

Abstract
Acute myeloid leukemia (AML) is the most common form of acute leukaemia and has the highest mortality rate. Screening for mutations in patients with AML has shown that in many cases genes carrying mutations are involved in the alternate splicing of mRNA. These include members of the Serine Arginine (SR) family of splicing factors, as well as components of the spliceosome. Mutations in associated genes also affect the function of members of the heterogeneous nuclear ribonucleoproteins (hnRNPs). These mutations in splicing factors can lead to changes in the expression of different isoforms whose splicing is controlled by these splicing factors. These different isoforms may have completely different functions, for example, members of the BCl-2 family are alternately spliced to give rise to pro and anti-apoptotic members. Mutations in the splicing factors that control the splicing of these mRNAs can lead to changes in the expression level of these isoforms. In this review we will examine the mechanics of the regulation of the various splice isoforms and how this drives the development of tumors. This information is pertinent for drug discovery, and the splicing factors with the most promise for pharmacological control will be discussed.
Keywords: SR proteins, SFRS2, U2AF1, SRSF1, hnRNPs, SPF-45, FLT3
Introduction
Acute myeloid leukaemia (AML) is a malignant disorder of the bone marrow. Its major characteristics are the clonal expansion of myeloid progenitor cells and the cessation of the differentiation of these cells. The abnormal differentiation of these myeloid stem cells, results from chromosomal rearrangements and genetic mutations. While it only accounts for approximately 25% of all leukaemia cases the high mortality rate associated with this cancer means that disproportionate amounts of effort are spent attempting to treat it [1]. Initially, AML develops from the pre-cancerous blood condition, Myelodysplastic Syndrome (MDS). A transformative event results in MDS developing into AML in about a third of MDS patients [2]. In the United States of America AML is the most common form of acute leukaemia, with nearly 20,000 cases recorded in 2019, making up 1% of all cancers and up to 80% of all acute leukaemia cases in the USA [3]. In South Africa it is difficult to establish accurate figures as the National Cancer Registry does not differentiate between subtypes of leukaemia. South Africa’s National Cancer Registry recorded 227 cases of leukaemia in females and 264 cases in males in 2016 (https://www.nicd.ac.za/centres/national-cancer-registry/). The incidence of AML cases increases with age, while the prognosis decreases. The prognosis in patients 65 years or older is very poor, it is estimated that up to 70% of patients die within a year of diagnosis. This high mortality rate in older patients becomes even greater when aggressive treatments are used. Treatment is made more difficult due to the development of drug resistance by AML, making treatments less effective.
One of the most important factors behind the molecular basis of the development and progression of AML is alternate splicing. Alternate gene splicing also plays a role in the development of drug resistance [4]. Here alternate splicing can be affected by changes to splicing factor sequence or activity through changes in the phosphorylation state of the splicing factor or mutations in the splicing factor. The resultant changes in splicing influences or is influenced by changes in gene expression. This result in changes in the populations of isoforms as the altered splicing factors selects for or favours different isoforms (Figure 1).
Figure 1.
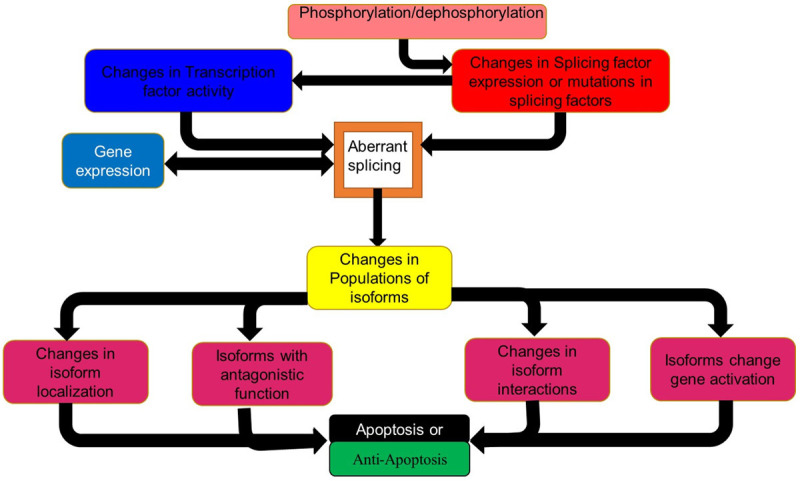
How splicing is affected in cancer and its effects. Schematic diagram of factors influencing aberrant splicing and drug resistance in AML. TF transcription factor, SF splicing factor.
Splicing and the spliceosome
The spliceosome complex is the molecular machinery responsible for alternate splicing. The spliceosome is formed by small nuclear ribonucleic acids (RNAs) in combination with splice factor proteins. The spliceosome complex is responsible for the splicing of mRNA into different transcripts that code for different protein isoforms by favouring or repressing the use of various splice sites [5,6]. There are two dominant families of splicing proteins. Firstly, there are the serine-rich (SR) proteins. Secondly, there are the heterogenous nuclear ribonucleoproteins (hnRNP) [7] (Table 1). The prevalence of aberrant alternative splicing events in MDS have been revealed through next generation sequencing (NGS) [8], with over 50% of AML patients having mutations in spliceosome components [9]. This is unsurprising as approximately 2022 genes were found to be mutated in MDS patients thus far. However, it has been found that mutations in splicing machinery that affects the splicing of multiple genes is not as common in AML than it is in MDS. Despite this, these mutations have been shown to contribute to the development of AML [10].
Table 1.
Splicing factors involved in AML and their domain structure

|
Splicing events and mutations in the various splicing factors increase or decrease the chances of MDS progressing to AML. Next Generation Sequencing and Genome Wide Association Studies of AML patients show that up to a third of all genes are aberrantly spliced compared to healthy control samples [11]. Most of these splicing changes were in the coding regions while those in the untranslated regions may affect the expression of the proteins coded for by these transcripts by affecting the transcripts stability or translation [11]. Commonly mutated splicing modulators include members of the serine (S) and arginine (R) family of splicing factors, such as SRSF2, SF3B1, U2AF1 and ZRSR2. These proteins contain at least one RNA recognition motif (RRM) domain and at least one SR domain [10,12] (Figure 2; Table 1). Mutations in other splicing factors such as SF3A1 and PRPF40B are rarely found [8]. The expression of some of these splice variants depends on the stage of the disease, with some only being detected at early stages when the disease was first diagnosed, while others were only present during remission or relapse [11].
Figure 2.
