitors are currently under clinical investigation as well (BEZ235, PF-04691502, XL765). However, given the fact that PI3K activates multiple targets, it is unlikely that the therapeutic efficacy of these dual inhibitors relies solely on inhibition of eIF4E hyperactivation. In addition, it is also possible that targeting mTOR is sufficient to induce the same anticancer effect as a dual kinase inhibitor with fewer associated toxicities.
eIF4E-eIF4G interaction inhibitor, 4EGI-1
The dorsal surface of eIF4E is a critical interface regulated by the recruitment and binding of eIF4G (Fig. 1A). Binding of eIF4G to eIF4E increases cap-dependent translation through recruitment of eIF4A and the eIF3–40S ribosomal subunit. Targeting the eIF4E/eIF4G protein-protein interaction is a plausible mechanism to inhibit cap-dependent translation. eIF4G possesses a Y(X)4LF motif, in which X is variable and F is hydrophobic, which binds to conserved hydrophobic residues on the dorsal surface of eIF4E (37). It has been shown that mutations to the Y(X)4LF motif prevent the formation of the eIF4F complex. Given the conserved residues that facilitate the binding of eIF4G to the dorsal surface of eIF4E, a chemical inhibitor that targets this interaction could potentially prevent the formation of the eIF4F complex, thereby decreasing aberrant protein synthesis in the setting of eIF4E hyperactivity.
Using a high-throughput fluorescence polarization assay, in which the investigators screened chemical libraries for candidates that could displace or prevent the binding of a fluorescently labeled peptide that contained the Y(X)4LF motif from eIF4E, a candidate compound was discovered. 4EGI-1 is able to inhibit the formation of the eIF4F complex at micromolar concentrations. Given that both eIF4G and 4EBP1 possess Y(X)4LF motifs, it was hypothe-sized that 4EGI-1 would also inhibit the 4EBP1/eIF4E complex formation; however, this was not observed and, on the contrary, 4EGI-1 only inhibits the eIF4E/eIF4G interaction. 4EGI-1 is both a cytotoxic and cytostatic agent across multiple cell lines and, importantly, inhibits proliferation and clonogenic growth of transformed cells more significantly than untransformed cells (38, 39). The 4EGI-1 compound is licensed to Eugenix, who are working toward an Investigational New Drug application for this compound.4
These findings are novel and exciting, but are tempered by several considerations. To date, it has not been shown conclusively that 4EGI-1 does not interrupt or inhibit other protein-protein interactions and, therefore, may elic-it antitumor effects through more general inhibition of important oncogenic pathways. This finding is supported by the recent finding that 4EGI-1 can induce apoptosis through an eIF4G/eIF4E–independent mechanism through proteasome-mediated degradation of the anti-apoptotic c-FLIP (40). In addition, micromolar amounts of 4EGI-1 are required to decrease cell proliferation and induce apoptosis, making this compound less likely to be considered for clinical use at such high concentrations. However, a forthcoming generation of increasingly potent inhibitors of the eIF4G/eIF4E protein-protein interaction may serve as a novel therapeutic approach to target eIF4E hyperactivation in cancer.
Antisense targeting of eIF4E: eIF4E antisense oligonucleotide
On the basis of the finding that eIF4E is commonly elevated in multiple human malignancies and has bona fide oncogenic potential when overexpressed, direct targeting of eIF4E mRNA by specific antisense oligonucleotide (ASO) therapy has been extensively studied and is currently under investigation as a possible anticancer agent. ASOs recognize and hybridize to target mRNA and trigger RNase H-mediated RNA destruction. The efficacy of first generation ASOs was severely limited by a lack of nuclease resistance, resulting in relatively short half-lives in vivo. However, second generation ASOs have been reengi-neered to improve potency, nuclease resistance, and tissue half-life. Given the short lengths of the ASOs, they are unlikely to induce a systemic interferon response. eIF4E ASO decreases eIF4E protein levels and is cytotoxic across a panel of human cancer cell lines at nanomolar concentrations (19, 41). In a tumor xenograft model in which mice were treated with 5, 12.5, or 25 mg/kg of eIF4E-ASO three times per week, significant inhibition of tumor growth was observed without any noticeable toxicity as measured by changes in body weight and liver function tests (41). A dose-escalation phase I clinical trial is currently underway with the eIF4E ASO LY2275796 (NCT00903708). It is uncertain if eIF4E-targeted degradation will be achieved in humans; however, eIF4E levels are being ascertained in enrolled patients. These findings have important implications for our understanding of the physiologic versus oncogenic role of eIF4E. In particular, xenograft mice treated with doses of eIF4E-ASO leading to greater than 50% reduction in eIF4E levels did not show any overt toxicities, indicating that low eIF4E levels are tolerated in normal tissues. However, cancer cells, which possess increased eIF4E activity, may be more sensitive to eIF4E inhibition.
