

Abstract
Anti-PD-1/PD-L1 immunotherapy, as a treatment for many tumors, has shown good efficacy. However, responses to immunotherapy did not always occur or last long., i.e. primary or acquired resistance, even tumors were PD-L1 positive. Several oncogenic pathways, including PI3K/AKT activation by PTEN loss and NF-κB activation, induce PD-L1 expression and PD-L1 inhibitor-resistance. They also induce expression of CCL2, an inhibitory chemokine that blocks T cell tracking into the tumor by binding to CCR2 on the T cell surface. In this study, we showed that transglutaminase 2 (TG2), a post-translational modification enzyme, induced ubiquitin-proteasome dependent degradation of tumor suppressors including PTEN and IκBα by peptide cross-linking, inducing CCL2 as well as PD-L1 expression via PI3K/AKT and NF-κB activation. It also induced PD-L1 inhibitor-resistance because CCL2 was expressed despite increased PD-L1, which was blocked by PD-L1 inhibitor. We also revealed that inhibition of TG2, instead of PD-L1, restored T cell-dependent killing effect by blocking expression of both PD-L1 and CCL2 in PD-L1(+) triple negative breast cancer (TNBC) cells. In addition, the TG2-expressing TNBC patient group showed higher PD-L1 expression incidence than did the TG2-negative TNBC patient group. In conclusion, TG2 induces primary PD-1/PD-L1 inhibitor-resistance by inducing CCL2 expression. TG2 blockade can be utilized as an excellent therapeutic strategy to overcome PD-L1 inhibitor-resistance in PD-L1(+) TNBC patients. Our study suggested that PD-L1 expression alone might not always be a predictive biomarker for PD-L1(+) TNBC, but TG2 could be a useful predictive marker to select PD-L1 inhibitor-resistant TNBC patients.
Keywords: Transglutaminase 2, TNBC, PD-1, PD-L1, drug resistance
Introduction
The immune checkpoint inhibitors (ICIs) eliminate cancer cells by restoring immune cell functions. The blockade of programmed death-protein 1 (PD-1) on the T cell surface and its co-inhibitory ligand (PD-L1) on the tumor surface enhances T cell immunity; hence, this process is considered one of the most successful cancer immunotherapy treatments. The binding of PD-L1 to PD-1 leads to T cell exhaustion. Thus, blockade of immune checkpoints, such as PD-1 and PD-L1, improves the therapeutic effect against cancers by maximizing cytotoxic T cell activity in the tumor microenvironment.
In the last decade, cancer immunotherapy using PD-1/PD-L1 inhibitors has spotlighted treatment of various tumors because no long-term therapeutic responses were observed on using standard cytotoxic anti-cancer agents.
Utilization of PD-L1 inhibitor in patients with different cancers, including melanoma [1], non-small cell lung cancer (NSCLC) [2], renal cell carcinoma (RCC) [3], Hodgkin’s lymphoma [4], bladder cancer [5], and head and neck squamous cell carcinoma (HNSCC) [6], has shown impressive clinical therapeutic effects. However, blockade of PD-1 and PD-L1 showed limitations in not only patients with low expression of PD-L1 but also those with high expression, demonstrating that the response rate to the inhibitors was only about 20% in most solid tumors. To overcome this limitation, we must understand the mechanisms of PD-1/PD-L1. In triple negative breast cancer (TNBC), objective response rates (ORRs) of PD-1/PD-L1 inhibitors were reportedly 20% in high PD-L1-expressed (+) tumors but less than 5% in lowly expressed or negative PD-L1(-) tumors. Some early responders with relapsed disease developed resistance to these therapies.
The several mechanisms of primary and acquired resistance to PD-1/PD-L1 inhibitors are well known. Representatively, mutations in STK11 and KEAP1 are associated with resistance to PD-1/PD-L1 blockade in NSCLC patients [7]. Currently, novel mechanisms of resistance to PD-1/PD-L1 blockade are being characterized, and new strategies are being suggested to overcome this resistance for improvement of patient outcomes.
As mentioned above, PD-L1 expression is currently utilized as a predictive biomarker to select anti-PD-1 or anti-PD-L1 therapy-sensitive patients [8]. PD-L1 immuno-histochemistry (IHC) is used as a representative predictive biomarker for various tumors including NSCLC and melanoma [9].
However, PD-1/PD-L1 inhibitor-resistant patients are still present even though these patients show PD-L1(+) tumors [10], indicating that PD-L1 is not the perfect biomarker to select patients for anti-PD1/PD-L1 therapies. In fact, some patients with both PD-L1(+) and PD-L1(-) tumors benefitted from PD-1/PD-L1 inhibitor treatment, but the benefit in PD-L1(+) patients was larger than that in PD-L1(-) patients [2]. Recently, tumor mutation burden (TMB) was also utilized as an additional predictable biomarker to select patients applicable for PD-1/PD-L1 inhibitor treatment [11]. However, the prediction accuracy of TMB used in clinical studies is still controversial [12]. Therefore, identification of novel exact biomarkers that can predict PD-1/PD-L1 inhibitor response is required.
TNBC constitutes nearly 15-20% of all breast tumors, and is negative for estrogen receptors, progesterone receptors, and excess HER2 protein. This indicates that TNBC cannot be treated by hormone restriction therapy and usage of HER2 inhibitors, but chemotherapy can be used effectively [12].
Many clinical trials using several PD-1/PD-L1 inhibitors, such as atezolizumab and avelumab, had been conducted for PD-L1(+) TNBC patients, and some PD-1/PD-L1 inhibitors were approved by the FDA in 2019. Even though a higher ORR was seen in PD-L1(+) TNBC patients than in PD-L1(-) TNBC patients (22.2% vs. 2.6%) [13,14], therapeutic effect of PD-1/PD-L1 inhibitors towards TNBC was low [15].
PD-L1 expression in the tumor is increased by activation of multiple oncogenic pathways and transcription factors, such as MYC, AP-1, STAT, IRF1, HIF, PI3K/AKT, and NF-κB [16]. Some oncogenic pathways contribute to PD-1/PD-L1 inhibitor-resistance. PTEN loss, which activates the PI3K/AKT pathway, induces PD-1 and PD-L1 inhibitor-resistance by promoting the release of anti-inflammatory cytokines that reduce infiltration and activation of CD8+ cytotoxic T cells in melanoma patients [17]. NF-κB activation usually increases not only inflammation through up-regulation of cytokines such as tumor necrosis factor (TNF)-α, but also immune response through activation of chemokines [18]. In contrast, NF-κB inhibition was reported to induce chemokine expression in lung adenocarcinoma [19]. In addition, NF-κB promotes angiogenesis and expression of anti-inflammatory cytokines in cancer patients [14,20]. According to a recent study, activation of m-TOR, the downstream signal of the PI3K/AKT pathway, and NF-κB increased CCL2 expression and release, inducing recruitment of tumor-associated macrophages [21]. CCL2 is a chemokine that inhibits trafficking of cytotoxic T cells by binding with CCR2 of the T cell receptor [22]. Therefore, when CCL2 and PD-L1 are induced simultaneously by PI3K/AKT and NF-κB activation, PD-1/PD-L1 inhibitor-resistance can occur due to release of CCL2, even though PD-L1 is completely blocked by the PD-L1 inhibitor.
Transglutaminase 2 (TG2) is a trans-peptidase, which is distributed widely in various tissues. It mediates cross-linking of proteins and participates in signal transduction by activating and hydrolyzing guanine triphosphate (GTP) enzyme. TG2 exerts diverse biological functions not only in normal tissues but also in both inflamed and cancer cells. TG2 is a multi-functional enzyme that demonstrates calcium dependent cross-linking activity and plays a vital role in inflammation mainly through modulation of the extracellular matrix (ECM) structure and stability. It also regulates tumor progression through these activities, attenuating cell adhesion and promoting ECM proteolysis. In normal breast tissues and those with benign hyperplasia, TG2 is rarely expressed, while in some breast cancer cells, TG2 expression is increased, contributing to tumor cell survival, invasion, and motility. In highly aggressive breast cancer cell lines, epithelial growth factor level is unimportant for migration and invasion but TG2 is already activated. Down-regulation of endogenous TG2 inhibits fibronectin-mediated cell attachment, survival, and invasion, while ectopic expression of TG2 augments attachment and invasion, suggesting that TG2 plays an important role in the development of metastases in breast cancer cells. TG2 causes cross-linking of the target molecule by transamidation, and the cross-linked target is then removed by ubiquitin-proteasome dependent degradation. Several tumor suppressors, including IκBα and PTEN, are target molecules of TG2 [23-26].
