

Abstract
ASK1 (Apoptosis Signal-regulating Kinase 1, also MEKK5) is known to mediate cellular stress signaling pathways through activating p38 kinase. We here observed that ectopically expression of ASK1, but not its kinase-dead mutant, impaired cell proliferation and migration in lung cancer A549 and NCI-H1975 cells. To our surprise, this inhibitory effect of ASK1 is independent on activation of p38 kinase. We further discovered that ASK1 interacts with the WW domain of YAP and TAZ (also WWTR1) that are transcriptional co-activators and the Hippo signaling effectors. Overexpression of wild type ASK1, but not the kinase-dead mutant, in the lung cancer cells down-regulated the expression of the YAP/TAZ target genes CYR61 and CTGF. It seems that ASK1 specifically inactivates TAZ, not YAP, as ASK1 blocked nuclear translocation of TAZ only, while had no effect on YAP. Furthermore, knockdown of TAZ in the lung cancer cells caused the same inhibitory effect on cell proliferation and migration as that of overexpression of ASK1. Thus, our studies have defined a new signaling pathway of ASK1 for regulation of lung cancer cell proliferation and migration via interacting with and inactivating TAZ.
Keywords: ASK1, TAZ, proliferation, migration, lung cancer
Introduction
ASK1 (also MEKK5) is a member of the MAP3K family and identified as an upstream kinase of the MKK3/6-p38 and the MKK4/7-JNK pathways [1,2]. Previous studies have shown that ASK1 is activated by a variety of cellular stress conditions, such as oxidative and endoplasmic reticulum stress, and plays important roles in intracellular stress and apoptotic signaling pathways [2-4]. It has been reported that activation of ASK1 induces cancer cell apoptosis in breast, liver, lung, colon, and osteosarcoma cells [5-9]. However, in some types of cancer, such as gastric, ovarian, and pancreatic cancers, ASK1 has been observed to promote cell proliferation and tumor growth [10-12]. The ASK1 inhibitor K811 has shown to suppress gastric tumor growth in a xenografted mouse model [13]. Accordingly, molecular mechanisms underlying these effects of ASK1 on tumor cell death or proliferation are diversified. For example, ASK1 induces apoptosis via activation of p38/JNK kinase pathways [1,2] and inhibits breast cancer cell migration through the GSK-3β/β-catenin signaling [14], while promotes gastric cancer cell proliferation through activation of AP1 and enhancement of expression of cyclin D1 [10]. These studies suggest that ASK1 can produce differential effects on cancer cell death, proliferation or migration via diversified signaling pathways, probably depending on cancer types and/or cellular signal context.
YAP and TAZ (also WWTR1), the two effectors of the Hippo pathway, are important transcriptional co-activators [15,16]. YAP and TAZ are known to play roles in organ development, homeostasis, and tumorigenesis [17,18]. In addition to the Hippo kinase cascade, YAP and TAZ are also regulated by the non-Hippo signals [19]. It has been shown that YAP and TAZ are overexpressed in multiple types of cancer and associated with tumor growth and progression [20-23]. Our previous studies have found that geranylgeranylation signaling activates the Hippo-YAP/TAZ pathway and promotes breast cancer cell proliferation and migration [24]. Furthermore, our immunohistochemical staining studies on the gastric cardia adenocarcinoma (GCA) tumors have revealed that TAZ and its target gene CRY61 are overexpressed in GCA and associated with metastasis and poor prognosis [25,26]. Knockdown of TAZ or CYR61 severely impaired migration and invasion of gastric cancer cells.
In this report, we demonstrated that overexpression of ASK1 inhibits lung cancer cell proliferation and migration through interaction with and inactivation of TAZ. Our studies have revealed a new ASK1 signaling pathway for regulation of cancer cell proliferation and migration.
Materials and methods
Materials
Antibodies: anti-ASK1 (F-9; SC-5294), anti-CYR61 (H78; SC-13100) and anti-YAP (G-6; SC-376830) were purchased from Santa Cruz Biotechnology; anti-p-p38 (9215S), anti-p38 (9212S), anti-LATS1 (9153S), anti-p-LATS1 (S909) (9157S) and anti-TAZ (4883S) from Cell Signaling Technology; anti-CTGF (D160212) from Sangon Biotech; anti-MYC (MMS-150R) from Covance; anti-tubulin (BS1699) from Bioworld Technology. SB203380 was purchased from Calbiochem. Epidermal growth factor (EGF) was purchased from Thermo Fisher Scientific. Tumor necrosis factor alpha (TNFα) was purchased from GenScript. All the cell lines were purchased from ATCC.
Cell culture and transfection
A549, NCI-H1975, MDA-MB-231, AGS, MCF7, DU145, and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2. For transfection, the HEK293T cells were seeded 1 day before transfection. The transfection procedures were the same as described previously [27,28].
Cell lysate preparation and immunoblotting
The cell lysate preparation was performed as described previously [28]. Briefly, the cells were rinsed with cold PBS after removal of culture medium and lysed using precooled mammalian cell lysis buffer or RIPA buffer by rocking plates at 4°C for 30 min. Cell lysates were cleared by centrifugation at 14,000 × g in a microcentrifuge for 15 min at 4°C before use.
The SDS-PAGE lysate samples were prepared by addition of 5 × SDS sample buffer directly to the lysates, followed by vortex and denatured at 100°C for 5 min. After electrophoresis on SDS-PAGE gels, the separated proteins on gels were transferred onto PVDF membranes (Millipore). The membranes were incubated with primary antibodies overnight at 4°C and with secondary antibodies for 1-2 h at room temperature. The target proteins were detected by the Western Lightning ECL Detection Kit (Beyotime).
GST-fusion protein pull-down assay
The GST-fusion protein pull-down assay was performed as described previously [28]. Briefly, the GST-fusion proteins were immobilized on glutathione-conjugated agarose beads. The GST-fusion protein beads (5-10 μg of protein) were incubated with the cell lysates for 3 h at 4°C with rotation, subsequently washed with mammalian cell lysis buffer, and resuspended in SDS-PAGE sample buffer for gel electrophoresis and immunoblotting.
Construction of plasmids and mutagenesis
Human ASK1 or its mutant cDNA was subcloned into the lentiviral expression vector pFUW for establishing stable cell lines in A549 and NCI-H1975 cells, and into the mammalian expression vector pcDNA3 for transient transfection in HEK293T cells. The WW domains of human YAP and TAZ were subcloned into GST fusion vector pGEX4T3 for expression of the GST-WW domains in bacteria.
Construction of TAZ shRNAs
The shRNA expression vector pLKO.1 was purchased from Addgene. A luciferase shRNA (shLUC) (5’-CGCTGAGTACTTCGAAATGTC-3’) was used as a control. Based on two targeting sequences in TAZ cDNA (5’-CTGTACGAGCTCATCGAGAAG-3’) and (5’-GATCCTGAAACTCCGCCACAT-3’), we synthesized two TAZ shRNA oligos: shTAZ-1 (Forward: 5’-CCGGCTGTACGAGCTCATCGAGAAGCT CGAGCTTCTCGATGAGCTCGTACAGTTTTTG-3’; Reverse: 5’-AATTCAAAAACTGTACGAGCTCATCGAGAAGCTCGAGCTTCTCGATGAGCTCGTACAG-3’) and shTAZ-2 (Forward: 5’-CCGGGATC CTGAAACTCCGCCACATCTCGAGATGTGGCGGAGTTTCAGGATCTTTTTG-3’; Reverse: 5’-AATT CAAAAAGATCCTGAAACTCCGCCACATCTCGAGATGTGGCGGAGTTTCAGGATC-3’). The shTAZ-1 and shTAZ-2 were sub-cloned into the vector pLKO.1 via AgeI/EcoRI sites.
