

Abstract
Protein fragmentation is a critical component of top-down proteomics, enabling gene-specific protein identification and full proteoform characterization. The factors that influence protein fragmentation include precursor charge, structure, and primary sequence, which have been explored extensively for collision-induced dissociation (CID). Recently, noticeable differences in CID-based fragmentation were reported for native versus denatured proteins, motivating the need for scoring metrics that are tailored specifically to native top-down mass spectrometry (nTDMS). To this end, position and intensity were tracked for 10,252 fragment ions produced by higher-energy collisional dissociation (HCD) of 159 native monomers and 70 complexes. We used published structural data to explore the relationship between fragmentation and protein topology and revealed that fragmentation events occur at a large range of relative residue solvent accessibility. Additionally, our analysis found that fragment ions at sites with an N-terminal aspartic acid or a C-terminal proline make up on average 40 and 27%, respectively, of the total matched fragment ion intensity in nTDMS. Percent intensity contributed by each amino acid was determined and converted into weights to (1) update the previously published C-score and (2) construct a native Fragmentation Propensity Score. Both scoring systems showed an improvement in protein identification or characterization in comparison to traditional methods and overall increased confidence in results with fewer matched fragment ions but with high probability nTDMS fragmentation patterns. Given the rise of nTDMS as a tool for structural mass spectrometry, we forward these scoring metrics as new methods to enhance analysis of nTDMS data.
Graphical Abstract
INTRODUCTION
Top-down mass spectrometry is the intact analysis and controlled fragmentation of proteins and large biomolecular complexes.1 In comparison to other proteomics techniques, top-down mass spectrometry directly characterizes proteoforms,2–4 or the exact molecular form of a protein, including all post-translational modifications (PTMs), isoform variants, or coding polymorphisms. Within top-down mass spectrometry, there are two major modes, known as denaturing top-down mass spectrometry (dTDMS) and native top-down mass spectrometry (nTDMS). In dTDMS, acid and organic solvents disrupt secondary and tertiary structures. In nTDMS, nondenaturing conditions are used to retain tertiary structure and quaternary protein composition, enabling characterization of noncovalently bound species and their stoichiometry in complex.5 A critical component of top-down experiments is the tandem mass spectrometric step, also known as MS/MS or MSn. In an MS/MS experiment, an analyte of interest is isolated and fragmented, yielding peptide fragment ions. Numerous fragmentation techniques have been developed,6–8 each offering unique levels of information. Fragment ions can be used to identify the precursor protein, localize modifications within the primary sequence,9–13 or probe higher-order structure in the case of natively folded proteins.
Mapping protein topology through tandem mass spectrometry is typically done using electron-based fragmentation methods such as electron capture (ECD) or electron transfer dissociation (ETD), as these methods produce fragments without substantial disruption of protein structure.8,14–16 Electron-based methods have been used to successfully map several aspects of protein structure, including cofactor binding sites17 and surface exposed or interfacial residues of native complexes.18–20 Similar approaches have also mapped dynamic processes such as protein unfolding.21,22 While ECD and ETD have proven fruitful in advancing structural mass spectrometry, fragmentation through collisions with neutral gas (collision-induced dissociation, CID) remains widely used and is accessible on many commercial instruments.23,24 While it is known that collisional activation induces protein unfolding and may be unamenable to structural studies,25–27 it is unclear if CID-based fragmentation is similarly correlated to protein topology and could thus be used to derive insights into protein structure.
The model for CID-based fragmentation of proteins and peptides posits that ionizing protons drive fragmentation.9,28,29 Depending on the experimental conditions and nature of the analyte, the driving proton may be mobile (able to migrate within the multiply charged gaseous cation) or localized (sequestered to a single residue side chain). CID exists in two major forms known as (1) ion-trap CID and (2) beam-type CID.30,31 Higher-energy collisional dissociation (HCD) is a subcategory of beam-type CID32 and generally is used in combination with Orbitrap mass analyzers, allowing for high resolution and mass accuracy measurements of fragment ions.33 The fragments produced by CID are far from stochastic and depend on several factors, including precursor structure,34 charge,35 and amino acid identity.36,37 In nTDMS, fragment ions tend to occur at residue pairs with N-terminal aspartic acid (DIX, X represents any other amino acid), C-terminal proline (XIP),36–38 and to a lesser extent sites with glutamic acid, alanine, leucine, isoleucine, valine, and lysine.34 This tendency to fragment at DIX and XIP sites is believed to arise from differing mechanisms. N-terminal aspartic acid cleavages are posited to result from proton sequestration by basic residues.39–41 For analytes in which the number of protons is equal to or less than the number of basic residues, acidic hydrogens in the carboxyl side chain donate the proton necessary for fragmentation. Given that native proteins ionized by electrospray ionization (ESI) carry far fewer charges as compared to denatured proteins,42 this is consistent with previous reports of DIX-type fragment ions being significantly more abundant in nTDMS versus dTDMS.34 C-terminal proline cleavages are believed to be the result of the increased basicity of proline’s unique secondary amine within the peptide backbone.43 Given this “proline effect” is highly dependent on the primary sequence and not the charge and structure of the precursor, XIP fragmentation is favored in both nTDMS and dTDMS.34
Understanding and quantification of fragmentation trends can be powerful, as known trends can inform scoring metrics used in top-down proteomics. Fragmentation information has been incorporated as weights (coefficients) at both the protein44 and proteoform45 level of scoring, both of which are necessary for confident protein identification and characterization by top-down mass spectrometry.46 For example, the McLuckey group has constructed and implemented a scoring metric which assigns extra weight to DIX, KIX, EIX, and XIP fragment ions over other “nonspecific” cleavages.44,47 Similar scoring methods have been successfully implemented into search tools such as ProSightPC (Thermo Fisher Scientific). Currently, these weights are informed by experimental data but have been set arbitrarily and are still largely qualitative with regards to nTDMS. Past studies into fragmentation by CID have either been (l) not tailored to nTDMS, (2) limited to a few model proteins, or (3) have failed to consider fragment site and intensity.34,48–50 Given the rise of nTDMS as a high-throughput and structurally informative technique,18,51–55 further detailed studies into nTDMS fragmentation and development of native-specific scoring is warranted.
To develop a better understanding of collisional fragmentation in nTDMS, we examine data from 159 native monomers and 70 complexes (also called “native multimers” here) fragmented by HCD.34,51 We track both the frequency (rate of occurrence) and intensity (abundance) of 10,252 fragment ions to (1) explore the relationship between HCD-based fragmentation and residue solvent accessibility, (2) determine the relationship between fragment ion intensity and primary sequence, and (3) construct nTDMS-tailored scoring metrics from these empirically derived intensities.
EXPERIMENTAL SECTION
Sample Preparation and Data Acquisition.
All data used within this study have been previously published.34,51,56 Briefly, the native data sets were derived from several cell lines including Ramos (Burkitt Lymphoma, B cell), Hg-3 (chronic lymphocytic leukemia, B cell), Jurkat (acute lymphoblastic leukemia, T cell), HEK-293T (human embryonic kidney), or CD-1 mouse hearts. Cells and tissue were lysed in hypotonic buffer, and the resulting lysates were fractionated using ion exchange chromatography (IEX) or native-GELFrEE.57,58 Fractions were exchanged into 100–200 mM ammonium acetate (aqueous) and concentrated using 3 kDa molecular weight cutoff centrifugal filters (Millipore). Fractions were ionized using a custom nanoelectrospray source59 and analyzed on a custom Q-Exactive HF (Thermo Fisher Scientific).60 For native monomers, a single charge state was quadrupole-isolated prior to HCD-based fragmentation. For native complexes, subunits were ejected at the source, quadrupole-isolated, and fragmented via HCD.60 A resolving power of >60,000 (at 200 m/z) was used for acquiring fragment-level information. Proteins were initially identified using ProSightPC 4.0 (Thermo Fisher Scientific).