TG2-dependent IκBα and PTEN degradation induces PI3K/AKT and NF-κB activation, contributing to the use of chemotherapeutic agents, taxol and doxorubicin, and targeted drugs, such as EGFR-TKIs, in various tumors [27]. Interestingly, TG2 activates PI3K/AKT and NF-κB, which in turn, activate PD-L1. In addition, as mentioned above, PI3K/AKT and NF-κB activation by TG2 induces both PD-L1 and CCL2. Therefore, in this study, we investigated whether CCL2 release induced by PI3K/AKT and NF-κB activation led to PD-1/PD-L1 inhibitor-resistance, even though up-regulated PD-L1 was blocked by the PD-1/PD-L1 inhibitor. In addition, we investigated whether PD-L1 inhibitor-resistance could be restored by TG2 inhibition or co-inhibition of PD-L1 and CCL2 in PD-L1(+) TNBC patients. Therefore, in this study, we investigated the relationship between TG2 and PD-L1 and their cellular mechanisms and optimized the use of PD-1/PD-L1 inhibitors in TNBC patients based on the expression levels of TG2 and/or PD-L1.
Materials and methods
Cell culture and reagents
The human breast cancer cell lines, BT20, MCF7 and MDA-MB-231, were obtained from the American Type Culture Collection (Manassas, VA, USA). To obtain cultures, cells were incubated in RPMI-1640 culture medium supplemented with 10% fetal bovine serum (ThermoFisher Scientific, Waltham, MA, USA) at 37°C in an atmosphere of 5% CO2. GK921, TG2 inhibitor, was purchased from MedChemExpress (Monmouth Junction, NJ, USA) and dissolved in dimethyl sulfoxide to obtain a final concentration of 10 mM.
Transfection
To prepare a stable cell with overexpressed TG2, the pcDNA3.1/TG2 vector was transfected into the MCF7 cell using Lipofectamine® 2000 Reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s protocol. MCF/TG2 cell was selected by G418 (500 µg/mL; Gibco, Carlsbad, CA, USA). To knockdown TG2 in the MDA-MB-231 cell line, we transfected short interfering RNA (siRNA) of TG2 (Sense: 5’-CCAAGUUCAUCAAGAACAUUU-3’, Anti-sense: 5’-AUGUUCUUGAUGAACUUGGUU-3’), CCL2 (ThermoFisher Scientific, Waltham, MA, USA, AM51331), NF-κB p65 (#6261, Cell signaling, Danvers, MA, USA), and AKT (#6211, Cell signaling, Danvers, MA, USA) using Lipofectamine RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol.
Western blot analysis
The harvested samples of the cells were lysed using RIPA buffer, which contained a protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor (1 mM sodium fluoride and 2 mM sodium orthovanadate). The lysed protein samples were stored on ice for 30 min and then centrifuged at 13,000 × g for 30 min. The whole cell lysate was collected from the supernatant, and total protein was determined. The total protein (10-20 µg) was collected with 8-15% SDS-PAGE and then transferred to a polyvinylidene fluoride membrane (Bio-Rad, Hercules, CA, USA). After blocking with 10% skim milk in Tris buffered saline-tween (TBS-T), the membrane was allowed to react with the primary antibody at 4°C overnight and then horseradish peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA, USA) in TBS-T, containing 1% bovine serum albumin, for 1 h at room temperature. The proteins were visualized using ECL Plus enhanced chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ, USA) and G-box Chemi Systems (SynGene, Bangalore, India). TG2 antibody was purchased from ThermoFisher Scientific (CUB 7402, Waltham, MA, USA). The other antibodies, including AKT, phosphorylated (p)-AKT, PTEN, cleaved Caspase 3, cleaved Caspase 7, cleaved PARP, IκBα, PD-L1, and β-Actin were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell IHC
Breast cancer cells (1 × 103) were seeded on an eight-well chamber slide (MERCK, Frankfurter, Germany). After leaving it overnight, the supernatant was removed, and the cells were fixed with 4% formaldehyde for 20 min. After fixation, IHC was performed using Ultra-Sensitive ABC Peroxidase Staining kits (ThermoFisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. The primary antibody on the fixed cells was stained with TG2 antibody (ThermoFisher Scientific, CUB 7402, Waltham, MA, USA) and PD-L1 antibody (Abcam, ab205921, Cambridge, United Kingdom), and the resultant samples were diluted to a concentration of 1 µg at 4°C overnight. Biotinylated secondary antibody and ABC Reagent were then sequentially added to the samples, and the resultant samples were allowed to react at room temperature for 30 min. Samples were then allowed to react with the substrate using AEC Substrate Chromogen (K3464; Dako, Carpinteria, CA, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the breast cancer cells using Allprep DNA/RNA mini kits (Qiazen, Hiden, Germany), following the manufacturer’s protocol. Complementary DNA (cDNA) from total RNA samples was prepared using cDNA Reverse Transcription Kits (Applied Biosystems, Waltham, Massachusetts, USA), following the manufacturer’s protocol. The real-time quantitative analysis of the below-mentioned genes was performed with the LightCycle 480 System (Roche, Basel, Switzerland) and SYBG Green real time PCR mix (TOYOBO, Osaka, Japan), following the manufacturer’s protocol. PD-L1 forward primer (5’-GCTATGGTGGTGCCGACTAC-3’), PD-L1 reverse primer (5’-TGGCTCCCAGAATTACCAAGT-3’), CCL2 forward primer (5’-AGATCTGTGCTGACCCCAAG-3’), and CCL2 reverse primer (5’-TCTTCGGAGTTTGGGTTTGCT-3’) were analyzed.
Jurkat T cell co-culture
Jurkat T cells were activated using Phorbol 12-myristate 13-acetate (PMA, 50 ng/mL) and Ionomycin (1 μg/mL) for 24 h. Breast cancer cells (5 × 105) were seeded on six-well plates. After leaving them overnight, siRNA transfection or drug treatment was performed. After 24 h of siRNA transfection or drug treatment, activated Jurkat T cells (3 × 106) were co-cultured with breast cancer cells. After 48 h, the supernatant was collected for harvesting the Jurkat T cells. PBS or free media washing was then performed three times thoroughly. Cancer cells or Jurkat T cells were harvested for western blot analysis and measurement of Caspase 3/7 by performing the Caspase-Glo 3/7 Luminescence Assay (Promega Corp. Madison, WI, USA). In order to make cancer cells alone remain on the each well, after co-culture with T cell and cancer cells, we conducted washing step with free media. As you know, T cells is suspended cells. As you can see in Supplementary Figure 1, most small suspended T cells are removed from attached cancer cells after three times washing step. Although some killed and suspended cancer cell are also removed in this washing step, this washing step makes damaged or killing cancer remain as attached status in the culture plate without suspended T cell.
Caspase 3/7 assay
Activated Caspase 3 and 7 were measured by performing the Caspase-Glo 3/7 Luminescence Assay (Promega Corp. Madison, WI, USA), following the manufacturers protocol. Protein samples from breast cancer cells or Jurkat T cells were prepared using RIPA buffer, in a manner similar to that for western blot sample preparation. Protein samples (10 µg), in a total volume of 100 µL, were transferred to white 96-well plates. Equilibrated Caspase-Glo 3/7 Reagent (100 µL) was added to the protein samples, and the resultant samples were incubated at room temperature for 1 h. Luminescence was measured using the Wallac 1420 apparatus (PerkinElmer, Waltham, MA, USA).