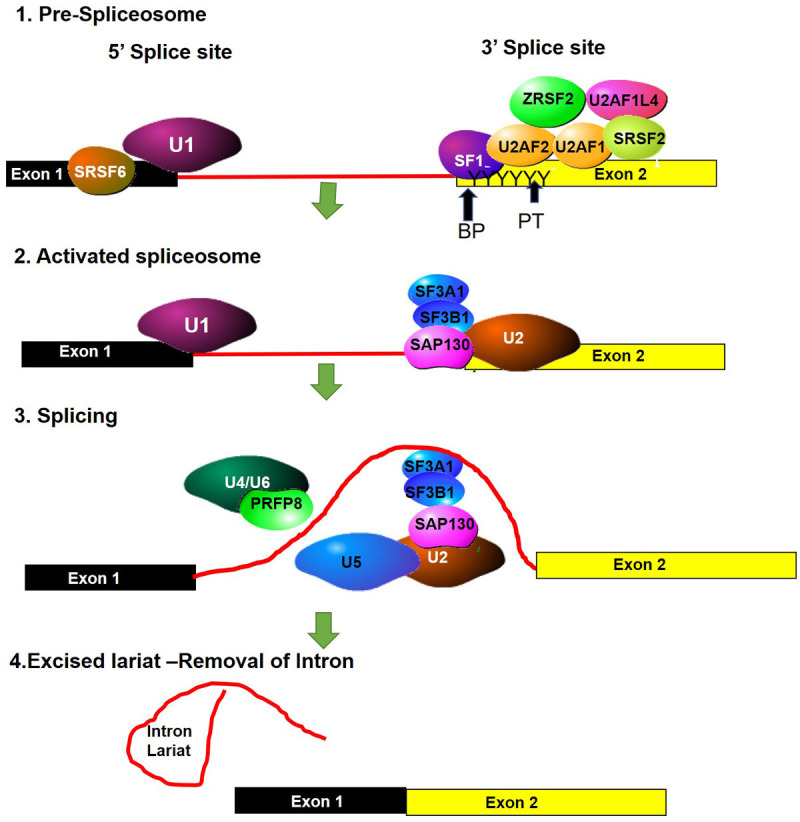
Spliceosome complex. (1) U1 snRNP binds to the 5’ splice site (SS) of the pre-mRNA. The SR rich splicing factor, SRSF6 binds near the 5’SS, where it regulates splicing events. At the 3’ splice site the SR proteins SF1, SRSF2 and ZESF2 as well as the large subunit of the U2 auxiliary factor U2AF2, bind to the splice site. These splicing regulators bind to the branch point (BP) sequence and the polypyrimidine tract (PT). The smaller subunit of U2AF (U2AF1) binds to the 3’SS, forming a complex with U2AF2 and SRSF2. Another two RNA-binding proteins interact with the U2AF complex (2). Once the 3’ splice site is recognised the next group of splicing factors bind to the splice site, these include U2 snRNP, SF3A1, SF3B1, and SAP130. (3) The U4/U6 and U5 complex, together with PRFP8 are recruited and localize on the BP and PT regions. (4) This catalyses the release of the intron lariat and ligation of the exons.
Serine arginine family of splicing factors
The U2 small nuclear RNA auxiliary factor 1 (U2AF1)
The U2-complex auxiliary factor 1 (U2AF1) splicing factor, also known as U2AF35, is a 35 kDa protein. The U2AF1 gene was found to be mutated in 3-4% of AML cases. These mutations have been found to play a role in 369 identified splicing changes [10]. By analysing exon usage in AML samples, it was found that there were many alternate splicing events taking place in immune related and inflammatory genes. It has also been established that mutations in U2AF1 leads to a poorer prognosis in AML patients [13]. The Interleukin-1 receptor-associated kinase 4 (IRAK4) was one of the most commonly spliced genes in AML. The splicing of this gene is controlled by U2AF1 [14]. One of the two isoforms of IRAK4 is named IRAK4-long (IRAK4-L) was the dominant isoform in AML (Figure 3). This isoform retains exon 4 which increases the activation of NF-κB [14] (Figure 2). Other genes involved in processes that may be relayed to malignancy in AML and whose splicing is affected by U2AF1 include H2AFY, STRAP, ATR, CEP164, EHMT1, WAC, PABPC4, PPWD1, PTBP1, STRAP, UPF3B [15].
Figure 3.
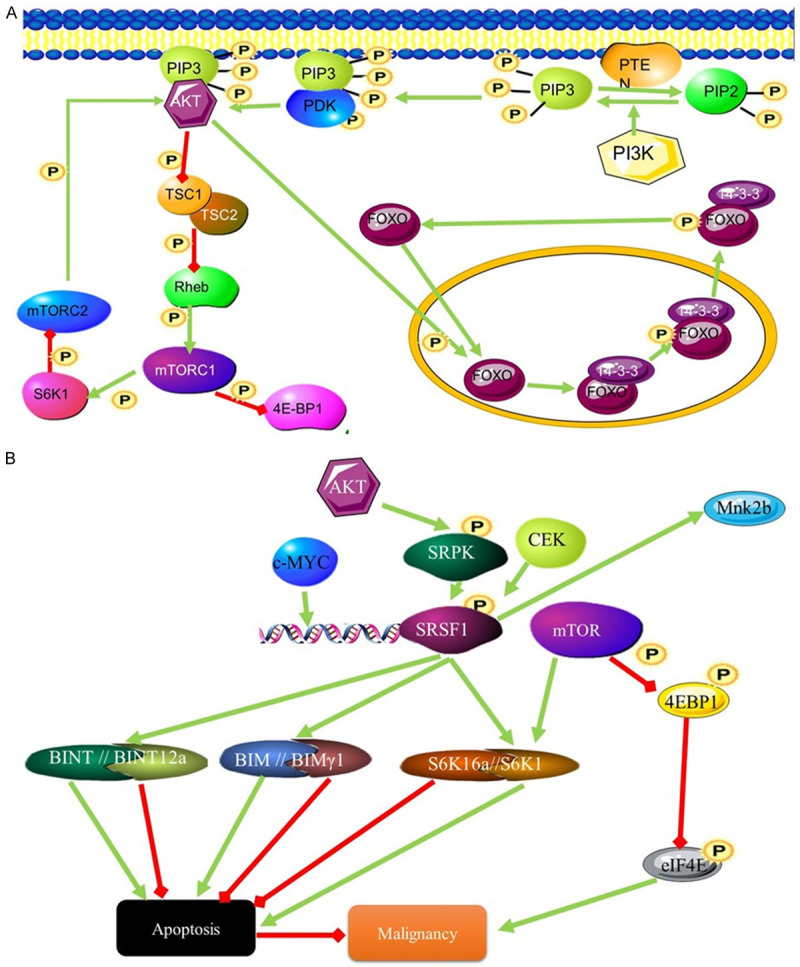
The influence of the SRSF1 splicing factor on malignancy through AKT signalling. (A) PIP3 activates AKT signalling through phosphorylation. This leads to the activation of transcription and inhibits the mTORC1 signalling pathway. mTORC1 activates AKT signalling through phosphorylation of AKT (B) SRFS1 influences splicing downstream of the AKT signalling pathway by interacting with mTORC1 pathway. mTOR and SRSF1 signalling converge at S6K. Different S6K isoforms can stimulate or inhibit apoptosis and contribute to malignancy.