The denatured data set was derived from NCI-H1299 cells (nonsmall cell lung carcinoma). Whole cell lysates or subcellular fractions were prepared as previously described.56 Samples were isoelectric focused prior to GELFrEE fractionation. Final fractions were precipitated using methanol, chloroform, and water,61 and resuspended into buffer A (95% water, 5% acetonitrile, 0.2% formic acid). Samples were then injected onto a reverse-phase liquid chromatography system and analyzed in line with an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Fragmentation was performed using a top-two data acquisition method with an isolation window of 15 m/z and a resolving power of 60,000 (at 400 m/z). For this study, only HCD-based fragmentation data were considered. Proteins were initially identified using ProSightPC 3.0 (Thermo Fisher Scientific).
In summary, fragmentation data were acquired for 159 native monomers, 70 native multimers, and 138 denatured proteins using HCD-based fragmentation. These data sets include 7,466, 2,786, and 4,859 individual matched fragment ions from native monomers, native multimers, and denatured proteins, respectively.
Fragment Ion Identification.
RAW files of supporting fragmentation spectra were curated for each identified protein. These .RAW files were processed using TDValidator 1.0 (Proteinaceous Inc.). TDValidator 1.0 matches and scores theoretical isotopic distributions of protein fragments to observed raw fragmentation spectra and outputs identified fragment ions above a threshold matching score. Internal fragments were not considered to minimize the number of falsely identified fragment ions. Fragments were identified in TDValidator 1.0 using a maximum tolerance of 10 ppm, a sub tolerance of 3 ppm, a minimum assigned score of 0.5, and a minimum S/N cutoff of 3. Fragment intensities were also measured in TDValidator 1.0; the intensity of unobserved fragments was defined as zero. Total fragment ion intensity is calculated by summing the intensity of all matched fragment ion isotopomers.
Structural Studies.
Structures were selected from the Protein Data Bank (PDB) based on identity to the protein sequences identified within the TDMS data sets. Thirty-eight structures corresponding to the monomeric nTDMS data set and thirty-one structures corresponding to the dTDMS data set were selected. Protein structures were allowed to differ from the TDMS data by terminal truncations extending only 5% or less of the identified TDMS sequence. No internal gaps or mismatches in sequence were allowed when selecting protein structures. Once structures were selected, average solvent accessible surface area (areaSAS) was calculated for each residue using UCSF Chimera.62–64 The probe radius was set to 1.4 Å. Relative areaSAS was calculated for each site by normalizing areaSAS values to the largest areaSAS for a given protein. Identified fragment ions were exported from TDValidator 1.0 and aligned with respective structures and relative areaSAS values with a custom code developed in R, using RStudio.65 Significant differences in the mean relative areaSAS were determined using a Wilcoxon–Mann–Whitney (WMW) test.
Fragment Ion Analysis and Z-Score Calculations.
Identified fragment ions and their intensities were exported from TDValidator 1.0 into a custom R script developed in RStudio as described above.65 For each fragment, the residues N-terminal (X|X′) and C-terminal (X|X′) to the fragmentation site were determined, and the percent of total observed fragment ions and total fragment intensity contributed by each amino acid were then calculated in the context of a single protein. These percentages were averaged across the entire respective data set. Significant differences in the mean percentage values were determined using a WMW test. Fragment intensity z-scores were then calculated for each residue pair for a given protein using the average and standard deviation of all fragment intensities within a single protein’s fragmentation spectrum. Average z-scores were calculated across the entire respective data set for each combination of two residues flanking a site of fragmentation. Significant differences in the average z-score relative to the base-mean were determined using a multiple pairwise WMW test, multiple-test corrected using the Bonferroni method.
An Updated C-Score for nTDMS Data.
The average intensity contributed by each N-terminal (X|X′) and C-terminal (X|X′) residue adjacent to a site of fragmentation was calculated as described above. These average intensities replaced the existing denatured propensity values present in the original C-score.45 The native monomeric and multimeric data were processed through an alpha version of ProSightPD 4.0 in Proteome Discoverer (Proteinaceous Inc. and Thermo Fisher Scientific) using either the original or intensity-based weights in the C-score calculation.
Native Fragmentation Propensity Score (nFPS) Calculation.
The nFPS is defined as the product of the forward matched fragment ion propensities divided by the geometric mean of the decoy fragmentation ion propensities product. The nFPS can be written as follows:
where P is the fragmentation propensity value defined by the fragmentation probabilities of the two residues on the N-terminal (X|X′) and C-terminal (X|X′) side of the fragmentation site. Both n and x represent the set of matched fragment ions for the forward and decoy hits, respectively. The denominator calculates the geometric mean for the set of y decoy instances.
To find the top nFPS and Poisson-based P-score66 values for a subset of the monomeric native proteoform data, 83 “true positive” proteoforms were searched via ProSightPC (Proteinaceous Inc. and Thermo Fisher Scientific) with a “no precursor” search methodology67 where the precursor mass tolerance was set to a value large enough that all forms in the search space were considered. A simple human database with N-Met on/off and N-terminal acetylation containing 222,844 forms was used to limit the possible search space. The top 100 results from each search were submitted to ProSight Native (Proteinaceous Inc.) to obtain nFPS metrics.
RESULTS AND DISCUSSION
Correlating Fragmentation with Residue Solvent Accessibility.
Thirty-eight PDB structures corresponding to native monomers were selected based on identity to sequences determined by mass spectrometry studies (gene names and structures are listed in Table S1). Relative solvent-accessible surface area (relative areaSAS) was calculated as described in the Experimental section and serves as a proxy for residue solvent exposure and overall protein topology. Relative areaSAS is calculated as values from 0 (solvent-inaccessible, most buried) to 1 (solvent-accessible, most exposed). For this analysis, proteins were also grouped according to the precursor charge. For globular proteins, the theoretical maximum number of charges that can be deposited during electrospray ionization (ESI) is defined by the Raleigh charge limit (ZR).68 Assuming proteins are globular before and during native ESI and carry charge ZActual, proteins can be divided into three classes: low charge (ZActual/ZR < 0.86), intermediate charge (0.86 ≤ ZActual/ZR ≤ 1.43), and high charge (ZActual/ZR ≤ 1.43).34,35 The data presented represent 29 low charge precursors, 7 intermediate charge precursors, and 2 high charge precursors. For low charge states, the average relative areaSAS was significantly higher (p ≤ 0.0001) for fragmented residues versus non-fragmented residues (Figure S1). Statistical significance was determined using a WMW test. While significant, this difference was small with the average relative areaSAS for non-fragmented sites being 0.26 and 0.32–0.33 for fragmented sites based on either N-terminal (X|X′) or C-terminal (X|X′) residue. Thirty-one structures corresponding to denatured proteins were similarly analyzed as a control (gene names and structures are listed in Table S2). There was no significant difference in average relative areaSAS for fragmented residues versus non-fragmented residues for denatured proteins (Figure S2), as to be expected given the disruption of secondary and tertiary structures post-denaturation.