Interleukin-2 (IL2) and interferon gamma (IFNγ) production assay
Jurkat T cells (1 × 107) were seeded on 6 well plates, and PMA (50 ng/mL) and Ionomycin (1 µg/mL) were added to activate the cells. The conditioned medium (CM) of the Jurkat T cells was harvested in a time-dependent manner to measure cytokines concentration. The CM was harvested and centrifuged at 2,000 rpm for 10 min to remove suspended cells, and each IL-2 and IFNγ concentration was measured using an IL-2 ELISA kit and IFNγ ELISA kit (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s protocol.
Immunohistochemistry
Serial sections (4 μm thickness) were mounted on glass slides coated with 10% polylysine. The sections were dewaxed in xylene and rehydrated in graded ethanol. The endogenous peroxidase activity was blocked by immersing the slides in 0.3% methanolic peroxide for 40 min. Immunoreactivity was enhanced by microwaving the tissue sections in 0.1 M citrate buffer for 10 min. Immunostaining was performed using the avidin-biotin-peroxidase complex method, and antigen-antibody reactions were visualized using chromogen diaminobenzidine. The TG2 and PD-L1 antibodies, used for IHC, were purchased from Sigma Aldrich (Limerick, PA, USA) and Cell Signaling Technology (Danvers, MA, USA), respectively. Overall staining intensity of the tissues was scored by a pathologist (Dr. Hee Jin Lee) using a scoring system of tumor cell staining intensity. A score of 0 indicated no staining of the tumor.
Immunocytochemistry
Cells seeded into 8-well glass (Millipore, 2.5 × 104 cells per well). The cells were fixed in 2% PFA (paraformaldehyde) at room temperature (RT) for 20 min. After treatment with blocking solution containing 5% FBS in PBS for 30 min, cells were stained with anti-human CD274 antibody (1:300; BioLegend, San Diego, California, USA, #329710) at 4°C overnight. Cells were washed three times with PBS and then stained with Alexa Fluor® 488 dye conjugated secondary antibody (1:2,000; invitrogen, Carlsbad, CA, USA, A-21202) in the dark at RT for 1 hr. After cells were washed once with PBS, nuclei were stained with DAPI (1:1,000; Sigma Aldrich, Limerick, PA, USA, D9542) at RT for 5 min. All images were analyzed using ZEISS Axio Vert.A1 FL microscopy (Carl Zeiss, Oberkochen, Germany).
Flow cytometry
Cells were detached from tissue culture dish by TrypLETM express (Gibco, Carlsbad, CA, USA). The cells were washed in FACS buffer containing 5% FBS (ThermoFisher Scientific, Waltham, MA, USA) in PBS. For immune staining, cells were resuspened in 100 ul of FACS buffer containing anti-human CD274 antibody (1:300; BioLegend, San Diego, California, USA, #329710) at 4°C for 30 min. After washing twice in FACS buffer, cells were resuspened in 100 ul of FACS buffer containing Alexa Fluor® 488 dye conjugated secondary antibody (1:2,000; Invitrogen, Carlsbad, CA, USA, #A-21202) at 4°C for 20 min. Data was acquired using FACS CantoTM II (BD Biosciences, San Jose, CA, USA) and A total of 10,000 events were collected per sample. Analysis was performed using FACSDiva software v 8.0 (BD Biosciences, San Jose, CA, USA).
Cell viability test
Cell viability was measured using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI, USA) following the manual instructions. Approximately 3 × 103 cancer cells were plated to white 96 well plates. The next day, the culture medium was removed and the desired concentrations of drug or siRNAs were treated. After 24 h, activated Jurkat T cells (1 × 106) were co-cultured with cancer cells. After 72 hours, 100 μl of CellTiter-Glo reagent was added and the plates were incubated for 10 minutes at room temperature. The luminescence was measured using a Wallac 1420 apparatus (PerkinElmer, Boston, MA, USA). For testing cytokine effect in conditioned medium (CM) of this co-culture, at 48 hr after adding of activated T cell in the wells, CM was harvested and centrifuged at 2,000 rpm for 10 min to remove suspended cells. CM were treated to each breast cancer cells, including MCF7/TG2 and MDA-MB-231, and after 72 hours, 100 μl of CellTiter-Glo reagent was added and the plates were incubated for 10 minutes at room temperature. The luminescence was measured using a Wallac 1420 apparatus (PerkinElmer, Boston, MA, USA).
Results
PD-L1 was an unpredictable biomarker of response to PD-1/PD-L1 inhibition
We first evaluated the activity of PD-1/PDL-1 inhibitor in selected PD-L1(+) TNBC cell lines, co-cultured with activated T cells, using avelumab (Figure 1). First of all, we selected one PD-L1(-) breast cancer cell (MCF7) and two PD-L1(+) breast cancer cells (BT20 and MDA-MB-231). PD-L1 expression and surface PD-L1 expression of each cells were evaluated by western blot (Figure 1A), Immunocytochemistry (Figure 1B) and FACS analysis (Figure 1C). We activated Jurkat T cells using PMA and ionomycin, as described in a previous study [28], and confirmed that activated Jurkat T cells induced PD-1 expression and IL-2 and IFNγ release (Figure 1D) [28]. Activated Jurkat T cells killed PD-L1(-) MCF7 cells (Figure 1E), but could not kill PD-L1(+) BT20 and MDA-MB-231 cells because PD-1 on activated Jurkat T cells binds to PD-L1 on BT20 and MDA-MB-231 cells (Figure 1F and 1G). We also observed that PD-1/PD-L1 inhibitor, Avelumab, rescues killing effect of activated Jurkat T cells in co-culture with PD-L1(+) BT20 cells, but not another PD-L1(+) MDA-MB-231 cells (Figure 1F and 1G). From these results, we double-checked the role of activated Jurkat T cells, which could kill both PD-L1(-) and PD-L1-blocked cancer cells. In addition, PD-1/PD-L1 inhibitor always not working on PD-L1-blocked cancer cells with activated T cells and induces intrinsic resistance to PD-1/PD-L1 blockade, suggesting that PD-L1 expression was not sufficient to predict the effect of PD-1/PD-L1 blockade.
Figure 1.