Virus packaging and transduction
The viral packaging was performed as described previously [28]. Briefly, the lentiviral plasmids were co-transfected with psPAX2 (Addgene) and pMD2.G (Addgene) packaging plasmids into actively growing HEK293T cells using Lipofectamine 2000 transfection reagent. Culture medium containing viral particles was collected every 24 hours for three times. The medium was cleared by centrifugation at 5000 × g for 5 minutes and used for infecting target cells in the presence of 6 μg/ml polybrene (Sigma-Aldrich).
RNA extraction and qRT-PCR
RNA was extracted using the RNAiso plus (Takara Bio). To produce cDNA, 2 μg of total RNA was processed with the PrimeScript RT Reagent kit (Takara Bio) according to the manufacturer’s instructions. The cDNAs were quantified by quantitative real-time polymerase chain reaction (PCR). qRT-PCR was performed using the comparative cycle threshold (CT) method with SYBR-Green PCR Master Mix (Takara Bio) according to the manufacturer’s instructions on a Bio-Rad CFX96 system (Bio-Rad Laboratories). The primer sequences were used in the qRT-PCR: for CYR61, 5’-TCCCAGTGCTCAAAGACC-3’ (forward) and 5’-CTCAAACATCCAGCGTAAG-3’ (reverse); for CTGF, 5’-TCCCTGCATCTTCGGTGGTA-3’ (forward) and 5’-TTTGGTCCTTGGGCTCGTCA-3’ (reverse); for GAPDH, 5’-AACGGATTTGGTCGTATTG-3’ (forward) and 5’-GGAAGATGGTGATGGGGAT-3’ (reverse). The qRT-PCR was performed with initial denaturation at 95°C for 30 seconds, followed by 40 cycles of denaturation at 95°C for 5 seconds, annealing at 60°C for 30 seconds and elongation at 72°C for 30 seconds. The relative results were analyzed using the comparative cycle threshold method (2-ΔΔCT) with GAPDH as the reference gene [29].
Immunofluorescent staining
Cells were cultured in glass coverslip-bottomed culture dishes to 50-70% confluence. After the culture medium was aspirated, the cells were rinsed with PBS twice, fixed with 4% paraformaldehyde at 25°C for 30 min, and permeabilized with 0.5% Triton X-100 in PBS at 25°C for 20 min. After washing with PBS, the cells were incubated with the primary antibody at 4°C overnight. The cells were washed with PBS three times and incubated with the secondary antibody conjugated with a fluorescent dye at 37°C for 1-2 h. After washing with PBS three times, fluorescent staining of the cells was visualized under a Zeiss LSM710 confocal microscope.
Cell proliferation assay
Lung cancer cells (3 × 104) were seeded in a 12-well culture plate. After cultured at indicated time points, the cells were trypsinized and counted under a phase microscope with a hemocytometer. The cell proliferation is evaluated by the cell number increased since seeded. The proliferation assay was repeated at least three times.
Cell migration assays
Cell migration was determined by the wound healing assay and the transwell assay. (1) The wound-healing assay. 4 × 105 cells were seeded on 12 well culture plates in DMEM supplemented with 10% FBS. 24 hours later, the cells reached to about 80-90% confluence in a monolayer. A pipette tip was used to make a straight scratch line in the cell monolayer. The cells were incubated for indicated times and treated as required. The area covered by the migrated cells was quantified with Image J software (from NIH) and the percentage of the covered area by migrated cells is used as the migration rate. (2) Transwell migration assay. Transwell chambers (Corning) were used for migration assays. The cells (2 × 105 cells/ml) in 200 μl serum-free DMEM were seeded in each transwell insert. DMEM medium with a migration attractant (10% FBS or/and 50 ng/ml EGF or/and 10 μM SB203380) (0.5 ml) was added to the lower chamber. After incubation for an indicated time, cells on the upper side of the membrane between the upper and the lower chambers were carefully removed. The cells migrated to the bottom side of the membrane were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet solution. The stained cells were washed with PBS three times, visualized under a phase microscope and counted under a microscope from three randomly selected fields. The migration rate is defined as the ratio of the migrated cell number in the indicated sample to the migrated cell number in the control sample.
Statistical analysis
The Student t-test was used in statistical analysis of experimental data. A p-value less than 0.05 is considered as statistically significant.
Results
Overexpression of ASK1 in lung cancer cells impairs cell proliferation and migration
To investigate the role of ASK1 in proliferation and migration of cancer cells, we examined the expression level in a panel of cancer cell lines. As shown in Figure 1A, the expression level of ASK1 was very low in the two non-small cell lung cancer (NSCLC) cell lines A549 and NCI-H1975 that have high proliferation and migration capacity. To determine the effect of ASK1 on proliferation and migration, we first overexpressed ASK1 in these two cell lines using a lentiviral expression system and established ASK1 wild-type (labeled as ASK1-WT) and the kinase-dead mutant ASK1-K709A (labeled as ASK1-KD) cell lines. The expression level of ASK1 and its kinase-dead mutant in these two cell lines is shown in Figure 1B and 1C. The level of phosphorylated p38 was significantly higher upon overexpression of ASK1 than that in the control and the kinase-dead mutant cells in both of the cell lines (Figure 1B and 1C), indicating that the overexpressed ASK1 is functioning in these two cell lines. As the ectopic expression level of the kinase-dead mutant is significantly lower than that of wild type ASK1 in A549 cells (the left panel in Figure 1B), we examined if the kinase-dead mutant has the dominant-negative function and disrupts the signaling of endogenous ASK1. It is known that TNFα activates the ASK1/p38 signaling [1]. We treated cells with TNFα in the control, the ASK1-WT, and the ASK1-KD cell lines for 0, 0.5, and 3 hours, and evaluated the activating effect on p38 by detecting the p38 phosphorylation level. As shown in the middle panel of Figure 1B, the kinase-dead mutant at a low expression level effectively blocked the TNFα-induced activation of p38 (lanes 7, 8 and 9) comparing to the control cells (lanes 1, 2, and 3), while ASK1-WT caused constitutive activation of p38 (lanes 4, 5 and 6). These data indicate that the kinase-dead mutant of ASK1 has the dominant-negative function even at the low expression level, probably due to the extremely low expression of endogenous ASK1 in A549 cells.
Figure 1.