Many fragment ions observed from nTDMS occurred at low relative areaSAS, especially for high charge state precursors. This is evident in Figure 1, which shows the distributions of relative areaSAS as probability density plots. By N-terminal residue (X|X′), 48–77% of fragments from low and intermediate charge state precursors fell between the relative areaSAS range of 0.3–0.7 (Figure 1a and Table S3). Similarly, by C-terminal residue (X|X′) 41–44% of fragments from low and intermediate charge state precursors fell within the relative areaSAS range of 0.3–0.7 (Figure 1b and Table S3); this region only encompassed 36% of non-fragmented residues. For high charge state precursors, the percent of fragments within the 0.3–0.7 range of relative areaSAS was only 38 or 31% by N-terminal (X|X′) or C-terminal (X|X′) residue, respectively. Instead, fragments from high charge state precursors concentrated within the relative areaSAS range of 0.0–0.3. Specifically, 60 or 65% of fragments concentrated to this region by N-terminal (X|X′) or C-terminal (X|X′) residue. This shift to fragment at lower relative areaSAS with increased charge state may be due to charge effects altering the native conformation of the protein precursor. While prior studies have suggested a correlation between HCD-based fragmentation and increased residue surface exposure,34 it is clear that overall fragment ions produced in nTDMS occurred at a wide range of relative areaSAS. Additionally, fragments occurred at a wide range of sequence depth for native proteins, especially for proteins below 200 amino acids in length (Figure S3). Analysis of residue identity versus relative areaSAS of a given fragment revealed that the established nTDMS fragmentation trends hold true across all charge states (Figures S4 and S5). Namely, most fragments were bordered by aspartic acid or proline, and to a lesser extent by isoleucine, leucine, valine, lysine, glycine, or glutamic acid.34
Figure 1.
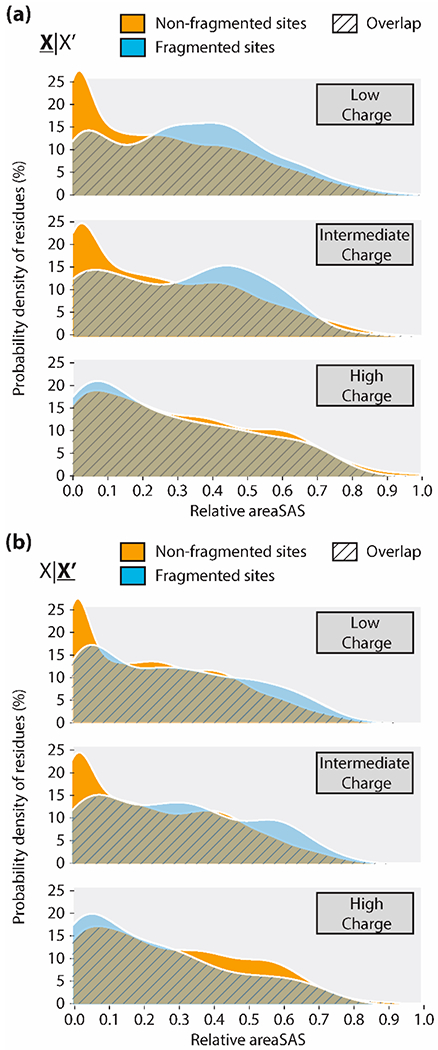
Ridgeline density plots of relative areaSAS for non-fragmented (gold) versus fragmented (blue) sites in native monomers. Plots are grouped by precursor charge state (low, intermediate, or high), and separated by the residue (a) N-terminal (X|X′) or (b) C-terminal (X|X′) to the site of interest.
Fragment Ion Occurrence and Intensity as a Function of Primary Sequence.
For proteins fragmented in nTDMS, D|X and X|P fragment ions constituted a large percentage of total fragments and total fragment intensity (Figure 2). Mean intensity values are available in Table S4; median values and standard interquartile ranges (IQR) corresponding to Figure 2 are available in Table S5. D|X sites contributed more to total fragment number and intensity than any other individual residue pair in nTDMS. Despite only constituting 4.2–5.9% of the primary sequence (Table S4), D|X sites constituted on average 26% of fragments (Figure 2a) and 37% of fragment intensity (Figure 2b) for native monomers. Similarly, for native multimers D|X sites constituted on average 31% of fragments and 43% of fragment intensity. In comparison, dTDMS fragmentation at D|X sites constituted on average only 6.9% of fragments and 7.0% of fragment intensity.
Figure 2.
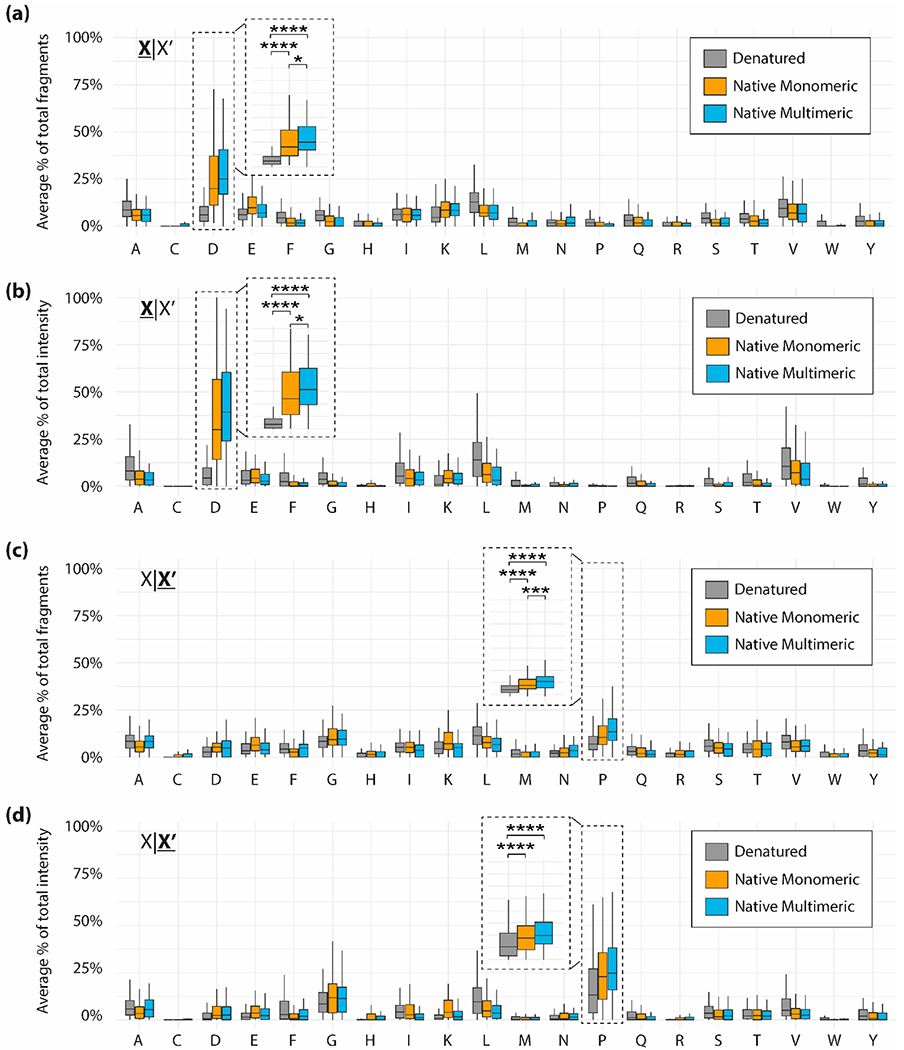
Average percent of total fragments (a) or total fragment intensity (b) contributed by each residue N-terminal (X|X′) to the fragmentation site. Average percent of total fragments (c) or total fragment intensity (d) contributed by each residue C-terminal (X|X′) to the fragmentation site. Data are divided into denatured proteins (gray), native monomeric proteins (gold), and native multimeric proteins (blue). Brackets indicate a comparison between two data points using a Wilcoxon–Mann–Whitney test. Significant differences are denoted as follows: p ≤ 0.05 is denoted by one asterisk, p ≤ 0.01 is denoted by two asterisks, p ≤ 0.001 is denoted by three asterisks, p ≤ 0.0001 is denoted by four asterisks.