Serine/arginine-rich splicing factor 1
The SRSF1 gene encodes the Serine/arginine-rich splicing factor (SRSF1) protein also called alternative splicing factor 1 (ASF1), Splicing Factor 2 (SF2) and SRp30. The 33 kDa protein is an absolute requirement for alternate splicing. Splice site selection is largely dependent on the levels of SRSF1 [16]. The SRSF1 splice factor is normally responsible for the inclusion of alternate exons and is responsible for the presence of alternate isoforms of proteins involved in apoptotic pathways. The overexpression of SRSF1 results in tumorigenesis due to the inhibition of apoptosis [17]. This is achieved through splicing that promotes the expression of pro-apoptotic isoforms of MCL-1S, BCL-XS, CASP9, and CASP2 variants [18]. AKT has been found to be expressed at higher levels in AML. This is probably due to its effects on splicing factors [19]. Caspase 9 is alternatively spliced to give rise to two isoforms, one pro apoptotic the other anti-apoptotic. The anti-apoptotic variant CASP-9b differs from the pro-apoptotic variant CASP-9a by the absence of four exons (Figure 3) [20]. The changes in the expression levels of these different Casp9 isoforms are controlled through AKT phosphorylation of SRSF1. SRSF1 controlled splicing of caspase 9 mRNA gives rise to the antiapoptotic isoform [20].
AKT and SRSF1 are also involved in the mTORC1 pathway. This pathway controls protein synthesis in response to growth and proliferation signals and does so by acting as an amino acid sensor. The PI3K-mTOR pathway is commonly activated in many cancers, with many components of this pathway being mutated in these cancers [21]. Higher expression of components of this pathway is associated with AML. Increased signalling activity of this pathway results in increased expression of oncogenes that promote growth and proliferation. Alternately, this could result from the inhibition of tumour suppressor genes that are controlled by the pathway such as, PTEN, LKB1, TSC1 and TSC2 [22]. Inhibition of this pathway leads to a reversal in transformation. The role of SRSF1 is also highlighted by the fact that overexpression of another splicing factor, RBM4, leads to the inhibition of SRSF1 and the inhibition of transformation sue to the initiation of apoptosis due to increased expression of the pro-apoptotic Bcl-Xs isoform [23]. mTORC1 is a complex of proteins that phosphorylates multiple substrates (Figure 3), including the translation inhibitors 4E-BP1 and 4E-BP2 (Figure 3) which are inactivated as a result of phosphorylation. Phosphorylation of AKT by mTORC leads to increased malignancy due to inhibition of apoptosis [24]. Blocking mTOR leads to inhibition of the ability of SRSF1 to change isoform populations and block apoptosis [25]. Although the mechanism behind the control of mTOR by SRSF1 splicing is not yet known, it is known that mTORC2 is not involved and this activation occurs downstream of AKT. The SRSF1 and mTOR signalling pathways converge on the mTOR substrate S6K1, where SRSF1 controls splicing of this substrate. The isoform favoured by SRFS1 promotes cell proliferation [25].
Splicing factor, arginine/serine-rich 2 SFRS2/SC35
The most common mutation in the SRSF2 gene occurs at proline 95 and alters the binding affinity of the RRM motif. Mutations in SRSF2 occur in 14% of MDS patients where they are associated with increase hemoglobin. Patients with SRSF2 mutations have a worse prognosis. The mutation involving a histidine to proline change at position 95 leads to an altered interaction in the second cytosine in the splice site. This results in a change in the splicing of multiple genes [15]. EZH2, is a gene known to be involved in the pathogenesis of MDS and AML, and is a histone methyltransferase. Normal splicing of this gene results in two isoforms with one isoform lacking one SANT domain due to the absence of exon 4. SFRS2 mutants promote the use of a premature termination codon leading to a truncated protein that is rapidly degraded [26,27] (Figure 3). The E2F transcription factor 1 (E2F1) is another transcription factor that is involved in promoting apoptosis. It achieves this through the regulation of the splicing of genes involved in apoptosis [28]. Some of these pro-apoptotic roles of E2F1 are dependent on E2F1 interacting with the splicing factor SFRS2 (SC35). This splicing factor leads to the alternate splicing of FLIP (FLICE like inhibitory Protein) and the increased expression of the pro apoptotic isoform of FLIP and the pro apoptotic form of Caspase 9 and BCL-X [29]. The tumour suppressor activity of E2F1 can be altered through the interaction of the transcription factor with different co-factors. These co-factors are commonly found to be expressed in tumour cells. The interaction of E2F1 with these cofactors results in tumours that behave more aggressively and have increased resistance to various chemotherapy treatments.
Serine/threonine-protein kinase (SRPK1)
SRPK1 regulates splicing by regulating the function of SR splicing proteins, SRSF1 and SRSF2. Knockdown or mutations that decrease the function of SRPK1 leads to changes in the splicing of genes that regulates the splicing of genes such as VEGF. Some isoforms of VEGF promote angiogenesis and can therefore promote tumorigenesis. The zinc finger transcription factor Wilms tumour 1 (WT1) is commonly used as a prognostic biomarker in AML. It is spliced to give rise to four isoforms. The SRPK1 splicing factor is responsible for the expression of one isoform which then affects the splicing of VEGF growth factor. It promotes the expression of the anti-angiogenic VEGF isoform [30]. In this way WT1 acts as tumour suppressor at least in kidney cancer. However, in AML it can act as an oncogene where it promotes resistance to death signals [31,32]. This is due to the different isoforms of the ability of WT to interact directly with the RBN4, WTAF and U2AF2 splicing factors. This leads to different splicing outcomes [30].
ZRSR2
This splicing factor and component of the U2 auxiliary factor heterodimer has been found to be mutated in approximately 5% of patients with MDS. These mutations are loss of function mutations that give rise to splicing errors [33].
RBM4
The splicing factor RBM4 controls splicing that leads to the generation of protein isoforms that inhibit proliferation and tumorigenesis. It acts as a competitor to SRSF1 for splice sites on mRNA and favours the expression of isoforms that DRSF1 does not, such as the Bcl-Xl isoforms which is pro-apoptotic [23]. As previously stated RBM1 also antagonises mTOR activity, working against SRAF1. In AML, like in many cancers there is thought to be an imbalance in the functional level of SRSF1:RBM1.