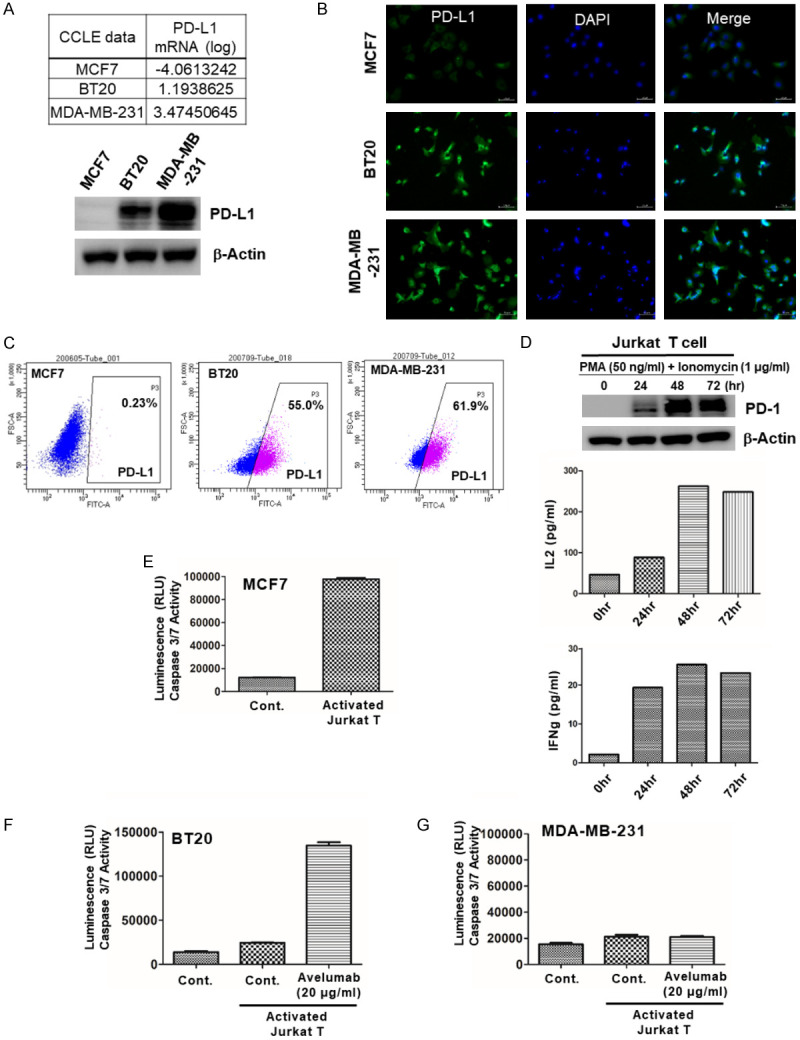
PD-L1 is an unpredictable biomarker of response to PD-1/PD-L1 Inhibition. (A) PD-L1 mRNA and protein expression levels and (B and C) surface PD-L1 expression in MCF7, BT20, and MDA-MB-231 cells; (D) western blot analysis for PD-1 expression and ELISA assay for IL-2 and IFNγ levels in activated Jurkat T cells after PMA (50 ng/mL) and Ionomycin (1 µg/mL) treatment; (E) Caspase 3/7 activity of PD-L1(-) MCF7 cells, co-cultured with activated Jurkat T cells; Caspase 3/7 activity of PD-L1(+) (F) BT20 and (G) MDA-MB-231 cells, co-cultured with activated Jurkat T cells, after treatment of PD-1/PD-L1 inhibitor for 24 h.
TG2 induced PD-L1 expression by blocking PTEN and NF-κB activation in TNBC patients
Using the RNA sequencing data obtained from a previous paper [29], we identified several differentially expressed genes (DEGs), and among them, TG2 was identified as a novel PD-1/PD-L1 inhibitor-resistant marker. First, we checked whether the expression of PD-L1 was correlated with TG2 in PD-L1(+) MDA-MB-231 and PD-L1(-) MCF7 cells. Only PD-L1(+) MDA-MB-231 cells showed high TG2 expression (Figure 2A). According to previous papers, both PI3K/AKT activation by PTEN loss and NF-κB activation by IκBα degradation induced PD-L1 in tumor cells, and TG2 induced both PI3K/AKT and NF-κB activation. Therefore, we tested whether PD-L1 expression was regulated by TG2 in these two cell lines. We confirmed that AKT and NF-κB activation in TG2(+) MDA-MB-231 cells induced PD-L1 expression (Figure 2A). To examine whether PD-L1 expression was directly regulated by TG2, we established a TG2(+) MCF7 cell, MCF7/TG2.
Figure 2.
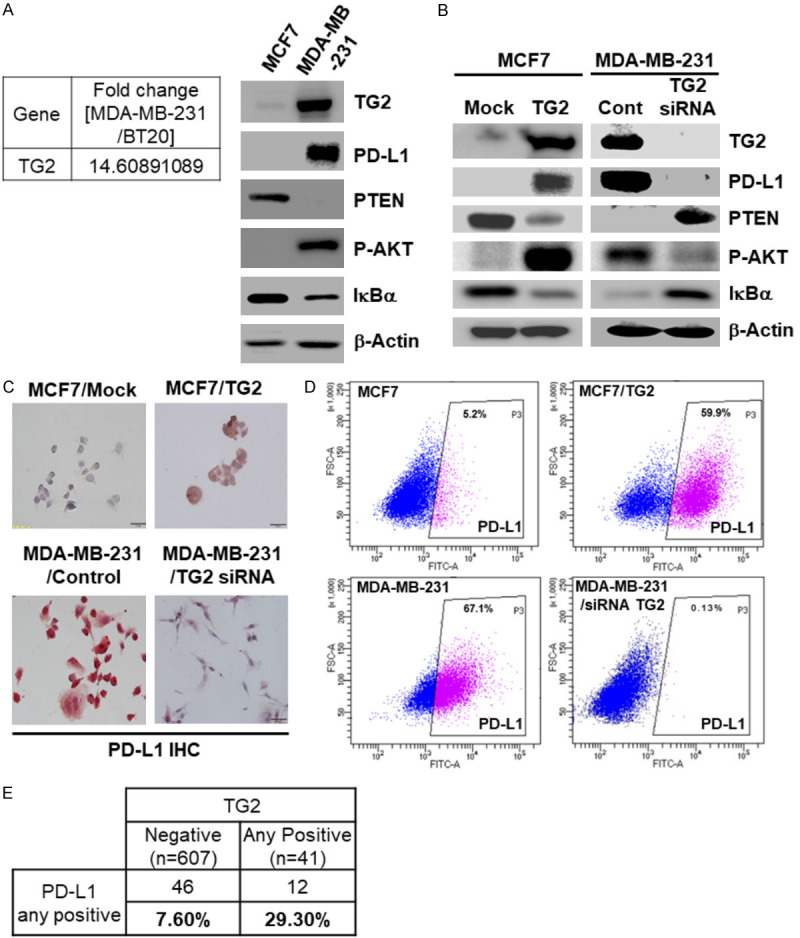
TG2 induces PD-L1 expression by blocking PTEN and activating NF-κB in TNBC cells. (A) Fold change in TG2 in MDA-MB-231 and BT20 cells, and western blot analysis of TG2, PD-L1, PTEN, pAKT, and IκBα in MCF7 and MDA-MB-231 cells, with β-Actin as a loading control. (B) Western blot analysis of TG2, PD-L1, PTEN, pAKT, and IκBα in MCF7/Mock, MCF7/TG2, MDA-MB-231, and MDA-MB-231/TG2 siRNA cells, with β-Actin as a loading control. (C) Cell Immunohistochemistry and (D) FACS analysis for surface PD-L1 expression level in MCF7/Mock, MCF7/TG2, MDA-MB-231 and MDA-MB-231/TG2 siRNA. (E) Incidence of TG2 and PD-L1 expression in Formalin-Fixed Paraffin-Embedded (FFPE) tissue samples of 648 TNBC patients.
In MCF7/TG2 cells, PD-L1 expression increased due to PI3K/AKT and NF-κB activation by TG2. Conversely, when TG2 was knocked-down in MDA-MB-231 cells, PD-L1 expression decreased due to blockade of AKT and NF-κB (Figure 2B-D). Even in the samples of 648 TNBC patients, the incidence of PD-L1 expression was much higher in TG2(+) TNBC samples than in TG2(-) TNBC samples (Figure 2E). These results suggested that PD-L1 could be induced by TG2-dependent signaling, involving PI3K/AKT and NF-κB activation.
TG2-induced PD-L1(+) TNBC cells showed PD-L1 inhibitor-resistance
Based on the finding that activated Jurkat T cells could not kill TG2(+)/PD-L1(+) MDA-MB-231 cells (Figure 1), activity of Jurkat T cells in the above-mentioned four cell lines was evaluated, and we found that activated T cells could kill TG2 down-regulated cells but not TG2 up-regulated MCF7/TG2 and MDA-MB-231 cells (Figure 3A).
Figure 3.
