

Abstract
DNA methyltransferase 1 (DNMT1) is scientifically validated as a molecular target to treat chemo-resistant pancreatic ductal adenocarcinoma (PDAC). Results of clinical studies of the pyrimidine nucleoside analog decitabine to target DNMT1 in PDAC have, however, disappointed. One reason is high expression in PDAC of the enzyme cytidine deaminase (CDA), which catabolizes decitabine within minutes. We therefore added tetrahydrouridine (THU) to inhibit CDA with decitabine. In this pilot clinical trial, patients with advanced chemorefractory PDAC ingested oral THU ~10 mg/kg/day combined with oral decitabine ~0.2 mg/kg/day, for 5 consecutive days, then 2X/week. We treated 13 patients with extensively metastatic chemo-resistant PDAC, including 8 patients (62%) with ascites: all had received ≥ 1 prior therapies including gemcitabine/nab-paclitaxel in 9 (69%) and FOLFIRINOX in 12 (92%). Median time on THU/decitabine treatment was 35 days (range 4-63). The most frequent treatment-attributable adverse event was anemia (n=5). No deaths were attributed to THU/decitabine. Five patients had clinical progressive disease (PD) prior to week 8. Eight patients had week 8 evaluation scans: 1 had stable disease and 7 PD. Median overall survival was 3.1 months. Decitabine systemic exposure is expected to decrease neutrophil counts; however, neutropenia was unexpectedly mild. To identify reasons for limited systemic decitabine effect, we measured plasma CDA enzyme activity in PDAC patients, and found a > 10-fold increase in those with metastatic vs resectable PDAC. We concluded that CDA activity is increased not just locally but also systemically in metastatic PDAC, suggesting a need for even higher CDA-inhibitor doses than used here.
Keywords: Pancreatic cancer, clinical trial, deaminase inhibitor
Introduction
Pancreatic ductal adenocarcinoma (PDAC), commonly referred to as pancreatic cancer, is a recalcitrant, deadly problem [1]. Most patients have advanced metastatic disease at presentation, and therefore receive aggressive multi-agent chemotherapy, either combinations of 5-fluorouracil, irinotecan and oxaliplatin or gemcitabine and nab-paclitaxel. Despite the substantial intensity and toxicity of such treatments, Median overall survival is < 1 year [2-4]. Several reasons for limited chemotherapy responsiveness have been proposed, but one well-established basis is mutation and deletion in most PDAC of the master regulator of apoptosis p53 [5,6]: absence of p53 undermines the ability of cytotoxic (apoptosis-intending) agents - all the agents listed above - to achieve tumor kill by apoptosis induction [7,8]. Meanwhile, normal dividing cells, with intact p53, are killed, causing onerous toxicity profiles [3,9]. Thus, alternative treatment modalities that do not rely on cell cycle exits via the p53-apoptosis pathway are needed.
Tumor cytoreduction despite absence of p53, e.g., via activation of terminal epithelial programs, can be achieved by inhibiting/deleting the key epigenetic regulator DNA methyltransferase 1 (DNMT1) that mediates repression of these programs in cancer cells [10-16]. Moreover, DNMT1 can be inhibited/depleted by the pyrimidine nucleoside analog pro-drugs decitabine or 5-azacytidine that are approved by the Food and Drug Administration (FDA) to treat myeloid cancers. In fact, succesful pre-clinical validation of DNMT1 as a molecular target for treating PDAC has motivated several clinical trials of decitabine, 5-azacytidine or analogs thereof to treat PDAC. Several of these trials have been completed, and more are ongoing as of April 2019 (Clinicaltrials.gov identifiers NCT02959164, NCT03257761, NCT03264404, NCT02961101) [17-29] (reviewed in [30]). Results from completed trials, however, have been discouraging [17-30]. Correlative studies in one prior trial of decitabine suggested a basis for disappointing clinical results: analyses of cancer tissue resected post-treatment revealed decitabine pharmacodynamic effect in solid cancer tissue in < 25% of the treated patients, despite continuous infusion of decitabine that sustained plasma Cmax > 40 nM for hours, enough to cause grade 3/4 myelotoxicity (particularly neutropenia) [31,32]. Decitabine and 5-azacytidine are pro-drugs that must be processed by pyrimidine metabolism in order to deplete DNMT1, and accordingly, tissue-differences in expression of key pyrimidine metabolism enzymes have been shown to underlie tissue-differences in pharmacodynamic effect. One key enzyme is cytidine deaminase (CDA) which rapidly catabolizes decitabine and 5-azacytidine into uridine counterparts that cannot deplete DNMT1 [33]. Naturally high CDA expression in gastro-intestinal tissues is the reason decitabine, 5-azacytidine and gemcitabine (first-line treatment for PDAC that is also a pyrimidine nucleoside analog) are not administered orally, and CDA is why the plasma half-lives of decitabine, 5-azacytidine or gemcitabine are ~15 minutes after parenteral administration [34,35]. Accordingly, the addition of a CDA inhibitor tetrahydrouridine (THU) to oral decitabine produced decitabine plasma pharmacokinetics comparable to continuous intravenous infusion of decitabine alone, to produce DNMT1-depleting pharmacodynamic effect in the myeloid compartment that was substantial and clinically significant [17,27,29].
Here, we examined contributions of CDA to PDAC resistance to decitabine and evaluated the use of THU to reverse this mechanism-of-resistance. We found that gemcitabine-resistance PDAC cells have upregulated CDA (observed also by others [36-39]), and that such cells are also resistant to decitabine. In pre-clinical in vitro and in vivo models, the addition of THU to inhibit CDA overcame this basis for resistance. Then, to translate the pre-clinical observations, we conducted a pilot clinical trial in 13 patients with advanced chemorefractory PDAC. The doses of THU and decitabine were derived from prior clinical studies in patients without PDAC. The frequent and distributed schedule of administration of THU/decitabine was intended to increase possibilities of overlap between malignant S-phase entries and drug exposures, needed for S-phase dependent DNMT1-depletion in PDAC tissue. Unfortunately, there was no clinical benefit, but correlative observations suggested why: there was surprisingly little neutropenia in this clinical trial, a sensitive indicator of decitabine systemic activity (the myeloid compartment is the most sensitive tissue compartment to systemic decitabine exposures [34,35,40-43]), and showing why, plasma CDA enzyme activity was > 10-fold higher in patients with metastatic vs resectable PDAC. That is, CDA activity was increased not just within PDAC tissue, but also systemically, and the THU dosages used, enough for CDA inhibition and decitabine exposure in patients without PDAC, were insufficient in patients with advanced PDAC. Since CDA contributes to resistance not just to decitabine/5-azacytidine, but also to resistance to standard first-line PDAC therapy with gemcitabine [38,39,44,45], this information is pertinent to future clinical evaluations of decitabine, 5-azacytidine or gemcitabine to treat PDAC.