Heterogeneous nuclear ribonucleoproteins (hnRNPs)
These splicing factors form complexes with RNA and they are responsible for suppressing splicing by blocking spliceosome access to mRNA resulting in splice sites being made either more or less accessible. Research in liver cancer implicated hnRNP L and lncRNA complexes associations affected Protein kinase B signalling and DNA damage sensing [34]. Protein kinase B (PKB) is also known as AKT, plays a role in multiple cellular processes including cell survival, cell cycle regulation, transcription, cell migration angiogenesis, metabolism and lysosomal biogenesis. AKT is also able to alter splicing in AML by selecting between two rival splicing enhancing factors hnRNP U and hnRNP L. These two splicing factors are also involved in the selection between pro and anti-apoptosis isoforms of caspase 9. One of the four exons absent from the anti-apoptotic isoform is the third exon. Both of these splicing factors bind to the third exon on the caspase 9 mRNA, specifically it interacts with the G rich region in exon 3 (GAGGG) (Figure 4). The displacement of one splicing factor in favour of the other results in changes in splicing leading to increased expression of one isoform and the decrease in the expression of another. This change in the binding of splicing factor is caused by phosphorylation and displacement of the bound splicing factor. AKT phosphorylates hnRNP L which is then able to remove hnRNP U which is currently bound to this third exons binding location. This allows for greater levels of expression of the anti-apoptotic isoform and increased levels of tumorigenesis [35].
Figure 4.
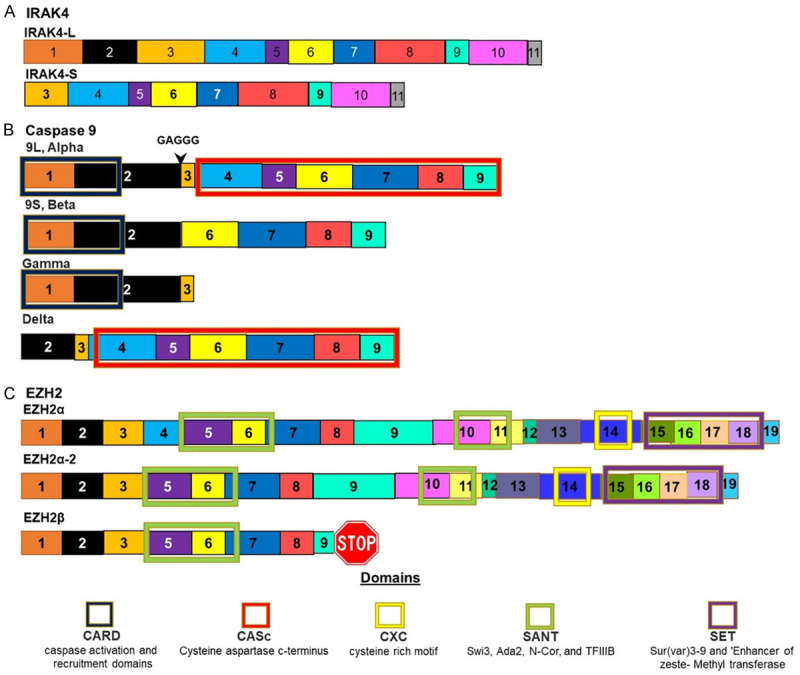
Isoforms of proteins whose splicing is controlled by SR proteins. A. IRAK4 is spliced to give rise to two isoforms. The splicing of this gene is controlled by U2AF1. In AML the longer isoform is favoured by mutations in this splicing factor. B. Caspase 9 is spliced to give rise to at least four isoforms. This splicing is regulated by SRSF1. Mutations in this splicing factor favours the shorter Beta isoform that lack of the CASc domain that has anti-apoptotic activity as this domain is responsible for Caspases peptidase activity. C. EZH2 is spliced to give rise to two isoforms. The inclusion of exon 4 leads to a longer isoform which favours AML.
hnRNP K
It was established that many AML patients have deletions at the 9q21.32 locus. One of the genes mapped to this location was Heterogeneous Nuclear Ribonuclear Protein K (HNRNPK) [36]. It is suspected that hnRNP K can act as a regulator of genes that are involved in hematopoietic maturation and differentiation. It is known that hnRNP K regulates p53 expression by acting on p21 [36]. hnRNP K also regulates the splicing of C.EBP to give rise to the isoforms C/EBPα p42 and p30 isoforms. The p42 isoform was downregulated when hnRNP K expression was decreased. The p42 isoform has anti-mitotic activity while the p30 isoform does not [37].
Other splicing factors
Splicing factor 3B subunit 1 (SF3B1)/spliceosome-associated protein 155 (Sap 155)
The spliceosomal machinery consists of small nuclear RNA associated with polypeptides (approximately 88 proteins). One of these associated proteins is Sap 155 also known as the 3β subunit 1 of the splicing factor (SF3B1). Next Generation Sequencing of AML samples revealed mutations in this splicing factor in 20% of patients [38]. Studies have indicated that mutations in the SF3B1 gene are factors which can act to initiate mutagenic events in lympho-myeloid hematopoietic stem cells [39]. Mutations have been identified as being common in four positions, the lysine to glutamic acid substitution at codon 700 and at amino acid positions 622, 625, 662, and 666 [40]. This splicing factor is a member of the U2 snRNP complex component and functions by binding pre-mRNA upstream on the branch site for the intron to be spliced [41]. The SRSF2 protein enhances the binding of the U1 snRNP complex to the branch point at the 5’ splice site of pre-mRNA. The BCL-2 family consists of both pro and anti-apoptotic members. Both of these groups are alternately spliced and, in many cases, these splicing events give rise to members with different functions in apoptosis. Aberrant splicing of members of the BCL-2 family has been observed in AML. MCL-1, BCL-X and BID are spliced to give rise to isoforms with this property [42]. The isoform of MCL-1 that is most commonly expressed is the anti-apoptotic long form MCL-1L. The same is true of BCL-X where the anti-apoptotic long form is known as BCL-XL. Both of these proteins bind the pro-apoptotic BAK protein and prevent it from forming an oligomer and permeabilising the mitochondrial membrane [43] (Figure 5). MCL-1L also binds other pro-apoptotic proteins such as BIM and BID. The splicing events that control the expression of these various isoforms are regulated by the splicing factor SF3B1. A decrease in the levels of this splicing factor increases cells sensitivity to apoptosis inducing treatments. Tumour growth can be blocked using drugs that downregulate the SRSF1 splicing factor [18].
Figure 5.