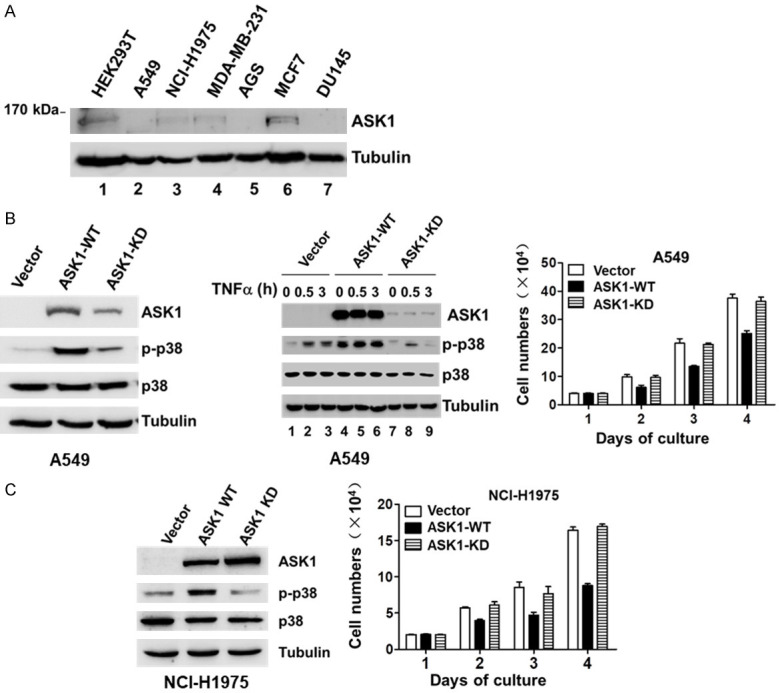
Overexpression of ASK1 inhibits lung cancer cell proliferation. A. The expression level of endogenous ASK1 in various cancer cell lines. B and C. The effect of overexpression of ASK1 on cell proliferation in the non-small cell lung cancer (NSCLC) cell lines A549 and NCI-H1975. The ASK1 (ASK1-WT) or its kinase-dead mutant ASK1-K709A (ASK1-KD) was stably expressed in the lung cancer cells using a lentiviral mammalian expression system. The expressed protein level of ASK1 or its kinase-dead mutant was detected by immunoblotting of the cell lysates with an anti-ASK1. The p38 and phosphor-p38 were also detected by immunoblotting. Treatment with TNFα (50 ng/ml) was employed for activation of ASK1 in A549 cells. The lung cancer cell proliferation was quantified by counting the cell number under a phase microscope with a hemocytometer. The data used for quantification were from three independent experiments.
Next, we examined the effect of expression of ASK1 and its kinase-dead mutant on cell proliferation. As shown in the right panels in Figure 1B and 1C, overexpression of ASK1 in both lung cancer cell lines significantly reduced the proliferation rate of the cells compared to that of the vector control cells, while overexpression of the kinase-dead mutant had no significant effect on the proliferation rate. This indicates that the kinase active ASK1 has an inhibitory effect on proliferation of the lung cancer cells. Thus, ASK1 is a negative regulator controlling lung cancer cell proliferation.
We further examined the effect of ASK1 on cell migration using the wound-healing and the transwell assays. As shown in Figure 2A and 2B, overexpression of ASK1 significantly inhibited cell migration determined by the wound-healing assay in both A549 and NCI-H1975 lung cancer cell lines, and also inhibited the EGF-stimulated cell migration in A549, while the kinase-dead mutant had no inhibitory effect. The same inhibitory effect of ASK1 on the cell migration was observed when determined by the transwell assay (Figure 2C and 2D). These results indicate that ASK1 is also a negative regulator controlling lung cancer cell migration. Taken together, we conclude that the kinase active ASK1, not the kinase-dead mutant, mediates the inhibitory signaling in both lung cancer cell proliferation and migration.
Figure 2.
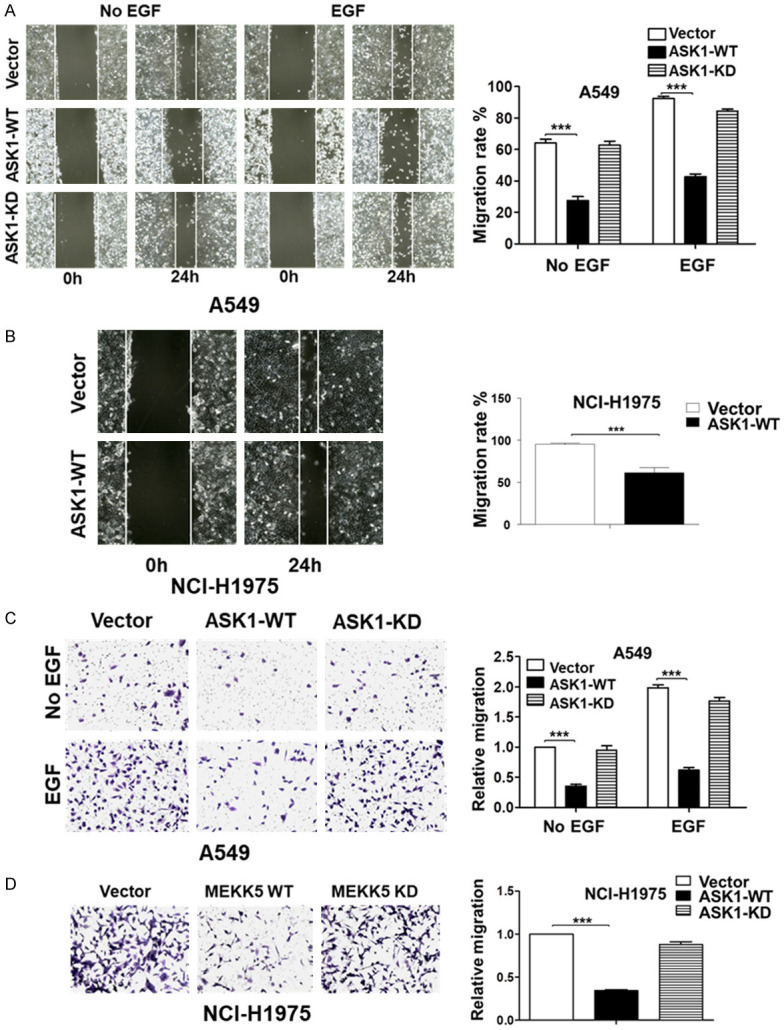
Overexpression of ASK1 inhibits lung cancer cell migration. The effect of overexpression of ASK1 (ASK1-WT) or the kinase-dead mutant (ASK1-KD) on cell migration determined by the wound-healing assay (A and B) and by the transwell assay (C and D). EGF (50 ng/ml) was used for stimulation of cell migration. The cell line is labeled in the Figure. For quantification in the wound-healing assay, the area covered by the migrated cells was quantified with Image J software (from NIH). The percentage of the covered area by migrated cells is used as the migration rate. The data used for quantification are from three independent experiments. For quantification in the transwell assay, the migrated cells were fixed, stained with crystal violet, and counted under a microscope from three randomly selected fields. **P < 0.01; ***P < 0.001.
ASK1 inhibits cell proliferation and migration independent of the p38 signaling pathway
p38 is a known downstream effector of ASK1 [1]. We examined whether the p38 mediates the effect of ASK1 on inhibition of lung cancer cell proliferation and migration. We observed that overexpression of ASK1 in both A549 and NCI-H1975 cell lines caused an increase in phosphorylation of p38 (Figure 1B), indicating that overexpression of ASK1 activates p38 in cells. We then treated A549 cells with a p38 kinase inhibitor SB203380 to determine the effect of p38 kinase activity on cell proliferation and migration. As shown in Figure 3A, inhibition of p38 kinase by SB203380 caused a minor reduction of proliferation rate in all the cell lines, including the vector, the ASK1, and the kinase-dead mutant cell lines in A549. However, treatment with SB203380 did not cause a change in the inhibitory effect of ASK1 on cell proliferation, suggesting that p38 does not mediate the inhibitory effect of ASK1. We further determined the effect of the p38 kinase inhibitor on ASK1-caused inhibition of cell migration. As shown in Figure 3B and 3C, treatment with SB203380 severely impaired cell migration in all the cell lines determined by both the wound-healing and transwell assays, indicating that p38 kinase activity promotes A549 cell migration. Furthermore, treatment with SB203380 did not cause a change in the inhibitory effect of ASK1 on cell migration. Taken together, we conclude that p38 kinase is not the downstream effector mediating the inhibitory effect of ASK1 on the lung cancer cell proliferation and migration.