Patients and methods
Study design and patients
This was an open-label, single-arm pilot clinical trial in patients with metastatic PDAC that had progressed on prior chemotherapy. The study was reviewed and approved by the Case Comprehensive Cancer Center Protocol Review and Monitoring Committee, and the Cleveland Clinic Institutional Review Board. The study was registered on clinicaltrials.gov (NCT02847000). Patients were enrolled at Cleveland Clinic and University Hospitals of the Case Western Reserve University from April to August 2017. Key inclusion criteria were: a confirmed pathologic diagnosis of pancreatic cancer (histologies other than carcinoma or adenocarcinoma were not eligible); one or more prior systemic chemotherapy regimens with treatment cessation due to disease progression or intolerable toxicity; adult patients with Eastern Cooperative Oncology Group (ECOG) Performance Score (PS) of 0, 1, or 2; with measurable disease per RECIST 1.1 criteria; adequate organ function (hemoglobin ≥ 8 g/dl; absolute neutrophil count (ANC) ≥ 1500/µl; platelets ≥ 75000/µl; serum creatinine ≤ 2.9 mg/dl; serum total bilirubin ≤ 2 times upper limit of laboratory normal; serum AST/ALT ≤ 2.5 times upper limit of laboratory normal; serum calcium ≤ 12 mg/dl); eligible and agreeable for baseline and week 16 percutaneous tumor biopsies. All prior cancer-directed therapies had to be completed/stopped at least 14 days prior to enrollment. Any uncontrolled comorbidities were exclusionary.
Study procedures
After written informed consent, patients underwent baseline screening, including a complete history and physical exam, laboratory evaluation (including organ function assessment, pregnancy test as applicable, CA19.9 levels), tumor imaging using CT or MRI scans, and a baseline tumor biopsy. After confirmation of eligibility, patients were started on treatment.
The doses were as follows: THU was supplied as 250 mg/capsules, and decitabine as 5 mg/capsules. Starting doses were by weight: Weight 40-60 kg: 2 capsules of each drug. Weight 61-80 kg: 3 capsules of each drug. Weight 81 kg or higher: 4 capsules of each drug. Patients were instructed to take the decitabine capsules ~60 minutes after taking the THU capsules, to generate sufficient time for the intended biological effect of THU of systemic CDA-inhibition.
The treatment schedule consisted of an induction cycle (4 weeks) and maintenance thereafter. During the induction cycle, the drugs were taken for 5 consecutive days (typically, Monday-Friday) in week 1. In week 2, the same doses were used if no grade 3 or higher hematologic toxicities were noted. Based on the absolute neutrophil count (ANC) at the start of week 2, treatments during this week were given for 2, 3, 4, or 5 consecutive days. During weeks 3 and 4, treatments were given for 2 consecutive days (typically, Monday and Tuesday) at the same doses, if no grade 3 or higher hematologic toxicities were noted. After the induction cycle, a maintenance phase was introduced, where treatments were given for 2 consecutive days (typically, Monday and Tuesday) of each week, for up to 52 weeks.
If hematologic toxicities precluded the above plan, then dose and schedule modifications were prespecified in the study protocol. If stable disease with absence of at least partial response by RECIST1.1 at week 8 or 16 staging scans was noted, and no treatment-related toxicities were seen, an increase in THU and Dec dosage by one capsule each was allowed.
Endpoints and statistics
The primary endpoint of this pilot study was to detect decitabine therapy-induced DNMT1 protein level decrease in tumor tissue. DNMT1 levels were to be assessed using quantitative immunofluorescence, on baseline tumor biopsy and the week 16 biopsy. We chose an effect size of 1, assessed using a paired t-test and alpha =0.05. The effect size was defined as the difference in mean DNMT1-protein levels between post-treatment and pre-treatment divided by the standard deviation and was thus a metric of change in the natural units of the distribution, its standard deviation. Our goal was thus to detect drops in DNMT1 of at least one standard deviation. To have 90% power to detect this change, a sample size of 12 patients was required.
Secondary endpoints included safety of the treatment regimen, response rate per RECIST 1.1 criteria, and overall survival, calculated from date of patient registration on study to date of death.
All patients enrolled into the study are included in the analyses.
Plasma CDA enzyme activity assay
Conversion of cytidine into uridine by plasma at 37°C was measured by high performance liquid chromatography (HPLC) based on published methods [46]. Reaction buffer 0.1 M Tris/HCL pH 7.5 (265 µl) was added to 25 µl of human plasma followed by addition of cytidine (Sigma-Aldrich C122106) to a final concentration of 4.1 mM and 5-flourouridine (Sigma-Aldrich F5130) 0.381 mM (not metabolized by CDA) as an internal control. After incubation at 37°C for 4 hours the reaction was terminated with 50 µl of hydrochloric acid 1 N. Blanks used in calculations consisted of the above but with cytidine substrate added at the end of the 60 minute incubation. After reaction termination, protein was precipitated with perchloric acid (TCA, 2%). 100 µl of supernatant was injected for HPLC using ammonium acetate (5 mM) as the mobile phase with a flow rate of 0.35 ml/min through ZORBAX eclipse XDB 80°A C18, 4.6 × 150 mm, 3.5 µM (Agilent 963967-902) High Performance Liquid Chromatography (HPLC) column on Dionex UltiMate 3000 µ-HPLC system supported with Chromeleon 7.1 data system (Dionex Corporation, Sunnyvale, CA). Retention time and peak area of uridine at 260 nm were compared to the internal control for each injection. The average net uridine peak area of test minus blank was calculated for each test sample. Known concentrations of uridine (0.0 to 95.8 µM) (Sigma-Aldrich U3750) were used to construct a standard curve to calculate the amount of uridine based on the net uridine peak area normalized to the net peak area of the internal standard 5-fluorouridine. One unit (U) of CDA enzyme activity is defined as the amount of enzyme needed to produce 1 µmole of uridine in 1 minute. Multiple runs with known concentrations of uridine were used to confirm accuracy and precision: between run variability was < 5%.
Pre-clinical in vitro studies of gemcitabine-resistant PDAC
MIA PaCa2 parental, MIA PaCa2 gemcitabine resistant and PANC-1 cells were a gift from John Pink (Case Western Reserve University). Gemcitabine resistant cells were kept under drug selection at 2 μM gemcitabine. The pancreatic cell lines and K562 cells (ATCC) were cultured in RPMI-1640 with 10% FBS at 37°C in a humidified atmosphere with 5% CO2 in air.