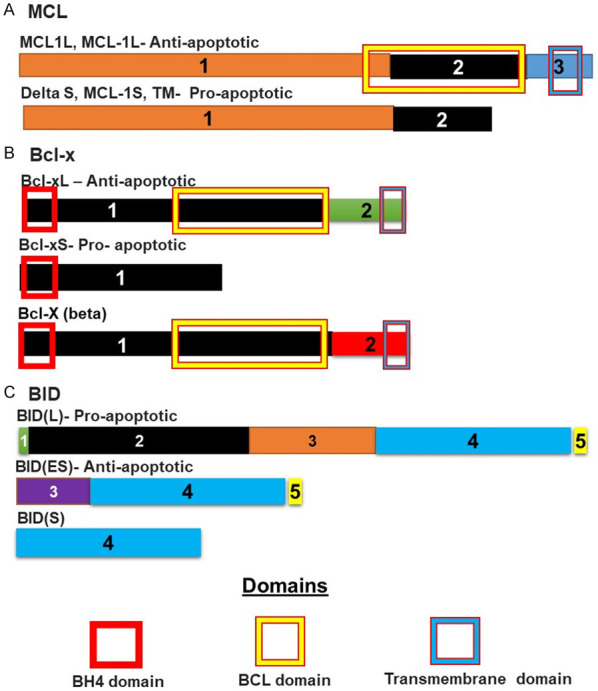
Isoforms of proteins involved in apoptosis and whose splicing is controlled by SPFB1: (A) MCL is spliced to give at least three different isoforms. The long isoform is anti-apoptotic and binds pro-apoptotic proteins such as BAk, BIM and BID, preventing them from dimerising and permeabilising the mitochondrial membrane. The shorter form lacks the sequence coding for the initial part of the BCL domain, preventing the formation of the BCL domain. This shorter protein cannot therefore, associate with these proteins. The same is true of (B) BCL-x. The shorter version lacks most of the sequence coding for the BCL domain. This prevents the proper formation of the BCL domain and the shorter protein can therefore not bind pro-apoptotic BH3 domain proteins such as (C) BID. BID is spliced to give at least three isoforms. Here the longer isoform is the pro-apoptotic isoform that has a BH3 domain coded for by exon 2. This domain allows the protein to oligomerise with other BH3 members leading to permeabilization of the mitochondrial membrane. This ability is lost in the shorter truncated form.
Mutation in SF3B1, affecting the function of the SF3B1 protein have been found to affect the splicing and isoform population of the ABCB7 (ATP Binding Cassette Subfamily B, Member 7) protein. The changes in the selection of isoforms is due to changes in the selection of the 3’ splice site, leading to early termination of protein [44]. This leads to decreased levels of ABCB7 protein levels and since this protein is involved in the transport of heme there are higher levels of heme deposition which is associated with malignancy in MDS [44]. Splicing changes as a result of SF3B1 associated splicing changes have also been found in tumor suppressor genes, where once again the expression of these proteins was decreased as a result. These include NF1, DICER1, PML, PDS5A, MAP3K7, and PPP2R5A [45].
SPF45
Splicing factors such as SPF45 are involved in splicing of components of the extrinsic apoptotic pathway. For example, the increased expression of this splicing factor in HeLa cells leads to increased resistance to chemotherapy treatments such as carboplatin, vinorelbine, doxorubicin, etoposide, mitoxantrone, and vincristine [46]. The Fas death receptor is alternatively spliced under control of the SPF45 splicing factor. SPF45 promotes the skipping of an exon, forming a Fas death receptor protein that alters the selection of differently spliced variants of Fast death receptor [47,48]. Alternate splicing of Caspase 8 can also inhibit Fas signalling, as one of the splice variants known as CASP8L, is an anti-apoptotic variant, upregulated in hematopoietic progenitor cells [49]. TNF-related apoptosis-inducing ligand (TRAIL) is another death receptor associated with AML. TRAIL signalling involves products of the IG20 gene. IG20 is alternately spliced to give rise to four isoforms. Some of these promote apoptosis and others inhibit apoptosis. This is due to differences in the ability of these splice variants to activate Caspase 8 [50].
RNA binding motif protein 25 (RBM25)
Studies in mouse AML models have indicated that RBM26 has tumour-suppressive properties. Here it was found that RBM25 delayed or halted cell cycle progression, increased apoptosis and led to myeloid differentiation. This splicing factor selects specific 5’ splice sites favouring the expression of the pro-apoptotic isoform of BCL2L1 (Figure 4). One of the genes RBM25 is involved in splicing is BIN1 [51]. RBM25 leads to the exclusion of exon 12 form BIN1 pre-mRNA. This results in an isoform which is unable to inhibit MYC, resulting in increased cell proliferation. It has also been established that downregulation of RBM25 in AML cells leads to high levels of MYC as a result of changes in BIN isoforms. The resulting high expression of MYC and downstream genes controlled by MYC can be used as diagnostic or prognostic markers in AML patients.
PRE-mRNA processing factor 8 (PRPF8)
PRE-mRNA processing factor 8 (PRPF8) is one of the splicing factors that acts in the catalytic core of the spliceosome. PRPF8 plays an important role in haematopoiesis and incorrect functioning of PRPF8 is implicated in AML [52]. PRPF8 mutations or hemizygous deletions occurred in 3.3% (15 out of 447 cases) and 5.3% (24 out of 450 cases) of myeloid neoplasms, respectively [53]. Notably, 50% of PRPF8 mutant and deletion of chromosome 17p cases were found in AML. Survival analysis confirmed that PRPF8 was associated with poor prognosis (91). Splicing factor proline and glutamine rich (SFPQ) is one of the three-member Drosophila behaviour/human splicing (DBHS) family in human [53]. The DBHS family shares highly conserved tandem N-terminal RRMs, a NonA/paraspeckle domain (NOPS) and a C-terminal.
DEAD-box polypeptide 41
DEAD-box polypeptide 41 (DDX41) is an RNA helicase that is involved in RNA spliceosome assembly and is also involved in translation initiation, pre-ribosomal RNA processing, small RNA biogenesis and chromosome condensation [54]. It has been identified as being mutated in about 3% of all blood malignancies. DDX41 functions as a tumour suppressor gene and mutations lead to the development and progression of AML [55].
Other pathways affecting mis-splicing in AML: FLT3
Another pathway which is de-regulated in AML is the FLT3 (CD135) pathway, which becomes constitutively activated in 25% of AML cases. FLT3 is also mis-spliced in AML altering the extracellular portion of the protein (Figure 6A)[56]. FLT3 is a MS-like tyrosine kinase that acts as a class III tyrosine kinase receptor. It stimulates proliferation and cell survival and also leads to the differentiation of hematopoietic stem/progenitor cells. CD135 is receptor tyrosine kinase that binds FLT3L in a one to one ratio, resulting in two FLT3 molecules bridged by a single FLT3L molecule (Figure 6B). This results in intracellular signalling. This signalling results in enhanced cell survival and proliferation of lymphocytes and T cells. FLT3 has been found to be one of the most highly mis-spliced genes in AML. Alternative splicing gives rise to four additional isoforms (Figure 6A). These aberrantly spliced isoforms have altered extracellular regions. These splice variants are not associated with mutant splice factors. However, they are the result of altered expression levels of other spliceosome components involved in splicing regulation. Many of the downstream signalling pathways regulated by FLT3 signalling are found to be disrupted in AML. These include AKT, STAT and ERK signalling [57].
Figure 6.