Figure 3.
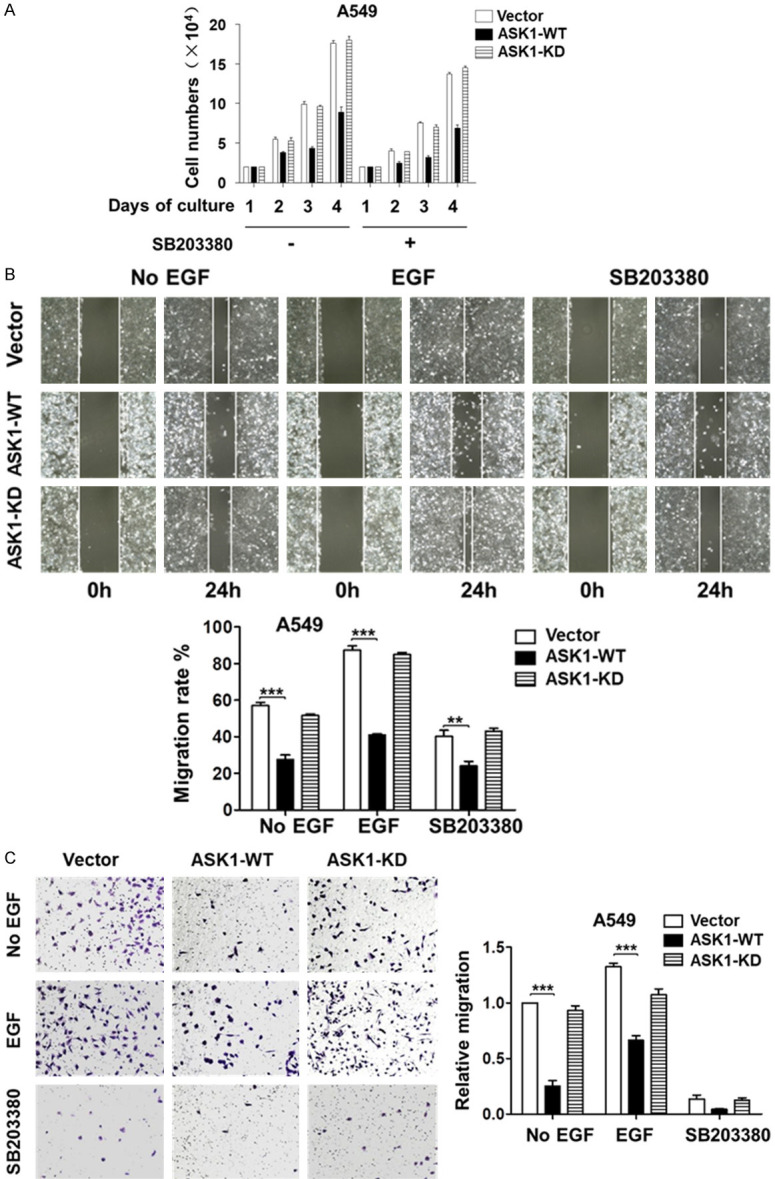
ASK1 inhibits cell proliferation and migration independent of the p38 pathway. A. The p38 kinase inhibitor SB203380 has a minor inhibitory effect on proliferation of lung cancer A549 cells and does not affect the inhibitory effect of ASK1 on cell proliferation. B and C. The p38 kinase inhibitor SB203380 inhibits migration of lung cancer A549 cells, but does not change the inhibitory effect of ASK1 on cell migration determined by both the wound-healing and the transwell assays. EGF (50 ng/ml) was used for stimulation of cell migration. SB203380 (10 μM) was used for inhibition of the p38 kinase. The data quantification was the same as described in Figure 2. **P < 0.01; ***P < 0.001.
ASK1 interacts with YAP and TAZ
Our previous studies have found that YAP/TAZ signaling plays an important role in breast cancer cell proliferation and migration [24]. It has been reported that ASK1 binds to the WW domain of NEDD4 [30]. Coincidently, both YAP and TAZ contain the WW domain similar to that of NEDD4. Thus, ASK1 may interact with and inhibit the YAP/TAZ, leading to inhibition of the lung cancer cell proliferation and migration. To test the hypothesis, we employed the GST-YAP-WW domain or GST-TAZ-WW domain pulldown assay to determine if ASK1 binds to YAP and TAZ through their WW domain. The YAP-WW domains and the TAZ-WW domain sketch are shown in Figure 4A. The GST-WW domain pulldown assay was performed by incubating the glutathione bead-bound GST-WW domain with Myc-tagged ASK1-overexpressed HEK293T cell lysates. As shown in Figure 4B, the bead-bound GST-YAP-WW2, GST-YAP-WWS, and GST-TAZ-WW precipitated Myc-ASK1 efficiently from the cell lysates, indicating that YAP-WW2 and TAZ-WW is the ASK1 binding domain. ASK1 contains a PPFY sequence, a putative PPXY motif known to interact with the WW domain [30], which is located in a proline-rich region from amino acid residue 879 to 882. To determine whether the PPFY is the site interacting with the WW domain of YAP/TAZ, we made the ASK1 mutant ASK1-Y882A, in which PPFY was mutated to PPFA, and performed an in vitro GST-WW domain pulldown assay. To our surprise, GST-YAP-WW2 or GST-TAZ-WW was able to precipitate the mutant ASK1-Y882A from the lysates to the same extent as wild type ASK1 (Figure 4C and 4D), demonstrating that the PPFY is not the site for ASK1 to bind to the WW domain of YAP or TAZ.
Figure 4.