Decitabine stock solution (5 mM) was generated by reconstituting lypholized decitabine in 100% DMSO. Stock solution aliquots were stored at -80°C for up to 3 weeks. Working solution was generated by diluting the stock solution 1:100 in phosphate-buffered saline (PBS) immediately before addition to the cells at a further dilution as per the intended final concentration. Similar amounts of PBS are added to untreated control cells. Cells were treated with increasing concentrations of decitabine (0-100 μM) to generate a dose-response curve or with 0.2 μM decitabine on day 1 and 2 of culture for the rest of the experiments. THU stock solution 12 mM was generated by reconstituting THU in 100% PBS. Cells were treated with 1 μM THU.
Approximately 60 μg of protein extracts, together with molecular weight markers, were subjected to 1D SDS-PAGE on 4-12% gradient gels (Invitrogen). After electrophoresis per manufacturer’s manual (Invitrogen), proteins were transferred to PVDF membranes (Millipore) at a constant voltage for 1 hour using Invitrogen’s semidry blotting apparatus. Western analyses of PVDF membranes utilized established protocols and antibodies for DNMT1 (Cell Signaling, #5032S), cMYC (Cell Signaling, #D84C12), and anti-β-Actin peroxidase (Sigma-Aldrich, #A3854).
mRNA levels were assayed by QRT-PCR using standard methods. Briefly, GAPDH was amplified as control. Primer sequences were as follow: CDA Forward 5’-AAGGGTACAAGGATTTCAGGG-3’ and CDA Reverse 5’-ACAATATACGTACCATCCGGC-3’. Real-time detection of the emission intensity of SYBR Green bound to double-stranded DNA was detected using the iCycler instrument (Bio-Rad). Data is reported as ‘relative expression value’ which was determined by raising 2 to the power of the negative value of delta-delta CT for each sample.
Cytospins of cells were fixed for 2 minutes in methanol, air-dried, and stained for 20 minutes with filtered modified solution of Giemsa stain (Sigma Aldrich, Cat #48900, St Louis, MO), diluted (1:20) with buffer solution pH6.5, rinsed with distilled water, air-dried and examined using low and high magnifications with a Leica DMR microscope (Leica Microsystems, Wetzlar GmbH, Germany) connected to Nuance multispectral imaging system FX using Nuance version 3.0.2 software (PerknElmer, Inc., Hopkinton, MA).
Pre-clinical in vivo studies of gemcitabine-resistant PDAC
All experiments were approved by the Cleveland Clinic IACUC and followed approved procedures. Gemcitabine Resistant MIA PaCa2 cells were subcutaneously injected into the right and left flanks of 6- to 8-week-old immunocompromised male BALB/c nu/nu mice (2 million cells per injection in 200 μl sterile control). When tumor volumes reached an average of 100 mm3, mice were randomized to the treatment groups, and tumor volume was assessed by caliper measurement twice weekly throughout the study using the equation: volume (mm3) = long (mm) × wide2 (mm)/2 (at least four mice with two tumors each for adequate power > 0.08). Mice were treated subcutaneously twice a week with vehicle control, twice a week with 60 mg/kg gemcitabine for two weeks followed by a week off, or a combination of THU (10 mg/kg) and decitabine (0.1 mg/kg) twice a week. The reagents were formulated in a PBS solution. Mice body weights were recorded weekly and percentage of mice body weights during treatment was calculated as: weight at each time point/initial weight × 100. Animals were observed for signs of toxicity (mucous diar-rhea, abdominal stiffness, and weight loss) and were euthanized when tumor volume reached 1000 mm3 using CO2 inhalation and followed by cervical dislocation. Statistical analysis was performed using ANOVA with significance reached at P < 0.05.
Results
PDAC cells resist gemcitabine and decitabine by upregulating CDA
Clinical trials are typically conducted in patients with PDAC that has progressed through earlier therapy with gemcitabine. Therefore, we evaluated sensitivity of gemcitabine-resistant patient-derived PDAC cells (MiaPaCa [37]) to decitabine 0-50 µM: the gemcitabine-resistant PDAC cells were also resistant to high concentrations of decitabine - the concentration of decitabine needed to produce 50% growth inhibition of the cells (IC50) was ~40 µM (Figure 1A). Gemcitabine and decitabine are both inactivated by CDA [47,48], and CDA expression was > 100-fold higher in gemcitabine-resistant PDAC cells compared to decitabine and gemcitabine-sensitive K562 cells, a model of decitabine-sensitive myeloid malignancy (Figure 1B). We also compared expression of CDA in several PDAC cancer cell lines (n=19) vs other cancer cell types (n=351) in the Cancer Cell Line Encyclopedia (CCLE): PDAC cells inherently express higher levels of CDA than almost any other cancer cell type (public RNA sequencing data) (Figure 1C).
Figure 1.