Instead, dTDMS fragmentation was dispersed across many residues with fragment number and intensity at these sites roughly proportional to their occurrence within the primary sequence (Table S5). As noted previously,34 abundant and frequently fragmented residues included alanine, glycine, glutamic acid, leucine, isoleucine, and lysine. X|P sites also contributed substantially to fragment number and intensity in nTDMS, however this difference between nTDMS and dTDMS is less pronounced than seen at D|X sites. Similarly, X|P sites constituted 4.4–4.8% of the primary sequence but constituted on average 12% of fragments (Figure 2c) and 25% of fragment intensity (Figure 2d) for native monomers; for native multimers, X|P sites constituted on average 16% of fragments and 29% of fragment intensity. This preference for fragmentation at X|P sites also extends into dTDMS, in which X|P sites constituted on average 8.2% of fragments and 17% of fragment intensity.
The large range of values presented in Figure 2 can be attributed to the many sample and instrument parameters that impact fragmentation such as initial precursor concentration, precursor charge, collisional energy, and collisional gas pressure. However, the mean percentages were determined to be significantly different (p ≤ 0.0001) between nTDMS and dTDMS fragmentation at D|X and X|P sites as determined using a Wilcoxon–Mann–Whitney test (Figure 2). Interestingly, the disparity between dTDMS fragmentation and nTDMS fragmentation is greater for native multimers than native monomers. Note that the data collated here are for subunits first ejected from native complexes prior to fragmentation (also referred to as “complex-down” mass spectrometry).69 Subunits ejected from native complexes often undergo asymmetric charge partitioning, with evidence showing that loss of tertiary structure may drive this phenomenon.70,71 Given this information, we initially hypothesized that subunits ejected from native complexes may fragment similar to denatured proteins. While this may be true for highly charged multimeric precursors, within the experimental parameters used here, fragmentation of native multimers is more similar to fragmentation of native monomers than to fragmentation of denatured proteins. Disulfide bridges are not a confounding factor, as only two native multimers within the data set contain disulfide bridges as determined from the complex’s intact mass and fragmentation spectrum. Given the putative mechanisms underlying fragmentation at D|X and X|P sites, it is not surprising that N-terminal aspartic acid cleavages and C-terminal proline cleavages are so highly favored in nTDMS as previously reported.34 However, tracking fragment number alone does not fully reveal the preference for D|X and X|P fragments in nTDMS. Instead, tracking fragment intensity shows an even further heightened preference for fragmentation at D|X and X|P sites and heightens the disparity between nTDMS and dTDMS.
Beyond total percentage of fragment ions and intensities, intensity z-scores reveal how abundant a fragment ion is in relation to the mean fragment intensity for a given protein. We used z-score calculations as a means of standardization to account for differences in intensity due to differing abundances of protein precursors within biological samples and differences in instrument sensitivity across time. Significant differences in the average z-score relative to the base-mean were determined for each residue combination using a multiple pairwise WMW test, multiple-test corrected using the Bonferroni method. While the distribution of z-scores was quite large for a given residue (Figure S6), the average z-scores for D|X and X|P fragments were significantly higher than the global average z-score for native fragments (p < 0.05, exact significance levels displayed in Figure 3 and Figure S6). The average z-score was nearly zero across all residues, while the average D|X z-scores were 1.2 ± 2.5 (mean ± SD) and 1.1 ± 2.7 for native monomers and multimers, respectively (Figure 3 and Table S6). Similarly, average X|P z-scores were 0.8 ± 2.6 and 0.7 ± 2.6 for native monomers and multimers. D|P fragments were consistently more intense than any other residue pair in the nTDMS data sets, with average z-scores of 4.8 ± 4.8 and 4.3 ± 5.0 for native monomers and multimers, respectively. These results recapitulate the preference to fragment frequently and with high intensity at D|X and X|P sites in nTDMS. Additionally, intensity z-scores revealed that other fragmentation propensity “hotspots”, including alanine, glycine, glutamic acid, leucine, isoleucine, and lysine, were at or below the global mean following intensity-based standardization. These sites are abundant and frequently fragmented (Figure S7), but do not show a heightened preference for fragmentation. All observed trends hold true when considering only b-type versus y-type ions (Figures S8–S11). Additionally, disulfide bridges do not impact the preference for fragmentation at D|X and X|P sites in native monomers. Seventeen of the native monomers contain disulfide bridges as determined from the intact mass and observed fragment ions; differences between this subset and the native monomeric data set as a whole were nonsignificant (Figures S12 and S13).
Figure 3.
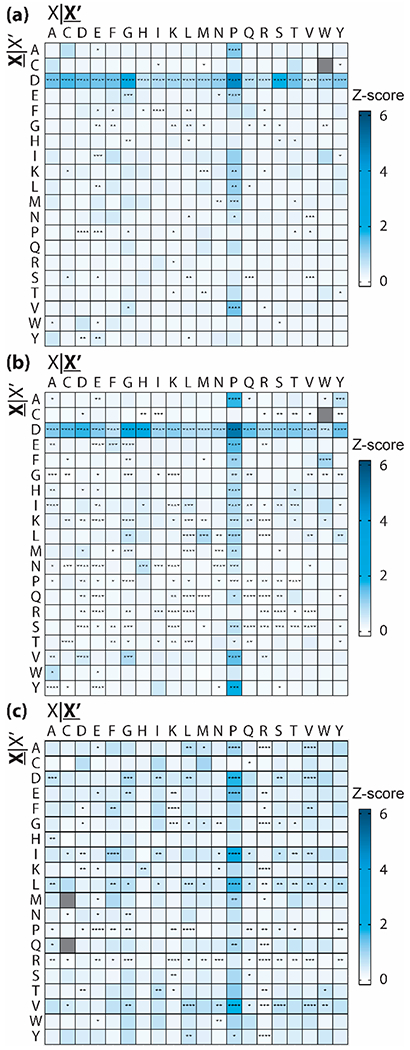
Average fragment intensity z-score by residue pair for (a) native multimers, (b) native monomers, and (c) denatured proteins. Residue pairs that are not observed in the data set are shown in gray. Asterisks denote significant differences from the base-mean as determined by a multiple pairwise WMW test, corrected using the Bonferroni method (p < 0.05 is denoted by one asterisk, p ≤ 0.01 is denoted by two asterisks, p ≤ 0.001 is denoted by three asterisks, p ≤ 0.0001 is denoted by four asterisks). For all panels, X|X′ refers to the residue N-terminal to the fragmentation site and X|X′ refers to the residue C-terminal to the fragmentation site.
Constructing a Native C-score Using Fragment Intensity-Based Coefficients.