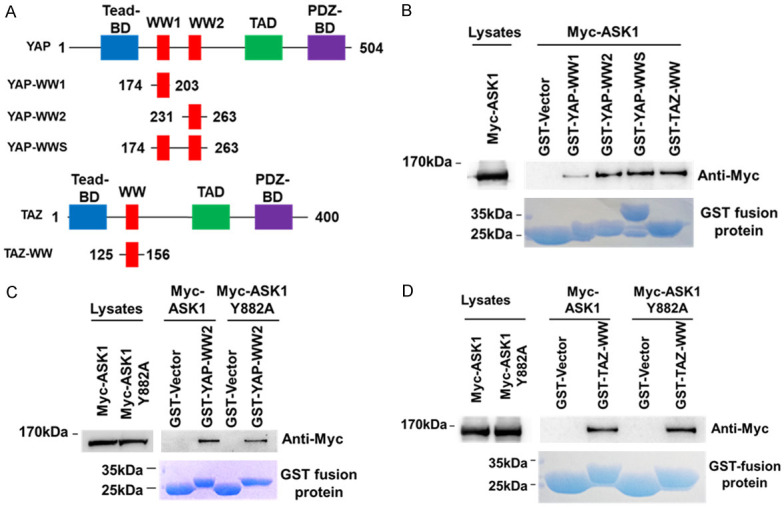
ASK1 binds to the WW domain of YAP and TAZ. (A) A structural sketch of the YAP and TAZ and their WW domains. Tead-BD, Tead binding domains; WW, WW domain; WW1, the first WW domain; WW2, the second WW domain; TAD, Tead activating domain; PDZ-BD, PDZ binding domain. The numbers labeled in YAP and TAZ constructs indicate the amino acid residue positions. (B) ASK1 binds to the WW domain of YAP and TAZ. The GST fused WW domain was expressed in bacteria, and purified with the glutathione-conjugated agarose beads. The bead-bound GST-WW domain was incubated with the ASK1-expressed HEK293T cell lysates. The co-precipitated ASK1 was detected by immunoblotting with an anti-ASK1 antibody. (C and D) The amino acid residues 799-PPFY-882 in ASK1 is not the YAP- or TAZ-binding site. The GST-WW domain pulldown assay was performed the same as in (B) except the bead-bound GST-WW domain was also incubated with the HEK293T cell lysates containing the ASK1 mutant ASK1-Y882A in which the 799-PPFY-882 is mutated into 799-PPFA-882. (C) The GST-YAP-WW domain pulldown assay; (D) The GST-TAZ-WW domain pulldown assay.
ASK1 inhibits transcription of the YAP/TAZ target genes
We further determined the effect of ASK1 on the transcriptional co-activator activity of YAP/TAZ. The transcriptional elevation of CYR61 and CTGF, two of the known YAP/TAZ target genes, has been used for indication of the activation of YAP/TAZ [15,16]. As shown in Figure 5A and 5B, overexpression of ASK1 in both A549 and NCI-H1975 cell lines significantly reduced the mRNA level of CYR61 and CTGF, while overexpression of the kinase-dead mutant ASK1-K709A either insignificantly changed or dramatically enhanced the mRNA level of CYR61 and CTGF. These results suggest that ASK1 inhibits the transcriptional co-activator activity of YAP/TAZ, and the kinase activity is required for the inhibition. We further confirmed the results by detection of the protein level of CYR61 in A549 cells as shown in Figure 5C. Overexpression of ASK1 significantly reduced the protein level of CYR61 (lanes 2, 5, and 8, Figure 5C), while overexpression of the kinase-dead mutant had a minor or no effect on the level of CYR61 (lanes 3, 6, and 9, Figure 5C). To confirm that the effect of ASK1 on the YAP/TAZ signaling is through direct interaction with YAP/TAZ, not the Hippo pathway, we detected the effect of overexpression of ASK1 or its kinase-dead mutant on activation of the Hippo signaling kinase LATS1 in A549 cells. As shown in Figure 5D, overexpression of ASK1 or its kinase-dead mutant did not cause a significant change in phosphorylation of LATS1, suggesting that ASK1 inhibits the YAP/TAZ transcriptional activity through direct interaction with YAP/TAZ, not by activation of the Hippo signaling.
Figure 5.
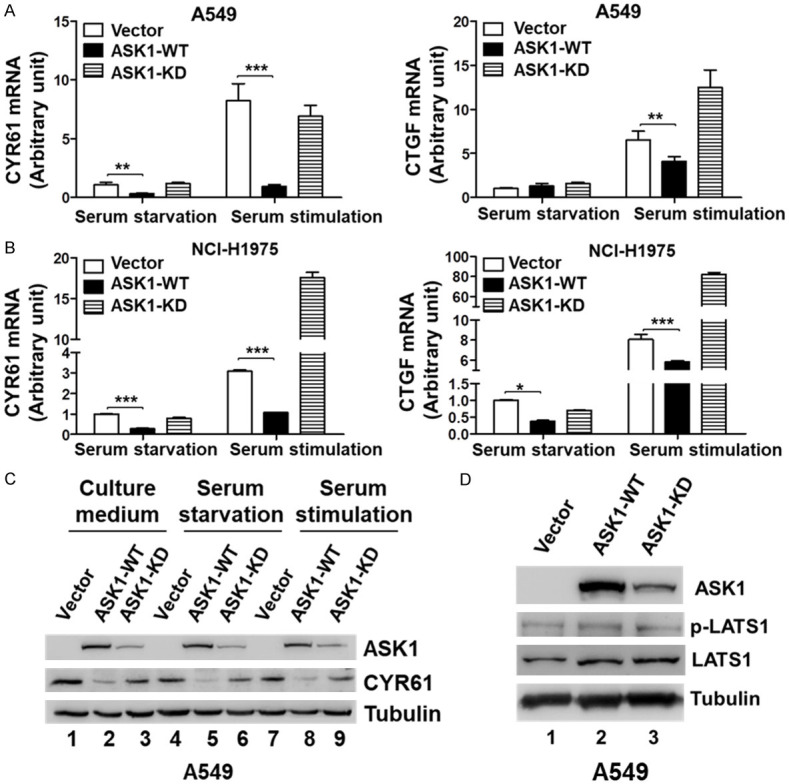
Overexpression of ASK1 inhibits the expression of the YAP/TAZ target genes CYR61 and CTGF. (A and B) The mRNA level of the YAP/TAZ target genes CYR61 and CTGF was dramatically reduced upon overexpression of ASK1, but not the kinase-dead mutant. The cells were serum-starved for 24 hrs followed by stimulation with 10% FBS for 30 min. Total RNA was purified and the mRNA level of CYR61 and CTGF was determined by the quantitative RT-PCR (qRT-PCR) assay. (A) In A549 cells; (B) In NCI-H1975 cells. (C) The protein level of CYR61 was reduced upon overexpression of ASK1, but not the kinase-dead mutant, in A549 cells. The serum starvation and stimulation were performed the same as in (A and B). The cells were lysed and CYR61 was detected by immunoblotting with an anti-CYR61 antibody. (D) Overexpression of ASK1 or the kinase-dead mutant in A549 cells had no significant effect on phosphorylation of the Hippo signaling kinase LATS1.
ASK1 specifically inactivates TAZ
Nuclear localization of YAP/TAZ is an indication of their activation, and the Hippo kinase cascade signaling is known to block YAP/TAZ nuclear localization to inhibit YAP/TAZ transcriptional co-activator activity [15,16]. We hypothesize that ASK1 inhibits the YAP/TAZ through a similar way to the Hippo kinase cascade by blocking the nuclear localization of YAP/TAZ. Thus, we examine the effect of ASK1 on nuclear localization of YAP/TAZ using immunofluorescent staining assay. As shown in Figure 6A, overexpression of ASK1 in A549 cells significantly enhanced amount of cytoplasmic TAZ and reduced amount of nuclear TAZ (panels k and k’), while overexpression of the kinase-dead mutant had an insignificant effect on the nuclear localization of TAZ (panels m and m’), suggesting that ASK1 is blocking the nuclear translocation of TAZ, and the kinase activity of ASK1 is required for the blockage.
Figure 6.