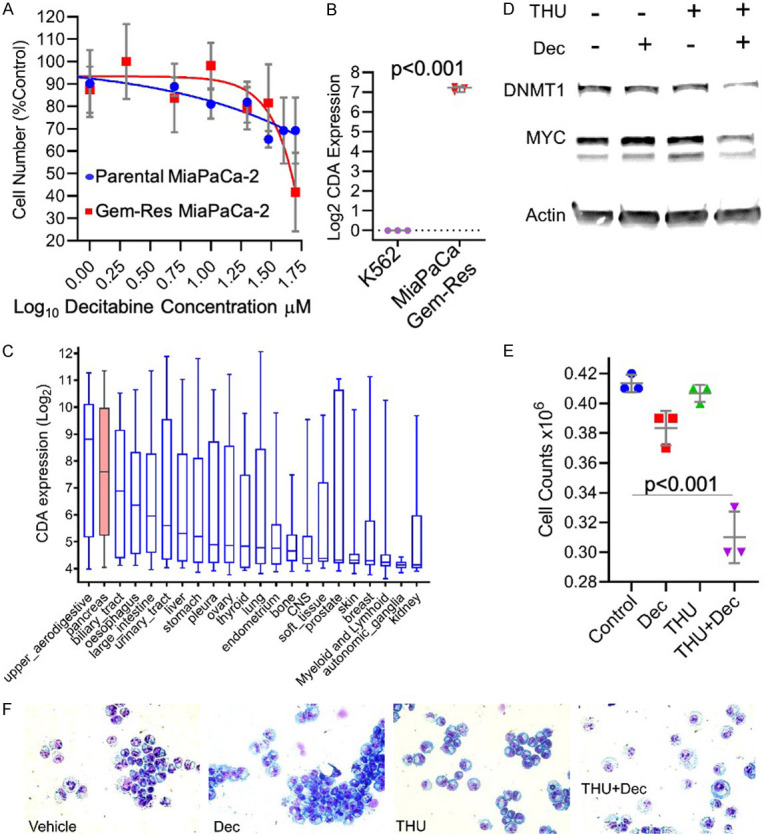
The addition of a CDA-inhibitor to decitabine enabled DNMT1-depletion by decitabine and hence cytoreduction of gemcitabine-resistant PDAC cells. A. Concentrations of decitabine needed to produce 50% growth inhibition (GI50) in parental and gemcitabine-resistant PDAC cells (MiaPaCa-2). Mean ± SD. B. CDA expression in gemcitabine-resistant PDAC cells (MiaPaCa-2) vs decitabine- and gemcitabine-resistant myeloid leukemia cells (K562). QRT-PCR. Mean ± SD. P-value 2-sided unpaired t-test. C. CDA expression is inherently higher in pancreatic cancer vs other cancers. CDA expression in cancer cell lines (n=370) grouped by histology of origin and ordered by median CDA expression level (analysis of public RNA-sequencing data from CCLE). Box-plots median ± IQR, whiskers range. D. The addition of the CDA-inhibitor THU (1 mM) to a clinically-relevant concentration of decitabine (0.2 mM) enabled depletion of DNMT1 and MYC from gemcitabine-resistant PDAC cells (MiaPaCa-2). Western blots 72 hours after a single addition of decitabine and/or THU. E. Cell counts of the cells 72 hours after a single addition of decitabine and/or THU as per panel D. Mean ± SD. P-value 2-sided unpaired t-test. F. Giemsa-stained cytospin preparations of the cells 72 hours after a single addition of decitabine and/or THU as per panel D.
We reasoned that if decitabine-resistance of the PDAC cells was mediated by CDA, then addition of the CDA-inhibitor THU might confer sensitivity to decitabine: addition of THU 1 µM enabled a clinically relevant concentration of decitabine 0.2 µM to deplete DNMT1 from the gemcitabine-resistant PDAC cells, decrease levels of the master regulator of proliferation MYC (Figure 1D), and substantially decrease proliferation of the gemcitabine-resistant PDAC cells (Figure 1E), accompanied by morphologic changes (e.g., decreased nuclear/cytoplasmic ratio) (Figure 1F). We then engrafted mice with gemcitabine-sensitive or gemcitabine-resistant PDAC cells and compared cytotoxic gemcitabine therapy with non-cytotoxic DNMT1-targeting regimens of decitabine: both a cytotoxic regimen of gemcitabine and non-cytotoxic, DNMT1-targeting regimens of decitabine or THU/decitabine controlled gemcitabine-sensitive PDAC tumor growth (Panc-1) (the non-cytotoxic, DNMT1-targeting properties of the decitabine regimens were documented previously [33,49,50]) (Figure 2A, 2B). However, the gemcitabine regimen was not able to control in vivo growth of gemcitabine-resistant PDAC cells (gemcitabine-resistant MiaPaCa-2 that were also resistant to decitabine), while the THU/decitabine regimen suppressed growth of these tumors (Figure 2C, 2D).
Figure 2.

Combination THU+Decitabine significantly cytoreduced gemcitabine-resistant PDAC in vivo. A. Experiment schema gemcitabine-sensitive Panc-1 PDAC cells: Once tumor volumes reached 100 mm3 (~Day 7), mice were randomized to the indicated treatments. The experiment was terminated on Day 18 for analysis. Mpk = mg/kg; SC = subcutaneous; IP = intra-peritoneal; Dec = decitabine. n=8/treatment group. B. Tumor volume measurements. Mean ± SE. ***P < 0.001 vs gemcitabine-treatment, 2-sided t-test. C. Experiment schema gemcitabine-resistant MiaPaCa-2 PDAC cells: Once tumor volumes reached 100 mm3 (~Day 7), mice were randomized to the indicated treatments. Mice were euthanized when their tumor volume measurement reached 1000 mm3 or when they exhibited signs of distress as defined in the Animal Protocol; D. Tumor volume measurements. Mean ± SE. ***P < 0.001 vs gemcitabine-treatment, 2-sided t-test.
Clinical trial study population
To translate pre-clinical observations into the clinic, we conducted a pilot clinical trial of THU/decitabine to treat advanced, chemorefractory PDAC. From April to August 2017, we enrolled 13 patients. Their median age was 65 years (range 44-74), and 7 were male (54%) (Table 1). The primary site of PDA within the pancreas was the head/neck in 4 (31%) and body/tail in 9 (69%) (Table 1). The median time from diagnosis was 13 months (range 3.9-53.5) (Table 1). Baseline ECOG PS was 0 or 1 in 12 (92%) of the patients. They had received a median of 2 prior lines of therapy (range 1-3), including FOLFIRINOX or its variants in 12 (92%) and gemcitabine/nab-paclitaxel in 9 (69%) patients. All patients had multiple sites of metastases, including 8 (62%) with ascites. Baseline CA19.9 levels were > 1000 U/ml in 8 (67%) patients (Table 1).
Table 1.
Baseline Characteristics of Study Patients
| Patient # | Age, Sex | ECOG PS | # Prior Rx* | Serum CA19.9 Level | Liver Lesions | Lung Lesions | Peritoneal Disease/Ascites |
|---|---|---|---|---|---|---|---|
| 1 | 44M | 0 | 3 | 9,187 | ✓ | ✓ | |
| 2 | 65M | 1 | 3 | NL | ✓ | ✓ | |
| 3 | 69M | 1 | 1 | 351 | ✓ | ✓ | |
| 4 | 59F | 1 | 2 | 1,699 | ✓ | ✓ | |
| 5 | 59F | 0 | 2 | 171,630 | ✓ | ✓ | |
| 6 | 74M | 1 | 2 | 9,747 | ✓ | ✓ | ✓ |
| 7 | 69F | 0 | 1 | 376 | ✓ | ||
| 8 | 65F | 1 | 1 | NL | ✓ | ✓ | ✓ |
| 9 | 64M | 1 | 2 | 618 | ✓ | ✓ | ✓ |
| 10 | 44F | 2 | 2 | 2,864 | ✓ | ✓ | |
| 11 | 67M | 1 | 2 | 3,824 | ✓ | ✓ | |
| 12 | 67F | 1 | 2 | 12,668 | ✓ | ✓ | ✓ |
| 13 | 60F | 1 | 1 | 3,707 | ✓ | ✓ |
Notes: ECOG PS: ECOG Performance Score (scale from 0 to 4; 0= best, with no impairments; 4= bedridden); # Prior Rx = number of prior lines of therapy;
prior therapy includes radiation if given separately from chemotherapy;
NL = normal; NA = not available.