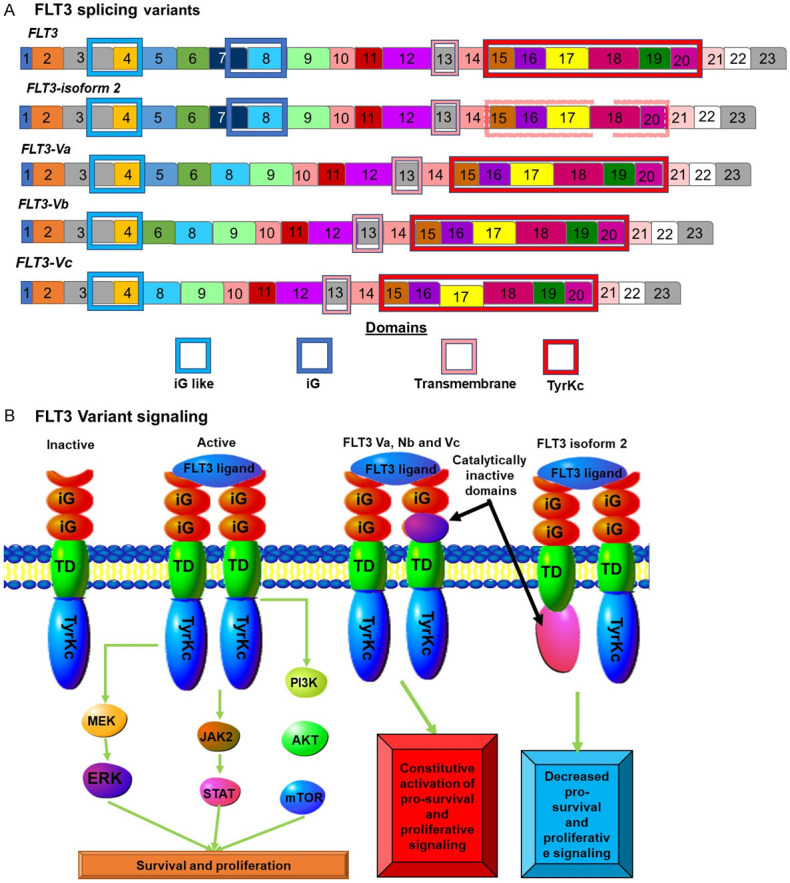
FLT3 isoforms in AML: (A) FLT3 splicing results in at least 4 different isoforms. One isoform known as isoform 2 is not involved in increased AML rates. This isoform is missing exon 19 resulting in a truncated non-functional tyrosine kinase domain. The other three isoforms are involved in AML and all contain deletions in the sequence coding for the extracellular region of the protein. (B) Normal ELF2 signalling results in increased proliferation and pro-survival signals. The exact nature of the signalling by the Va, Vb and Vc variants is not known but the absence of the second iG domain results in increased proliferative and survival signalling.
Treatment of AML based on splicing
The large number of ways in which changes in splicing factor function can alter the expression of various isoforms of proteins which can themselves either increase or decrease malignancy, has led to these various splicing proteins being targeted for therapy. These include proteins that regulate splicing catalysis and specific splicing events [15]. The wide variety of splicing factors involved in this process also means that there are multiple targets which could serve as targets for drug development.
One of the most attractive targets for drug development has been the SF3B splicing factor. Compounds have been isolated from bacteria that can inhibit the SF3B component of the spliceosome. This led to changes in splicing resulting in a decrease in tumour growth and progression [58]. One of these is the compound FR901464 and its synthetic derivative meayamycin B [59]. Other agents that are also being investigated for their ability to bind SF3B and disrupt the spliceosome include herboxidienes, pladienolides, analogs of pladienolide B, spliceostatin A and sudemycins [58]. A pladienolide derivative H3B-8800 has shown the ability to inhibit normal and mutant SF3b complexes and has the ability to arrest the growth of MDS/AML cells with SF mutations. This compound entered a phase 1 trial in 2017 to evaluate its use in AML treatment [60]. The results of this trial showed that this compound was able to decrease the requirement for RBC transfusion in 14% of patients. This is a common requirement to maintain patients with AML. The drug was also shown to be safe despite prolonged use [61].
A compound that is in relatively advanced stages of development chain, is the fusion protein luspatercept (ACE-536). This compound was originally designed to target the TGF-β and SMAD family proteins for cancer treatment. However, it was found to produce positive responses in people with SF3B1 mutations. This protein drug is a fusion of the extracellular domain of human activin receptor type IIB and human IgG1 Fc domain [62].
Conclusion
Mutations in genes for splicing factors and spliceosome components are some of the most common mutations found in AML. These mutated splicing “controllers” result in changes in the splicing of genes and thereby change the expression levels of various protein isoforms. This results in changes that may affect the phenotype and progression of AML. Targeting these mutated splicing factors and spliceosome components, presents novel targets for the design of new drugs. Higher expression levels of normally rare pro-malignant protein isoforms can also be used as diagnostic and prognostic biomarkers.
Acknowledgements
The authors would like to thank the South African Medical Research Council (SAMRC) for funding this research.
Disclosure of conflict of interest
None.