The C-score incorporates fragmentation propensities as priors (coefficients or weights) in its probability distribution function which determines the likelihood of a set of fragment ions being observed;45 previously, these coefficients were not tailored to nTDMS. Using the calculated average intensities shown in Figure 2b and Figure 2d, we created new nTDMS-tailored coefficients for a more specific C-score to be applied to native monomeric or native multimeric fragmentation data (coefficients are listed in Tables S7 and S8); we created unique coefficients for monomeric and multimeric data given the statistical differences shown in Figure 2. For many of the native proteoforms analyzed, using the intensity-based weights led to an increase in the final assigned score (Figure 4). For native monomers and multimers respectively, 77 and 66% were assigned a higher C-score when using the intensity-based weights derived herein (Figure 4a). Of particular interest is the impact of the intensity-based weighting on C-scores below 50, as a C-score of >40 has been used as an arbitrary cutoff point to designate a proteoform as “highly characterized”.45 For proteoforms assigned C-scores in the 0–50 range (Figure 4b), 55% of native monomers and 67% of native multimers were assigned larger C-scores using the intensity-based weights. Additionally, several proteoforms crossed the >40 threshold using the intensity-based weights. Similarly, 45% of proteoforms assigned C-scores in the 0–4 range (considered partially characterized) were assigned higher scores or remained unchanged using the native C-scores (Figure 4c). Importantly, a C-score of 3 denotes that two or more proteoforms are equally likely given a particular fragmentation data set used to search a proteoform database. Therefore, moving across a threshold from <3 to >3 is a notable improvement in proteoform characterization confidence; one proteoform crossed this specific threshold (Figure 4c). Proteoforms assigned higher C-scores using the intensity-based weights were generally supported by a lower number of matching fragment ions, but with a majority of those matching fragment ions occurring at D|X and X|P sites. A fragmentation map of N-terminally acetylated alpha-enolase (P06733) is shown as an example case (Figure 4d, at left), where 8 of 14 matching fragment ions belonged to D|X or X|P sites. By utilizing the new native fragmentation weights, the C-score was increased from 1,400 to 1,715.
Figure 4.
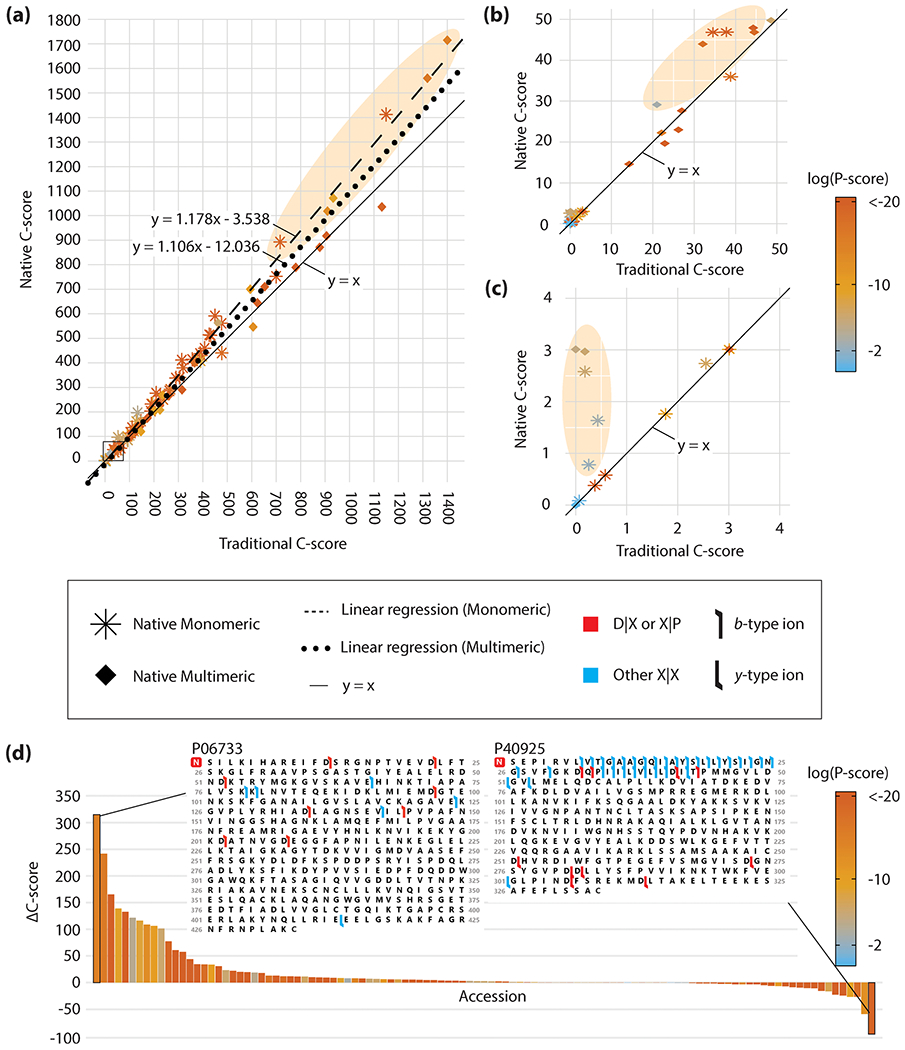
(a) Native C-score versus traditional C-score assigned to proteoforms within the native monomeric (asterisk) and native multimeric (diamond) data sets. Insets of (a) ranging from C-scores of (b) 0–50 and (c) 0–4 are provided. Linear regressions are shown for native monomers (dashed line) and native multimers (dotted line). Proteoforms with noticeably higher native C-scores are shown in shaded ellipses. P-scores for each proteoform are shown on a base ten logarithmic scale via color gradient. (d) ΔC-score (native C-score – traditional C-score) for all native monomers and multimers plotted by accession number and ranked from highest to lowest ΔC-score. Fragmentation maps are shown for the highest and lowest ΔC-score; the red box denotes N-terminal acetylation. Flags represent identified matching fragment ions and are colored red to denote a D|X or X|P fragment ion or blue for a fragment ion between any other residue pair. Flag directionality denotes a b-type (left to right) or y-type (right to left) ion.
Interestingly, proteoforms assigned lower C-scores using the nTDMS intensity-based weights generally exhibited fragmentation patterns similar to dTDMS data. These proteoforms were assigned more matching fragment ions, many of which occur at sites other than D|X and X|P. A fragmentation map of N-terminally acetylated malate dehydrogenase (P40925) is shown as an extreme example of this behavior (Figure 4d, at right). This case and others may be instances of unfolding during subunit ejection or an unstructured terminus, resulting in more “dTDMS-esque” fragmentation. Additional fragmentation maps demonstrating dTDMS- versus nTDMS-esque fragmentation are available in the Supporting Information (Figures S14 and S15). The native C-score was tested in an alpha version of a node within Proteome Discoverer (Thermo Fisher Scientific) for future release.
Creating a nFPS Using Fragment Intensity-Based Coefficients.
While updating C-scores with fragmentation intensities from nTDMS generally improved assigned C-scores for native proteoforms and will be useful in future proteomic analyses, the C-score cannot be universally accessed for all applications. Most current practitioners of nTDMS perform targeted applications and may not have forward and decoy databases from which to calculate a C-score. In these cases, a Poisson-based P-score66 is often used to assess confidence of matching a set of fragment ions to a known sequence. However, because nTDMS often produces significantly lower numbers of fragmentation channels for proteoforms than dTDMS, the standard P-score may mislead users by giving worse than expected values for particular proteoforms with only a handful (e.g., <~20) of matching fragment ions.66 However, as seen previously in Figure 4d with alpha-enolase (P06733), there exists cases where nearly all matched fragment ions correspond to highly probable fragmentation events according to the empirically determined fragmentation trends presented here. Because the P-score uses a Poisson model and places equal weight on all fragment ions, nTDMS results are disadvantaged using it alone. To address these concerns, we developed a new scoring routine named the Native Fragmentation Propensity Score (nFPS). The nFPS leverages the nTDMS fragmentation trends defined here to construct a metric complementary to the P-score. The derivation of this score is described in the Experimental Section and is graphically depicted in Figure S16. The nFPS weighs all fragment ion pairs using the native intensity-based weights reported here (Tables S7 and S8). This concept is akin to the McLuckey score,44 where D|X and X|P fragment ion matches were given the highest weighting over all other fragmentation sites. Because we consider every single possible amino acid combination (X|X′), the proposed nFPS presents a more general weighting of fragmentation data obtained from nTDMS experiments. For example, the new weighting system also favors to a lesser extent isoleucine, leucine, valine, lysine, glycine, and glutamic acid (Table S7 and S8). Additionally, the system “penalizes” (gives a weight of <5%) residues such as cysteine and tyrosine which typically do not enhance local fragmentation. We believe this better-informed weighting system is valuable particularly when identifying proteins with marginal fragmentation data or when localizing modifications, parsing polymorphisms, and defining isoforms.