Clinical safety
All enrolled patients started planned treatment. Median time on treatment was 35 days (range 4-63); median time on study was 72 days (range 25-105). The most frequent adverse events deemed to be possibly, probably, or definitely related to treatment were anemia (n=5), and in 3 patients each, increased alkaline phosphatase, anorexia, dehydration, nausea, fatigue, febrile neutropenia, decreased lymphocyte count, hyponatremia, and hypokalemia (Table 2). There were no deaths (grade 5 toxicity) attributed to the study drugs. Six patients required study drug holds per protocol for laboratory abnormalities (grade 3 alkaline phosphatase rise, n=2; grade 3 hyperbilirubinemia, n=1; thrombocytopenia, n=1). One patient required study drug hold for grade 3 nausea and vomiting, and another for atrial fibrillation (n=1).
Table 2.
Most Frequent Treatment-Related Adverse Events (N=13)
| Grade 1 or 2 | Grade 3 or 4 | Total | |
|---|---|---|---|
| Anemia | 4 | 1 | 5 |
| Febrile neutropenia | 0 | 3 | 3 |
| Decreased lymphocyte count | 1 | 2 | 3 |
| Fatigue | 3 | 0 | 3 |
| Nausea | 3 | 0 | 3 |
| Anorexia | 2 | 1 | 3 |
| Dehydration | 1 | 2 | 3 |
| Increased alkaline phosphatase | 2 | 1 | 3 |
| Hyponatremia | 1 | 2 | 3 |
| Hyperkalemia | 3 | 0 | 3 |
Clinical efficacy
Eight (62%) patients underwent evaluation scans at week 8. The best response was stable disease in 1 patient; the other 7 had progressive disease. Reasons for coming off of study drug treatment before or after week 8 were progressive disease (n=6), treating physician discretion (clinical progressive disease) (n=3), adverse events (n=2), and others (n=2). During follow-up, 6 patients died. Median overall survival in this cohort of patients from time of enrollment was 3.1 months.
Limited systemic pharmacodynamic effect of the THU/decitabine therapy could be from systemically elevated CDA enzyme activity
Toxicological studies have demonstrated that the myeloid compartment is the most sensitive tissue compartment to decitabine (with or without THU) systemic effect - neutropenia concurrent with thrombocytosis are expected side-effects of non-cytotoxic DNMT1-targeting regimens [34,35,40,42,43]. Most of the patients in this study, however, developed neither neutropenia nor thrombocytosis (Figure 3A) [34,35,42,43]. To identify a possible reason for limited systemic decitabine effect, we compared plasma CDA enzyme activity in patients with resectable vs advanced/metastatic PDAC, and found a significant increase in the patients with advanced disease (Figure 3B). Moreover, PDAC metastatic to liver or peritoneum expresses significantly higher (~2-fold) CDA levels than primary PDAC or normal pancreas tissue (analysis of public data [51] (Figure 3C).
Figure 3.

The THU/decitabine therapy had minimal effects on blood counts in most patients, most likely because of systemically elevated CDA enzyme activity in patients with advanced PDAC. A. Neutropenia and increase platelet counts expected with systemic decitabine exposure and DNMT1-depletion mostly did not occur. Serial blood counts during therapy. B. Plasma CDA enzyme activity is increased in patients with metastatic vs resectable PDAC. Plasma CDA enzyme activity was measured using an HPLC-based assay in a separate cohort of patients with PDAC enrolled in a sample collection protocol, grouped by resectable vs metastatic disease (n=17). Median ± IQR, Mann-Whitney test, 2-sided. C. CDA expression is significantly higher in PDAC metastases in the liver and peritoneum than in primary tumor. Analysis of public gene expression data GeoDatabase GSE71729, gene expression by microarray (n=223). Median ± IQR, Mann-Whitney test, 2-sided.
Discussion
DNMT1 has been scientifically validated by several research groups as a molecular target for cytoreduction of PDAC, importantly, offering a p53-independent pathway for cell cycle exits, distinct from that used by intensive chemotherapy. Encouraged by pre-clinical science, several clinical trials have been conducted with the pyrimidine nucleoside analog decitabine to target DNMT1 in PDAC, but with disappointing results. The pyrmidine metabolism enzyme CDA rapidly catabolizes decitabine into a uridine analog that does not deplete DNMT1. CDA also catabolizes the pyrimidine nucleoside analog gemcitabine that is a first-line agent for treatment of PDAC, and CDA in PDAC cells [37-39], or from other cells, and even from bacteria in the PDAC micro-environment [36,52], has been implicated as in PDAC resistance to first-line gemcitabine therapy. As expected with CDA as the cause of resistance, we found here that PDAC cells resistant to gemcitabine were also resistant to decitabine. Accordingly, the addition of a CDA-inhibitor THU to clinically relevant concentrations/doses of decitabine enabled cytoreduction of gemcitabine/decitabine-resistant PDAC in pre-clinical models. To translate these observations, we used doses of oral THU and oral decitabine that we had previously identified as being sufficient for clinically meaningful engagement of the DNMT1-target in the myeloid compartment in human beings without PDAC [35]: that is, these doses of oral decitabine, administered after oral THU, successfully distributed through CDA-enriched intestines and liver in order to produce clinically significant pharmacodynamic effect in the myeloid compartment. Since the oral decitabine (administered with THU) successfully distributed through CDA-rich intestines and liver, we inferred that the decitabine could also distribute through CDA-enriched PDAC. To increase the fraction of PDAC subject to S-phase dependent DNMT1-depletion, these doses of oral THU/decitabine were administered frequently and in distributed fashion (for 5 consecutive days then 2X/week). Unfortunately, no clinical benefit was observed. Neither did we observe neutropenia concurrent with an increase in the platelet count, the side-effect expected with systemic exposure to non-cytotoxic, DNMT1-depleting concentrations of decitabine (the myeloid compartment is the most sensitive compartment to systemic decitabine exposures [35,40,42]). A reason for this was indicated by measurements of serum CDA enzyme activity: in 17 PDAC patients, we found ~10-fold higher levels in those with advanced versus resectable PDAC. Additionally, in analyses of public datasets, we found that PDAC cells express higher CDA levels than almost every other type of cancer, and CDA expression is especially high in PDAC metastatic to liver or peritoneum vs primary PDAC. Corroborating our findings are published observations from others: in 40 patients with PDAC, serum CDA activity was more than 2-fold increased in those with progressive vs controlled disease [45]; differences in systemic CDA activity between individual patients [53] has been implicated as contributing to PDAC resistance to first-line gemcitabine; and generally higher serum CDA enzyme activity in solid tumor versus other patients has been documented [53, 54). Also worth noting, decitabine is used at several-fold lower doses than gemcitabine (5-20 mg/m2/day versus 1000 mg/m2/day), suggesting it would be even more vulnerable to CDA upregulation [55].