Inhibition of eIF4E phosphorylation: MNK kinase inhibitors
eIF4E phosphorylation is, theoretically, an attractive antitumor target, given the minimal effect of phosphorylation on basic cellular functions, but with a significant effect on transformation in vitro. However, chemical inhibitors of the putative kinase of eIF4E S209, MNK1/2, have shown only limited preclinical efficacy in cell-based systems. For instance, although targeted mutations of the eIF4E S209 site significantly inhibit the transforming potential of eIF4E in NIH3T3 cells, it has not been shown whether using a targeted inhibitor of MNK1/2 has a similar effect (6). Furthermore, the MNK kinase inhibitor CGP57380 has shown limited antiproliferative efficacy in human chronic myelogenous leukemia; however, it does improve the antiproliferative and apoptotic effects of imatinib when used in combination (42). Although multiple MNK kinase inhibitors are available, no clinical trials are underway, at present, to test the efficacy of these compounds in human cancer. The absence of trials is likely due to the fact that the oncogenic potential of eIF4E phosphorylation still requires rigorous validation both in vitro and in vivo. In addition, it is uncertain if the inhibition of MNK1/2 also inhibits the phosphorylation of additional downstream targets that are necessary for oncogenesis and cancer progression.
Targeting the eIF4E–5′ cap interaction
Ribavirin, an antiviral therapy, is a guanosine ribo-nucleoside analog that functions in part by mimicking the 5′ 7-methyl guanosine cap structure to compete with endogenous mRNAs for binding with eIF4E. It has been shown to decrease the formation of the eIF4F complex in vitro and has shown preclinical efficacy in acute myelogenous leukemia (AML) and squamous-cell carcinoma cell–based models (43). Furthermore, in a phase I dose-escalation trial with ribavirin in patients with AML, 1 complete response, 2 partial responses, and 4 patients with stable disease were reported out of 11 enrolled in the study (44). On the basis of these promising results, ribavirin is currently being studied in phase I-II clinical trials for patients with metastatic breast cancer and M4/M5 AML (NCT01056757, NCT01056523). Although these finding are significant for the clinical efficacy of ribavirin in human cancers, the specific mechanism of ribavirin as a cap-mimetic has been called into question by two independent groups (45–47). Thus, it is uncertain if the therapeutic efficacy of ribavirin is mediated primarily through inhibition of cap-dependent translation.
Future Developments and Considerations
eIF4E and translation initiation is an attractive target for anticancer therapeutics because it is a common downstream node on which multiple oncogenic signaling pathways converge. In addition to the inhibitory modalities already mentioned, other inhibitors of different components of the initiation machinery should be considered, including, for instance, Silvesterol, which inhibits the RNA helicase eIF4A leading to antitumor effects in xenograft models (48). With regards to future clinical trials with inhibitors of eIF4E, ideally, a prospective analysis of patient eIF4E status as determined by total eIF4E levels, 4EBP1 phosphorylation, and eIF4E phosphorylation should be ascertained to correlate with treatment response.
Although multiple putative mRNAs deregulated by increased eIF4E activity have been described (Fig. 1B), their impact on eIF4E-mediated cancer formation and progression is poorly understood. It is uncertain which of the aforementioned eIF4E targets may be necessary and/or sufficient for the development of cancer. In fact, it is likely that the relevant interaction among the several eIF4E targets would provide an oncogenic network that will differ dramatically depending on tumor type, tumor grade, and the presence or absence of metastasis. Therefore, a comprehensive map of mRNAs deregulated by eIF4E oncogenic activity will be imperative to elucidate the eIF4E translational signatures from patient samples that will serve as novel biomarkers or future therapeutic targets.