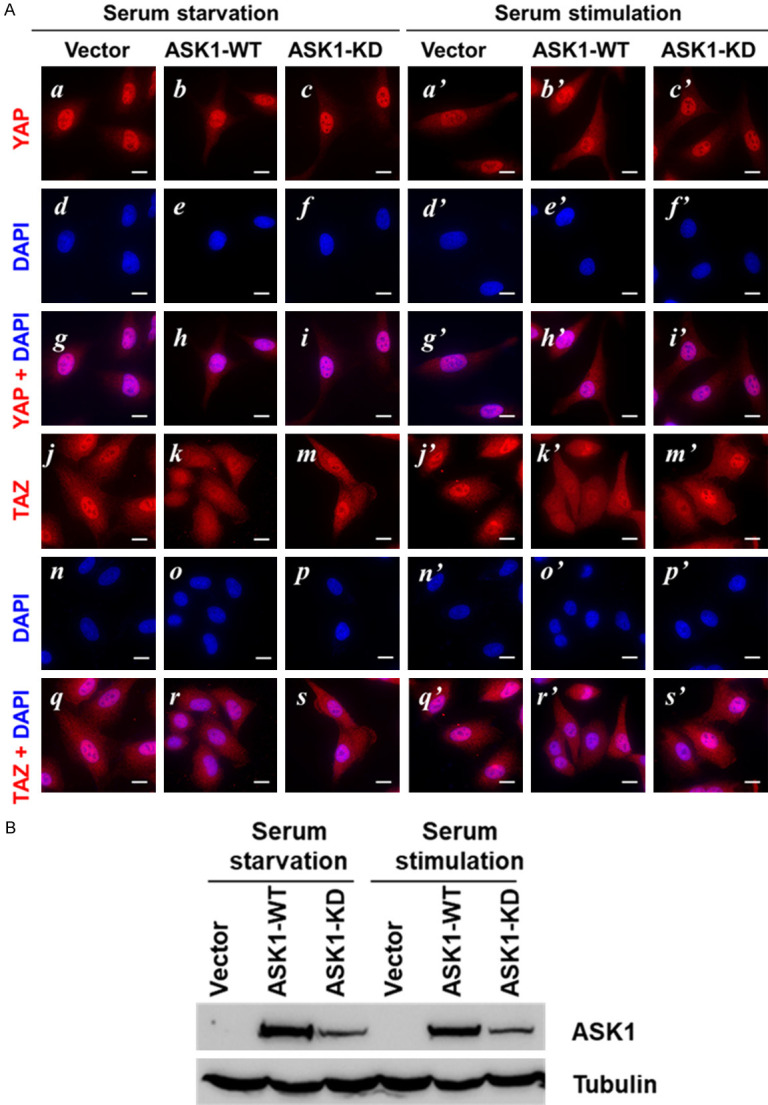
Overexpression of ASK1 inhibits nuclear localization of TAZ, but not YAP. Lung cancer A549 cells stably expressed ASK1 or its kinase-dead mutant were cultured on glass cover-slips, serum-starved 24 hrs followed by stimulation with 10% serum for 30 min. A. The cells were fixed and immuno-stained with the anti-YAP or the anti-TAZ antibody, and the nuclei were stained with fluorescent dye DAPI. B. The expression of ASK1 and its kinase-dead mutant was detected by immunoblotting of the cell lysates with an anti-ASK1 antibody. Bar, 20 mm.
To our surprise, overexpression of either wild type ASK1 or the kinase-dead mutant in A549 cells had no significant effect on nuclear localization of YAP (panels a-c and a’-c’ in Figure 6A). It seems that the majority of YAP is constitutively localized in nuclear and has no response to ASK1. The expression level of ASK1 or the kinase-dead mutant was confirmed by immunoblotting (Figure 6B). These results indicate that ASK1 is specifically inactivation of TAZ in these lung cancer cells.
Knockdown of TAZ produces a similar inhibitory effect on cell proliferation and migration to overexpression of ASK1
To verify that inactivation of TAZ is the cause for the inhibitory effect of ASK1 on lung cancer cell proliferation and migration, we examined the effect of TAZ knockdown on the lung cancer cell proliferation and migration. We established two shTAZ cell lines in A549. More than 80% of TAZ was depleted in both shTAZ cell lines (Figure 7A). Depletion of TAZ down-regulated expression of its target gene product CYR61 and CTGF (Figure 7A), confirming that knockdown of TAZ disables its transcriptional function. Consistent with the effect of overexpression of ASK1, knockdown of TAZ significantly inhibited the proliferation of A549 cells (Figure 7B). Next, we determined the effect of TAZ knockdown on cell migration in the shTAZ cell lines by the wound-healing and the transwell assays. As shown in Figure 7B and 7C, knockdown of TAZ, similar to overexpression of ASK1, inhibited both the basal and the EGF-promoted cell migration of the lung cancer cells, indicating that TAZ plays a pivotal driving role in lung cancer cell migration.
Figure 7.
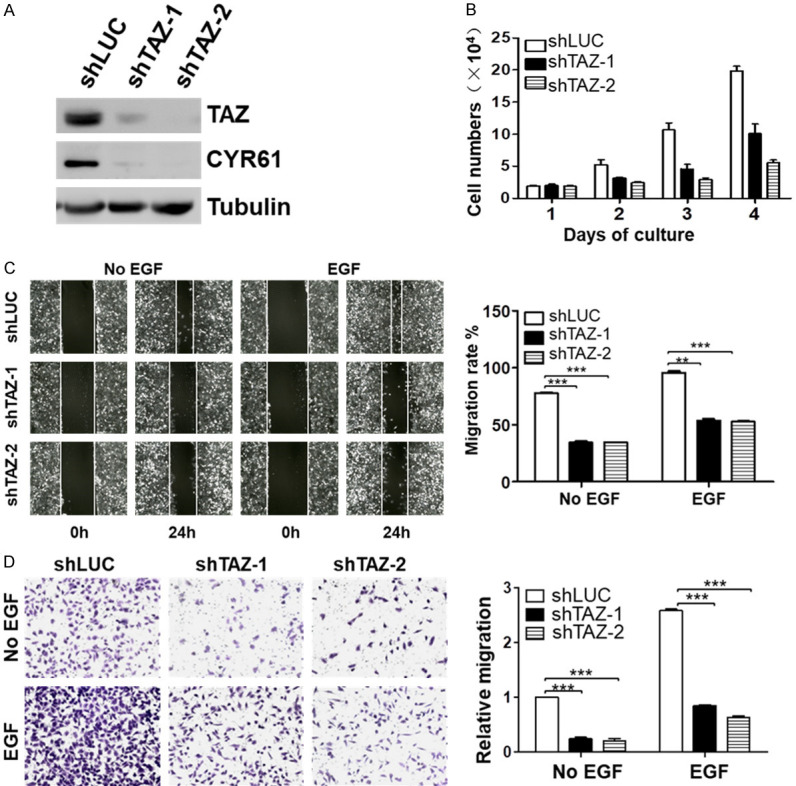
Depletion of TAZ in lung cancer inhibits cell proliferation and migration. A. Knockdown of TAZ by shTAZ-1 and shTAZ-2 in the NSCLC cell line A549. The effect of shTAZs on the protein level of TAZ and the target gene product CYR61 was determined by immunoblotting of the cell lysates with anti-TAZ and anti-CYR61 antibodies. B. The knockdown of TAZ by shTAZs inhibits A549 cell proliferation. The data used for quantitation were from three independent experiments. C and D. Knockdown of TAZ by shTAZs inhibits A549 cell migration. C. The wound-healing cell migration assay; D. The transwell cell migration assay. EGF (50 ng/ml) was used for stimulation of cell migration. The data quantification was the same as described in Figure 2. **P < 0.01; ***P < 0.001.