Deoxycytidine kinase (DCK), which executes the initial, rate-limiting modification of decitabine towards the nucleotide form that depletes DNMT1, is another pyrimidine metabolism enzyme essential to DNMT1-depletion by decitabine. DCK also executes the initial rate-limiting modification of gemcitabine, and thus, gemcitabine-therapy can select for DCK-low as well as CDA-high PDAC cells [56]. Thus, it is conceivable that even with adequate CDA inhibition to permit decitabine distribution through PDAC tissue, that DNMT1-depletion may not be achieved if the gemcitabine-resistant PDAC does not express sufficient DCK. Nevertheless, inhibiting CDA to prevent rapid systemic catabolism of decitabine is a needed first-step for decitabine to have even the possibility of being captured by DCK in PDAC cells.
In sum, CDA activity is increased not just within PDAC tissue, but also systemically in patients with advanced PDAC, likely requiring higher THU dosages then found enough for CDA inhibition in other clinical contexts. Since CDA contributes to resistance not just to decitabine/5-azacytidine, but also to standard first-line PDAC therapy gemcitabine [38,39,44,45], this information is pertinent to future clinical evaluations of decitabine, 5-azacytidine or gemcitabine to treat PDAC.
Acknowledgements
D.S. is supported by a 2019 VeloSano Award; Y.S. is supported by NCI P30 CA043703, NCI RO1CA204373, philanthropic funds from Robert and Jennifer McNeil, Leszek and Jolanta Czarnecki, and Dane and Louise Miller, and the James Oberle family, and NIH Shared Instrument award S10OD018205.
Disclosure of conflict of interest
None.
References
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sohal DP, Mangu PB, Khorana AA, Shah MA, Philip PA, O’Reilly EM, Uronis HE, Ramanathan RK, Crane CH, Engebretson A, Ruggiero JT, Copur MS, Lau M, Urba S, Laheru D. Metastatic pancreatic cancer: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2016;34:2784–96. doi: 10.1200/JCO.2016.67.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grützmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA Australian Pancreatic Cancer Genome Initiative. Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV, Grimmond SM. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu; Cancer Genome Atlas Research Network. Integrated Genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185–203. e13. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 8.Romano FJ, Rossetti S, Conteduca V, Schepisi G, Cavaliere C, Di Franco R, La Mantia E, Castaldo L, Nocerino F, Ametrano G, Cappuccio F, Malzone G, Montanari M, Vanacore D, Quagliariello V, Piscitelli R, Pepe MF, Berretta M, D’Aniello C, Perdonà S, Muto P, Botti G, Ciliberto G, Veneziani BM, De Falco F, Maiolino P, Caraglia M, Montella M, De Giorgi U, Facchini G. Role of DNA repair machinery and p53 in the testicular germ cell cancer: a review. Oncotarget. 2016;7:85641–9. doi: 10.18632/oncotarget.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997;15:2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 10.Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, Wang TC, Szabolcs M, Tycko B, Ludwig T. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res. 2013;73:885–96. doi: 10.1158/0008-5472.CAN-12-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cecconi D, Astner H, Donadelli M, Palmieri M, Missiaglia E, Hamdan M, Scarpa A, Righetti PG. Proteomic analysis of pancreatic ductal carcinoma cells treated with 5-aza-2’-deoxycytidine. Electrophoresis. 2003;24:4291–303. doi: 10.1002/elps.200305724. [DOI] [PubMed] [Google Scholar]
- 12.Kumagai T, Wakimoto N, Yin D, Gery S, Kawamata N, Takai N, Komatsu N, Chumakov A, Imai Y, Koeffler HP. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121:656–65. doi: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 13.Yamada T, Ohwada S, Saitoh F, Adachi M, Morishita Y, Hozumi M. Induction of Ley antigen by 5-aza-2’-deoxycytidine in association with differentiation and apoptosis in human pancreatic cancer cells. Anticancer Res. 1996;16:735–40. [PubMed] [Google Scholar]
- 14.Velcheti V, Radivoyevitch T, Saunthararajah Y. Higher-level pathway objectives of epigenetic therapy: a solution to the p53 problem in cancer. Am Soc Clin Oncol Educ Book. 2017;37:812–24. doi: 10.1200/EDBK_174175. [DOI] [PubMed] [Google Scholar]
- 15.Mottini C, Tomihara H, Carrella D, Lamolinara A, Iezzi M, Huang JK, Amoreo CA, Buglioni S, Manni I, Robinson FS, Minelli R, Kang Y, Fleming JB, Kim MP, Bristow CA, Trisciuoglio D, Iuliano A, Del Bufalo D, Di Bernardo D, Melisi D, Draetta GF, Ciliberto G, Carugo A, Cardone L. Predictive signatures inform the effective repurposing of decitabine to treat KRAS-dependent pancreatic ductal adenocarcinoma. Cancer Res. 2019;79:5612–25. doi: 10.1158/0008-5472.CAN-19-0187. [DOI] [PubMed] [Google Scholar]
- 16.Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, Liesa M, Hammerman PS, Wong KK, Hayes DN, Shirihai OS, Dyson NJ, Haas W, Meissner A, Bardeesy N. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–5. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Pishchik V, Figg WD, Murgo AJ, Steinberg SM. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12:5777–85. doi: 10.1158/1078-0432.CCR-06-0669. [DOI] [PubMed] [Google Scholar]
- 18.Steven A, Fisher SA, Robinson BW. Immunotherapy for lung cancer. Respirology. 2016;21:821–33. doi: 10.1111/resp.12789. [DOI] [PubMed] [Google Scholar]