Acknowledgments
We would like to thank M. Barna, K. Tong, and A. Sher for critical discussion and reading of the manuscript.
Grant Support
A.C. Hsieh is a postdoctoral fellow of the American Cancer Society and a recipient of a Prostate Cancer Foundation Young Investigator Award and an ASCO Young Investigator Award. D. Ruggero is a Leukemia and Lymphoma Society Scholar and is currently funded by the CRCC.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
N. Sonenberg, personal communication; and eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression, submitted for publication.
G. Wagner, personal communication.
References
- 1.Sonenberg N, Rupprecht KM, Hecht SM, Shatkin AJ. Eukaryotic mRNA cap binding protein: purification by affinity chromatography on sepharose-coupled m7GDP. Proc Natl Acad Sci U S A 1979;76: 4345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tahara SM, Morgan MA, Shatkin AJ. Two forms of purified m7G-cap binding protein with different effects on capped mRNA translation in extracts of uninfected and poliovirus-infected HeLa cells. J Biol Chem 1981;256:7691–4. [PubMed] [Google Scholar]
- 3.Pyronnet S, Imataka H, Gingras AC, Fukunaga R, Hunter T, Sonenberg N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J 1999;18: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheper GC, Morrice NA, Kleijn M, Proud CG. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol Cell Biol 2001;21:743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol 2004;24:6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topisirovic I, Ruiz-Gutierrez M, Borden KL. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res 2004;64: 8639–42. [DOI] [PubMed] [Google Scholar]
- 7.Wendel HG, Silva RL, Malina A, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev 2007;21:3232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konicek BW, Dumstorf CA, Graff JR. Targeting the eIF4F translation initiation complex for cancer therapy. Cell Cycle 2008;7:2466–71. [DOI] [PubMed] [Google Scholar]
- 9.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer 2010;10:254–66. [DOI] [PubMed] [Google Scholar]
- 10.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 1990;345:544–7. [DOI] [PubMed] [Google Scholar]
- 11.Avdulov S, Li S, Michalek V, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 2004;5: 553–63. [DOI] [PubMed] [Google Scholar]
- 12.Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med 2004;10:484–6. [DOI] [PubMed] [Google Scholar]
- 13.Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004;428: 332–7. [DOI] [PubMed] [Google Scholar]
- 14.Sorrells DL, Black DR, Meschonat C, et al. Detection of eIF4E gene amplification in breast cancer by competitive PCR. Ann Surg Oncol 1998;5:232–7. [DOI] [PubMed] [Google Scholar]
- 15.Sorrells DL, Ghali GE, Meschonat C, et al. Competitive PCR to detect eIF4E gene amplification in head and neck cancer. Head Neck 1999; 21:60–5. [DOI] [PubMed] [Google Scholar]
- 16.Jones RM, Branda J, Johnston KA, et al. An essential E box in the promoter of the gene encoding the mRNA cap-binding protein (eukaryotic initiation factor 4E) is a target for activation by c-myc. Mol Cell Biol 1996;16:4754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh AC, Costa M, Zollo O, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell 2010;17:249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell 2003;12:889–901. [DOI] [PubMed] [Google Scholar]
- 19.Graff JR, Konicek BW, Lynch RL, et al. eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res 2009;69:3866–73. [DOI] [PubMed] [Google Scholar]
- 20.Coleman LJ, Peter MB, Teall TJ, et al. Combined analysis of eIF4E and 4E-binding protein expression predicts breast cancer survival and estimates eIF4E activity. Br J Cancer 2009;100:1393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holm N, Byrnes K, Johnson L, et al. A prospective trial on initiation factor 4E (eIF4E) overexpression and cancer recurrence in node-negative breast cancer. Ann Surg Oncol 2008;15:3207–15. [DOI] [PubMed] [Google Scholar]
- 22.Chen CN, Hsieh FJ, Cheng YM, Lee PH, Chang KJ. Expression of eukaryotic initiation factor 4E in gastric adenocarcinoma and its association with clinical outcome. J Surg Oncol 2004;86:22–7. [DOI] [PubMed] [Google Scholar]