To assess the performance of the nFPS versus the P-score across a wide range of proteoforms, 83 single scan fragmentation cases with known true-positive proteoform results were searched against the entire human database and assigned a nFPS and P-score. Sixty-eight of the searched scans produced a match between the true positive and the forward result with the nFPS calculated. While the rank order of P-score was not able to differentiate between correct and incorrect results for nearly 50% of the lowest scoring results, the rank order of nFPS was able to discern between correct and incorrect for all except one of the results (Figure 5a versus 5b). As can be seen in Figure 5c, several cases with relatively low confidence P-scores (i.e., >1 × 10−10, region highlighted in blue) still produced high nFPS scores. An example case is shown in Figure 5d, which shows the fragmentation map for identification of N-terminally acetylated parathymosin (P20962). The 16 matching fragment ions are relatively sparse, particularly if compared to robust dTDMS results (Figure S14), and the P-score of 1.78 × 10−10 reflects this lower confidence assignment. However, 14 of the 16 matching fragment ions occur at D|X and X|P sites, thereby leading to a robust nFPS of 13.7 (i.e., the observed fragmentation propensity product is 13.7 orders of magnitude higher than the geometric mean of the decoy fragmentation propensity products). This example illustrates that although the P-score was unable to provide a high degree of confidence in the assignment for this proteoform, the nFPS incorporated the sites of fragmentation to bring a higher level of confidence to the assignment. A combination of P-score and nFPS provides complementary metrics to nTDMS researchers, particularly for those that may be working on targeted applications and are unable to produce C-scores for their results. However, the nFPS can also be an important metric for proteoform identification, alongside both the P-score and C-score. A developing application, ProSight Native, is underway to assign P-score and nFPS values for targeted studies of native proteoforms and protein complexes.
Figure 5.
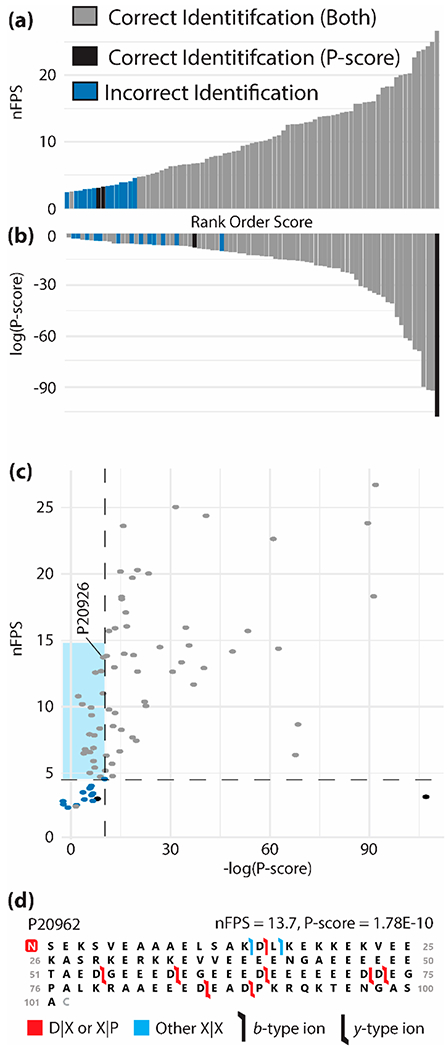
(a) nFPS and (b) P-score assigned to searched “true positive” native monomeric proteoforms in rank order of score value. (c) nFPS versus P-score, each point represents a single proteoform. Proteoforms are divided into those identified correctly by both scores (gray), by only the P-score (black), or incorrectly identified by both scores (blue). (d) Fragmentation map for parathymosin (P20962); the red box denotes N-terminal acetylation. Flags represent identified fragment ions and are colored red to denote a D|X or X|P fragment ion or blue for a fragment ion between any other residue pair. Flag directionality denotes a b-type (left to right) or y-type (right to left) ion.
CONCLUSION
We have expanded upon the relationship between primary sequence and HCD-based fragmentation for a large set of fragmentation data obtained from native monomers and protein subunits ejected from complexes. Our results demonstrate that known “hotspots” for collisional fragmentation (sites with N-terminal aspartic acid or C-terminal proline) are not only frequent, but significantly more intense than other preferential fragmentation sites in nTDMS. We have leveraged these fragment ion intensity data to (1) update the parameter set in a Bayesian model underlying the previously publishedC-score and (2) construct a native-specific fragmentation score which will increase the value and confidence of results obtained in both targeted and high throughput nTDMS applications. Overall, this work is another step in scoring evolution, which often begins with raw scores like the Xcorr from the 1994 SeQuest classic paper from the Yates lab.72 Raw metrics are typically then augmented with probability-based scores66,73 and mature with the use of Bayesian methods and prior knowledge embedded in an expert system.45 Continued refinement of scoring will be critical as automated data production becomes routine for native proteomics, as this transition will greatly expand the volume of tandem mass spectra and range of data quality.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Awards T32GM105538 and R43GM130262 as well as the National Institute of Aging Award 1RF1AG063903-01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00026.
Referenced supplemental figures and tables (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.0c00026
The authors declare the following competing financial interest(s): K.R.D., R.T.F., and N.L.K. are involved in ProSight commercialization.
Contributor Information
Ashley N. Ives, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Taojunfeng Su, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Kenneth R. Durbin, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States; Proteinaceous Inc., Evanston, Illinois 60204, United States
Bryan P. Early, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States
Henrique dos Santos Seckler, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Ryan T. Fellers, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States; Proteinaceous Inc., Evanston, Illinois 60204, United States
Richard D. LeDuc, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Luis F. Schachner, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States
Steven M. Patrie, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States.
Neil L. Kelleher, Departments of Chemistry and Molecular Biosciences, the Chemistry of Life Processes Institute, and the Proteomics Center of Excellence, Northwestern University, Evanston, Illinois 60208, United States; Proteinaceous Inc., Evanston, Illinois 60204, United States.