- 19.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, Harris J, Yen RW, Ahuja N, Brock MV, Stearns V, Feller-Kopman D, Yarmus LB, Lin YC, Welm AL, Issa JP, Minn I, Matsui W, Jang YY, Sharkis SJ, Baylin SB, Zahnow CA. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–46. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oki Y, Jelinek J, Shen L, Kantarjian HM, Issa JP. Induction of hypomethylation and molecular response after decitabine therapy in patients with chronic myelomonocytic leukemia. Blood. 2008;111:2382–4. doi: 10.1182/blood-2007-07-103960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. 2010;2:71–86. doi: 10.2217/epi.09.44. [DOI] [PubMed] [Google Scholar]
- 22.Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, Sebree R, Rodgers K, Hooker CM, Franco N, Lee B, Tsai S, Delgado IE, Rudek MA, Belinsky SA, Herman JG, Baylin SB, Brock MV, Rudin CM. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benton CB, Thomas DA, Yang H, Ravandi F, Rytting M, O’Brien S, Franklin AR, Borthakur G, Dara S, Kwari M, Pierce SR, Jabbour E, Kantarjian H, Garcia-Manero G. Safety and clinical activity of 5-aza-2’-deoxycytidine (decitabine) with or without Hyper-CVAD in relapsed/refractory acute lymphocytic leukaemia. Br J Haematol. 2014;167:356–65. doi: 10.1111/bjh.13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke MJ, Lamba JK, Pounds S, Cao X, Ghodke-Puranik Y, Lindgren BR, Verneris MR, Miller JS. A therapeutic trial of decitabine and vorinostat in combination with chemotherapy for relapsed/refractory acute lymphoblastic leukemia. Am J Hematol. 2014;89:889–95. doi: 10.1002/ajh.23778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stathis A, Hotte SJ, Chen EX, Hirte HW, Oza AM, Moretto P, Webster S, Laughlin A, Stayner LA, McGill S, Wang L, Zhang WJ, Espinoza-Delgado I, Holleran JL, Egorin MJ, Siu LL. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin Cancer Res. 2011;17:1582–90. doi: 10.1158/1078-0432.CCR-10-1893. [DOI] [PubMed] [Google Scholar]
- 26.Blum KA, Liu Z, Lucas DM, Chen P, Xie Z, Baiocchi R, Benson DM, Devine SM, Jones J, Andritsos L, Flynn J, Plass C, Marcucci G, Chan KK, Grever MR, Byrd JC. Phase I trial of low dose decitabine targeting DNA hypermethylation in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: dose-limiting myelosuppression without evidence of DNA hypomethylation. Br J Haematol. 2010;150:189–95. doi: 10.1111/j.1365-2141.2010.08213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart DJ, Issa JP, Kurzrock R, Nunez MI, Jelinek J, Hong D, Oki Y, Guo Z, Gupta S, Wistuba II. Decitabine effect on tumor global DNA methylation and other parameters in a phase I trial in refractory solid tumors and lymphomas. Clin Cancer Res. 2009;15:3881–8. doi: 10.1158/1078-0432.CCR-08-2196. [DOI] [PubMed] [Google Scholar]
- 28.Malik A, Shoukier M, Garcia-Manero G, Wierda W, Cortes J, Bickel S, Keating MJ, Estrov Z. Azacitidine in fludarabine-refractory chronic lymphocytic leukemia: a phase II study. Clin Lymphoma Myeloma Leuk. 2013;13:292–5. doi: 10.1016/j.clml.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, Banerjee S, Vasanthakumar A, Culjkovic B, Scott DW, Wyman S, Leser M, Shaknovich R, Chadburn A, Tabbo F, Godley LA, Gascoyne RD, Borden KL, Inghirami G, Leonard JP, Melnick A, Cerchietti L. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013;3:1002–19. doi: 10.1158/2159-8290.CD-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saunthararajah Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematology Am Soc Hematol Educ Program. 2013;2013:511–21. doi: 10.1182/asheducation-2013.1.511. [DOI] [PubMed] [Google Scholar]
- 31.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Pishchik V, Figg WD, Murgo AJ, Steinberg SM. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12:5777–85. doi: 10.1158/1078-0432.CCR-06-0669. [DOI] [PubMed] [Google Scholar]
- 32.Saunthararajah Y, Sekeres M, Advani A, Mahfouz R, Durkin L, Radivoyevitch T, Englehaupt R, Juersivich J, Cooper K, Husseinzadeh H, Przychodzen B, Rump M, Hobson S, Earl M, Sobecks R, Dean R, Reu F, Tiu R, Hamilton B, Copelan E, Lichtin A, Hsi E, Kalaycio M, Maciejewski J. Evaluation of noncytotoxic DNMT1-depleting therapy in patients with myelodysplastic syndromes. J Clin Invest. 2015;125:1043–55. doi: 10.1172/JCI78789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebrahem Q, Mahfouz R, Ng KP, Saunthararajah Y. High cytidine deaminase expression in the liver provides sanctuary for cancer cells from decitabine treatment effects. Oncotarget. 2012;3:1137–45. doi: 10.18632/oncotarget.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavelle D, Vaitkus K, Ling Y, Ruiz MA, Mahfouz R, Ng KP, Negrotto S, Smith N, Terse P, Engelke KJ, Covey J, Chan KK, Desimone J, Saunthararajah Y. Effects of tetrahydrouridine on pharmacokinetics and pharmacodynamics of oral decitabine. Blood. 2012;119:1240–7. doi: 10.1182/blood-2011-08-371690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molokie R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, Ibanez V, Ruiz MA, Ng KP, Woost P, Radivoyevitch T, Pacelli D, Fada S, Rump M, Hsieh M, Tisdale JF, Jacobberger J, Phelps M, Engel JD, Saraf S, Hsu LL, Gordeuk V, DeSimone J, Saunthararajah Y. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med. 2017;14:e1002382. doi: 10.1371/journal.pmed.1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, Hong HS, Kremer DM, Nelson BS, Kemp S, Zhang L, Chang D, Biankin A, Shi J, Frankel TL, Crawford HC, Morton JP, Pasca di Magliano M, Lyssiotis CA. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29:1390–9. e6. doi: 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samulitis BK, Pond KW, Pond E, Cress AE, Patel H, Wisner L, Patel C, Dorr RT, Landowski TH. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol Ther. 2015;16:43–51. doi: 10.4161/15384047.2014.986967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, Tuveson DA. Nab-paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2:260–9. doi: 10.1158/2159-8290.CD-11-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Funamizu N, Okamoto A, Kamata Y, Misawa T, Uwagawa T, Gocho T, Yanaga K, Manome Y. Is the resistance of gemcitabine for pancreatic cancer settled only by overexpression of deoxycytidine kinase? Oncol Rep. 2010;23:471–5. [PubMed] [Google Scholar]