References
- 1.Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70–87. doi: 10.1016/j.blre.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Montalban-Bravo G, Garcia-Manero G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am J Hematol. 2018;93:129–147. doi: 10.1002/ajh.24930. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997-2002. Cancer Causes Control. 2008;19:379–390. doi: 10.1007/s10552-007-9097-2. [DOI] [PubMed] [Google Scholar]
- 4.Meyers J, Yu Y, Kaye JA, Davis KL. Medicare fee-for-service enrollees with primary acute myeloid leukemia: an analysis of treatment patterns, survival, and healthcare resource utilization and costs. Appl Health Econ Health Policy. 2013;11:275–286. doi: 10.1007/s40258-013-0032-2. [DOI] [PubMed] [Google Scholar]
- 5.Chen HC, Cheng SC. Functional roles of protein splicing factors. Biosci Rep. 2012;32:345–359. doi: 10.1042/BSR20120007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos NR, Mo CC, Karp JE, Hourigan CS. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J Clin Med. 2015;4:665–695. doi: 10.3390/jcm4040665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3:1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsson CA, Cote G, Quintas-Cardama A. The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol Cancer Res. 2013;11:815–827. doi: 10.1158/1541-7786.MCR-12-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boultwood J, Dolatshad H, Varanasi SS, Yip BH, Pellagatti A. The role of splicing factor mutations in the pathogenesis of the myelodysplastic syndromes. Adv Biol Regul. 2014;54:153–161. doi: 10.1016/j.jbior.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, Gundem G, Van Loo P, Martincorena I, Ganly P, Mudie L, McLaren S, O’Meara S, Raine K, Jones DR, Teague JW, Butler AP, Greaves MF, Ganser A, Döhner K, Schlenk RF, Döhner H, Campbell PJ. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, Lode L, Verselis S, Fox EA, Burke J, Galinsky I, Dagogo-Jack I, Wadleigh M, Steensma DP, Motyckova G, Deangelo DJ, Quackenbush J, Stone R, Griffin JD. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res. 2014;20:1135–1145. doi: 10.1158/1078-0432.CCR-13-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, Sekeres MA, Padgett RA, Tiu RV, Maciejewski JP. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao TY, Guan LX, Zheng WS, Wang ML, Cheng LC, Hu YL, Xu YY, Gu ZY, Dou LP. Clinical characteristics and prognosis of U2AF1 mutation in patients with acute myeloid leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2020;28:7–11. doi: 10.19746/j.cnki.issn.1009-2137.2020.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Smith MA, Choudhary GS, Pellagatti A, Choi K, Bolanos LC, Bhagat TD, Gordon-Mitchell S, Von Ahrens D, Pradhan K, Steeples V, Kim S, Steidl U, Walter M, Fraser IDC, Kulkarni A, Salomonis N, Komurov K, Boultwood J, Verma A, Starczynowski DT. U2AF1 mutations induce oncogenic IRAK4 isoforms and activate innate immune pathways in myeloid malignancies. Nat Cell Biol. 2019;21:640–650. doi: 10.1038/s41556-019-0314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visconte V, Nakashima MO, Rogers HJ. Mutations in splicing factor genes in myeloid malignancies: significance and impact on clinical features. Cancers (Basel) 2019;11:1844. doi: 10.3390/cancers11121844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sokół E, Bogusławska J, Piekiełko-Witkowska A. The role of SRSF1 in cancer. Postepy Hig Med Dosw (Online) 2017;71:422–430. doi: 10.5604/01.3001.0010.3825. [DOI] [PubMed] [Google Scholar]
- 17.Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore MJ, Wang Q, Kennedy CJ, Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010;142:625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Necochea-Campion R, Diaz Osterman CJ, Hsu HW, Fan J, Mirshahidi S, Wall NR, Chen CS. AML sensitivity to YM155 is modulated through AKT and Mcl-1. Cancer Lett. 2015;366:44–51. doi: 10.1016/j.canlet.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shultz JC, Goehe RW, Wijesinghe DS, Murudkar C, Hawkins AJ, Shay JW, Minna JD, Chalfant CE. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010;70:9185–9196. doi: 10.1158/0008-5472.CAN-10-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 22.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Chen D, Qian H, Tsai YS, Shao S, Liu Q, Dominguez D, Wang Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell. 2014;26:374–389. doi: 10.1016/j.ccr.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karimi M, Nilsson C, Dimitriou M, Jansson M, Matsson H, Unneberg P, Lehmann S, Kere J, Hellström-Lindberg E. High-throughput mutational screening adds clinically important information in myelodysplastic syndromes and secondary or therapy-related acute myeloid leukemia. Haematologica. 2015;100:e223–225. doi: 10.3324/haematol.2014.118034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, Cho H, Kim MK, Zebari AS, Aumann S, Park CY, Buonamici S, Smith PG, Deeg HJ, Lobry C, Aifantis I, Modis Y, Allain FH, Halene S, Bradley RK, Abdel-Wahab O. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masaki S, Ikeda S, Hata A, Shiozawa Y, Kon A, Ogawa S, Suzuki K, Hakuno F, Takahashi SI, Kataoka N. Myelodysplastic syndrome-associated SRSF2 mutations cause splicing changes by altering binding motif sequences. Front Genet. 2019;10:338. doi: 10.3389/fgene.2019.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korotayev K, Ginsberg D. Many pathways to apoptosis: E2F1 regulates splicing of apoptotic genes. Cell Death Differ. 2008;15:1813–1814. doi: 10.1038/cdd.2008.155. [DOI] [PubMed] [Google Scholar]
- 29.Merdzhanova G, Edmond V, De Seranno S, Van den Broeck A, Corcos L, Brambilla C, Brambilla E, Gazzeri S, Eymin B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ. 2008;15:1815–1823. doi: 10.1038/cdd.2008.135. [DOI] [PubMed] [Google Scholar]
- 30.Mohamed AM, Thénoz M, Solly F, Balsat M, Mortreux F, Wattel E. How mRNA is misspliced in acute myelogenous leukemia (AML)? Oncotarget. 2014;5:9534–9545. doi: 10.18632/oncotarget.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andersson C, Li X, Lorenz F, Golovleva I, Wahlin A, Li A. Reduction in WT1 gene expression during early treatment predicts the outcome in patients with acute myeloid leukemia. Diagn Mol Pathol. 2012;21:225–233. doi: 10.1097/PDM.0b013e318257ddb9. [DOI] [PubMed] [Google Scholar]
- 32.Mohamed AM, Balsat M, Thenoz M, Koering C, Payen-Gay L, Cheok M, Mortada H, Auboeuf D, Pinatel C, El-Hamri M, Dumontet C, Cros E, Flandrin-Gresta P, Nibourel O, Preudhomme C, Michallet M, Thomas X, Nicolini F, Solly F, Guyotat D, Campos L, Wattel E, Mortreux F. Oncogene- and drug resistance-associated alternative exon usage in acute myeloid leukemia (AML) Oncotarget. 2016;7:2889–2909. doi: 10.18632/oncotarget.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, Miyano S, Thol F, Ganser A, Yang H, Haferlach T, Ogawa S, Koeffler HP. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun. 2015;6:6042. doi: 10.1038/ncomms7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klingenberg M, Groß M, Goyal A, Polycarpou-Schwarz M, Miersch T, Ernst AS, Leupold J, Patil N, Warnken U, Allgayer H, Longerich T, Schirmacher P, Boutros M, Diederichs S. The long noncoding RNA cancer susceptibility 9 and RNA binding protein heterogeneous nuclear ribonucleoprotein L form a complex and coregulate genes linked to AKT signaling. Hepatology. 2018;68:1817–1832. doi: 10.1002/hep.30102. [DOI] [PubMed] [Google Scholar]