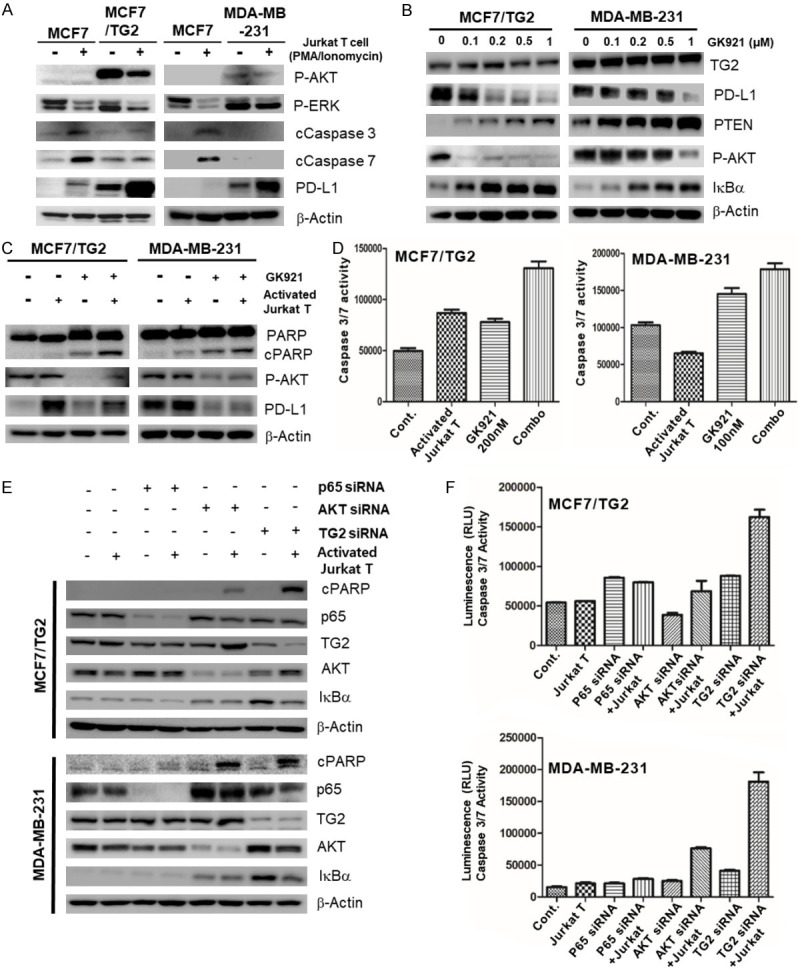
TG2-induced PD-L1(+) TNBC cells show PD-L1 inhibitor-resistance. (A) Expression levels of pAKT, pERK, cleaved Caspase 3, cleaved Caspase 7, PD-L1, and β-Actin in MCF7, MCF7/TG2, and MDA-MB-231 cells with or without activated Jurkat T cell co-culture, measured by western blot analysis; (B) western blot analysis to assess the expressions of TG2, PD-L1, PTEN, pAKT, IκBα, and β-actin in MCF7/TG2 and MDA-MB-231 cells after GK921 (TG2 inhibitor) treatment for 24 h in a dose-dependent manner; (C) western blot analysis and (D) Caspase 3/7 activity assay performed using protein samples from MCF7/TG2 and MDA-MB-231 cells, co-cultured with activated Jurkat T cells, after GK921 treatment for 24 h; (E) western blot analysis and (F) Caspase 3/7 activity assay implemented using protein samples from MCF7/TG2 and MDA-MB-231 cells, co-cultured with or without activated Jurkat T cells for 24 h, after siRNA transfection of p65, AKT, or TG2 for 24 h.
We investigated whether TG2 inhibitor, GK921, could regulate PD-L1 expression in two TG2-induced PD-L1(+) TNBC cell lines, MCF7/TG2 and MDA-MB-231. GK921 decreased PD-L1 expression by restoring PTEN, blocking NF-κB activation, and inhibiting the PI3K/AKT pathway (Figure 3B). Using the co-cultures with activated Jurkat T cells, we observed that GK921 restored T cell-mediated apoptosis of TG2(+) MDA-MB-231 and MCF7/TG2 cells (Figure 3C and 3D). We have conducted MTS assay (Supplementary Figure 2) as well to evaluate IFNγ-induced killing effect of activated Jurkat T cells. Like Casepase 3/7 assay, TG2 inhibitor (GK921), instead of PD-1/PD-L1 inhibitor, restored activated T cell-mediated killing effect (Supplementary Figure 2A and 2B). Especially, conditioned-media (CM) of co-culture condition with GK921 also directly killed cancer cells because CM includes IFNγ by activated Jurkat T cells (Supplementary Figure 2A and 2B). Suspended cells in CM are removed by centrifuge before treatment. However, we also found that blocking AKT or p65, a downstream signal, was not sufficient to restore sensitivity to T cell immunity (Figure 3E and 3F). These results suggested that TG2 inhibitor or the blocking-signal pathway, instead of the blocking PD-1/PD-L1 interaction, could restore cancer-killing T cell immunity if the initiating mechanism that induced PD-1/PD-L1 expression was known.
CCL2 induction by TG2 contributed to PD-L1 inhibitor-resistance via regulation of negatively cytotoxic T cells in TNBC cells
The mechanism of induction of PD-1/PD-L1 remains unknown. Therefore, we explored the mechanism that could be involved in intrinsic resistance to PD-1/PD-L1 inhibitors. Among the many T cell associated chemokines including endothelin-1, CCL2, CCL3, fractalkine, CXCL9/10/11, CCL4/5, CCL20, S1P, and CXCL16, CCL2 was checked, and based on recent findings, we found that CCL2 expression was increased by mTOR, the downstream signal of PI3K/AKT, and NF-κB [21]. CCL2 is a well-known chemokine that binds to the T cell receptor CCR2 to reduce T cell activation and inhibit cytotoxicity of T cells [22]. Fortunately, we found that only TG2-induced PD-L1(+) breast cancer MDA-MB-231 and MCF7/TG2 cells showed high CCL2 expression. Interestingly, low CCL2 expression was observed in the BT20 cell line, which also showed low expression of TG2 and relatively high expression of PD-L1 and could be killed by activated Jurkat T cells. CCL2 was not expressed in TG2(-)/PD-L1(-) MCF7 cells (Figure 4A-C). When TG2 was knocked-down or down-regulated by TG2 siRNA, both PD-L1 and CCL2 expression levels greatly decreased (Figure 4D), demonstrating that CCL2 expression was also TG2-dependent. We then confirmed that CCR2 expression increased in the activated Jurkat T cells (Figure 4E). To investigate whether co-inhibition or dual blockade of CCL2 and PD-L1 could overcome intrinsic resistance to PD-1/PD-L1 inhibition, we down-regulated CCL2 expression using siRNA and added PD-L1 inhibitor, avelumab, to MCF7/TG2 and MDA-MB-231 cells. Then, by performing western blot analysis and Caspase 3/7 assay, we observed that dual blockade of CCL2 by siRNA and PD-L1 by avelumab restored T cell-mediated apoptosis of TG2-induced PD-L1(+) TNBC MDA-MB-231 and MCF7/TG2 cells. The dual blockade of CCL2 and PD-L1 showed more apoptosis than did blockade of either CCL2 by siRNA or PD-L1 by avelumab (Figure 4F and 4G).
Figure 4.

CCL2 induction by TG2 contributes to PD-L1 inhibitor-resistance via regulation of negatively cytotoxic T cells in TNBC cells. The level of (A) PD-L1 and (B) CCL2 mRNA expression in MCF7, BT20, MCF7/TG2, and MDA-MB-231 cells measured by qRT-PCR analysis; (C) western blot analysis to check expression levels of TG2, P-AKT, IκBα, PD-L1, CCL2, and β-Actin in MCF7, BT20, MCF7/TG2, and MDA-MB-231 cells; (D) the expression of TG2, PD-L1, and CCL2 in MCF7/TG2 and MDA-MB-231 cells tested by western blotting analysis after TG2 siRNA transfection; (E) expression of PD-1 and CCR2 in Jurkat T cells with or without treatment of PMA (50 ng/mL) and Ionomycin (1 µg/mL) for 24 h measured by western blotting; (F and G) the protein levels of cPARP, IκBα, PD-L1, and CCL2 in MCF7/TG and MDA-MB-231 cells with or without activated Jurkat T cell co-culture for 24 h measured by western blotting after CCL2 siRNA transfection, Avelumab (20 µg/mL) treatment, or dual combination treatment and Caspase 3/7 activity measured by Caspase 3/7 assay using proteins from MCF7/TG and MDA-MB-231 cells.