REFERENCES
- (1).Cui W; Rohrs HW; Gross ML Top-down mass spectrometry: recent developments, applications and perspectives. Analyst 2011, 136, 3854–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Catherman AD; Skinner OS; Kelleher NL Top Down proteomics: facts and perspectives. Biochem. Biophys. Res. Commun. 2014, 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith LM; Kelleher NL Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Skinner OS; Havugimana PC; Haverland NA; Fornelli L; Early BP; Greer JB; Fellers RT; Durbin KR; Do Vale LHF ; Melani RD; Seckler HS; Nelp MT; Belov ME; Horning SR; Makarov AA; LeDuc RD; Bandarian V; Compton PD; Kelleher NL An informatic framework for decoding protein complexes by top-down mass spectrometry. Nat. Methods 2016, 13, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sleno L; Volmer DA Ion activation methods for tandem mass spectrometry. J. Mass Spectrom. 2004, 39, 1091–1112. [DOI] [PubMed] [Google Scholar]
- (7).Brodbelt JS Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2016, 88, 30–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lermyte F; Valkenborg D; Loo JA; Sobott F Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based analysis of protein structure. Mass Spectrom. Rev. 2018, 37, 750–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Paizs B; Suhai S Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 2005, 24, 508–548. [DOI] [PubMed] [Google Scholar]
- (10).McLafferty F Tandem mass spectrometry. Science 1981, 214, 280–287. [DOI] [PubMed] [Google Scholar]
- (11).Taylor GK; Kim Y-B; Forbes AJ; Meng F; McCarthy R; Kelleher NL Web and Database Software for Identification of Intact Proteins Using “Top Down” Mass Spectrometry. Anal. Chem. 2003, 75, 4081–4086. [DOI] [PubMed] [Google Scholar]
- (12).Roth MJ; Parks BA; Ferguson JT; Boyne MT; Kelleher NL Proteotyping”: Population Proteomics of Human Leukocytes Using Top Down Mass Spectrometry. Anal. Chem. 2008, 80, 2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Amunugama R; Hogan JM; Newton KA; McLuckey SA Whole Protein Dissociation in a Quadrupole Ion Trap: Identification of an a Priori Unknown Modified Protein. Anal. Chem. 2004, 76, 720–727. [DOI] [PubMed] [Google Scholar]
- (14).Breuker K; Oh H; Horn DM; Cerda BA; McLafferty FW Detailed Unfolding and Folding of Gaseous Ubiquitin Ions Characterized by Electron Capture Dissociation. J. Am. Chem. Soc. 2002, 124, 6407–6420. [DOI] [PubMed] [Google Scholar]
- (15).Horn DM; Breuker K; Frank AJ; McLafferty FW Kinetic Intermediates in the Folding of Gaseous Protein Ions Characterized by Electron Capture Dissociation Mass Spectrometry. J. Am. Chem. Soc. 2001, 123, 9792–9799. [DOI] [PubMed] [Google Scholar]
- (16).Oh H; Breuker K; Sze SK; Ge Y; Carpenter BK; McLafferty FW Secondary and tertiary structures of gaseous protein ions characterized by electron capture dissociation mass spectrometry and photofragment spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15863–15868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Breuker K; McLafferty FW Native Electron Capture Dissociation for the Structural Characterization of Noncovalent Interactions in Native Cytochrome c. Angew. Chem., Int. Ed. 2003, 42, 4900–4904. [DOI] [PubMed] [Google Scholar]
- (18).Li H; Nguyen HH; Ogorzalek Loo RR; Campuzano IDG ; Loo JA An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem 2018, 10, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lermyte F; Konijnenberg A; Williams JP; Brown JM; Valkenborg D; Sobott F ETD Allows for Native Surface Mapping of a 150 kDa Noncovalent Complex on a Commercial Q-TWIMS-TOF Instrument. J. Am. Soc. Mass Spectrom. 2014, 25, 343–350. [DOI] [PubMed] [Google Scholar]
- (20).Lermyte F; Sobott F Electron transfer dissociation provides higher-order structural information of native and partially unfolded protein complexes. Proteomics 2015, 15, 2813–2822. [DOI] [PubMed] [Google Scholar]
- (21).Breuker K; McLafferty FW The Thermal Unfolding of Native Cytochrome c in the Transition from Solution to Gas Phase Probed by Native Electron Capture Dissociation. Angew. Chem. Int. Ed. 2005, 44, 4911–4914. [DOI] [PubMed] [Google Scholar]
- (22).Schennach M; Breuker K Probing Protein Structure and Folding in the Gas Phase by Electron Capture Dissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mitchell Wells J; McLuckey SA Collision-Induced Dissociation (CID) of Peptides and Proteins; Academic Press, 2005. [DOI] [PubMed] [Google Scholar]
- (24).Hayes RN; Gross ML Collision-Induced Dissociation; Academic Press, 1990. [DOI] [PubMed] [Google Scholar]
- (25).Hopper JTS; Oldham NJ Collision induced unfolding of protein ions in the gas phase studied by ion mobility-mass spectrometry: The effect of ligand binding on conformational stability. J. Am. Soc. Mass Spectrom. 2009, 20, 1851–1858. [DOI] [PubMed] [Google Scholar]
- (26).Dixit SM; Polasky DA; Ruotolo BT Collision induced unfolding of isolated proteins in the gas phase: past, present, and future. Curr. Opin. Chem. Biol. 2018, 42, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Allison TM; Reading E; Liko I; Baldwin AJ; Laganowsky A; Robinson CV Quantifying the stabilizing effects of protein-ligand interactions in the gas phase. Nat. Commun 2015, 6, 8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dongré AR; Jones JL; Somogyi Á; Wysocki VH Influence of Peptide Composition, Gas-Phase Basicity, and Chemical Modification on Fragmentation Efficiency: Evidence for the Mobile Proton Model. J. Am. Chem. Soc. 1996, 118, 8365–8374. [Google Scholar]
- (29).Wysocki VH; Tsaprailis G; Smith LL; Breci LA Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 2000, 35, 1399–1406. [DOI] [PubMed] [Google Scholar]
- (30).Xia Y; Liang X; McLuckey SA Ion Trap versus Low-Energy Beam-Type Collision-Induced Dissociation of Protonated Ubiquitin Ions. Anal. Chem. 2006, 78, 1218–1227. [DOI] [PubMed] [Google Scholar]
- (31).Chanthamontri C; Liu J; McLuckey SA Charge state dependent fragmentation of gaseous α-synuclein cations via ion trap and beam-type collisional activation. Int. J. Mass Spectrom. 2009, 283, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Olsen JV; Macek B; Lange O; Makarov A; Horning S; Mann M Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. [DOI] [PubMed] [Google Scholar]
- (33).Michalski A; Neuhauser N; Cox J; Mann M A Systematic Investigation into the Nature of Tryptic HCD Spectra. Journal of Proteome Research. 2012, 11, 5479–5491. [DOI] [PubMed] [Google Scholar]
- (34).Haverland NA; Skinner OS; Fellers RT; Tariq AA; Early BP; LeDuc RD; Fornelli L; Compton PD; Kelleher NL Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Reid GE; Wu J; Chrisman PA; Wells JM; McLuckey SA Charge-State-Dependent Sequence Analysis of Protonated Ubiquitin Ions via Ion Trap Tandem Mass Spectrometry. Anal. Chem. 2001, 73, 3274–3281. [DOI] [PubMed] [Google Scholar]
- (36).Schaaff TG; Cargile BJ; Stephenson JL; McLuckey SA Ion Trap Collisional Activation of the (M + 2H)2+ – (M + 17H)17+ Ions of Human Hemoglobin β-Chain. Anal. Chem. 2000, 72, 899–907. [DOI] [PubMed] [Google Scholar]
- (37).Cobb JS; Easterling ML; Agar JN Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. J. Am. Soc. Mass Spectrom. 2010, 21, 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Loo JA; Edmonds CG; Smith RD Tandem mass spectrometry of very large molecules. 2. Dissociation of multiply charged proline-containing proteins from electrospray ionization. Anal. Chem. 1993, 65, 425–438. [DOI] [PubMed] [Google Scholar]