- 35.Vu NT, Park MA, Shultz JC, Goehe RW, Hoeferlin LA, Shultz MD, Smith SA, Lynch KW, Chalfant CE. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J Biol Chem. 2013;288:8575–8584. doi: 10.1074/jbc.M112.443333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallardo M, Lee HJ, Zhang X, Bueso-Ramos C, Pageon LR, McArthur M, Multani A, Nazha A, Manshouri T, Parker-Thornburg J, Rapado I, Quintas-Cardama A, Kornblau SM, Martinez-Lopez J, Post SM. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015;28:486–499. doi: 10.1016/j.ccell.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400. doi: 10.1038/nrc1363. [DOI] [PubMed] [Google Scholar]
- 38.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, Campbell PJ. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mortera-Blanco T, Dimitriou M, Woll PS, Karimi M, Elvarsdottir E, Conte S, Tobiasson M, Jansson M, Douagi I, Moarii M, Saft L, Papaemmanuil E, Jacobsen SEW, Hellström-Lindberg E. SF3B1-initiating mutations in MDS-RSs target lymphomyeloid hematopoietic stem cells. Blood. 2017;130:881–890. doi: 10.1182/blood-2017-03-776070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 41.Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akgul C, Moulding DA, Edwards SW. Alternative splicing of Bcl-2-related genes: functional consequences and potential therapeutic applications. Cell Mol Life Sci. 2004;61:2189–2199. doi: 10.1007/s00018-004-4001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bose P, Grant S. Mcl-1 as a therapeutic target in acute myelogenous leukemia (AML) Leuk Res Rep. 2013;2:12–14. doi: 10.1016/j.lrr.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dolatshad H, Pellagatti A, Liberante FG, Llorian M, Repapi E, Steeples V, Roy S, Scifo L, Armstrong RN, Shaw J, Yip BH, Killick S, Kušec R, Taylor S, Mills KI, Savage KI, Smith CW, Boultwood J. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia. 2016;30:2322–2331. doi: 10.1038/leu.2016.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiozawa Y, Malcovati L, Gallì A, Sato-Otsubo A, Kataoka K, Sato Y, Watatani Y, Suzuki H, Yoshizato T, Yoshida K, Sanada M, Makishima H, Shiraishi Y, Chiba K, Hellström-Lindberg E, Miyano S, Ogawa S, Cazzola M. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat Commun. 2018;9:3649. doi: 10.1038/s41467-018-06063-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perry WL 3rd, Shepard RL, Sampath J, Yaden B, Chin WW, Iversen PW, Jin S, Lesoon A, O’Brien KA, Peek VL, Rolfe M, Shyjan A, Tighe M, Williamson M, Krishnan V, Moore RE, Dantzig AH. Human splicing factor SPF45 (RBM17) confers broad multidrug resistance to anticancer drugs when overexpressed--a phenotype partially reversed by selective estrogen receptor modulators. Cancer Res. 2005;65:6593–6600. doi: 10.1158/0008-5472.CAN-03-3675. [DOI] [PubMed] [Google Scholar]
- 47.Aratake K, Kamachi M, Iwanaga N, Kawasaki E, Izumi Y, Ida H, Tanaka F, Tamai M, Arima K, Nakamura H, Origuchi T, Kawakami A, Eguchi K. A cross-talk between RNA splicing and signaling pathway alters Fas gene expression at post-transcriptional level: alternative splicing of Fas mRNA in the leukemic U937 cells. J Lab Clin Med. 2005;146:184–191. doi: 10.1016/j.lab.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 48.Inaba H, Komada Y, Li QS, Zhang XL, Tanaka S, Azuma E, Yamamoto H, Sakurai M. mRNA expression of variant Fas molecules in acute leukemia cells. Am J Hematol. 1999;62:150–158. doi: 10.1002/(sici)1096-8652(199911)62:3<150::aid-ajh4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 49.Mohr A, Zwacka RM, Jarmy G, Büneker C, Schrezenmeier H, Döhner K, Beltinger C, Wiesneth M, Debatin KM, Stahnke K. Caspase-8L expression protects CD34+ hematopoietic progenitor cells and leukemic cells from CD95-mediated apoptosis. Oncogene. 2005;24:2421–2429. doi: 10.1038/sj.onc.1208432. [DOI] [PubMed] [Google Scholar]
- 50.Prabhakar BS, Mulherkar N, Prasad KV. Role of IG20 splice variants in TRAIL resistance. Clin Cancer Res. 2008;14:347–351. doi: 10.1158/1078-0432.CCR-07-0493. [DOI] [PubMed] [Google Scholar]
- 51.Ge Y, Schuster MB, Pundhir S, Rapin N, Bagger FO, Sidiropoulos N, Hashem N, Porse BT. The splicing factor RBM25 controls MYC activity in acute myeloid leukemia. Nat Commun. 2019;10:172. doi: 10.1038/s41467-018-08076-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grainger RJ, Beggs JD. Prp8 protein: at the heart of the spliceosome. RNA. 2005;11:533–557. doi: 10.1261/rna.2220705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, Nakashima M, Hsi ED, Yoshida K, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Ogawa S, Boultwood J, Makishima H, Maciejewski JP, Padgett RA. PRPF8 defects cause missplicing in myeloid malignancies. Leukemia. 2015;29:126–136. doi: 10.1038/leu.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang Y, Zhu Y, Liu ZJ, Ouyang S. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell. 2017;8:83–89. doi: 10.1007/s13238-016-0303-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, Schreiber AW, Feng J, Babic M, Chong CE, Lee Y, Yong A, Suthers GK, Poplawski N, Altree M, Phillips K, Jaensch L, Fine M, D’Andrea RJ, Lewis ID, Medeiros BC, Pollyea DA, King MC, Walsh T, Keel S, Shimamura A, Godley LA, Hahn CN, Churpek JE, Scott HS. Novel germ line DDX41 mutations define families with a lower age of MDS/AML onset and lymphoid malignancies. Blood. 2016;127:1017–1023. doi: 10.1182/blood-2015-10-676098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–89. doi: 10.1016/j.drup.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adamia S, Bar-Natan M, Haibe-Kains B, Pilarski PM, Bach C, Pevzner S, Calimeri T, Avet-Loiseau H, Lode L, Verselis S, Fox EA, Galinsky I, Mathews S, Dagogo-Jack I, Wadleigh M, Steensma DP, Motyckova G, Deangelo DJ, Quackenbush J, Tenen DG, Stone RM, Griffin JD. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): new potential targets in AML. Blood. 2014;123:2816–2825. doi: 10.1182/blood-2013-02-481507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonnal S, Vigevani L, Valcárcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 59.Wojtuszkiewicz A, Assaraf YG, Jansen G, Koide K, Bressin RK, Basu U, Sonneveld E, Giovannetti E, Kaspers G, Cloos J. Spliceosome inhibitor meayamycin B as a novel potential chemotherapeutic agent in ALL and AML. Blood. 2014;124:924. [Google Scholar]
- 60.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, Nijhawan D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356:eaal3755. doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- 61.Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS, Yang J, Brunner AM, Carraway HE, Ades L, Al-Kali A, Alonso Dominguez JM, Alonso A, Coombs CC, Deeg HJ, Donnellan WB, Foran JM, Garcia-Manero G, Maris MB, McMasters M, Micol JB, Perez De Oteyza J, Thol F, Wang ES, Watts JM, Buonamici S, Kim A, Gourineni V, Marino AJ, Rioux N, Schindler J, Smith S, Yao H, Yuan X, Yu K, Platzbecker U. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML) Blood. 2019;134:673–673. [Google Scholar]
- 62.Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, Radsak M, Wolff T, Zhang X, Laadem A, Sherman ML, Attie KM, Giagounidis A. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18:1338–1347. doi: 10.1016/S1470-2045(17)30615-0. [DOI] [PubMed] [Google Scholar]