Discussion
PD-1/PD-L1 inhibitors have recently gained attention, and are being used as major cancer treatments in patients with various cancers. However, some cancer patients show intrinsic resistance to PD-1/PD-L1 inhibitors even though the tumor cell surfaces express PD-L1. In recent years, evidence on the prognostic effects of PD-L1 expression, which causes immune evasion of tumor, on TNBC has been revealed [30]. Therefore, the effective therapeutic approaches using ICIs, such as PD-1/PD-L1 inhibitors, are being continuously tested for treatment of TNBC. Over the last year, novel and significant evidence on the perfection of immune checkpoint-based treatments in TNBC, based on the results of the IMpassion130 trial, has been revealed, driving breast cancer into the immunotherapy age [31]. In addition, interestingly, PD-L1 expression assessment of immune cells in tumors also utilizes a novel CDx marker for PD-L1 expression on the tumor cell surface [31]. In the Impassion130 biomarker subgroup analysis [32], PD-L1 expression in immune cells positively correlated with the CD8+ T cell number in the tumor. This result implicated that PD-L1 expression in both tumor and immune cells should be assessed to select PD-1/PD-L1 inhibitor-sensitive TNBC patients.
Several PD-1/PD-L1 inhibitors selectively inhibit PD-L1 interaction with PD-1 or B7-1 (a co-stimulatory cell-surface protein), relieving T-cell suppression. Some of the inhibitors have already been approved for the treatment of melanoma [33] and NSCLC [9], and have shown good treatment effects with a safety profile in patients with other solid tumors [1,6], including TNBC [34].
However, in another clinical trial, only a few TNBC patients showed a response to PD-1/PD-L1 inhibitor because the number of PD-L1(+) TNBC patients was low [15] and some of them showed intrinsic resistance to PD-1/PD-L1 inhibitors.
Therefore, the effective therapeutic approaches of ICIs in TNBC have been continuously researched. However, as with patients with other carcinomas, only a few TNBC patients showed effective intrinsic resistance. In particular, when PD-L1 inhibitor was provided alone in PD-L1(+) TNBC patients, ORR of avelumab was 5.2% and of atezolizumab was about 10%, indicating a very low response [13]. This indicated that PD-L1 expression alone was an unpredictable marker to select patients that are applicable for PD-1/PD-L1 inhibitor treatment.
Recently, combined treatment of chemotherapy and PD-L1 antibody therapy has been widely used in TNBC patients with FDA approval [35]. In particular, the combination treatment of Nab-paclitaxel and atezolizumab shows a very high treatment rate with 56% ORR in PD-L1(+) TNBC patients [36]. However, many critical targets and mechanisms of inducing immunotherapy resistance remain unclear. Therefore, many studies on the mechanism of intrinsic resistance of immunotherapy agents are being actively conducted to uncover predictive markers and therapeutic targets, and instead of PD-L1 expression, multifarious potential biomarkers including tumor infiltrating lymphocytes (TILs), TMB, microsatellite instability (MSI), and mismatch repair (MMR) deficiency are now being assessed worldwide to predict exact immunotherapeutic efficacy in breast cancer.
Recently, the importance of TILs has attracted attention due to rise in the concept of “hot tumor” and “cold tumor” in relation to immunotherapy resistance. “Hot tumor” is a tumor that shows a very high response rate to ICIs, a high rate of T cell infiltration, and an interferon signature. In contrast, “cold tumor” refers to low immune infiltration and low response rate to ICIs [37]. In TNBC patients that are treated with combined atezolizumab and nab-paclitaxel, higher PD-L1 expression and CD8+ cytotoxic T cell infiltration indicate higher progression free survival (PFS) and overall survival (OS) [37]. Therefore, low infiltration of cytotoxic T cells and a low immunogenic tumor micro-environment are emerging as important causes of immunotherapy resistance. Indeed, several studies have reported them to cause resistance to ICIs by inhibiting priming and trafficking of T cells [38]. In particular, chemokine release by several oncogenic pathways is one of the major mechanisms that cause ICI resistance by inactivating cytotoxic T cells. Among the pathways, PI3K/AKT activation by PTEN loss is the most representative of ICI resistance. When PTEN loss occurs, the expression of immunosuppressive cytokines, such as VEGF, increases, in turn causing immunotherapy resistance by inhibition of T cell activity and trafficking of cytotoxic T cell tracking towards the tumor [17]. NF-κB activation increases the expression of immunosuppressive cytokines such as VEGF or TGF-beta [14,20]. A recent report revealed that CCL2 expression increased by activation of both mTOR, the downstream protein of the PI3K/AKT pathway, and NF-κB [21]. Unlike other chemokines, CCL2 is the chemokine that inhibits cytotoxic T cell activity by binding to the T cell receptor CCR2 [22]. In addition, PI3K/AKT and NF-κB activation are oncogenic pathways that increase PD-L1 expression in tumors [38].
Therefore, even if the binding of PD-L1 with PD-1 is effectively blocked by PD-1/PD-L1 inhibitors, increased CCL2 expression by PI3K/AKT and NF-κB activation may lead to PD-1/PD-L1 inhibitor-resistance by inhibition of cytotoxic T cell tracking towards the tumor.
CCL2 induces recruitment of macrophages in the tumor [39]. According to a recent research, macrophages reduce the effect of the PD-1/PD-L1 inhibitor by delaying infiltration of T cells, such as CD8+ cytotoxic T cells, in the tumor [40]. Therefore, the increase in macrophage recruitment by CCL2 is likely to induce resistance to immunotherapy by reducing infiltration of cytotoxic T cells into the tumor. Thus, CCL2 is considered an important target in cancer immune-treatment because it not only promotes cancer growth by angiogenesis in the tumor microenvironment, but also reduces infiltration of cytotoxic T cells, reducing the efficiency of immunotherapy treatment.
Indeed, we don’t know whether activated Jurkat T cell effect is antigen or MHC dependent. In this study, we focused IFNγ-induced killing effect of activated Jurkat T cell. In Figure 1D, we showed cytokines, including IL2 and IFN gamma (IFN-γ), release by activation of T cells. Especially, IFN-γ is well known to decrease tumor growth by acting not only directly on cancer cells via STAT1 activation, but also indirectly on endothelial cells and immune cells in the tumor microenvironments [41]. Therefore, we have conducted MTS assay (Supplementary Figure 2) as well as Caspase-3/7 assay (Figure 3) to evaluate IFNγ-induced killing effect of activated Jurkat T cells. Especially, conditioned-media (CM) of co-culture condition also directly killed cancer cells because CM includes IFNγ by activated Jurkat T cells (Supplementary Figure 2). Suspended cells in CM are removed by centrifuge.
TG2 is a calcium-dependent multifunctional enzyme, and is overexpressed in many cancers [26]. In fact, TG2 is an oncogene that contributes to drug resistance and metastasis in cancer patients [27]. TG2 causes ubiquitin-proteasome dependent degradation through cross-linking of several target molecules. In particular, TG2 degrades several tumor suppressors, such as PTEN and IκBα, through cross-linking [23-25]. Therefore, in this study, we investigated whether PI3K/AKT and NF-κB pathways were activated by PTEN and IκBα loss and TG2, and whether these two activated pathways induced CCL2 and PD-L1 expression. In particular, we investigated whether CCL2 induction by TG2 could be the main factor of PD-1/PD-L1 inhibitor-resistance in PD-L1(+) TNBC patients.
In this study, we proved that intrinsic PD-1/PD-L1 inhibitor-resistance was induced by TG2 in TNBC cells. As mentioned in the results, we presented that TG2 was highly expressed in PD-L1(+) TNBC cell lines and confirmed that PD-L1 expression increased due to both PI3K/AKT and NF-κB activation by TG2.