- 40.Terse P, Engelke K, Chan K, Ling Y, Sharpnack D, Saunthararajah Y, Covey JM. Subchronic oral toxicity study of decitabine in combination with tetrahydrouridine in CD-1 mice. Int J Toxicol. 2014;33:75–85. doi: 10.1177/1091581814524994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milhem M, Mahmud N, Lavelle D, Araki H, DeSimone J, Saunthararajah Y, Hoffman R. Modification of hematopoietic stem cell fate by 5aza 2’ deoxycytidine and trichostatin A. Blood. 2004;103:4102–10. doi: 10.1182/blood-2003-07-2431. [DOI] [PubMed] [Google Scholar]
- 42.Saunthararajah Y, Hillery CA, Lavelle D, Molokie R, Dorn L, Bressler L, Gavazova S, Chen YH, Hoffman R, DeSimone J. Effects of 5-aza-2’-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood. 2003;102:3865–70. doi: 10.1182/blood-2003-05-1738. [DOI] [PubMed] [Google Scholar]
- 43.Saunthararajah Y, Sekeres M, Advani A, Mahfouz R, Durkin L, Radivoyevitch T, Englehaupt R, Juersivich J, Cooper K, Husseinzadeh H, Przychodzen B, Rump M, Hobson S, Earl M, Sobecks R, Dean R, Reu F, Tiu R, Hamilton B, Copelan E, Lichtin A, Hsi E, Kalaycio M, Maciejewski J. Evaluation of noncytotoxic DNMT1-depleting therapy in patients with myelodysplastic syndromes. J Clin Invest. 2015;125:1043–55. doi: 10.1172/JCI78789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, Gil Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33:3812–9. doi: 10.1038/onc.2013.357. [DOI] [PubMed] [Google Scholar]
- 45.Serdjebi C, Seitz JF, Ciccolini J, Duluc M, Norguet E, Fina F, Lacarelle B, Ouafik L, Dahan L. Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine-based regimen. Pharmacogenomics. 2013;14:1047–51. doi: 10.2217/pgs.13.93. [DOI] [PubMed] [Google Scholar]
- 46.Richards DA, Sherwood RA, Ndebele D, Rocks BF. Determination of plasma cytidine deaminase activity by HPLC. Biomed Chromatogr. 1987;2:148–51. doi: 10.1002/bmc.1130020404. [DOI] [PubMed] [Google Scholar]
- 47.Chabner BA, Johns DG, Coleman CN, Drake JC, Evans WH. Purification and properties of cytidine deaminase from normal and leukemic granulocytes. J Clin Invest. 1974;53:922–31. doi: 10.1172/JCI107633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker JA, Wickremsinhe ER, Li CH, Oluyedun OA, Dantzig AH, Hall SD, Qian YW, Ring BJ, Wrighton SA, Guo Y. Pharmacogenomics of gemcitabine metabolism: functional analysis of genetic variants in cytidine deaminase and deoxycytidine kinase. Drug Metab Dispos. 2013;41:541–5. doi: 10.1124/dmd.112.048769. [DOI] [PubMed] [Google Scholar]
- 49.Ng KP, Ebrahem Q, Negrotto S, Mahfouz RZ, Link KA, Hu Z, Gu X, Advani A, Kalaycio M, Sobecks R, Sekeres M, Copelan E, Radivoyevitch T, Maciejewski J, Mulloy JC, Saunthararajah Y. p53 independent epigenetic-differentiation treatment in xenotransplant models of acute myeloid leukemia. Leukemia. 2011;25:1739–50. doi: 10.1038/leu.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu X, Ebrahem Q, Mahfouz RZ, Hasipek M, Enane F, Radivoyevitch T, Rapin N, Przychodzen B, Hu Z, Balusu R, Cotta CV, Wald D, Argueta C, Landesman Y, Martelli MP, Falini B, Carraway H, Porse BT, Maciejewski J, Jha BK, Saunthararajah Y. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J Clin Invest. 2018;128:4260–79. doi: 10.1172/JCI97117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, Bentrem DJ, Talamonti MS, Iacobuzio-Donahue CA. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–78. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, Thaiss CA, Reuben A, Livny J, Avraham R, Frederick DT, Ligorio M, Chatman K, Johnston SE, Mosher CM, Brandis A, Fuks G, Gurbatri C, Gopalakrishnan V, Kim M, Hurd MW, Katz M, Fleming J, Maitra A, Smith DA, Skalak M, Bu J, Michaud M, Trauger SA, Barshack I, Golan T, Sandbank J, Flaherty KT, Mandinova A, Garrett WS, Thayer SP, Ferrone CR, Huttenhower C, Bhatia SN, Gevers D, Wargo JA, Golub TR, Straussman R. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156–60. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serdjebi C, Gagnière J, Desramé J, Fein F, Guimbaud R, François E, André T, Seitz JF, Montérymard C, Arsene D, Volet J, Abakar-Mahamat A, Lecomte T, Guerin-Meyer V, Legoux JL, Deplanque G, Guillet P, Ciccolini J, Lepage C, Dahan L. FFCD-1004 clinical trial: impact of cytidine deaminase activity on clinical outcome in gemcitabine-monotherapy treated patients. PLoS One. 2015;10:e0135907. doi: 10.1371/journal.pone.0135907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fanciullino R, Farnault L, Donnette M, Imbs DC, Roche C, Venton G, Berda-Haddad Y, Ivanov V, Ciccolini J, Ouafik L, Lacarelle B, Costello R. CDA as a predictive marker for life-threatening toxicities in patients with AML treated with cytarabine. Blood Adv. 2018;2:462–9. doi: 10.1182/bloodadvances.2017014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahfouz RZ, Jankowska A, Ebrahem Q, Gu X, Visconte V, Tabarroki A, Terse P, Covey J, Chan K, Ling Y, Engelke KJ, Sekeres MA, Tiu R, Maciejewski J, Radivoyevitch T, Saunthararajah Y. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin Cancer Res. 2013;19:938–48. doi: 10.1158/1078-0432.CCR-12-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saiki Y, Yoshino Y, Fujimura H, Manabe T, Kudo Y, Shimada M, Mano N, Nakano T, Lee Y, Shimizu S, Oba S, Fujiwara S, Shimizu H, Chen N, Nezhad ZK, Jin G, Fukushige S, Sunamura M, Ishida M, Motoi F, Egawa S, Unno M, Horii A. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem Biophys Res Commun. 2012;421:98–104. doi: 10.1016/j.bbrc.2012.03.122. [DOI] [PubMed] [Google Scholar]