- (39).Gu C; Tsaprailis G; Breci L; Wysocki VH Selective Gas-Phase Cleavage at the Peptide Bond C-Terminal to Aspartic Acid in Fixed-Charge Derivatives of Asp-Containing Peptides. Anal. Chem. 2000, 72, 5804–5813. [DOI] [PubMed] [Google Scholar]
- (40).Tsaprailis G; Somogyi Á; Nikolaev EN; Wysocki VH Refining the model for selective cleavage at acidic residues in arginine-containing protonated peptides22Dedicated to Bob Squires for his many seminal contributions to mass spectrometry and ion chemistry. Int. J. Mass Spectrom. 2000, 195–196, 467–479. [Google Scholar]
- (41).Gu C; Somogyi A; Wysocki VH; Medzihradszky KF Fragmentation of protonated oligopeptides XLDVLQ (X = L, H, K or R) by surface induced dissociation: additional evidence for the ‘mobile proton’ model. Anal. Chim. Acta 1999, 397, 247–256. [Google Scholar]
- (42).Compton PD; Zamdborg L; Thomas PM; Kelleher NL On the Scalability and Requirements of Whole Protein Mass Spectrometry. Anal. Chem. 2011, 83, 6868–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bleiholder C; Suhai S; Harrison AG; Paizs B Towards Understanding the Tandem Mass Spectra of Protonated Oligopeptides. 2: The Proline Effect in Collision-Induced Dissociation of Protonated Ala-Ala-Xxx-Pro-Ala (Xxx = Ala, Ser, Leu, Val, Phe, and Trp). J. Am. Soc. Mass Spectrom. 2011, 22, 1032–1039. [DOI] [PubMed] [Google Scholar]
- (44).Foreman DJ; Dziekonski ET; McLuckey SA Maximizing Selective Cleavages at Aspartic Acid and Proline Residues for the Identification of Intact Proteins. J. Am. Soc. Mass Spectrom. 2019, 30, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).LeDuc RD; Fellers RT; Early BP; Greer JB; Thomas PM; Kelleher NL The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. Journal of proteome research. 2014, 13, 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Dang X; Scotcher J; Wu S; Chu RK; Tolić N; Ntai I; Thomas PM; Fellers RT; Early BP; Zheng Y; Durbin KR; LeDuc RD; Wolff JJ; Thompson CJ; Pan J; Han J; Shaw JB; Salisbury JP; Easterling M; Borchers CH; Brodbelt JS; Agar JN; Paša-Tolić L; Kelleher NL; Young NL The first pilot project of the consortium for top-down proteomics: A status report. Proteomics 2014, 14, 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Reid GE; Shang H; Hogan JM; Lee GU; McLuckey SA Gas-Phase Concentration, Purification, and Identification of Whole Proteins from Complex Mixtures. J. Am. Chem. Soc. 2002, 124, 7353–7362. [DOI] [PubMed] [Google Scholar]
- (48).Durbin KR; Skinner OS; Fellers RT; Kelleher NL Analyzing Internal Fragmentation of Electrosprayed Ubiquitin Ions During Beam-Type Collisional Dissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hogan JM; McLuckey SA Charge state dependent collision-induced dissociation of native and reduced porcine elastase. J. Mass Spectrom. 2003, 38, 245–256. [DOI] [PubMed] [Google Scholar]
- (50).Engel BJ; Pan P; Reid GE; Wells JM; McLuckey SA Charge state dependent fragmentation of gaseous protein ions in a quadrupole ion trap: bovine ferri-, ferro-, and apo-cytochrome c. Int. J. Mass Spectrom. 2002, 219, 171–187. [Google Scholar]
- (51).Skinner OS; Haverland NA; Fornelli L; Melani RD; Do Vale LHF; Seckler HS; Doubleday PF; Schachner LF; Srzentic K; Kelleher NL; Compton PD Top-down characterization of endogenous protein complexes with native proteomics. Nat. Chem. Biol. 2018, 14, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Park YJ; Kenney GE; Schachner LF; Kelleher NL; Rosenzweig AC Repurposed HisC Aminotransferases Complete the Biosynthesis of Some Methanobactins. Biochemistry 2018, 57, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Heck AJR Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods 2008, 5, 927–933. [DOI] [PubMed] [Google Scholar]
- (54).Duijn E Current limitations in native mass spectrometry based structural biology. J. Am. Soc. Mass Spectrom. 2010, 21, 971–978. [DOI] [PubMed] [Google Scholar]
- (55).Marcoux J; Cianférani S Towards integrative structural mass spectrometry: Benefits from hybrid approaches. Methods 2015, 89, 4–12. [DOI] [PubMed] [Google Scholar]
- (56).Catherman AD; Durbin KR; Ahlf DR; Early BP; Fellers RT; Tran JC; Thomas PM; Kelleher NL Large-scale Top-down Proteomics of the Human Proteome: Membrane Proteins, Mitochondria, and Senescence. Mol. Cell. Proteomics 2013, 12, 3465–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Skinner OS; Do Vale LHF; Catherman AD; Havugimana PC; Sousa M.V.d.; Compton PD; Kelleher NL Native GELFrEE: A New Separation Technique for Biomolecular Assemblies. Anal. Chem. 2015, 87, 3032–3038. [DOI] [PubMed] [Google Scholar]
- (58).Melani RD; Seckler HS; Skinner OS; Do Vale LHF; Catherman AD; Havugimana PC; Valle de Sousa M; Domont GB; Kelleher NL; Compton PD CN-GELFrEE - Clear Native Gel-eluted Liquid Fraction Entrapment Electrophoresis. J. Visualized Exp. 2016, 53597–53597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wojcik R; Dada OO; Sadilek M; Dovichi NJ Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid Commun. Mass Spectrom. 2010, 24, 2554–2560. [DOI] [PubMed] [Google Scholar]
- (60).Belov ME; Damoc E; Denisov E; Compton PD; Horning S; Makarov AA; Kelleher NL From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem. 2013, 85, 11163–11173. [DOI] [PubMed] [Google Scholar]
- (61).Wessel D; Flugge UI A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [DOI] [PubMed] [Google Scholar]
- (62).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (63).Meng EC; Pettersen EF; Couch GS; Huang CC; Ferrin TE Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinf. 2006, 7, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Rodríguez-Guerra Pedregal J; Maréchal J-D PyChimera: use UCSF Chimera modules in any Python 2.7 project. Bioinformatics 2018, 34, 1784–1785. [DOI] [PubMed] [Google Scholar]
- (65).Racine JS RStudio: A Platform-Independent IDE for R and Sweave. Journal of Applied Econometrics. 2012, 27, 167–172. [Google Scholar]
- (66).Meng F; Cargile BJ; Miller LM; Forbes AJ; Johnson JR; Kelleher NL Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat. Biotechnol 2001, 19, 952–957. [DOI] [PubMed] [Google Scholar]
- (67).Tran JC; Zamdborg L; Ahlf DR; Lee JE; Catherman AD; Durbin KR; Tipton JD; Vellaichamy A; Kellie JF; Li M; Wu C; Sweet SMM; Early BP; Siuti N; LeDuc RD; Compton PD; Thomas PM; Kelleher NL Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Fernandez de la Mora J Electrospray ionization of large multiply charged species proceeds via Dole’s charged residue mechanism. Anal. Chim. Acta 2000, 406, 93–104. [Google Scholar]
- (69).Lermyte F; Tsybin YO; O’Connor PB; Loo JA Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. J. Am. Soc. Mass Spectrom. 2019, 30, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Jurchen JC; Williams ER Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J. Am. Chem. Soc. 2003, 125, 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Jurchen JC; Garcia DE; Williams ER Further studies on the origins of asymmetric charge partitioning in protein homodimers. J. Am. Soc. Mass Spectrom. 2004, 15, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Eng JK; McCormack AL; Yates JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- (73).Sadygov RG; Yates JR A Hypergeometric Probability Model for Protein Identification and Validation Using Tandem Mass Spectral Data and Protein Sequence Databases. Anal. Chem. 2003, 75, 3792–3798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.