In clinical practice, the correlation between TG2 and PD-L1 expression was also confirmed through a higher incidence of PD-L1 expression in TG2(+) TNBC patients than in TG2(-) TNBC patients. In addition, PD-L1 and CCL2 expression levels were up-regulated by TG2, and PD-L1 inhibitor-resistance was only exhibited in TNBC cell lines with high TG2 and PD-L1 expression levels. If the initiating molecule that induces immune escape (TG2 in this study) is known, we can use an agent that targets the molecule (TG2 inhibitor) to restore T cell immunity in tumor cells. Among the 648 clinical samples tested in this study, we found 12 TNBC patients (1.8%) who co-expressed both TG2 and PD-L1.
Taken together, our results suggested that PD-L1 and CCL2 expression could be increased simultaneously by the TG2-dependent PTEN/NF-κB and PI3K/AKT signaling pathways. In this case, even if PD-L1 was blocked by PD-1/PD-L1 inhibitor, intrinsic resistance to PD-1/PD-L1 inhibitors could occur by weakening cytotoxicity of T cells via TG2-induced CCL2 activation (Figure 5). Above all, PD-1/PD-L1 inhibitor-resistant individuals among PD-L1(+) TNBC patients could be selected based on their TG2 expression level, which was found to be a novel companion diagnostic marker, and these patients could benefit by overcoming intrinsic PD-1/PD-L1 inhibitor-resistance via “TG2 inhibition alone” or “dual inhibition of PD-L1 and CCL2 by TG2-induced immune suppressors” (Figure 5). In addition, our findings suggest that TG2 can be used as a novel exact CDx marker to select intrinsic PD-1/PD-L1 inhibitor-resistant PD-L1(+) TNBC patients.
Figure 5.
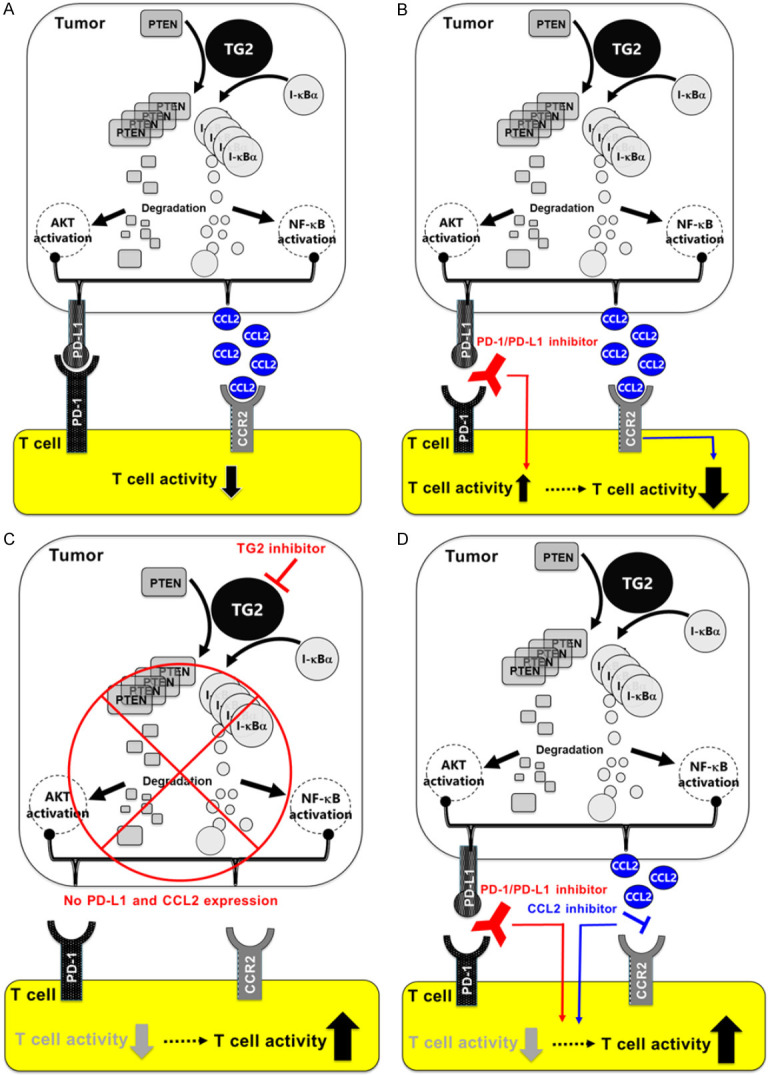
Diagrams illustrate the mechanisms of action of TG2 in intrinsic resistance to PD-1/PD-L1 inhibitors. (A) High TG2 expression induces CCL2 and PD-L1 in TNBC cells via PI3K/AKT and NF-κB activation pathways. (B) Therefore, TG2-induced PD-L1(+) TNBC patients cannot be treated with PD-1/PD-L1 inhibitors, and show intrinsic PD-1/PD-L1 inhibitor-resistance because TG2-induced CCL2 negatively regulates cytotoxic T cells despite complete blockade of binding of PD-L1 with PD-1 by PD-1/PD-L1 inhibitor. In conclusion, (C) TG2 inhibition or (D) dual inhibition of PD-L1 and CCL2 can be effective treatment options to overcome intrinsic PD-1/PD-L1 inhibitor-resistance in TG2(+)/PD-L1(+) TNBC patients. In addition, TG2 can be used as a novel CDx marker to select PD-1/PD-L1 inhibitor-resistant patients in a clinical set-up.
Acknowledgements
This manuscript was supported the Post-Genome Technology Development Program [10067758, Business model development driven by clinico-genomic database for precision immuno-oncology] funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea) and the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. NRF-2017 R1D1A1B03033550) and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science & ICT (NRF-2018M3A9E2024004) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2020R1A2C1012506).
Disclosure of conflict of interest
Dae Ho Lee declares honoraria from AstraZeneca, Boehringer-Ingelheim, Bristol-Myers Squibb, CJ Healthcare, ChongKunDang, Eli Lilly, Janssen, Merck, MSD, Mundipharma, Novartis, Ono, Pfizer, Roche, Samyang Biopharm, and ST Cube. The other authors declare that they have no conflict of interest.