Taken together, these results suggest that the inhibitory effect of ASK1 on the lung cancer cell proliferation and migration is through suppressing the transcriptional co-activator activity of TAZ.
Discussion
ASK1 (also ASK1) has been demonstrated to be activated by multiple cellular stress signals and transduce these stress signals to the MAP kinase family members, such as JNK and p38, for transcriptional activation [2-4]. Here we have shown a novel ASK1 signaling pathway in lung cancer cells, in which ASK1 functions as an inhibitor of cell proliferation and migration through inactivation of the Hippo signaling effector TAZ. ASK1 physically interacts with the WW domain of TAZ and effectively impairs the transcriptional co-activator activity by blocking the nuclear localization of TAZ. Depletion of TAZ by shRNA produced a similar effect on cell proliferation and migration to that of ASK1, suggesting that inactivation of TAZ is likely to be the cause for the inhibitory effect of ASK1 on lung cancer cell proliferation and migration. This novel ASK1 signaling pathway may provide a new strategy for lung cancer therapy.
We have shown that inhibition of p38 with its kinase inhibitor SB223080 caused a partial inhibition of proliferation and severe impairment of the lung cancer cell migration (Figure 3), suggesting that p38 kinase promotes the lung cancer cell migration. Interestingly, we observed that overexpression of ASK1 activates p38 (Figure 1B and 1C). However, overexpression of ASK1 did not produce the effect of p38 activation on cell migration, rather, it caused inhibition of the lung cancer cell migration via inactivation of TAZ. The results suggest that the ASK1/TAZ signaling pathway is dominant in controlling cell proliferation and migration and overrides the ASK1/p38 kinase signaling pathway in the lung cancer cells.
We demonstrated that ASK1 is physically interacting with YAP/TAZ through the WW domain (Figure 4). There is a PPFY sequence in ASK1 that matches the PPXY motif, a known motif interacting with WW domains. However, this PPFY sequence is not the site for ASK1 to bind to the WW domain of YAP/TAZ (Figure 5). Currently, we do not know the region in ASK1 that binds to the WW domain of YAP/TAZ. As the WW domain of YAP/TAZ directly binds to ASK1 and the kinase activity of ASK1 is essential for the effect on the lung cancer cell proliferation and migration, we propose that ASK1 directly interacts with and phosphorylates TAZ in a similar way to that of LATS1/2, thus blocks translocation of TAZ to nuclei, inactivates the transcriptional co-activator activity of TAZ, and inhibits the cell proliferation and migration. We will investigate whether ASK1 phosphorylates TAZ and how ASK1 blocks nuclear translocation of TAZ in future studies.
To our surprise, ASK1 only inactivates TAZ although it binds to both YAP and TAZ (Figure 6). The molecular base for this preference of ASK1 toward to TAZ currently is not clear. It seems that YAP is constitutively localized in nuclei and its nuclear localization has no response to ASK1 (Figure 6). A possible cause is that YAP is not the phosphorylation substrate of ASK1 or phosphorylation of YAP by ASK1 does not block its nuclear translocation. Furthermore, despite YAP is constitutively localized in nuclei, it did not compensate for the inhibitory effect on the target gene expression and cell proliferation and migration caused by ASK1 overexpression or TAZ knockdown. This raises the possibility that the transcriptional co-activator activity of YAP may need to partner with the active TAZ in these lung cancer cells. Our previous studies also observed that knockdown of TAZ alone impaired cell migration in gastric cancer AGS cells [25], suggesting that lack of TAZ in cells could cause defects in the YAP/TAZ signaling. A recent study found that expression of a group of genes was inhibited by knockout of either YAP or TAZ [31], indicating that partnership between YAP and TAZ is required for partial, if not all, of their transcriptional co-activator activity.
Screening cancer cell lines by immunoblotting with anti-ASK1, we found that most of the cancer cell lines express a very low level of ASK1 (Figure 1A). This suggests that ASK1 may function as a tumor suppressor in cancers. ASK1 has been found to mediate apoptotic signaling in multiple cancer cell lines [2-4]. Many studies observed that chemo drugs or cytotoxic chemicals induces activation of ASK1 and its downstream signaling kinases, such as p38 and JNK, and mediates apoptosis in lung cancer cells [32-34]. This report is the first to demonstrate that ASK1 inhibits lung cancer cell proliferation and migration via binding to and inactivation of TAZ. Currently, only a few studies reported that the YAP/TAZ signaling in lung cancer cells. Our studies have shown that TAZ plays a pivotal role in lung cancer cell proliferation and migration, suggesting that the YAP/TAZ signaling might be dominant in controlling tumorigenesis and progression in lung cancer. It has been reported that TAZ renders resistance of the EGFR-T790M lung cancer cells to gefitinib [35], suggesting that TAZ is a key mediator for cell proliferation signaling initiated by EGFR-T790M. Therefore, the molecular mechanism underlying the TAZ-mediated signaling in lung cancer is particularly interesting to investigate in future studies. Utilizing a drug to induce expression of ASK1 or directly inhibiting the TAZ signaling could be a promising strategy for lung cancer targeted therapy.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81372208 and No. 81871888 to Q. L. and No. 81472558 to W.Y.), the Innovation and Entrepreneurship Team Program of Jiangsu Province (to H. L. and Q. L.), and Phase III Programs of the University Advanced Subject Development Project of Jiangsu Province (to Q.L.).
Disclosure of conflict of interest
None.