- 23.Rosenwald IB, Chen JJ, Wang S, Savas L, London IM, Pullman J. Upregulation of protein synthesis initiation factor eIF-4E is an early event during colon carcinogenesis. Oncogene 1999;18:2507–17. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Geng J, Wang JH, Chu XY, Geng HC, Chen LB. Overexpression of eukaryotic initiation factor 4E (eIF4E) and its clinical significance in lung adenocarcinoma. Lung Cancer 2009;66:237–44. [DOI] [PubMed] [Google Scholar]
- 25.Salehi Z, Mashayekhi F, Shahosseini F. Significance of eIF4E expression in skin squamous cell carcinoma. Cell Biol Int 2007;31:1400–4. [DOI] [PubMed] [Google Scholar]
- 26.Wang S, Rosenwald IB, Hutzler MJ, et al. Expression of the eukaryotic translation initiation factors 4E and 2alpha in non-Hodgkin’s lymphomas. Am J Pathol 1999;155:247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiller DJ, Chu Q, Meschonat C, Panu L, Burton G, Li BD. Predictive value of eIF4E reduction after neoadjuvant therapy in breast cancer. J Surg Res 2009;156:265–9. [DOI] [PubMed] [Google Scholar]
- 28.Flowers A, Chu QD, Panu L, et al. Eukaryotic initiation factor 4E overexpression in triple-negative breast cancer predicts a worse outcome. Surgery 2009;146:220–6. [DOI] [PubMed] [Google Scholar]
- 29.Rojo F, Najera L, Lirola J, et al. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clin Cancer Res 2007;13:81–9. [DOI] [PubMed] [Google Scholar]
- 30.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A 2008;105: 17414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 2009;7:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mTOR inhibitor reveals rapamycin-insensitive functions of mTORC1. J Biol Chem 2009;284:8023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu K, Shi C, Toral-Barza L, et al. Beyond rapalog therapy: preclinical pharmacology and antitumor activity of WYE-125132, an ATP-competitive and specific inhibitor of mTORC1 and mTORC2. Cancer Res 2010;70:621–31. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Martinez JM, Moran J, Clarke RG, et al. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J 2009;421:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue Q, Hopkins B, Perruzzi C, Udayakumar D, Sherris D, Benjamin LE. Palomid 529, a novel small-molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res 2008;68:9551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010;70:288–98. [DOI] [PubMed] [Google Scholar]
- 37.Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell 1999;3:707–16. [DOI] [PubMed] [Google Scholar]
- 38.Moerke NJ, Aktas H, Chen H, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007;128:257–67. [DOI] [PubMed] [Google Scholar]
- 39.Tamburini J, Green AS, Bardet V, et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009;114:1618–27. [DOI] [PubMed] [Google Scholar]
- 40.Fan S, Li Y, Yue P, Khuri FR, Sun SY. The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP Down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia 2010;12:346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graff JR, Konicek BW, Vincent TM, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest 2007;117:2638–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M, Fu W, Prabhu S, et al. Inhibition of polysome assembly enhances imatinib activity against chronic myelogenous leukemia and overcomes imatinib resistance. Mol Cell Biol 2008;28:6496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kentsis A, Topisirovic I, Culjkovic B, Shao L, Borden KL. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap. Proc Natl Acad Sci U S A 2004;101:18105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Assouline S, Culjkovic B, Cocolakis E, et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood 2009;114:257–60. [DOI] [PubMed] [Google Scholar]
- 45.Westman B, Beeren L, Grudzien E, et al. The antiviral drug ribavirin does not mimic the 7-methylguanosine moiety of the mRNA cap structure in vitro. RNA 2005;11:1505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan Y, Svitkin Y, Lee JM, Bisaillon M, Pelletier J. Ribavirin is not a functional mimic of the 7-methyl guanosine mRNA cap. RNA 2005; 11:1238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kentsis A, Volpon L, Topisirovic I, et al. Further evidence that ribavirin interacts with eIF4E. RNA 2005;11:1762–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cencic R, Carrier M, Galicia-Vazquez G, et al. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS ONE 2009;4:e5223. [DOI] [PMC free article] [PubMed] [Google Scholar]