Supporting Information
References
- 1.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob JJ, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 2.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 3.Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, Vaishampayan UN, Drabkin HA, George S, Logan TF, Margolin KA, Plimack ER, Lambert AM, Waxman IM, Hammers HJ. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J. Clin. Oncol. 2015;33:1430–1437. doi: 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meti N, Esfahani K, Johnson NA. The role of immune checkpoint inhibitors in classical hodgkin lymphoma. Cancers (Basel) 2018;10:204. doi: 10.3390/cancers10060204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plimack ER, Bellmunt J, Gupta S, Berger R, Chow LQ, Juco J, Lunceford J, Saraf S, Perini RF, O’Donnell PH. Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-012): a non-randomised, open-label, phase 1b study. Lancet Oncol. 2017;18:212–220. doi: 10.1016/S1470-2045(17)30007-4. [DOI] [PubMed] [Google Scholar]
- 6.Larkins E, Blumenthal GM, Yuan W, He K, Sridhara R, Subramaniam S, Zhao H, Liu C, Yu J, Goldberg KB, McKee AE, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of recurrent or metastatic head and neck squamous cell carcinoma with disease progression on or after platinum-containing chemotherapy. Oncologist. 2017;22:873–878. doi: 10.1634/theoncologist.2016-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, Ali SM, Elvin JA, Singal G, Ross JS, Fabrizio D, Szabo PM, Chang H, Sasson A, Srinivasan S, Kirov S, Szustakowski J, Vitazka P, Edwards R, Bufill JA, Sharma N, Ou SI, Peled N, Spigel DR, Rizvi H, Aguilar EJ, Carter BW, Erasmus J, Halpenny DF, Plodkowski AJ, Long NM, Nishino M, Denning WL, Galan-Cobo A, Hamdi H, Hirz T, Tong P, Wang J, Rodriguez-Canales J, Villalobos PA, Parra ER, Kalhor N, Sholl LM, Sauter JL, Jungbluth AA, Mino-Kenudson M, Azimi R, Elamin YY, Zhang J, Leonardi GC, Jiang F, Wong KK, Lee JJ, Papadimitrakopoulou VA, Wistuba II, Miller VA, Frampton GM, Wolchok JD, Shaw AT, Janne PA, Stephens PJ, Rudin CM, Geese WJ, Albacker LA, Heymach JV. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835. doi: 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 9.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, Leiby MA, Lubiniecki GM, Shentu Y, Rangwala R, Brahmer JR KEYNOTE-024 Investigators. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 10.Buttner R, Gosney JR, Skov BG, Adam J, Motoi N, Bloom KJ, Dietel M, Longshore JW, Lopez-Rios F, Penault-Llorca F, Viale G, Wotherspoon AC, Kerr KM, Tsao MS. Programmed death-ligand 1 immunohistochemistry testing: a review of analytical assays and clinical implementation in non-small-cell lung cancer. J. Clin. Oncol. 2017;35:3867–3876. doi: 10.1200/JCO.2017.74.7642. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Liu Q, Chen Z, Wang Y, Yang W, Hu Y, Han W, Zeng H, Ma H, Dai J, Zhang H. PD-L1 expression and tumor mutational burden status for prediction of response to chemotherapy and targeted therapy in non-small cell lung cancer. J Exp Clin Cancer Res. 2019;38:193. doi: 10.1186/s13046-019-1192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, Zaidi N, Azad NS, Laheru DA, Frampton GM, Jaffee EM. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight. 2019;4:e126908. doi: 10.1172/jci.insight.126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, Boccia R, Lippman ME, Somer R, Smakal M, Emens LA, Hrinczenko B, Edenfield W, Gurtler J, von Heydebreck A, Grote HJ, Chin K, Hamilton EP. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat. 2018;167:671–686. doi: 10.1007/s10549-017-4537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rameshwar P, Narayanan R, Qian J, Denny TN, Colon C, Gascon P. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: an inflammatory response beyond the realm of homeostasis. J Immunol. 2000;165:2271–2277. doi: 10.4049/jimmunol.165.4.2271. [DOI] [PubMed] [Google Scholar]
- 15.Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, Liu MC, Iwata H, Ryvo L, Wimberger P, Rugo HS, Tan AR, Jia L, Ding Y, Karantza V, Schmid P. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase II KEYNOTE-086 study. Ann Oncol. 2019;30:405–411. doi: 10.1093/annonc/mdy518. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Wang H, Yao H, Li C, Fang JY, Xu J. Regulation of PD-L1: emerging routes for targeting tumor immune evasion. Front Pharmacol. 2018;9:536. doi: 10.3389/fphar.2018.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen PL, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget MA, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL Jr, Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muthuswamy R, Berk E, Junecko BF, Zeh HJ, Zureikat AH, Normolle D, Luong TM, Reinhart TA, Bartlett DL, Kalinski P. NF-kappaB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012;72:3735–3743. doi: 10.1158/0008-5472.CAN-11-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, Jacks T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie TX, Xia Z, Zhang N, Gong W, Huang S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol Rep. 2010;23:725–732. [PubMed] [Google Scholar]
- 21.Nakatsumi H, Matsumoto M, Nakayama KI. Noncanonical pathway for regulation of CCL2 expression by an mTORC1-FOXK1 axis promotes recruitment of tumor-associated macrophages. Cell Rep. 2017;21:2471–2486. doi: 10.1016/j.celrep.2017.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J. 2019;17:1–13. doi: 10.1016/j.csbj.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DS, Park SS, Nam BH, Kim IH, Kim SY. Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-kappaB inactivation. Cancer Res. 2006;66:10936–10943. doi: 10.1158/0008-5472.CAN-06-1521. [DOI] [PubMed] [Google Scholar]
- 24.Jang GY, Jeon JH, Cho SY, Shin DM, Kim CW, Jeong EM, Bae HC, Kim TW, Lee SH, Choi Y, Lee DS, Park SC, Kim IG. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-kappaB activity in hypoxic tumor cells. Oncogene. 2010;29:356–367. doi: 10.1038/onc.2009.342. [DOI] [PubMed] [Google Scholar]
- 25.Verma A, Guha S, Wang H, Fok JY, Koul D, Abbruzzese J, Mehta K. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin Cancer Res. 2008;14:1997–2005. doi: 10.1158/1078-0432.CCR-07-1533. [DOI] [PubMed] [Google Scholar]
- 26.Park KS, Kim DS, Jeong KC, Kim SY. Increase in transglutaminase 2 expression is associated with NF-kappaB activation in breast cancer tissues. Front Biosci (Landmark Ed) 2009;14:1945–1951. doi: 10.2741/3354. [DOI] [PubMed] [Google Scholar]
- 27.Park KS, Kim HK, Lee JH, Choi YB, Park SY, Yang SH, Kim SY, Hong KM. Transglutaminase 2 as a cisplatin resistance marker in non-small cell lung cancer. J Cancer Res Clin Oncol. 2010;136:493–502. doi: 10.1007/s00432-009-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brignall R, Cauchy P, Bevington SL, Gorman B, Pisco AO, Bagnall J, Boddington C, Rowe W, England H, Rich K, Schmidt L, Dyer NP, Travis MA, Ott S, Jackson DA, Cockerill PN, Paszek P. Integration of kinase and calcium signaling at the level of chromatin underlies inducible gene activation in T cells. J Immunol. 2017;199:2652–2667. doi: 10.4049/jimmunol.1602033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, Bertucci F. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Li M, Lian Z, Zhu H, Kong L, Wang P, Yu J. Prognostic role of programmed death ligand-1 expression in breast cancer: a systematic review and meta-analysis. Target Oncol. 2016;11:753–761. doi: 10.1007/s11523-016-0451-8. [DOI] [PubMed] [Google Scholar]
- 31.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA IMpassion130 Trial Investigators. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 32.Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Henschel V, Molinero L, Chui SY, Maiya V, Husain A, Winer EP, Loi S, Emens LA IMpassion130 Investigators. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59. doi: 10.1016/S1470-2045(19)30689-8. [DOI] [PubMed] [Google Scholar]
- 33.Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764–782. doi: 10.1016/j.clinthera.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cortes J, Andre F, Goncalves A, Kummel S, Martin M, Schmid P, Schuetz F, Swain SM, Easton V, Pollex E, Deurloo R, Dent R. IMpassion132 phase III trial: atezolizumab and chemotherapy in early relapsing metastatic triple-negative breast cancer. Future Oncol. 2019;15:1951–1961. doi: 10.2217/fon-2019-0059. [DOI] [PubMed] [Google Scholar]
- 35.Cyprian FS, Akhtar S, Gatalica Z, Vranic S. Targeted immunotherapy with a checkpoint inhibitor in combination with chemotherapy: a new clinical paradigm in the treatment of triple-negative breast cancer. Bosn J Basic Med Sci. 2019;19:227–233. doi: 10.17305/bjbms.2019.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmid P, Chui SY, Emens LA. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. Reply. N Engl J Med. 2019;380:987–988. doi: 10.1056/NEJMc1900150. [DOI] [PubMed] [Google Scholar]
- 37.Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer. 2018;6:157. doi: 10.1186/s40425-018-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, Liu LN, Gills JJ, Dennis PA. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 39.Low-Marchelli JM, Ardi VC, Vizcarra EA, van Rooijen N, Quigley JP, Yang J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. 2013;73:662–671. doi: 10.1158/0008-5472.CAN-12-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, Bercovici N, Guerin M, Biton J, Ouakrim H, Regnier F, Lupo A, Alifano M, Damotte D, Donnadieu E. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. 2018;115:E4041–E4050. doi: 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Zhu J, Shan S, Qin Y, Kong Y, Liu J, Wang Y, Xie Y. Repression of interferon-gamma expression in T cells by Prospero-related homeobox protein. Cell Res. 2008;18:911–920. doi: 10.1038/cr.2008.275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.