References
- 1.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 2.Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136:261–5. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- 3.Shiizaki S, Naguro I, Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv Biol Regul. 2013;53:135–44. doi: 10.1016/j.jbior.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Matsuzawa A, Ichijo H. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. J Biochem. 2001;130:1–8. doi: 10.1093/oxfordjournals.jbchem.a002947. [DOI] [PubMed] [Google Scholar]
- 5.Dyari HRE, Rawling T, Chen Y, Sudarmana W, Bourget K, Dwyer JM, Allison SE, Murray M. A novel synthetic analogue of ω-3 17,18-epoxyeicosatetraenoic acid activates TNF receptor-1/ASK1/JNK signaling to promote apoptosis in human breast cancer cells. FASEB J. 2017;31:5246–5257. doi: 10.1096/fj.201700033R. [DOI] [PubMed] [Google Scholar]
- 6.Jiang CF, Wen LZ, Yin C, Xu WP, Shi B, Zhang X, Xie WF. Apoptosis signal-regulating kinase 1 mediates the inhibitory effect of hepatocyte nuclear factor-4α on hepatocellular carcinoma. Oncotarget. 2016;7:27408–21. doi: 10.18632/oncotarget.8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuo CT, Chen BC, Yu CC, Weng CM, Hsu MJ, Chen CC, Chen MC, Teng CM, Pan SL, Bien MY, Shih CH, Lin CH. Apoptosis signal-regulating kinase 1 mediates denbinobin-induced apoptosis in human lung adenocarcinoma cells. J Biomed Sci. 2009;16:43. doi: 10.1186/1423-0127-16-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qu L, Liu FX, Cao XC, Xiao Q, Yang X, Ren KQ. Activation of the apoptosis signal-regulating kinase 1/c-Jun N-terminal kinase pathway is involved in the casticin-induced apoptosis of colon cancer cells. Exp Ther Med. 2014;8:1494–1500. doi: 10.3892/etm.2014.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou CH, Fong YC, Chen JT, Liu JF, Lin MS, Chang CS, Tang CH. The novel isoflavone 7-hydroxy-3’,4’-benzoisoflavone induces cell apoptosis in human osteosarcoma cells. Cancer Lett. 2008;271:117–28. doi: 10.1016/j.canlet.2008.05.037. [DOI] [PubMed] [Google Scholar]
- 10.Hayakawa Y, Hirata Y, Nakagawa H, Sakamoto K, Hikiba Y, Kinoshita H, Nakata W, Takahashi R, Tateishi K, Tada M, Akanuma M, Yoshida H, Takeda K, Ichijo H, Omata M, Maeda S, Koike K. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc Natl Acad Sci U S A. 2011;108:780–5. doi: 10.1073/pnas.1011418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin M, Zhou HJ, Zhang J, Lin C, Li H, Li X, Li Y, Zhang H, Breckenridge DG, Ji W, Min W. ASK1-dependent endothelial cell activation is critical in ovarian cancer growth and metastasis. JCI Insight. 2017;2:e91828. doi: 10.1172/jci.insight.91828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo Y, Gao S, Hao Z, Yang Y, Xie S, Li D, Liu M, Zhou J. Apoptosis signal-regulating kinase 1 exhibits oncogenic activity in pancreatic cancer. Oncotarget. 2016;7:75155–75164. doi: 10.18632/oncotarget.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayakawa Y, Hirata Y, Sakitani K, Nakagawa H, Nakata W, Kinoshita H, Takahashi R, Takeda K, Ichijo H, Maeda S, Koike K. Apoptosis signal-regulating kinase-1 inhibitor as a potent therapeutic drug for the treatment of gastric cancer. Cancer Sci. 2012;103:2181–5. doi: 10.1111/cas.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noh KT, Cho SG, Choi EJ. Knockdown of apoptosis signal-regulating kinase 1 modulates basal glycogen synthase kinase-3β kinase activity and regulates cell migration. FEBS Lett. 2010;584:4097–101. doi: 10.1016/j.febslet.2010.08.029. [DOI] [PubMed] [Google Scholar]
- 15.Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20:638–46. doi: 10.1016/j.ceb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong W, Guan KL. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23:785–93. doi: 10.1016/j.semcdb.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94:1287–312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 19.Dobrokhotov O, Samsonov M, Sokabe M, Hirata H. Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin Transl Med. 2018;7:23. doi: 10.1186/s40169-018-0202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803. doi: 10.1016/j.ccell.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, Jeong KJ, Tsang YH, Moreno D, Bhavana VH, Peng X, Ge Z, Chen H, Li J, Chen Z, Zhang H, Han L, Du D, Creighton CJ, Mills GB Cancer Genome Atlas Research Network. Camargo F, Liang H. Comprehensive molecular characterization of the hippo signaling pathway in cancer. Cell Rep. 2018;25:1304–1317. doi: 10.1016/j.celrep.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Shi S, Guo Z, Zhang X, Han S, Yang A, Wen W, Zhu Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS One. 2013;8:e65539. doi: 10.1371/journal.pone.0065539. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Melucci E, Casini B, Ronchetti L, Pizzuti L, Sperati F, Pallocca M, De Nicola F, Goeman F, Gallo E, Amoreo CA, Sergi D, Terrenato I, Vici P, Di Lauro L, Diodoro MG, Pescarmona E, Barba M, Mazzotta M, Mottolese M, Fanciulli M, Ciliberto G, De Maria R, Buglioni S, Maugeri-Saccà M. Expression of the Hippo transducer TAZ in association with WNT pathway mutations impacts survival outcomes in advanced gastric cancer patients treated with first-line chemotherapy. J Transl Med. 2018;16:22. doi: 10.1186/s12967-018-1385-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi W, Lin Q, Childress C, Sudol M, Robishaw J, Berlot CH, Shabahang M, Yang W. Geranylgeranylation signals to the Hippo pathway for breast cancer cell proliferation and migration. Oncogene. 2015;34:3095–106. doi: 10.1038/onc.2014.251. [DOI] [PubMed] [Google Scholar]
- 25.Wei J, Wang L, Zhu J, Sun A, Yu G, Chen M, Huang P, Liu H, Shao G, Yang W, Lin Q. The Hippo signaling effector WWTR1 is a metastatic biomarker of gastric cardia adenocarcinoma. Cancer Cell Int. 2019;19:74. doi: 10.1186/s12935-019-0796-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei J, Yu G, Shao G, Sun A, Chen M, Yang W, Lin Q. CYR61 (CCN1) is a metastatic biomarker of gastric cardia adenocarcinoma. Oncotarget. 2016;7:31067–78. doi: 10.18632/oncotarget.8845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Q, Wang J, Childress C, Sudol M, Carey DJ, Yang W. HECT E3 ubiquitin ligase Nedd4-1 ubiquitinates ACK and regulates epidermal growth factor (EGF)-induced degradation of EGF receptor and ACK. Mol Cell Biol. 2010;30:1541–54. doi: 10.1128/MCB.00013-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun A, Wei J, Childress C, Shaw JH 4th, Peng K, Shao G, Yang W, Lin Q. The E3 ubiquitin ligase NEDD4 is an LC3-interactive protein and regulates autophagy. Autophagy. 2017;13:522–537. doi: 10.1080/15548627.2016.1268301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Persaud A, Alberts P, Amsen EM, Xiong X, Wasmuth J, Saadon Z, Fladd C, Parkinson J, Rotin D. Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol Syst Biol. 2009;5:333. doi: 10.1038/msb.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plouffe SW, Lin KC, Moore JL 3rd, Tan FE, Ma S, Ye Z, Qiu Y, Ren B, Guan KL. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem. 2018;293:11230–11240. doi: 10.1074/jbc.RA118.002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuo CT, Chen BC, Yu CC, Weng CM, Hsu MJ, Chen CC, Chen MC, Teng CM, Pan SL, Bien MY, Shih CH, Lin CH. Apoptosis signal-regulating kinase 1 mediates denbinobin-induced apoptosis in human lung adenocarcinoma cells. J Biomed Sci. 2009;16:43. doi: 10.1186/1423-0127-16-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han J, Lv W, Sheng H, Wang Y, Cao L, Huang S, Zhu L, Hu J. Ecliptasaponin A induces apoptosis through the activation of ASK1/JNK pathway and autophagy in human lung cancer cells. Ann Transl Med. 2019;7:539. doi: 10.21037/atm.2019.10.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Zhao X, Gong X. Costunolide induces lung adenocarcinoma cell line A549 cells apoptosis through ROS (reactive oxygen species)-mediated endoplasmic reticulum stress. Cell Biol Int. 2016;40:289–97. doi: 10.1002/cbin.10564. [DOI] [PubMed] [Google Scholar]
- 35.Xu W, Wei Y, Wu S, Wang Y, Wang Z, Sun Y, Cheng SY, Wu J. Up-regulation of the Hippo pathway effector TAZ renders lung adenocarcinoma cells harboring EGFR-T790M mutation resistant to gefitinib. Cell Biosci. 2015;5:7. doi: 10.1186/2045-3701-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]